
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-delivery of anti-PLK-1 siRNA and camptothecin by nanometric polydiacetylenic micelles results in a synergistic cell killing†
Manon Ripoll a,
Marie Pierdantb,
Patrick Neuberga,
Dominique Bagnardb,
Alain Wagnera,
Antoine Kichlera and
Jean-Serge Remy*a
a,
Marie Pierdantb,
Patrick Neuberga,
Dominique Bagnardb,
Alain Wagnera,
Antoine Kichlera and
Jean-Serge Remy*a
aUniversity of Strasbourg, CNRS, UMR7199, Labex Medalis, icFRC, 67400 Illkirch, France. E-mail: remy@unistra.fr
bMN3T Lab, Fédération de Médecine Translationnelle, Labex Medalis, INSERM U1109, University of Strasbourg, 67400 Illkirch, France
First published on 6th June 2018
Abstract
Recently, it has been shown that the efficiency of antitumoral drugs can be enhanced when combined with therapeutic siRNAs. In the present study, an original platform based on polydiacetylenic micelles containing a cationic head group able to efficiently deliver a small interfering RNA (siRNA) targeting the PLK-1 gene while offering a hydrophobic environment for encapsulation of lipophilic drugs such as camptothecin is developed. We demonstrate that the co-delivery of these two agents with our micellar system results in a synergistic tumor cell killing of cervical and breast cancer cell lines in vitro. The combined drugs are active in a subcutaneous in vivo cancer model. Altogether, the results show that our nanometric micellar delivery system can be used for the development of new drug–siRNA combo-therapies.
Introduction
By allowing post-transcriptional gene silencing, small interfering RNAs (siRNAs) have proved to be extremely useful and powerful tools in fundamental research. In addition, RNA interference, and siRNAs in particular, have shown therapeutic potential against numerous diseases, including cancer, viral infection and hypercholesterolaemia.1 However, safe and efficient delivery of therapeutic siRNAs remains a major hurdle toward their clinical applications, despite enormous efforts that were made in the last years on the design of specific delivery devices.Recent research showed that therapeutic effect of small interfering RNAs in the anticancer field could be enhanced by their combination with classical antitumoral drugs.2–6 Hence, drug–nucleic acid combination approaches can successfully overcome drug resistance.7 Nevertheless, co-delivery of an antitumoral drug (often hydrophobic) and a siRNA (negatively charged and hydrophilic) into the same target cell remains a tough challenge because of their different physicochemical properties.
We recently reported the optimization of a new promising family of nanostructured carriers based on photopolymerized polydiacetylenic (PDA) micelles or nanofibers for addressing the specific requirements of the delivery of diverse biomolecules such as hydrophobic drugs,8 plasmid DNA9 and siRNAs.10,11 The last generation of PDA-micelles stabilized by UV-photopolymerization, are self-organized objects in the size range of 10 nm, containing a histidine head group (Hist-PMs, Scheme 1). The micelles allowed for a 90% silencing of a reporter gene using 10 nM siRNA concentration.10 In parallel, E. Doris and H. Tian's groups demonstrated the potential of polydiacetylene nanocarriers for antitumoral drug delivery.12–14 Considering the properties of the photopolymerized diacetylenic micelles, we wanted to explore for the first time the capacities of this nanomaterial for a simultaneous delivery of both, small interfering RNA of therapeutic interest and poorly water-soluble antitumoral drugs in vitro and in vivo. We choose to silence Polo-like kinase 1 (PLK-1), an endogenous gene involved in cell division. PLK-1 is a target gene of interest in the therapeutic field as it plays a key role in cell division while being often overexpressed in cancers.15 It is well described in the literature that silencing of PLK-1 expression using siRNA leads to a decrease of cell viability with induction of apoptosis and defects in mitosis processes.16 Notably, diverse systems such as polymers or mesoporous silica nanoparticles demonstrate high efficiency to deliver siPLK-1.17,18
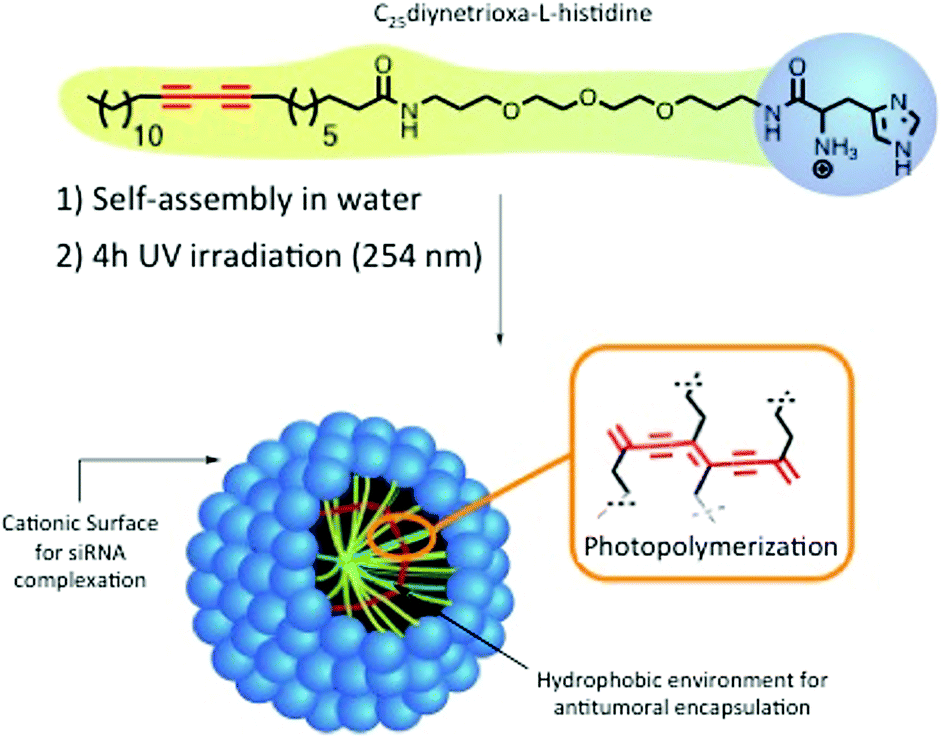 | ||
| Scheme 1 Structure and formulation of the Hist-PMs. | ||
Results and discussion
The nanocarrier abbreviated Hist-PMs (Scheme 1) was formulated and characterized, as described previously.10 The siRNA delivery capacity was optimized using a luciferase expressing cell line.In this work, transfection assays were performed on HeLa (cervical cancer cell line) and MDA-MB-231 (breast cancer cell line) cells using concentrations of siRNA targeting the PLK-1 mRNA (abbreviated siPLK-1) ranging from 10 to 200 nM. As control, we used a siRNA that does not target any mRNA (abbreviated siCTL). The complexation of the siRNAs was performed by adding the Hist-PMs to the diluted siRNA while keeping the N/P ratio (protonated primary amines versus phosphate groups of siRNA) constant and equal to 5.6. The cationic nature of the micelle is necessary to achieve complete complexation of the negatively charged siRNAs (see our previous work10) and to permit efficient cell penetration (Fig. 1A). After 48 h of transfection, cytotoxicity assays were performed in both cell lines, in order to evaluate the efficiency of our system. The results show that siPLK-1 transfection induces more than 50% of cell death with a siRNA concentration of 100 nM. Thereby, it is reflecting a significant silencing of the target gene, as compared to the results obtained with the control siRNA that induces no toxicity (Fig. 1B).
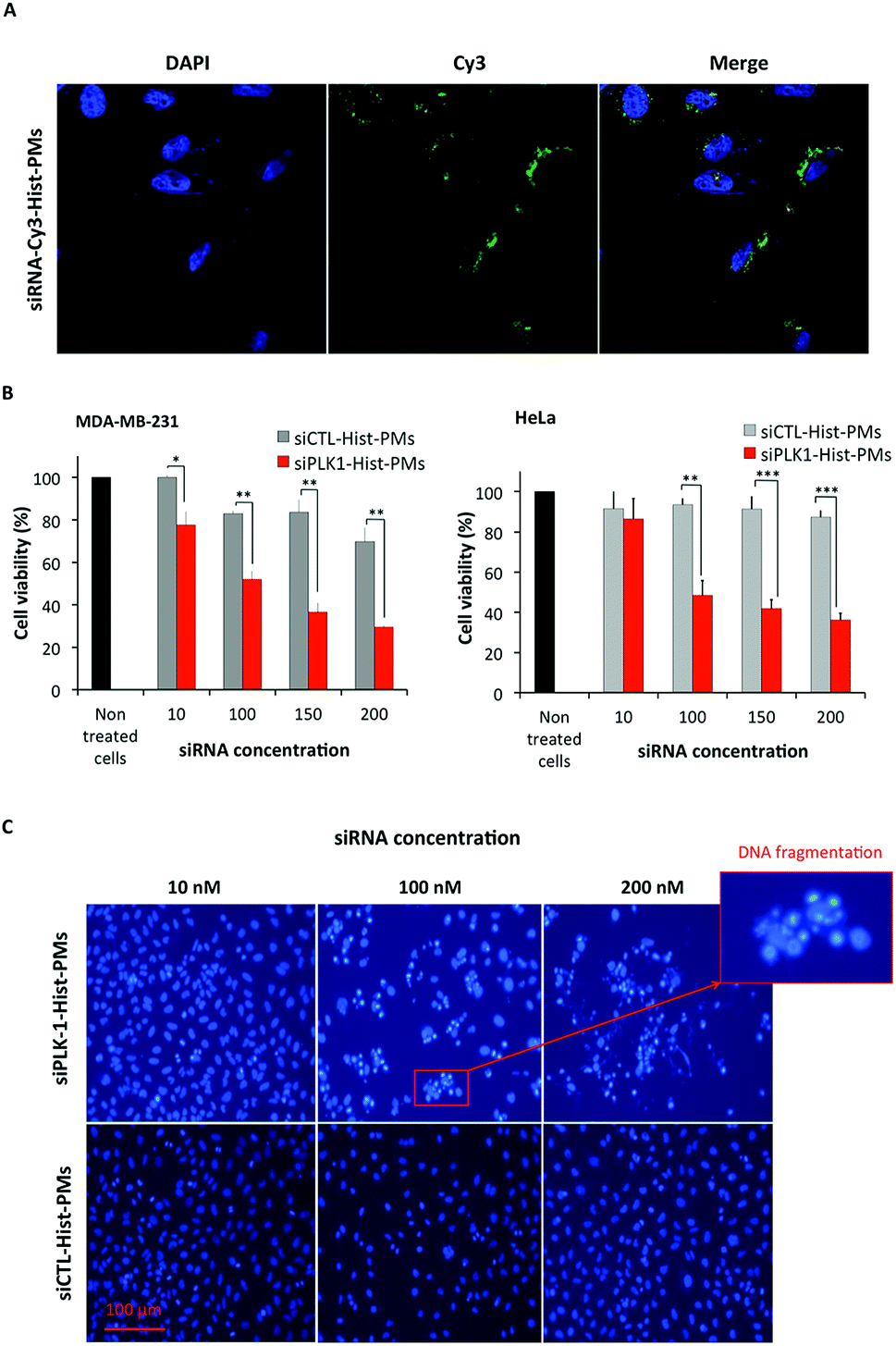 | ||
Fig. 1 (A) Confocal fluorescence images of siRNA-Cy3-Hist-PMs internalization in MDA-MB-231 cells. Cells were treated with complexes (50 nM siRNA-Cy3, N/P = 5.6) at 37 °C for 3 h. Lasers: DAPI 405 nm and Cy3 561 nm. Objective: 63×. Image size: 367.83 μm × 367.83 μm. (B) Cell viability results after anti-PLK-1 siRNA delivery performed on MDA-MB-231 and HeLa cells using various concentrations of siPLK-1, with a N/P ratio of 5.6. The data are shown as the mean standard deviation (n = 3). Statistical significance: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***) were considered to be statistically significant. (C) Representative Epi-fluorescence microscopy images of HeLa cells incubated with Hist-PMs micelles complexed with either the siRNA targeting PLK-1 (siPLK-1-Hist-PMs) or a control siRNA (siCTL-Hist-PMs). The cells were incubated at 37 °C for 48 h in the presence of 3 different concentrations of siRNA. Nucleus staining was performed with Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 342 (blue color). Scale bar = 100 μm. 342 (blue color). Scale bar = 100 μm. | ||
These results could be confirmed by microscopy (Fig. 1C). Indeed, when HeLa cells were incubated with siPLK-1-Hist-PMs or siCTL-Hist-PMs at 37 °C for 48 h using various concentration of siRNA, we observed lower cell proliferation followed by nucleic acid fragmentation with ≥100 nM siRNA concentration, while nuclei exhibited normal shape with siCTL. Similar results could be obtained on MDA-MB-231 cells, although a higher concentration of siRNA was required (150 nM) (Fig. 1B). Altogether, these results show that (i) the Hist-PMs based delivery system can silence an endogenous gene, and (ii) silencing of PLK-1 in HeLa and MDA-MB-213 cells results in cell death. The mechanism of the siRNA release into the cytoplasm is probably made possible thanks to the imidazole protonation of the micelle that acts like a proton sponge enabling endosome disruption that leads to the endosomal siRNA release.10
Next, we measured the loading efficacy of an antitumoral drug into the micelles. We chose the hydrophobic and poorly water-soluble drug camptothecin (CPT), an inhibitor of DNA topoisomerase I which inhibits DNA re-ligation and leads to apoptosis.19 First, various amounts of CPT were encapsulated in Hist-PMs by using sonication. The suspension, named CPT-Hist-PMs, was then filtered using a 0.45 μm filter to remove the free and insoluble CPT.14 The loading efficacy of CPT in the micelles was determined by fluorescence measurements (Fig. 2). The results show a linear correlation between 1 and 10% of CPT loading, before reaching a plateau reflecting the maximal quantity of CPT that can be encapsulated. From these results, we concluded that the maximal weight ratio of CPT/Hist-PMs was 1:10 w/w. We used this ratio for all the subsequent experiments. We also investigated the in vitro drug release by fluorescence at different pH and time intervals (see ESI, Fig S1†). Briefly, data showed that the release profiles were time and pH dependent probably due to different states of protonation of the micelles. After 5 hours, at pH = 4.5 and 6, the drug release was slower than at pH = 7.5 meaning that once in the endosome, the drug release is slowed down, leaving time to the drug to reach its site of action, the cytoplasm.
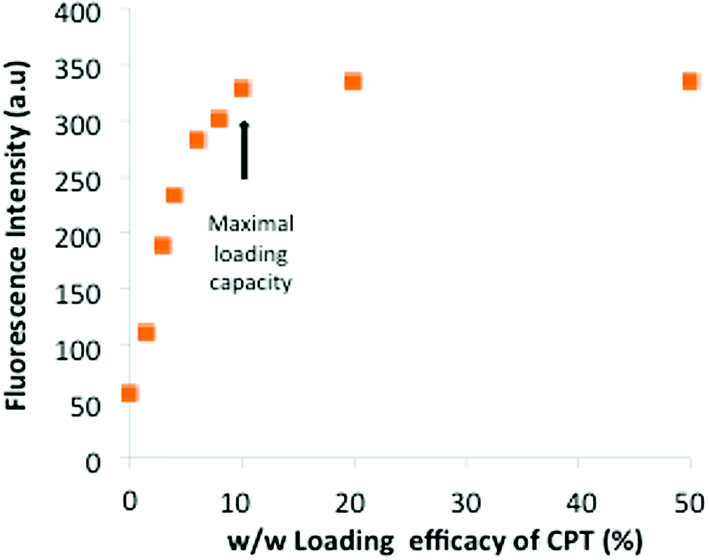 | ||
| Fig. 2 Fluorescence at λ = 430 nm of CPT encapsulated in Hist-PMs at various ratios. | ||
Before performing co-delivery experiments, we had to determine whether the activity of CPT is maintained after encapsulation. The cell viability assays done on HeLa showed that CPT-Hist-PMs (IC50 = 1 μM) were almost 2 times more toxic than free-CPT (IC50 = 1.9 μM), which confirmed the conservation of the CPT activity (see ESI, Fig S2A†). On MDA-MB-231 cells, we also observed a slightly higher efficiency reflected by a reduction of IC50 with CPT-Hist-PMs (4.1 μM), as compared to free-CPT (4.8 μM) (see ESI, Fig. S2B†). Strikingly, at concentrations above 4 μM, cell death reached a plateau with free-CPT, while CPT-Hist-PMs induced further cytotoxicity. The plateau observed with free CPT is probably due to a lack of solubility of free CPT at this concentration. In turn, this highlighted that the drug solubility was enhanced by encapsulation into micelles.
The sizes of the different objects were controlled by Dynamic Light Scattering (DLS) to ensure that all formulations remain below 100 nm. Indeed, a small size is suitable because bigger sizes are less prone to tumor accumulation via an EPR effect.20 The diameter of the micelles without drug and siRNA in HBG is 17 nm (see ESI, Fig. S3A†). The micelles loaded with siPLK-1 or CPT show increased average hydrodynamic diameters of 60 and 25 nm, respectively (see ESI, Fig. S3C and S3B†). Notably, the size of the complexes formed with the two drugs simultaneously loaded remained around 60 nm (see ESI, Fig. S3D†).
The next step consisted in evaluating in vitro the biological activity of Hist-PMs micelles loaded with the combination of the siRNA and CPT. CPT-Hist-PMs were formed as previously described, and then incubated with siPLK-1 in HBG. After 1 hour of complexation at 37 °C, these complexes were added to HeLa and MDA-MB-231 cells (Fig. 3A). In the same experiment, the activity of the siRNA/CPT formulation was compared to that of CPT-Hist-PMs and siPLK-1-Hist-PMs. After 48 h of transfection, a MTT assay was performed. In parallel, a blank experiment was run with the empty vehicle that showed no effect on the two cell lines at these concentrations.
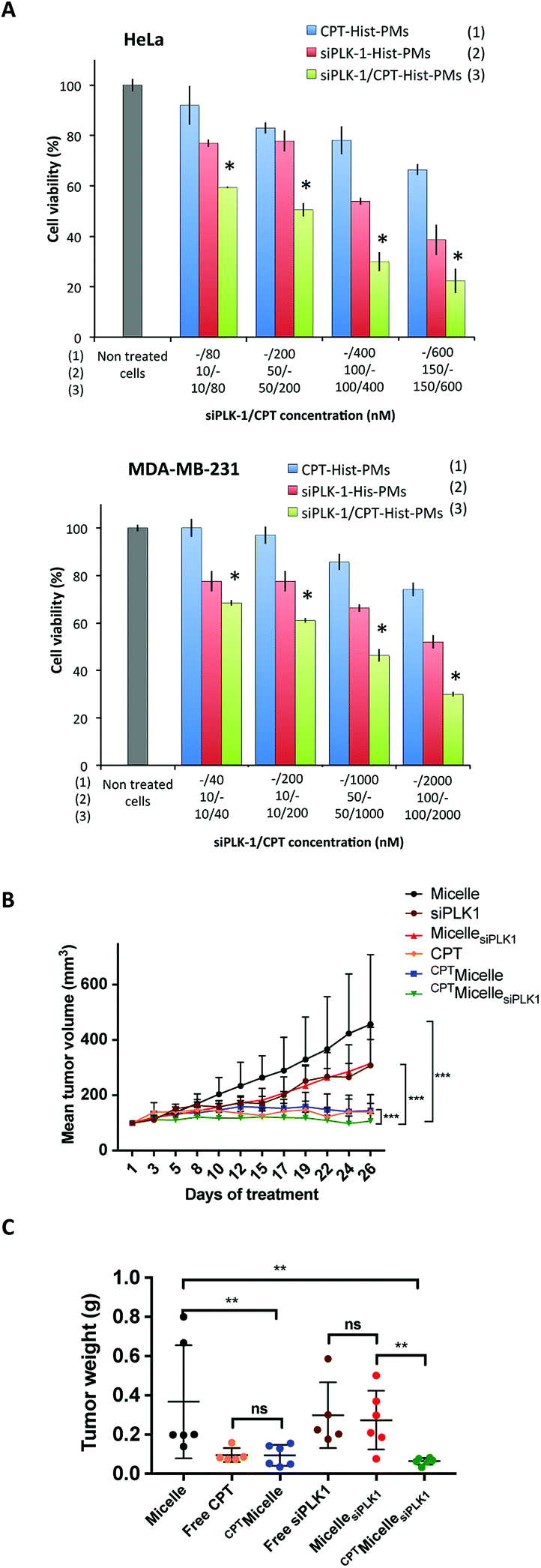 | ||
| Fig. 3 (A) MTT cytotoxicity assay performed 48 h after addition of CPT encapsulated into Hist-PMs (CPT-Hist-PMs; blue bars), siPLK1-Hist-PMs (red bars) and the combination of drugs (siPLK-1/CPT-Hist-PMs; green bar) on HeLa and MDA-MB-231 cells at various siPLK-1/CPT ratios. Cell viabilities were compared to non-treated cells (white bar). *A combination index (CI) < 1.0 indicates a synergistic effect. (B) In vivo anticancer activity. Changes in the tumor volume after peritumoral injections of Micelle (black curve), free siPLK1 (orange curve), MicellesiPLK1 (brown curve), CPT (green curve), CPTMicelle (red curve) and CPTMicellesiPLK1 (blue curve) three times a week during 4 weeks in MDA-MB-231 tumor bearing mice. (C) Mean tumor weight after treatment of Micelle, free siPLK1, MicellesiPLK1, CPT, CPTMicelle and CPTMicellesiPLK1. For each graph, data are represented as average ± standard deviation (n = 6 or 5). Statistical significance: p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***) were considered to be statistically significant. | ||
From the cell viability, the percentage of inhibition was calculated. The combination indexes were determined with Compusyn software.21 In both cell lines, the calculated combination indexes were below 1. This permitted to conclude that there was indeed an improvement of their respective effects showing cooperativity (see ESI, Fig. S4 and S5†). Thus, with the combination of the two therapeutic agents, we did not only observe additive but even synergistic effects probably by inducing cell cycle arrest and apoptosis. At higher concentrations of CPT and/or siRNA, we observed complete cell death (data not shown).
To further confirm the potential of these Hist-PMs (abbreviated Micelle in this part for greater clarity) for siRNA and drug co-delivery, we evaluated the in vivo therapeutic efficacy of CPTMicellesiPLK1 in female nude mice bearing subcutaneous tumors upon MDA-MB-231 breast carcinoma cells grafting (Fig. 3B).
After tumors were established (reaching a minimum size of 100 mm3), mice were treated by peritumoral injections three times per week with the vehicle, free CPT, free siPLK1, CPTMicelle, MicellesiPLK1 and the combination CPTMicellesiPLK1. The tumor volume and body weight were monitored during four weeks. While mice treated with siPLK-1 or MicellesiPLK1 showed a slight reduction of tumor growth as compared to the control group treated with the vehicle, mice treated with CPT, CPTMicelle and CPTMicellesiPLK1 presented almost complete stop of tumor growth. Moreover, no significant difference was observed between, in one hand free siPLK1 and MicellesiPLK1 and in the other hand free CPT and CPTMicelle. These results demonstrated that when encapsulated into the micelles, the therapeutic agents retained their efficiency. Most importantly, we observe a significant difference between mice treated with CPTMicelle and the CPTMicellesiPLK1. These observations suggest an enhanced therapeutic effect by combined treatments. After the end of the treatment, the mice were sacrificed and tumors were weighted (Fig. 3C). The comparison of tumor weights obtained are in accordance with the previous observations of tumor growth curves.
Lastly, body weights of treated mice remained stable, reflecting that treatments were well tolerated (Fig. S6†).
Experimental
Materials and methods
All reagents used for the surfactant synthesis, camptothecin and solvents were obtained from Sigma-Aldrich. 1H and 13C NMR spectra were recorded at room temperature on a Bruker Avance 400 spectrometer. Recorded shifts are reported in parts per million (δ) and calibrated using residual undeuterated solvent. Data are represented as follows: chemical shift, multiplicity (s = singlet, t = triplet and m = multiplet), coupling constant (J in Hz) and integration. High-resolution mass spectra (HRMS) were obtained using an Agilent Q-TOF (time of flight) 6520. Sonication of the micellar solutions was realized in an ultrasound bath (Fisherbrand) and photopolymerization was carried out with a UV light (48 W, 254 nm) cross-linker Bio-link 254 (Fisher Bioblock).The siRNA targeting PLK-1, obtained from Eurogentec, has the following sequence: PLK-1 sense: 5′-AGA UCA CCC UCC UUA AAU AUU-3′; PLK-1 antisense: 5′-UAU UUA AGG AGG GUG AUC UUU-3′ in bold, the 2′-O-methylated modified nucleotides.
The control siRNA was obtained from Qiagen.
Surfactant synthesis
C25diynetrioxa-L-Histidine was synthesized as described in our previous work.101H NMR (400 MHz, MeOD, δ) 8.97 (s, 1H), 7.52 (s, 1H), 4.17 (t, J = 7 Hz, 1H), 3.65–3.55 (m, 8H), 3.53–3.44 (m, 4H), 3.35–3.22 (m, 6H), 2.24 (t, J = 6.5 Hz, 4H), 2.18 (t, J = 7.5 Hz, 2H), 1.77–1.69 (m, 4H), 1.64–1.57 (m, 2H), 1.54–1.47 (m, 4H), 1.43–1.28 (m, 26H, alkyl chain), 0.9 (t, J = 7 Hz, 3H) ppm.
13C NMR (75 MHz, MeOD, δ) 174.9, 161.6, 135.5, 132.6, 118.4, 115.9, 115.4, 76.6, 76.5, 70.1, 69.9, 69.8, 68.5, 68.3, 65.0, 53.5 38.7, 36.6 36.4, 35.8, 31.7, 29.4–28.1, 25.6, 22.3, 18.3, 13.1 ppm.
HRMS (ESI) m/z: 714.554 [M + H]+ calculated for C41H72N5O5, 714.554; found 714.554.
Formulations
:10 ratio (w/w) and the mixture was sonicated for 2 hours. The solution was finally filtered through a 0.45 μm filter to provide CPT-Hist-PMs.Loading efficacy
Various volumes of a solution of camptothecin (CPT; 0.2 mg mL−1) in DMSO were freeze-dried under vacuum in order to obtain the desired amounts of drug. Then, 100 μL of Hist-PMs (0.5 mg mL−1) were added into each sample, and the mixtures were sonicated for 2 hours before being filtrated through a 0.45 μm filter to remove the non-encapsulated CPT.14 Fluorescence was quantified before and after filtration on a RF-5301 PC spectrofluorophotometer (Shimadzu) with the following wavelength: λEx = 370 nm, λEm = 440 nm.Size measurement by dynamic light scattering (DLS)
The hydrodynamic diameters of the CPT-Hist-PMs, siPLK-1-Hist-PMs and siPLK-1/CPT-Hist-PMs were determined using a Zetasizer Nano ZS system (Malvern Instruments) with the following specifications: material index of refraction: 1.43; sampling time = 55 s; refractive index of HBG (Hepes Buffered Glucose (5%)) 1.337; viscosity of HBG (Hepes buffered glucose): 1.1557; temperature = 25 °C by using micro-cuvettes of 90 μL. The values correspond to average size ± standard deviation of three runs.Cell culture
Human cervical cancer cells (HeLa) and human breast cancer cells (MDA-MB-231), purchased from ECACC, were cultured in Dulbecco's modified Eagle medium (DMEM, high glucose, Gibco–Invitrogen) supplemented with 10% FBS (Eurobio), 1% antibiotic solution (penicillin–streptomycin, Gibco–invitrogen) in a 5% CO2 humidified atmosphere at 37 °C. Cells were passaged using trypsin when they reached 90% confluency.Analysis of cellular uptake of complexes by confocal microscopy analysis
Twenty-four hours prior to transfection, MDA-MB-231 cells were seeded at a density of 1 × 104 cells per well in Lab-tek® II chamber slide from Thermo Fisher. Complexes were formed by adding micelle on siRNA-Cy3 diluted in HBG in order to obtain a final concentration of siRNA of 50 nM at the optimal N/P ratio. According to the preliminary in vitro experiments, the optimal N/P ratio was 5.6. Samples were mixed and incubated one hour at room temperature. 40 μL of the complexes were added onto the cells in 200 μL DMEM medium supplemented by 10% of serum. After 3 h of incubation, the medium was removed and the cells washed twice with PBS. Cells were fixed with a 4% formaldehyde solution in PBS. After 30 min of incubation at room temperature, cells were rinsed with cold PBS and then deionized water. The chamber was removed and cells were coverslipped using an aqueous mounting medium containing DAPI (fluoroshield from Sigma). Confocal microscopy was performed on a Leica SPE confocal microscope 11506513 (lasers used: DAPI 405 nm, Cy3 561 nm and Cy5 635 nm, objective 63×), Image size: 367.83 μm × 367.83 μm.In vitro experiments
Twenty-four hours prior to transfection, 1 × 104 HeLa or MDA-MB-231 cells were seeded per well in 96-well tissue culture plates. Complexes were prepared with siPLK-1 or a control siRNA (siCTL) and the cationic micelle (Hist-PMs) (N/P = 5.6) by adding the micelles to different amounts of siRNA pre-diluted in HBG to obtain final concentrations of siRNA-PMs ranging between 10 and 200 nM. CPT or CPT-Hist-PMs (1:10 w/w ratio) were diluted in HBG in order to have final concentrations of CPT ranging between 0.04 to 10 μM in cell culture medium. As a control, the same concentrations of Hist-PMs in absence of CPT were also tested. Finally, the siPLK-1/CPT-Hist-PMs formulation was generated by adding CPT-Hist-PMs (1:10 w/w ratio) to different amounts of siPLK-1, pre-diluted in HBG to obtain final concentrations ranging from 10 to 200 nM (and a N/P of 5.6). Twenty microliters of each complex were then added onto the cells, in absence of serum, in 100 μL DMEM medium. After 4 h of incubation, 10 μL of fetal bovine serum (Eurobio) were added to obtain 10% serum final concentration. 10% DMSO were also added on three control wells to provide the blanks for absorbance readings representing total cell death. Cell viability was measured after 48 h, by quantifying the mitochondrial succinate dehydrogenase, using an MTT assay (Invitrogen) as described below. All experiments were done at least 3 times using triplicates.MTT assay
One hundred microliters of MTT solution (1 mg mL−1 in serum-free DMEM) were added to the medium for 2 h, allowing viable cells to reduce the yellow tetrazolium salt (MTT) into dark blue formazan, which precipitates as crystalline solids. After removal of the medium, formazan solubilization was performed with 100 μL DMSO and quantified by spectrophotometry at 570 nm on a Multiskan FC (Thermo scientific). Cell viability (%) = (mean of abs. value of treatment group/mean abs. value of control)×100%.Microscopy
Twenty-four hours prior to transfection, 2.5 × 104 HeLa cells were seeded per well in 24-multiwell tissue culture plates. Complexes were formed between siPLK-1 or siCTL and Hist-PMs (N/P = 5.6), by adding the micelles to different amounts of siPLK-1 pre-diluted in HBG to obtain final concentrations of 10, 100 and 200 nM. Samples were mixed and incubated one hour at room temperature. One hundred microliters of the complexes were added onto the cells in absence of serum in 500 μL DMEM. After 4 h of incubation, 60 μL of fetal bovine serum were added to obtain 10% serum concentration. Forty-eight hours after transfection, the medium was removed and the cells were washed 3 times in PBS. Ice-cold MeOH to cover the cells was added to fix the cells. Fifteen minutes after, cells were washed again and 5 μg mL−1 of a solution of Hoechst 33342 in deionized water (5 mg mL−1) were added. After 30 min of incubation at room temperature, fluorescence microscopy was performed on an Axio Vert A1 microscope (Zeiss) using DAPI standard filter.Combination index determination
Combination drug analyses were performed with CompuSyn software (http://www.combosyn.com).21,22 Percentage of inhibition of the drug, the siRNA and the combination of the two therapeutic agents are modeled by the software to determine IC50 and the combination index.If CI < 1, there is a synergistic effect.
If CI = 1, there is an additive effect.
If CI > 1, there is an antagonistic effect.
In vivo experiments
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Animal Care Facility at the Faculty of Pharmacy at the University of Strasbourg (France) and were approved by the Animal Ethics Committee of CREMAS (Comité régional d’éthique en matière d'expérimentation animale de Strasbourg). The experiments were validated by the regional ethical committee and received agreement by the French Minister of Research (APAFIS#5945-2016063017352801). Subcutaneous grafting of cells (106 of MDA-MB-231 suspended in 100 μL of PBS) was performed in the flank of 8 week old females.When the tumors reached a mean volume of 100 mm3, the mice were randomly separated into six groups: vehicle (Micelle 220 μg per mouse in 0.1 mL HBG, n = 6), Free CPT (15 μg per mouse, diluted in 0.1 mL of 3% DMSO/HBG solution, n = 5), free siPLK1 (7 μg per mouse in 0.1 mL HBG, n = 5), CPTMicelle (15 μg per mouse CPT, 220 μg Micelle in 0.1 mL HBG, n = 6), MicellesiPLK1 (7 μg per mice with 220 μg of Micelle in HBG, N/P = 10, n = 6) and CPTMicellesiPLK1 (7 μg siPLK1, 15 μg CPT, 220 μg Micelle in 0.1 mL HBG, N/P = 10, n = 6). The mice received peritumoral injections 3 times a week for 4 weeks (total 12 injections, 100 μL per injection). Length and width of tumors were measured individually using a Vernier caliper. Tumor volumes were calculated using the following formula:
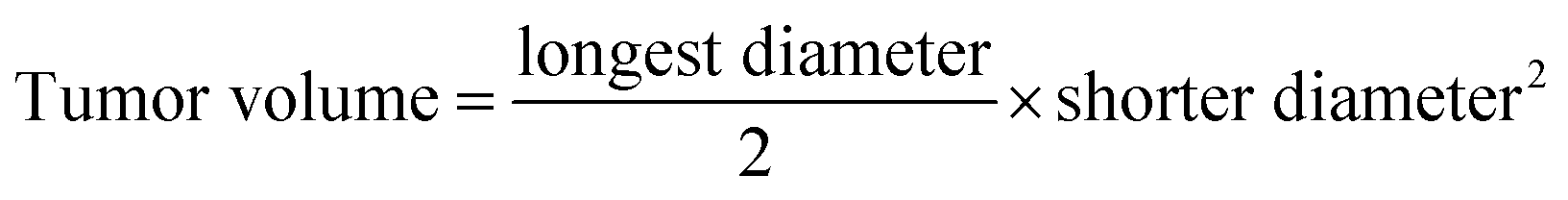
All along the treatment, weight of the mice was monitored once per week. The mice were sacrificed 30 days after the first injection according to institutional guidelines. Tumors were resected and weighted.
Statistical analysis
The statistical significance of the in vitro data was evaluated using the Student's t-test. For in vivo experiment, statistical analyses were performed using Mann Whitney test (for sample n < 30) using GraphPad software (USA). For all tests, p-values are given in the figure legends, and values of p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***) were considered to be statistically significant. All data were presented as means ± S.D.Conclusions
We demonstrate for the first time that polydiacetylenic micelles can co-deliver into tumour cells a hydrophilic siRNA and a hydrophobic antitumoral drug. As with siRNA delivery, the histidine head group functionalized PDAs also show great promise in co-delivery with hydrophobic anticancer drug, CPT. We showed that this co-delivery results in an induced synergistic cell death in vitro. The combined treatment results in a remarkable tumor growth inhibition in peritumoral injection in a subcutaneous mouse tumor model.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Labex Medalis and by the FRM (fondation pour la recherche médicale). M. R. has a financial support from the University of Strasbourg.Notes and references
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef PubMed.
- T. Liu, M. Wang, T. Wang, Y. Yao and N. Zhang, Colloids Surf., B, 2015, 126, 531–540 CrossRef PubMed.
- M. Saad, O. B. Garbuzenko and T. Minko, Nanomedicine, 2008, 3, 761–776 CrossRef PubMed.
- C. Zhu, S. Jung, S. Luo, F. Meng, X. Zhu, T. G. Park and Z. Zhong, Biomaterials, 2010, 31, 2408–2416 CrossRef PubMed.
- Y. H. Yu, E. Kim, D. E. Park, G. Shim, S. Lee, Y. B. Kim, C.-W. Kim and Y.-K. Oh, Eur. J. Pharm. Biopharm., 2012, 80, 268–273 CrossRef PubMed.
- Q. Tang, B. Cao and G. Cheng, Chem. Commun., 2014, 50, 1323–1325 RSC.
- H. Yu, Z. Xu, X. Chen, L. Xu, Q. Yin, Z. Zhang and Y. Li, Macromol. Biosci., 2014, 14, 100–109 CrossRef PubMed.
- P. Neuberg, A. Perino, E. Morin-Picardat, N. Anton, Z. Darwich, D. Weltin, Y. Mely, A. S. Klymchenko, J.-S. Remy and A. Wagner, Chem. Commun., 2015, 51, 11595–11598 RSC.
- E. Morin, M. Nothisen, A. Wagner and J.-S. Remy, Bioconjugate Chem., 2011, 22, 1916–1923 CrossRef PubMed.
- M. Ripoll, P. Neuberg, A. Kichler, N. Tounsi, A. Wagner and J.-S. Remy, ACS Appl. Mater. Interfaces, 2016, 8, 30665–30670 Search PubMed.
- P. Neuberg, I. Hamaidi, S. Danilin, M. Ripoll, V. Lindner, M. Nothisen, A. Wagner, A. Kichler, T. Massfelder and J.-S. Remy, Nanoscale, 2018, 10, 1587–1590 RSC.
- E. Gravel, J. Ogier, T. Arnauld, N. Mackiewicz, F. Ducongé and E. Doris, Chem.–Eur. J., 2012, 18, 400–408 CrossRef PubMed.
- N. Mackiewicz, E. Gravel, A. Garofalakis, J. Ogier, J. John, D. M. Dupont, K. Gombert, B. Tavitian, E. Doris and F. Ducongé, Small, 2011, 7, 2786–2792 CrossRef PubMed.
- D. Yao, S. Li, X. Zhu, J. Wu and H. Tian, Chem. Commun., 2017, 53, 1233–1236 RSC.
- K. Strebhardt and A. Ullrich, Nat. Rev. Cancer, 2006, 6, 321–330 CrossRef PubMed.
- A. D. Judge, M. Robbins, I. Tavakoli, J. Levi, L. Hu, A. Fronda, E. Ambegia, K. McClintock and I. MacLachlan, J. Clin. Invest., 2009, 119, 661–673 Search PubMed.
- N. P. Truong, W. Gu, I. Prasadam, Z. Jia, R. Crawford, Y. Xiao and M. J. Monteiro, Nat. Commun., 2013, 4, 1902 CrossRef PubMed.
- S. B. Hartono, N. T. Phuoc, M. Yu, Z. Jia, M. J. Monteiro, S. Qiao and C. Yu, J. Mater. Chem. B, 2014, 2, 718–726 RSC.
- V. J. Venditto and E. E. Simanek, Mol. Pharm., 2010, 7, 307–349 CrossRef PubMed.
- A. M. Jhaveri and V. P. Torchilin, Front. Pharmacol., 2014, 5, 77 Search PubMed.
- T.-C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef PubMed.
- T. T. Chang, S. C. Gulati, T.-C. Chou, R. Vega, L. Gandola, S. M. E. Ibrahim, J. Yopp, M. Colvin and B. D. Clarkson, Cancer Res., 1985, 45, 2434–2439 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03375g |
| This journal is © The Royal Society of Chemistry 2018 |