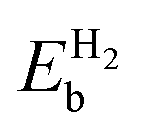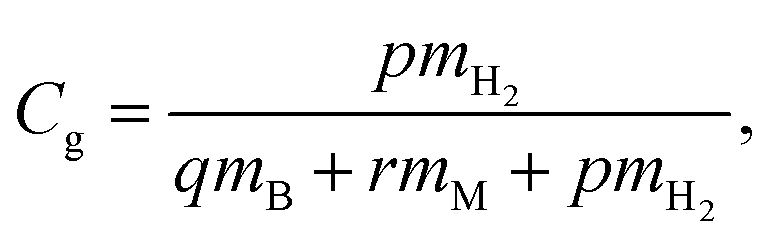DOI:
10.1039/C7RA12512G
(Paper)
RSC Adv., 2018,
8, 20748-20757
Hydrogen storage in Li, Na and Ca decorated and defective borophene: a first principles study
Received
17th November 2017
, Accepted 10th May 2018
First published on 6th June 2018
Abstract
Recently synthesized two-dimensional (2D) borophene possesses unique structural, mechanical, electrical and optical properties. Herein, we present a comprehensive study of H2 storage in alkali metal decorated and defect containing 2D borophene using density functional theory calculations. While the adsorption of H2 over pristine borophene was found to be weak with a binding energy of −0.045 eV per H2, metal decoration and point defects enhanced the adsorption strength significantly. Interestingly, the magnitudes of binding energy for a single H2 molecule over Li, Na and Ca decorated borophene were found to increase up to −0.36, −0.34, and −0.12 eV per H2, respectively. On the other hand, while the binding energy of one H2 molecule over the borophene substrate containing a single vacancy (SV) was only −0.063 eV per H2, similar to that of phosphorene, the binding energy increased to an enormous −0.69 eV per H2 over borophene containing a double vacancy (DV). To gain further insight into the H2 adsorption process and identify sources of charge transfer, differential charge densities and projected density of states were calculated. Significant charge accumulation and depletion caused strong polarization of the H2 molecules. Finally, Na, Li and Ca decorated borophene yielded the gravimetric densities 9.0%, 6.8%, and 7.6%, respectively. The gravimetric density of the borophene containing a DV was found to be the highest, a staggering 9.2%, owing to increased interactions between DV borophene and the H2 molecules. These results suggest that borophene can be an effective substrate for H2 storage by carefully engineering it with metal decoration and point defects.
1. Introduction
Borophene, a two-dimensional (2D) form of boron, offers intriguing structural, electronic, and optical properties. For example, the monolayer is inherently metallic, has a puckered structure, possesses excellent in-plane mechanical properties, with a Young’s modulus of up to 398 GPa nm, and exhibits Dirac transport properties and superconductivity.1–3 Due to these unique properties, monolayer borophene can be used for heterogeneous catalysis and H2 evolution,4 as electrodes in rechargeable metal-ion batteries,5 and in photoelectronic devices.6 In 2015 Mannix et al. reported for the first time the successful synthesis of borophene by physical vapor deposition on a Ag(111) surface.7,8 Ab initio molecular dynamics (AIMD) based simulations have confirmed its thermodynamic stability.9 Recently, Pekoz et al.10 have shown that borophene is both dynamically and thermodynamically stable. The phonon dispersion curve at the ground state does not contain imaginary frequencies and the structure is also intact at temperatures as high as 1000 K. These facts raise the following question: can borophene be used for H2 storage?
Molecular hydrogen is a renewable and green energy source which is used in fuel cells and for chemical hydrogenation. While it is an excellent candidate as a fuel, conventional storage methods like high pressure cylinders or cryogenic tanks pose practical limits on its efficient and safe storage and transportation.11 The primary targets for effective H2 storage based on a physisorption mechanism are (a) a high gravimetric density; the target of 7.5 wt% system gravimetric density has been decided as the revised target by the United States Department of Energy (US DOE)12 for ultimate full fleet applications, and (b) easy recycling of hydrogen at ambient pressure and temperature.13,14 Since the discovery of graphene, 2D materials, by virtue of their high surface to volume ratio, have been identified as providing an advanced route for hydrogen storage by physical adsorption.11,15 In the recent past, first principles based density functional theory (DFT) calculations have been extensively used to computationally investigate H2 storage over 2D substrates. For reversible H2 storage (i.e. adsorption and release under near ambient conditions), a binding energy in the range between −0.1 and −0.2 eV per H2 is considered acceptable.16,17
Experimental advances in fabricating 2D materials have expedited their practical realization in current day progress in H2 storage and the potential to adopt modern methods for H2 storage applications. During the quest for potential substrates for H2 storage several 2D materials have been studied e.g. boron nitride sheets,18–20 graphene,21–25 phosphorene,26–28 C2N29 and MOS2.17 Among 2D materials, graphene has been predicted to offer a 7% gravimetric density in H2 storage and phosphorene has shown a gravimetric density of more than 5%. In the studies of H2 storage over 2D materials, it has been found that metal decoration and defects enhance the H2 storage capacity compared to that over pristine materials. Decorating 2D surfaces using metallic adatoms is an effective way to increase the interactions between the substrate and the H2 molecules, thereby increasing the gravimetric density. For example, Li decorated graphene has been reported to have a binding energy of −0.35 eV per H2 compared to −0.114 eV per H2 in pristine graphene.30 Yu et al.27 reported that Li decoration in pristine phosphorene increased the gravimetric density to 4.4% compared to the negligible storage capacity of pristine phosphorene. Hashmi et al.29 reported a gravimetric density of 13 wt% over Li decorated C2N. Putungan et al.17 reported a gravimetric density of 4.4 wt% over Li decorated MOS2. Another avenue for improving the storage capacity of potential substrates is defect engineering.31 Yadav et al. demonstrated that using defective graphene, it was possible to achieve a gravimetric density of 7%. Recently, Haldar et al.28 combined these two methods, i.e. Li decoration and defect engineering, to improve the adsorption energy and gravimetric density of H2 over phosphorene. They demonstrated that defective phosphorene possessed a binding energy of <0.1 eV per H2, while Li-decorated single vacancy phosphorene had an enhanced adsorption energy of −0.48 eV per H2 leading to a gravimetric density of 5.3 wt%.
In the recent past several authors have attempted to study the H2 storage capacity of pristine and metal decorated monolayer borophene. For example, Li et al.32 reported that Li decorated borophene possessed a gravimetric density of 13.7 wt%. Chen et al.33 reported a gravimetric density of 1.3 wt% in Ca decorated borophene. However, these studies have approached the metal decoration aspect on a case by case basis. Therefore, there is a need to carry out studies with several alkali atoms for better comparison and an in-depth understanding. Additionally, to the best of our knowledge, the effect of point defects on the H2 storage capacity of borophene has not been studied yet. Only a few reports are available on the H2 storage capacity of borophene and they are limited to metal decoration.33–35 In this manuscript, we attempt to fill the gaps in our understanding with a comprehensive study of H2 storage over metal decorated and defective borophene using extensive first principles based density functional theory (DFT) calculations. We have investigated H2 storage in Li, Na and Ca decorated and single and double vacancy defective borophene. All the cases have been simulated in the same DFT-D framework with a similar type of pseudopotential to compare the performance of different decorations and defects. The results from our simulations have been compared with the data available in the literature. To gain insight into H2 adsorption onto borophene, projected density of states (PDOS) have been computed to show the charge transfers and interactions among the elements.
2. Material and computational details
First principles based DFT calculations were performed using Quantum Espresso, an open source package available under a GNU license.36 For all the elements, a Vanderbilt ultrasoft type pseudopotential was used along with the Perdew–Burke–Ernzerhof (PBE) exchange correlation functional.37,38 The van der Waals correction was accounted for by adopting a dispersion corrected DFT (DFT-D) framework.39 A spacing of around 20 Å was used in the direction normal to the 2D substrate to avoid interlayer long range van der Waals interactions between the periodic images. The cut-off energies for the wave functions and charge densities were set to be 60 Ry and 480 Ry, respectively. Self-consistent field (SCF) calculations were performed using a convergence threshold of le5 Ry for the total system energy to obtain the charge densities of the systems. A Monkhorst–Pack grid with 4 × 4 × 1 k-points was used for the Brillouin zone integrations.40 The simulations were performed using Methfessel–Paxton smearing with a degauss value of 0.02.41
The binding energy of H2 over borophene can be calculated as:
| | |
Eb = (Eboro+iH2 − Eboro − EiH2)/i,
| (1) |
and
| | |
Eb = (Eboro+M+iH2 − Eboro+M − EiH2)/i,
| (2) |
where
Eb represents the binding energy of the hydrogen (eV per H
2).
Eboro+iH2,
Eboro, and
EiH2 are the total energies (eV) of the borophene supercell and the hydrogen molecules, only the borophene substrate, and only the H
2 molecules, respectively. Similarly,
Eboro+M+iH2 and
Eboro+M represent the total energy of the full metal (M) decorated H
2 adsorbed borophene, and the metal decorated borophene, respectively. As per the above relation, a negative binding energy implies that adsorption is energetically favorable and spontaneous, while a positive binding energy indicates desorption. Smaller values of
Eb imply stronger binding of H
2 with the substrate. The projected density of states (PDOS) has also been calculated for the systems to understand the charge transfer among the elements in the system.
For studying the H2 adsorption capacity, at first the borophene substrate was relaxed at its ground state. To calculate the binding energy of hydrogen molecules over adatom decorated borophene, first, metal adatoms were relaxed over borophene for a range of initial positions. Most of the energetically favorable adatom configurations were chosen for further H2 adsorption. Several initial positions for the H2 molecules were considered over metal decorated borophene and the structures were energetically optimized.
3. Results and discussions
3.1. H2 adsorption over pristine borophene
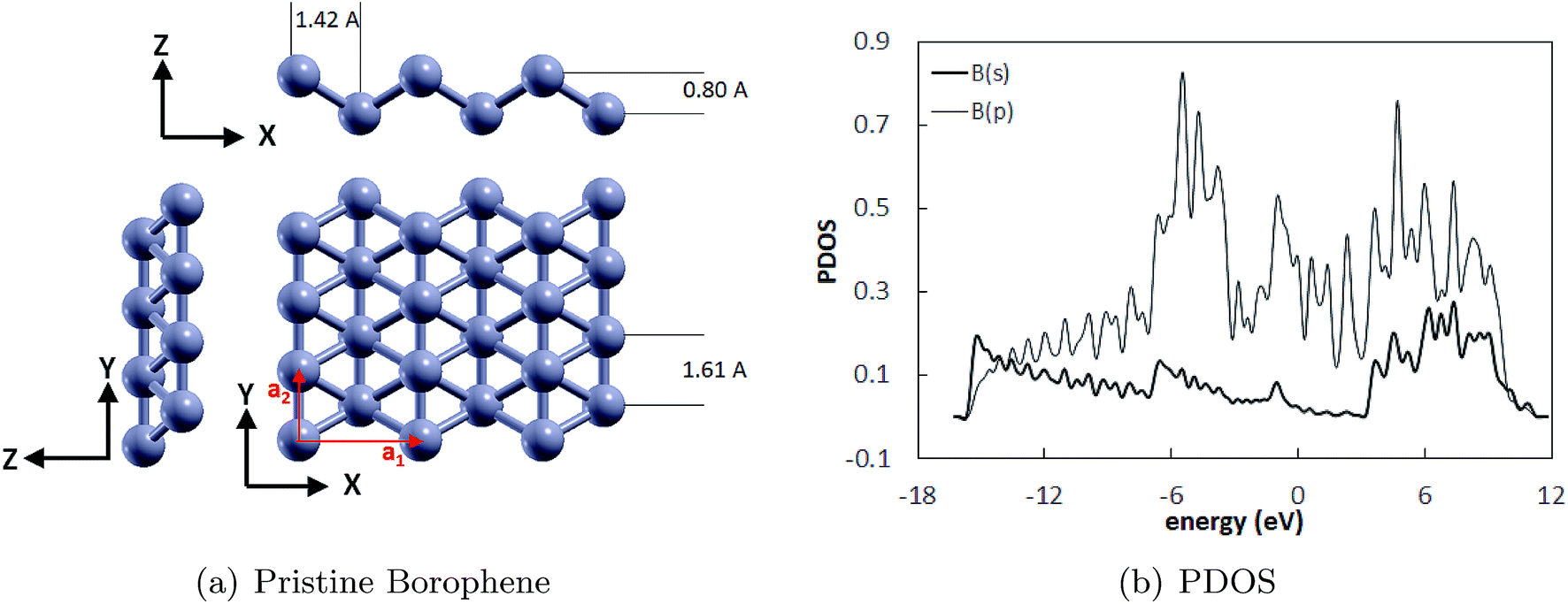
The optimized structure of the monolayer borophene supercell (4·3) is shown in Fig. 1(a) and the projected density of states (PDOS) is shown in Fig. 1(b). It can be seen that borophene has a puckered structure wherein the B atoms belong to two different atomic planes in the out-of-plane direction. The lattice parameters of the unit cell are a1 = 2.85 Å and a2 = 1.61 Å, and are shown by the red arrows. The lattice parameters reported here are in excellent agreement with those reported before. For example, DFT calculations performed by Jiang et al.5 predicted lattice parameters of 2.861 Å and 1.641 Å for borophene. Similarly, the lattice parameters of the borophene supercell reported by Xiang et al.,42 i.e., 2.85 Å and 1.62 Å, also match with our calculations. The pucker height of 0.8 Å reported here is in excellent agreement with the results obtained by Mannix et al.7 Additionally, the PDOS shows that borophene is inherently metallic at the ground state due to the absence of a finite bandgap, which agrees well with that previously reported in ref. 32.
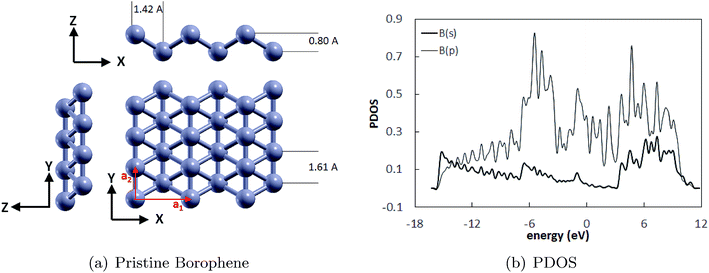 |
| | Fig. 1 (a) The pristine borophene substrate with its unit cell vectors and dimensions (a1 = 2.85 Å and a2 = 1.61 Å), and (b) PDOS of the unit cell of borophene with the Fermi energy shifted to zero. | |
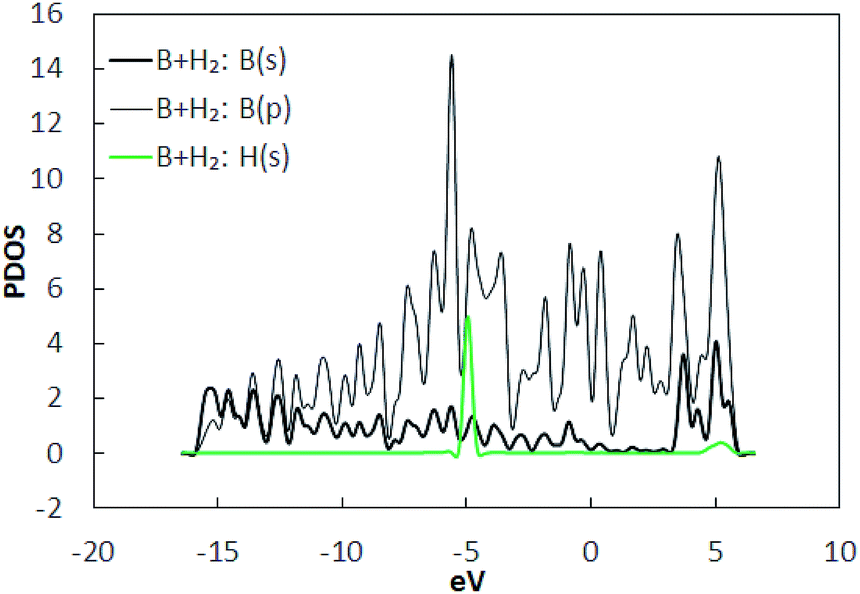
To find the adsorption energies, as shown in Fig. 2(a), several geometrically unique sites in borophene were selected. H2 molecules were placed at a distance of 3 Å away from the borophene substrate along the out-of-plane direction and the structures were optimized. The binding energies obtained from the DFT calculations are listed in Table 1. It was observed that the binding energies were in the range −0.032 to −0.045 eV per H2, with position A (Fig. 2(b)) the best site with a binding energy of −0.045 eV per H2. The stable position of the H2 molecule was measured at a vertical distance of around 2.95 Å. However, this binding energy of well below −0.1 eV per H2 is not sufficient for hydrogen storage. The PDOS of the system with the H2 molecule at position A is shown in Fig. 3. A comparison of the projected density of states presented in Fig. 1(b) and those in 3 shows that the electrons in the p shells of boron and the s shell of the H2 molecule are hybridized close to −5 eV and 5 eV.
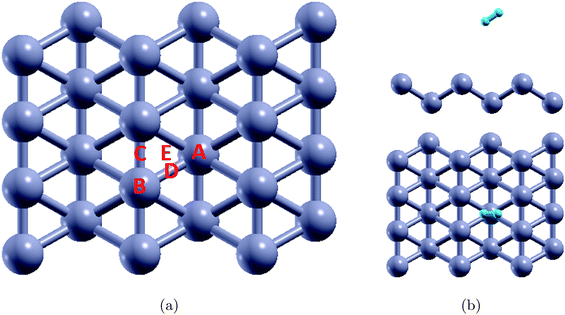 |
| | Fig. 2 (a) Different initial locations of the H2 molecules over pristine borophene substrate and (b) the best adsorption site in terms of binding energy. | |
Table 1 Binding energies of H2 over pristine borophene for different adsorption sites (Fig. 2(a))
| H2 position |
A |
B |
C |
D |
E |
| Eb (eV per H2) |
−0.045 |
−0.032 |
−0.034 |
−0.039 |
−0.045 |
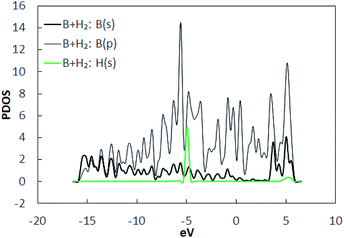 |
| | Fig. 3 Projected density of states (PDOS) of the constituents in H2 adsorbed pristine borophene. | |
3.2. H2 adsorption over Li, Na and Ca decorated borophene
To evaluate the effect of alkali metal decoration on the hydrogen adsorption capability of borophene, first, isolated metallic adatoms (Li, Na and Ca) were adsorbed over borophene. Several initial positions for the adatoms were chosen at a distance of 3 Å away from borophene on both sides, and the structures were energetically minimized. The stability of borophene decorated (by physisorption) with alkali metal atoms on both sides has been studied in a host of research articles.5,42–45 These studies have performed ground state DFT and finite temperature AIMD simulations and have reported these structures to be stable. Additionally, the gravimetric density of H2 storage for various 2D materials (graphene and phosphorene) has been evaluated for double-sided metal decoration.
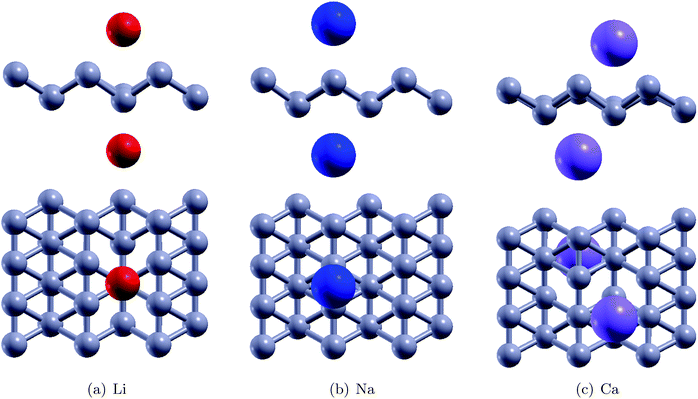
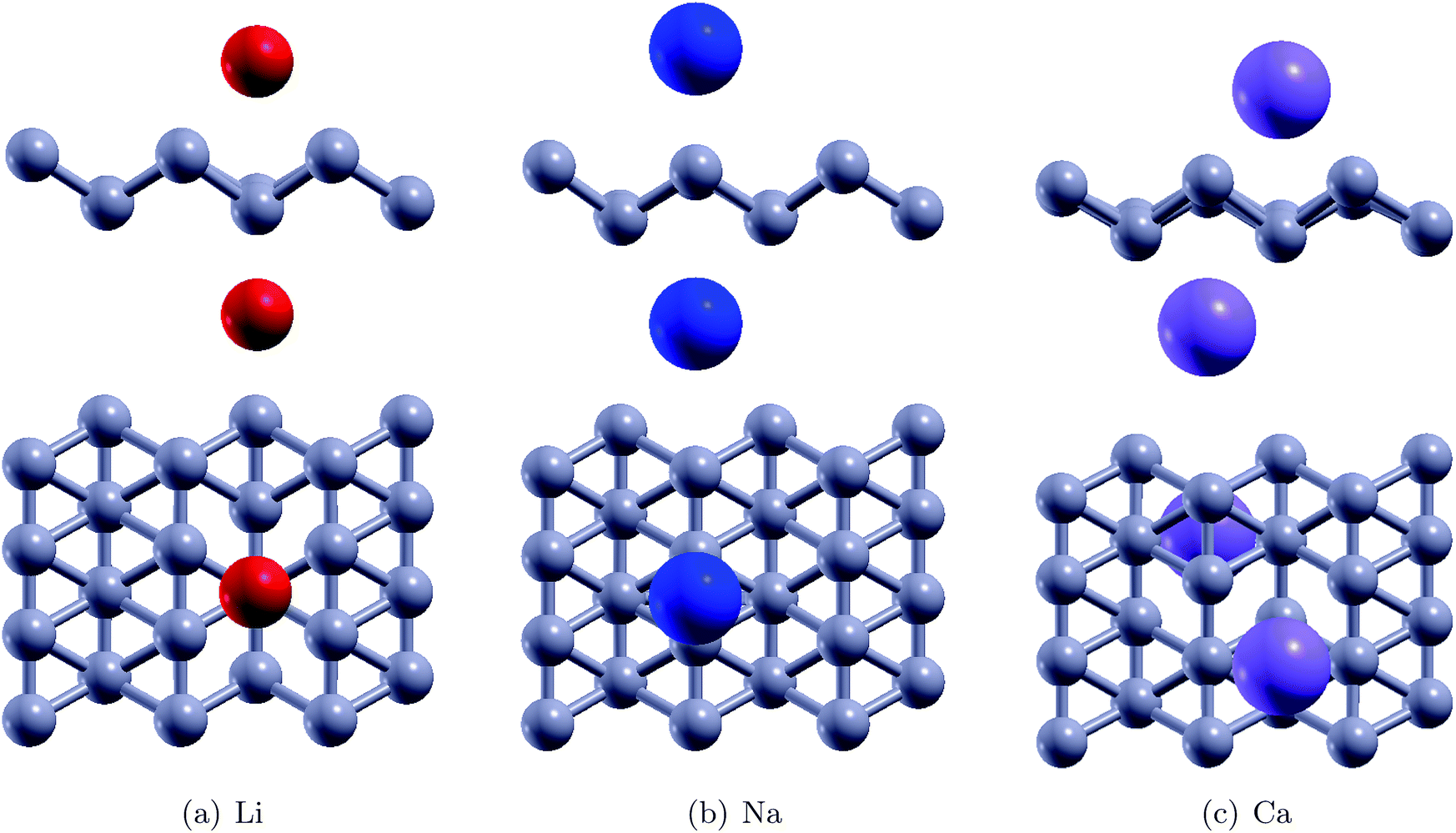
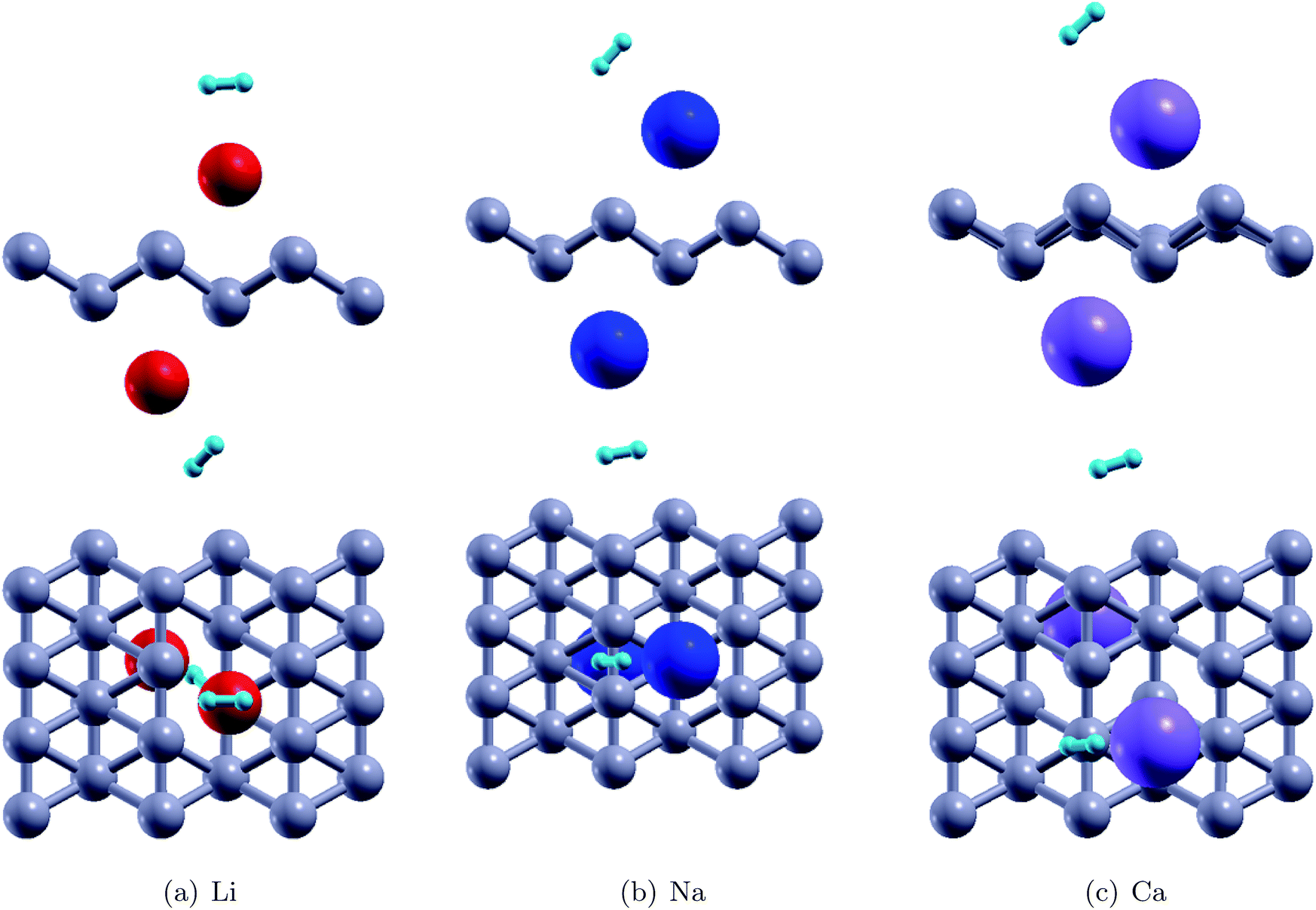
Fig. 4 shows the relaxed configurations after adatom decoration on both sides, which are consistent with those reported in ref. 32. It can be noted that Li and Na are stable at symmetric positions with respect to borophene, however, the Ca atoms have stable positions which are oblique with respect to the borophene sheet. This fact can be attributed to the electronegativity of the adatoms: due to the Ca atom being more electronegative, the two Ca atoms interact with each other. The binding energies of the Li, Na and Ca atoms onto the substrate were determined using the relation EMb = EB+2M − EB − E2M and were found to be 2.47 eV, 2.50 eV, and 3.64 eV, respectively (Table 2). In the fully relaxed state, the vertical distances of Li, Na and Ca from the substrate were measured to be 1.80 Å (top) and 2.0 (bottom); 2.0 Å (bottom) and 2.35 Å (top); and 1.80 Å (both top and bottom), respectively, in the relaxed configurations. The cohesive energies of bulk Li, Na and Ca are 1.63 eV per atom, 1.11 eV per atom, and 1.84 eV per atom, respectively.46 The binding energies of the adatoms over the borophene substrate are stronger than the cohesive energy of the bulk material and thus, can be adsorbed as single atoms over borophene instead of coalescing into their bulk forms.
 |
| | Fig. 4 The relaxed configurations of Li, Na and Ca decorated pristine borophene | |
Table 2 The binding energies of the different elements (EMb) over pristine borophene and the binding energies of H2 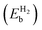 over metal decorated borophene
over metal decorated borophene
| Adatom |
Atomic mass (amu) |
Zsub-M (Å) |
EMb (eV per atom) |
RM−H2 (Å) |
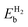
(eV per H2) |
| 2Li |
6.94 |
1.80 |
−2.47 |
1.95 |
−0.36 |
| 2Na |
22.99 |
2.35 |
−2.50 |
2.35 |
−0.34 |
| 2Ca |
40.08 |
1.80 |
−3.64 |
2.65 |
−0.12 |
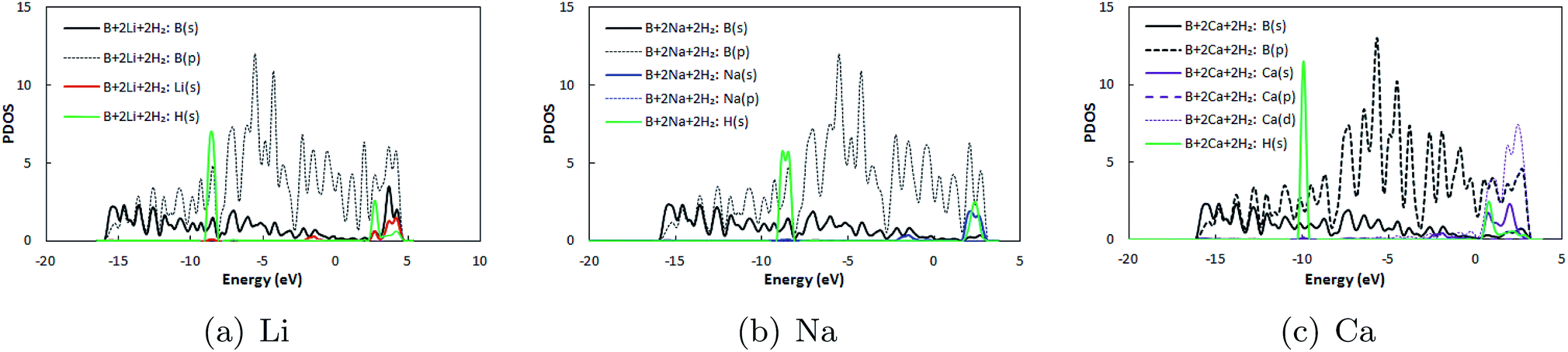
Thereafter, H2 molecules were adsorbed over metal decorated borophene by placing the H atoms facing the adatoms (Fig. 5). In the stable configuration, the H2 molecules were at a distance of 1.95 Å, 2.35 Å, and 2.65 Å away from the Li, Na, and Ca adatoms, respectively. The binding energies of the H2 molecules were calculated to be −0.36 eV per H2, −0.34 eV per H2, and, −0.12 eV per H2 for Li, Na and Ca decoration, respectively. It can be deduced that Li decoration offers better adsorption of the H2 molecule to the substrate. The projected density of states (PDOS) of pristine borophene for the substrate alone and those of borophene in the H2 adsorbed system are shown in Fig. 6 to elucidate the interactions. It can be observed that the peak in the PDOS for borophene is reduced in the H2 adsorbed system compared to that in the PDOS for borophene with the substrate alone. Furthermore, in metal decorated systems significant hybridization can be observed between the metal atoms and the boron and H atoms. For example, in Li-decorated borophene, an additional H peak is seen at 3 eV, and electrons in the valence band of hydrogen are pulled down to the −9 eV level by the electrons in the valence band of Li and hybridization can be observed between the Li(s) electrons and the H(s) electrons. In the case of Na, the density of the H peak in the valence band is significantly smaller than that for the Li and Ca decorated borophene samples. Additionally, the valence band peak for H can be seen to be split into two peaks, unlike that for Li and Ca decorated borophene. In the case of Ca decorated borophene, the H peak in the valence band is pulled to the −10 eV energy level, and hybridization can be seen to take place between the H(s) and Ca(s) and Ca(f) electrons.
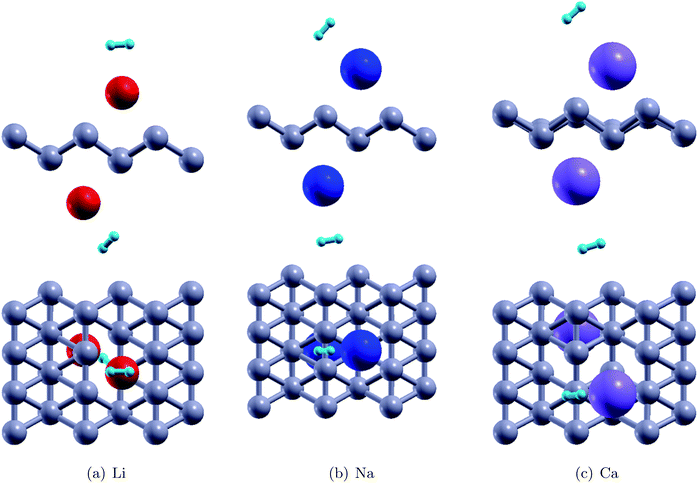 |
| | Fig. 5 The stable configurations of H2 adsorbed Li, Na and Ca decorated borophene | |
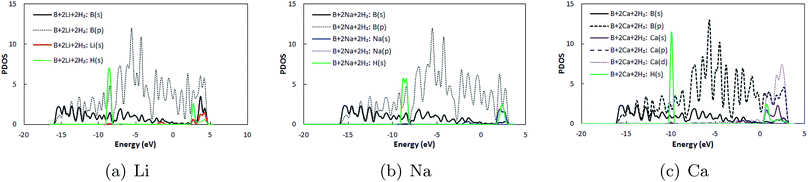 |
| | Fig. 6 Projected density of states (PDOS) of borophene in H2 adsorbed (a) Li, (b) Na, and (c) Ca decorated pristine borophene. In the legends, B represents the borophene substrate only and B + 2Li + 2H2 represents the H2 adsorbed Li decorated borophene system. | |
3.3. H2 adsorption over defective borophene
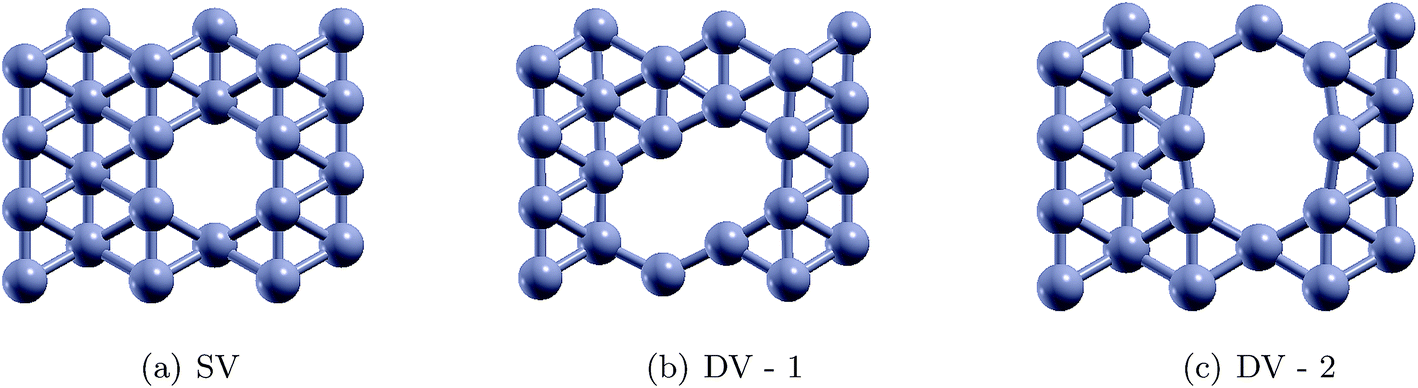
Single vacancy (SV) and double vacancy (DV) defects were created by removing atoms from the supercell. Different unique SV and DV defects created by removing the atoms are shown in Fig. 7. It should be noted that one SV defect and two unique DV defects could be created (Fig. 7). The defective borophene substrates were then relaxed at their ground state by energy minimization. The formation energies of the defects were calculated using the relation, 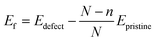 , where Edefect and Epristine are the total energies of defective and pristine borophene, respectively, N is the number of atoms in the pristine substrate, and n is the number of atoms removed from the pristine supercell to create the defects (n = 1 for SV, and n = 2 for DV). As summarized in Table 3, the calculated formation energy for the SV defect is 0.12 eV and those for the DV defects are 1.31 eV (DV-1) and 1.26 eV (DV-2).
, where Edefect and Epristine are the total energies of defective and pristine borophene, respectively, N is the number of atoms in the pristine substrate, and n is the number of atoms removed from the pristine supercell to create the defects (n = 1 for SV, and n = 2 for DV). As summarized in Table 3, the calculated formation energy for the SV defect is 0.12 eV and those for the DV defects are 1.31 eV (DV-1) and 1.26 eV (DV-2).
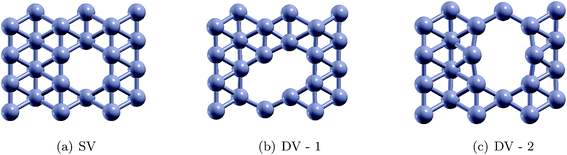 |
| | Fig. 7 Schematics for the single vacancy (SV) and double vacancy (DV) defects in borophene. | |
Table 3 Formation energies for the different single vacancy (SV) and double vacancy (DV) defects in borophene
| Defect |
SV |
DV-1 |
DV-2 |
| Formation energy (eV) |
0.12 |
1.31 |
1.26 |
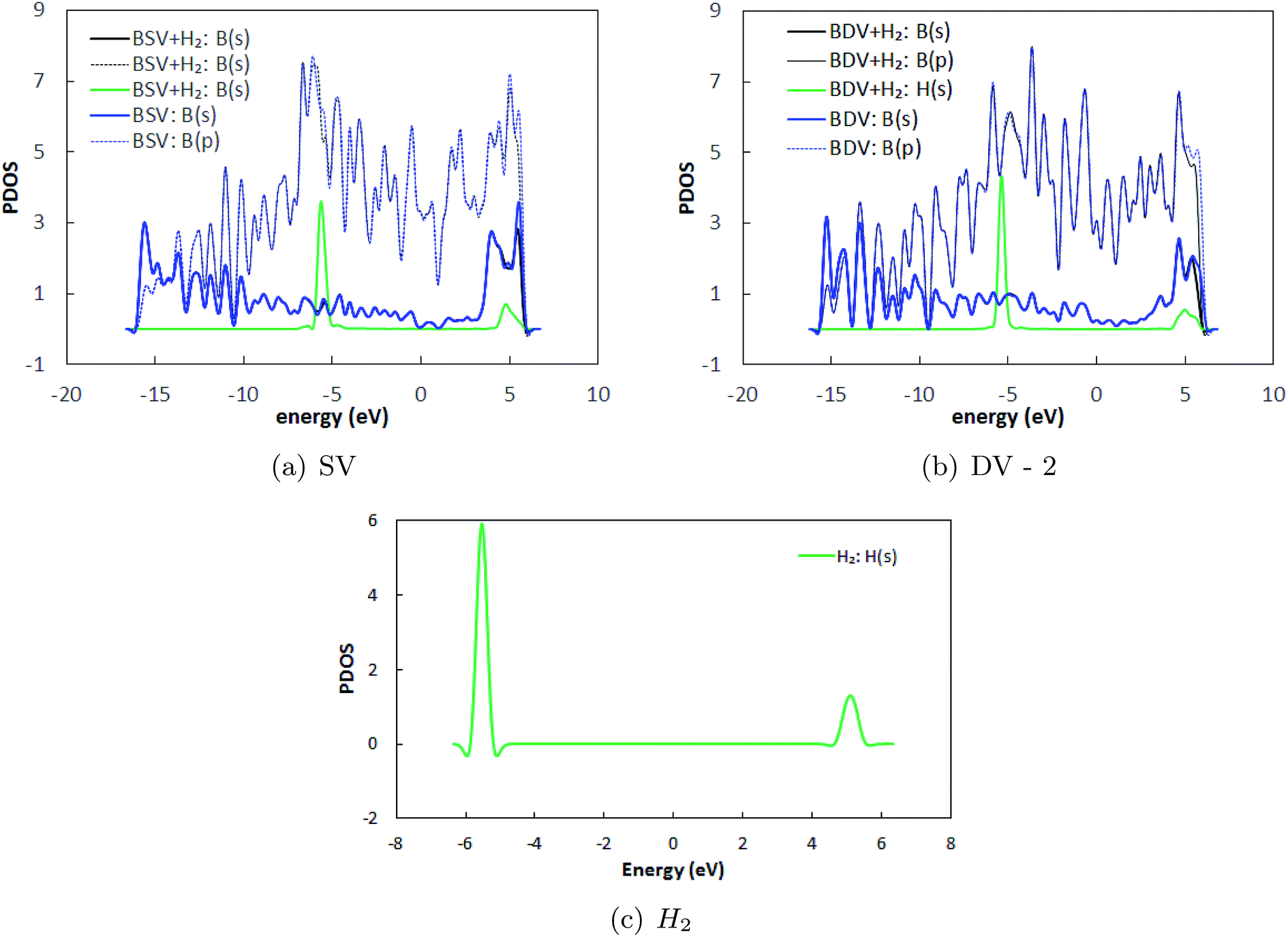
Herein, we have studied H2 adsorption over borophene containing SV and DV-2 defects (since the formation energy of DV-2 is lower than that of DV-1). H2 molecules were placed at several locations facing the defects at a distance of 3 Å away from the substrate and the systems were energetically optimized. The atomic configurations corresponding to the best adsorption energies are shown in Fig. 8. The stable distances for the H2 molecules from the defective substrate were measured as 3.08–3.09 Å for SV and DV defective borophene. The binding energies of the H2 molecule over defective borophene were calculated to be −0.063 eV per H2 for the SV defect and a staggeringly large −0.69 eV per H2 for the DV defect (Table 4). It is worth noting that the binding energy of the H2 molecule over DV defective borophene is better than those obtained by decorating borophene with alkali metals. To visualize the interactions between the H2 molecules and the borophene substrate, the projected density of states (PDOS) was calculated, as shown in Fig. 9. The PDOS indicates the interaction of the H2 molecules with the defective substrate around the peak at 5 eV above the Fermi energy level. A comparison between Fig. 3 and 9 indicates that the density of the H peak close to the 5 eV level is much more pronounced in the defective substrates than in the pristine substrate. This indicates a transfer of charge from the defective substrates to the H2 molecules leading to greater interaction and a larger binding energy for the H2 molecules.
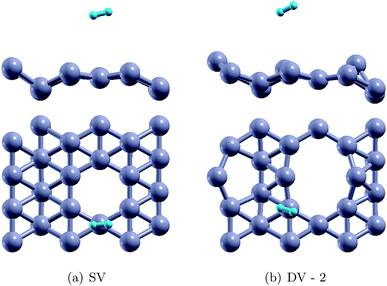 |
| | Fig. 8 The stable configurations of H2 adsorbed SV and DV defective borophene. | |
Table 4 The binding energy 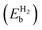 , distance (Rsub-H2) of H2 from the substrate, and bond length (RH–H) of H2 in borophene with different defects. The binding energies correspond to the adsorption of one H2 molecule. The distance is measured from the closest boron atom in defective borophene
, distance (Rsub-H2) of H2 from the substrate, and bond length (RH–H) of H2 in borophene with different defects. The binding energies correspond to the adsorption of one H2 molecule. The distance is measured from the closest boron atom in defective borophene
| System |
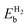
(eV) |
Rsub-H2 (Å) |
RH–H (Å) |
| SV |
−0.063 |
3.08 |
0.7523 |
| DV |
−0.690 |
3.09 |
0.7519 |
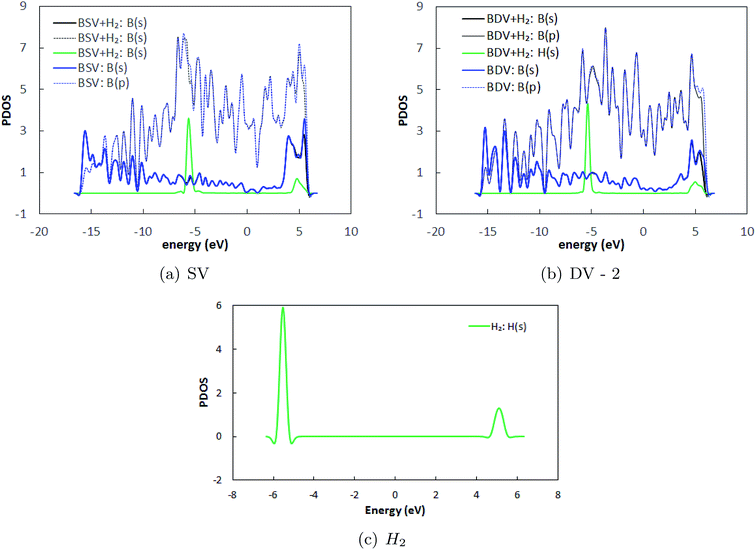 |
| | Fig. 9 Projected density of states (PDOS) of borophene in (a) SV and (b) DV defective borophene, and (c) only H2 with the Fermi energy shifted to zero. BSV and BDV represent single vacancy and double vacancy defective borophene. Similarly BSV + H2 and BDV + H2 represent H2 adsorbed systems. | |
3.4. Gravimetric densities for H2 storage in borophene
In order to estimate the maximum capacity for H2 storage, gravimetric densities have been calculated for the metal decorated and DV defective borophene. The gravimetric density of H2 storage is given by:| |
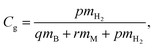 | (3) |
where mH2, mB, mM and p, q and r represent the atomic mass and the total number of H2, B, and the metal, respectively. To study the gravimetric density, the number of H2 molecules was sequentially increased and the binding energy was calculated using eqn (1) and (2). To determine the energy of the system, the structures with the three initial positions have been relaxed. It was observed that the total energies of the systems were very close to each other. The values obtained from the best case among the different initial positions have been reported. In all cases, the number of H2 molecules was increased till the binding energy had decreased to around −0.1 eV per H2.
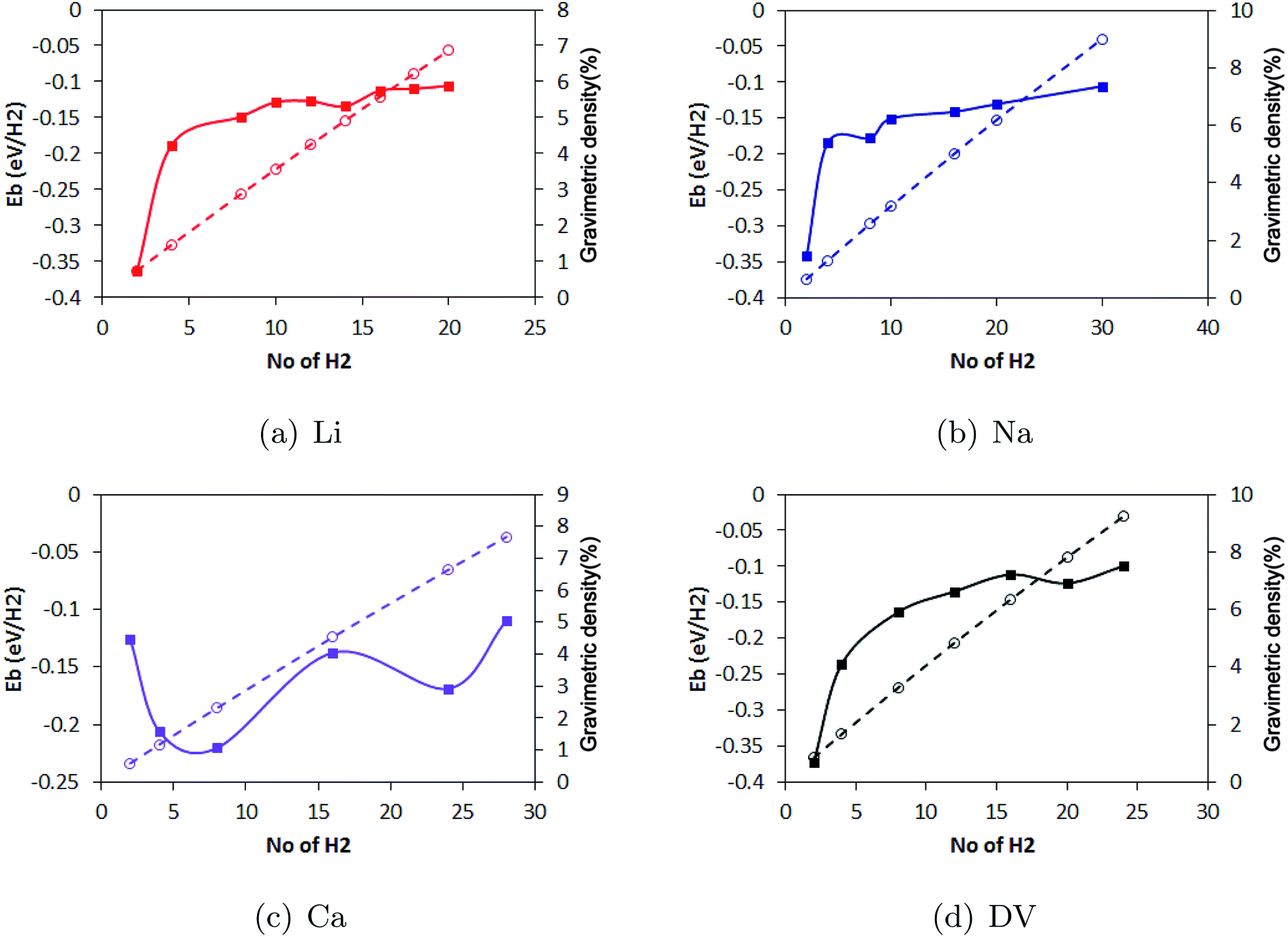
Metal decoration on both sides of the substrate significantly enhanced the H2 storage capacity of pristine borophene. While Li yielded the highest binding energy of −0.36 eV per H2, for Na and Ca, the binding energies of −0.34 eV per H2 and −0.12 eV per H2 were calculated. Among the decorating elements Li, Na and Ca, Li yielded the best binding energy of −0.36 eV per H2. To show the relation between the H2 coverage and binding energy, the evolution in the binding energy of the H2 molecules during the sequential adsorption of the H2 molecules over the borophene substrate is depicted in Fig. 10. The average binding energy of the H2 molecules with respect to the number of H2 molecules adsorbed over the substrate is shown. As can be observed in the figure image, the binding of the H2 molecules became weaker when more H2 molecules were adsorbed. The results show that the Li decoration on both sides could adsorb 10 H2 molecules per Li atom yielding a gravimetric density of 6.8%. In the case of Na decoration, the number of adsorbed H2 molecules was 15 per Na atom and the gravimetric density was 9.0%. Furthermore, 14 H2 molecules could be adsorbed per Ca atom on both sides of borophene while the binding energy per H2 molecule was around −0.1 eV. This yielded a gravimetric density of 7.6%. The gravimetric density of H2 in DV defective borophene is shown in Fig. 11. Since DV defective borophene showed a better binding energy than SV defective borophene, the gravimetric density was only evaluated for DV defective borophene. It was found from the simulations that DV defective borophene could adsorb as many as 14 H2 molecules on both sides of the substrate leading to a gravimetric density of 9.2%. It should be noted that the gravimetric density obtained was for one value of defect density. A decrease in the defect density in the borophene substrate may alter the gravimetric density. Therefore, the values presented in this work can be considered as the upper limit for borophene with a great number of defects. The gravimetric density of DV defective borophene was found to be better than that of metal decorated pristine borophene (Fig. 11).
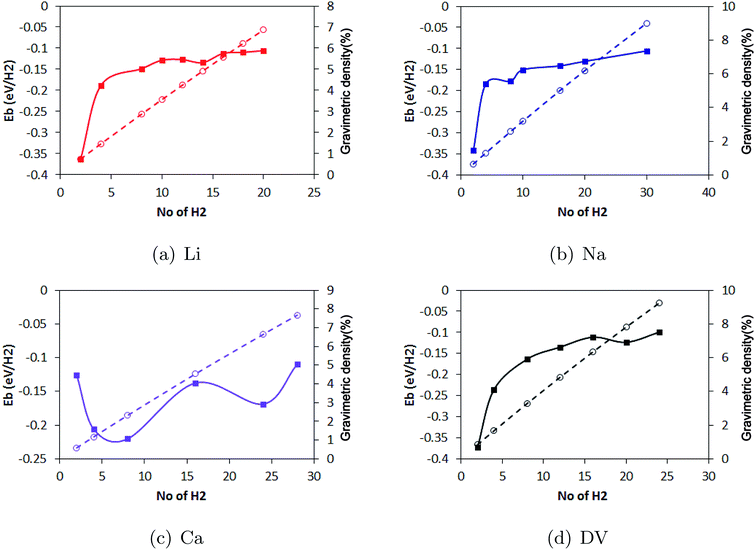 |
| | Fig. 10 The evolution in binding energy (continuous line) and gravimetric density (dashed line) during the sequential addition of H2 molecules to (a) Li, (b) Na and (c) Ca decorated pristine borophene and (d) DV defective borophene. | |
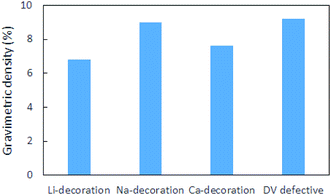 |
| | Fig. 11 Gravimetric densities for Li, Na and Ca decorated and defective borophene | |
In the recent past, first principles based calculations have predominantly been used to screen 2D materials for H2 storage purposes by characterising their adsorption strengths, gravimetric densities, etc. In all these studies, the methods used for calculation have varied, (for example, density functional theory, ab initio and molecular dynamics). Additionally, the choice of pseudopotential (PAW or norm-conserving) and computational parameters (van der Waals corrections and energy and force cutoffs) is also erratic. We start our discussion by comparing the performance of borophene with that of graphene. Yadav et al.23,31 reported that Ni decorated pristine and defective graphene (with Stone–Wales and single vacancies) can achieve gravimetric densities of 5.81% and 7.02%, respectively. Wang et al.47 and Muthu et al.48 found that while h-BN possesses a gravimetric density of 2.6%, h-BN nanoparticle modified MWCNTs can store 2.3% H2. On the other hand, while metal decorated pristine phosphorene had a gravimetric density of 3.6%,27 calculations performed by Haldar et al.28 predicted a gravimetric density of 5.3% for metal decorated phosphorene containing single vacancies. Additionally, another 2D material, Cr2C, also possesses a gravimetric density of 7.6%.49 Therefore, the results of this first principles based study show that borophene can be a very competitive system for H2 storage.
4. Conclusions
First principles based density functional theory (DFT) calculations have been performed to study H2 storage in Li, Na and Ca decorated pristine and single vacancy (SV) and double vacancy (DV) defective borophene. The DFT calculations were performed using the Vanderbilt ultrasoft pseudopotential along with the PBE exchange correlation functional in a DFT-D framework accounting for van der Waals interactions. While the binding energy of the H2 molecule over pristine borophene was −0.045 eV per H2, alkali metal decoration significantly enhanced the binding energy to be in the range −0.12 to −0.36 eV per H2, increasing the binding energy to acceptable values for reversible H2 storage. The binding energies of the alkali metal were also found to be higher than their cohesive energies, thus decoration over the substrate was favored. The gravimetric densities of H2 were found to be 6.8%, 9.0%, and 7.6% in Li, Na and Ca decorated borophene, respectively. Next, single vacancy (SV) and double vacancy (DV) defects were created in the borophene substrate. The formation energies of the SV and DV defects were determined to be 0.12 eV and 1.26 eV. Though the SV defect had a very low formation energy compared to that of the DV defect, the binding energy of the H2 molecule over SV defective borophene was only −0.063 eV per H2. On the other hand, DV defective borophene offered a binding energy of −0.69 eV per H2 for the first H2 molecule. DV defective borophene yielded a gravimetric density of 9.2%, which was higher than that of metal decorated pristine borophene. Our simulation results show that borophene could be used as a substrate for H2 storage. Defective borophene can offer competing H2 storage capacity compared to that obtained with metal decoration.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Computations were performed with support from the Compute Canada supercomputing facilities SciNet HPC Consortium50 and Calcul Quebec. Financial support for this work was provided by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Connaught New Researcher Award and the University of Toronto. The authors gratefully acknowledge their support.
References
- V. V. Kulish, Surface reactivity and vacancy defects in single-layer borophene polymorphs, Phys. Chem. Chem. Phys., 2017, 19(18), 11273–11281 RSC.
- E. S. Penev, A. Kutana and B. I. Yakobson, Can two-dimensional boron superconduct?, Nano Lett., 2016, 16(4), 2522–2526 CrossRef PubMed.
- L.-C. Xu, A. Du and L. Kou, Hydrogenated borophene as a stable two-dimensional dirac material with an ultrahigh fermi velocity, Phys. Chem. Chem. Phys., 2016, 18(39), 27284–27289 RSC.
- L. Shi, C. Ling, Y. Ouyang and J. Wang, High intrinsic catalytic activity of two-dimensional boron monolayers for the hydrogen evolution reaction, Nanoscale, 2017, 9(2), 533–537 RSC.
- H. Jiang, Z. Lu, M. Wu, F. Ciucci and T. Zhao, Borophene: a promising anode material offering high specific capacity and high rate capability for lithium-ion batteries, Nano Energy, 2016, 23, 97–104 CrossRef.
- J. Xu, Y. Chang, L. Gan, Y. Ma and T. Zhai, Ultrathin single-crystalline boron nanosheets for enhanced electro-optical performances, Adv. Sci., 2015, 2(6), 1500023 CrossRef PubMed.
- A. J. Mannix, X.-F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago and J. R. Guest, et al., Synthesis of borophenes: anisotropic, two-dimensional boron polymorphs, Science, 2015, 350(6267), 1513–1516 CrossRef PubMed.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Experimental realization of two-dimensional boron sheets, Nat. Chem., 2016, 8(6), 563–568 CrossRef PubMed.
- B. Peng, H. Zhang, H. Shao, Z. Ning, Y. Xu, G. Ni, H. Lu, D. W. Zhang and H. Zhu, Stability and strength of atomically thin borophene from first principles calculations, Mater. Res. Lett., 2017, 5(6), 399–407 CrossRef.
- R. Peköz, M. Konuk, M. E. Kilic and E. Durgun, Two-dimensional fluorinated boron sheets: mechanical, electronic, and thermal properties, ACS Omega, 2018, 3(2), 1815–1822 CrossRef.
- K. L. Lim, H. Kazemian, Z. Yaakob and W. W. Daud, Solid-state materials and methods for hydrogen storage: a critical review, Chem. Eng. Technol., 2010, 33(2), 213–226 CrossRef.
- Z. Ao and F. Peeters, High-capacity hydrogen storage in Al-adsorbed graphene, Phys. Rev. B, 2010, 81(20), 205406 CrossRef.
- S. Shevlin and Z. Guo, Density functional theory simulations of complex hydride and carbon-based hydrogen storage materials, Chem. Soc. Rev., 2009, 38(1), 211–225 RSC.
- U. Sahaym and M. G. Norton, Advances in the application of nanotechnology in enabling a hydrogen economy, J. Mater. Sci., 2008, 43(16), 5395–5429 CrossRef.
- A. Züttel, Materials for hydrogen storage, Mater. Today, 2003, 6(9), 24–33 CrossRef.
- S. K. Bhatia and A. L. Myers, Optimum conditions for adsorptive storage, Langmuir, 2006, 22(4), 1688–1700 CrossRef PubMed.
- D. B. Putungan, S.-H. Lin, C.-M. Wei and J.-L. Kuo, Li adsorption, hydrogen storage and dissociation using monolayer MoS2: an ab initio random structure searching approach, Phys. Chem. Chem. Phys., 2015, 17(17), 11367–11374 RSC.
- N. S. Venkataramanan, M. Khazaei, R. Sahara, H. Mizuseki and Y. Kawazoe, First-principles study of hydrogen storage over Ni and Rh doped BN sheets, Chem. Phys., 2009, 359(1), 173–178 CrossRef.
- A. Seif and K. Azizi, A new strategy for hydrogen storage using BNNS: simultaneous effects of doping and charge modulation, RSC Adv., 2016, 6(63), 58458–58468 RSC.
- M. Samolia and T. D. Kumar, A conceptual DFT study of the hydrogen trapping efficiency in metal functionalized BN system, RSC Adv., 2014, 4(58), 30758–30767 RSC.
- V. Tozzini and V. Pellegrini, Prospects for hydrogen storage in graphene, Phys. Chem. Chem. Phys., 2013, 15(1), 80–89 RSC.
- S. Yadav, J. Tam and C. V. Singh, A first principles study of hydrogen storage on lithium decorated two dimensional carbon allotropes, Int. J. Hydrogen Energy, 2015, 40(18), 6128–6136 CrossRef.
- J. Wong, S. Yadav, J. Tam and C. Veer Singh, A van der Waals density functional theory comparison of metal decorated graphene systems for hydrogen adsorption, J. Appl. Phys., 2014, 115(22), 224301 CrossRef.
- M. Seydou, K. Lassoued, F. Tielens, F. Maurel, F. Raouafi and B. Diawara, A DFT-D study of hydrogen adsorption on functionalized graphene, RSC Adv., 2015, 5(19), 14400–14406 RSC.
- R. E. Ambrusi, C. R. Luna, A. Juan and M. E. Pronsato, DFT study of Rh-decorated pristine, B-doped and vacancy defected graphene for hydrogen adsorption, RSC Adv., 2016, 6(87), 83926–83941 RSC.
- Q.-F. Li, X. G. Wan, C.-G. Duan and J.-L. Kuo, Theoretical prediction of hydrogen storage on Li-decorated monolayer black phosphorus, J. Phys. D: Appl. Phys., 2014, 47(46), 465302 CrossRef.
- Z. Yu, N. Wan, S. Lei and H. Yu, Enhanced hydrogen storage by using lithium decoration on phosphorene, J. Appl. Phys., 2016, 120(2), 024305 CrossRef.
- S. Haldar, S. Mukherjee, F. Ahmed and C. V. Singh, A first principles study of hydrogen storage in lithium decorated defective phosphorene, Int. J. Hydrogen Energy, 2017, 42(36), 23018–23027 CrossRef.
- A. Hashmi, M. U. Farooq, I. Khan, J. Son and J. Hong, Ultra-high capacity hydrogen storage in a Li decorated two-dimensional C2N layer, J. Mater. Chem. A, 2017, 5(6), 2821–2828 Search PubMed.
- S. Seenithurai, R. K. Pandyan, S. V. Kumar, C. Saranya and M. Mahendran, Li-decorated double vacancy graphene for hydrogen storage application: a first principles study, Int. J. Hydrogen Energy, 2014, 39(21), 11016–11026 CrossRef.
- S. Yadav, Z. Zhu and C. V. Singh, Defect engineering of graphene for effective hydrogen storage, Int. J. Hydrogen Energy, 2014, 39(10), 4981–4995 CrossRef.
- L. Li, H. Zhang and X. Cheng, The high hydrogen storage capacities of Li-decorated borophene, Comput. Mater. Sci., 2017, 137, 119–124 CrossRef.
- X. Chen, L. Wang, W. Zhang, J. Zhang and Y. Yuan, Ca-decorated borophene as potential candidates for hydrogen storage: a first-principle study, Int. J. Hydrogen Energy, 2017, 42(31), 20036–20045 CrossRef.
- L. Wang, X. Chen, H. Du, Y. Yuan, H. Qu and M. Zou, First-principles investigation on hydrogen storage performance of Li, Na and K decorated borophene, Appl. Surf. Sci., 2018, 427, 1030–1037 CrossRef.
- J. Li, H. Zhang and G. Yang, Ultrahigh-capacity molecular hydrogen storage of a lithium-decorated boron monolayer, J. Phys. Chem. C, 2015, 119(34), 19681–19688 Search PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, Quantum espresso: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21(39), 395502 CrossRef PubMed.
- D. Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenvalue formalism, Phys. Rev. B, 1990, 41(11), 7892 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for brillouin-zone integrations, Phys. Rev. B, 1976, 13(12), 5188 CrossRef.
- M. Methfessel and A. Paxton, High-precision sampling for brillouin-zone integration in metals, Phys. Rev. B, 1989, 40(6), 3616 CrossRef.
- P. Xiang, X. Chen, W. Zhang, J. Li, B. Xiao, L. Li and K. Deng, Metallic borophene polytypes as lightweight anode materials for non-lithium-ion batteries, Phys. Chem. Chem. Phys., 2017, 19(36), 24945–24954 RSC.
- X. Zhang, J. Hu, Y. Cheng, H. Y. Yang, Y. Yao and S. A. Yang, Borophene as an extremely high capacity electrode material for Li-ion and Na-ion batteries, Nanoscale, 2016, 8(33), 15340–15347 RSC.
- D. Rao, L. Zhang, Z. Meng, X. Zhang, Y. Wang, G. Qiao, X. Shen, H. Xia, J. Liu and R. Lu, Ultrahigh energy storage and ultrafast ion diffusion in borophene-based anodes for rechargeable metal ion batteries, J. Mater. Chem. A, 2017, 5(5), 2328–2338 Search PubMed.
- J. Liu, X. Chen, X. Deng, W. Zhang, J. Li, B. Xiao and M. Pu, How to boost the sluggish lithium-ion hopping dynamic in borophene?, Appl. Surf. Sci., 2018, 441, 356–363 CrossRef.
- C. Kittel, Introduction to solid state physics, Wiley, 2005 Search PubMed.
- P. Wang, S. Orimo, T. Matsushima, H. Fujii and G. Majer, Hydrogen in mechanically prepared nanostructured h-BN: a critical comparison with that in nanostructured graphite, Appl. Phys. Lett., 2002, 80(2), 318–320 CrossRef.
- R. N. Muthu, S. Rajashabala and R. Kannan, Hexagonal boron nitride (h-BN) nanoparticles decorated multi-walled carbon nanotubes (MWCNT) for hydrogen storage, Renewable Energy, 2016, 85, 387–394 CrossRef.
- A. Yadav, A. Dashora, N. Patel, A. Miotello, M. Press and D. Kothari, Study of 2D MXene Cr2C material for hydrogen storage using density functional theory, Appl. Surf. Sci., 2016, 389, 88–95 CrossRef.
- C. Loken, D. Gruner, L. Groer, R. Peltier, N. Bunn, M. Craig, T. Henriques, J. Dempsey, C.-H. Yu and J. Chen, et al., SciNet: lessons learned from building a power-efficient top-20 system and data centre, in: Journal of Physics: Conference Series, IOP Publishing, 2010, vol. 256, p. 012026 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab

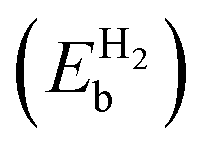 over metal decorated borophene
over metal decorated borophene
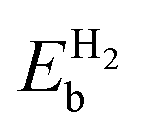
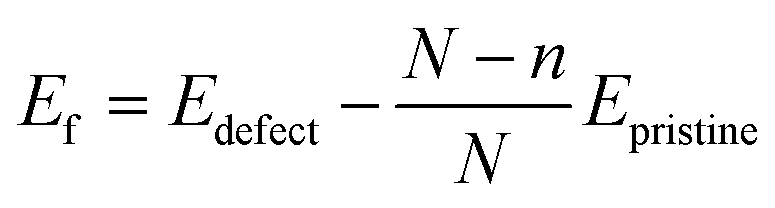 , where Edefect and Epristine are the total energies of defective and pristine borophene, respectively, N is the number of atoms in the pristine substrate, and n is the number of atoms removed from the pristine supercell to create the defects (n = 1 for SV, and n = 2 for DV). As summarized in Table 3, the calculated formation energy for the SV defect is 0.12 eV and those for the DV defects are 1.31 eV (DV-1) and 1.26 eV (DV-2).
, where Edefect and Epristine are the total energies of defective and pristine borophene, respectively, N is the number of atoms in the pristine substrate, and n is the number of atoms removed from the pristine supercell to create the defects (n = 1 for SV, and n = 2 for DV). As summarized in Table 3, the calculated formation energy for the SV defect is 0.12 eV and those for the DV defects are 1.31 eV (DV-1) and 1.26 eV (DV-2).

 , distance (Rsub-H2) of H2 from the substrate, and bond length (RH–H) of H2 in borophene with different defects. The binding energies correspond to the adsorption of one H2 molecule. The distance is measured from the closest boron atom in defective borophene
, distance (Rsub-H2) of H2 from the substrate, and bond length (RH–H) of H2 in borophene with different defects. The binding energies correspond to the adsorption of one H2 molecule. The distance is measured from the closest boron atom in defective borophene