
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMgH2–CoO: a conversion-type composite electrode for LiBH4-based all-solid-state lithium ion batteries†
Abdelouahab El Kharbachi *a,
Hiroki Uesatob,
Hironori Kawaib,
Sigurd Wennerc,
Hiroki Miyaokab,
Magnus H. Sørbya,
Helmer Fjellvågd,
Takayuki Ichikawab and
Bjørn C. Haubacka
*a,
Hiroki Uesatob,
Hironori Kawaib,
Sigurd Wennerc,
Hiroki Miyaokab,
Magnus H. Sørbya,
Helmer Fjellvågd,
Takayuki Ichikawab and
Bjørn C. Haubacka
aInstitute for Energy Technology, P. O. Box 40, NO-2027 Kjeller, Norway. E-mail: abdele@ife.no
bInstitute for Advanced Materials Research, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima 739-8530, Japan
cSINTEF Materials and Chemistry, P. O. Box 4760 Sluppen, NO-7465 Trondheim, Norway
dCentre for Materials Science and Nanotechnology (SMN), University of Oslo, P. O. Box 1126 Blindern, NO-0318 Oslo, Norway
First published on 27th June 2018
Abstract
Several studies have demonstrated that MgH2 is a promising conversion-type anode toward Li. A major obstacle is the reversible capacity during cycling. Electrochemical co-existence of a mixed metal hydride-oxide conversion type anode is demonstrated for lithium ion batteries using a solid-state electrolyte. 75MgH2·25CoO anodes are obtained from optimized mixing conditions avoiding reactions occurring during high-energy ball-milling. Electrochemical tests are carried out to investigate the cycling capability and reversibility of the on-going conversion reactions. The cycling led to formation of a single-plateau nanocomposite electrode with higher reversibility yield, lowered discharge–charge hysteresis and mitigated kinetic effect at high C-rate compared to MgH2 anodes. It is believed that reduced diffusion pathways and less polarized electrodes are the origin of the improved properties. The designed composite-electrode shows good preservation and suitability with LiBH4 solid electrolyte as revealed from electron microscopy analyses and X-ray photoelectron spectroscopy.
Introduction
Development of beyond-intercalation systems is of utmost importance for achieving novel Li-ion battery concepts.1–3 Magnesium hydride has been demonstrated to be a candidate for conversion-type anodes,4,5 where the (de)lithiation processes are governed by a conversion mechanism, MgH2 + 2Li ⇆ 2LiH + Mg. This material features a high theoretical capacity of 2037 mA h g−1 toward lithium and low discharge–charge hysteresis.4,6 However, the reversible capacity is still limited owing to the poor conductivity of the involved entities (MgH2/LiH), along with transient aging issues of LiH in carbonate-based liquid electrolytes.7–9 Using amorphous 80Li2S–20P2S5 as a solid electrolyte (SE), Ikeda et al.10 reported ∼31% reversible capacity for MgH2 for the 1st cycle at room temperature, compared to ∼75% for liquid electrolytes.6 Possible interaction at the electrode/SE (80Li2S–20P2S5) interface is pointed out as a cause for the low reversible capacity. When LiBH4 is used as SE at 120 °C, better performance has been reported with high initial reversibility yield (∼90%) in the presence of Nb2O5 catalyst,11,12 which decreases during cycling. Single phase LiBH4 as SE, operating above its phase-transition temperature (Tortho–hexatrans ∼ 113 °C)13 with the highly ion-conducting hexagonal phase, has been demonstrated for other electrode materials with many advantages;14–21 including flexible mechanical properties, strain-induced diffusion activation energy and formation of a stable composite with the MgH2 anode.22–25 Furthermore, it has been reported that LiBH4-SE – in the presence of metal hydride anode – contributes to the enhancement of the mobility of Li+ ions by H− exchange effects which seems to play a key role during the electrochemical cycling processes.11,26,27 CoO negative electrode (∼700 mA h g−1) has been reported to undergo a conversion reaction similar to MgH2, with formation of Co nanoparticles dispersed in the Li2O matrix.28 Its cycling performance is unrivalled to any other conversion-type anode over hundreds of cycles owing to privileged nanoparticles distribution with exalted reactivity.29 However, the high working potential and large cycling hysteresis hinders its application in Li-ion batteries.3,28 Pure CoO adopts a rock-salt type structure with the lattice parameter a = 4.258 Å. A few reports exist on oxide additions to MgH2 anodes, but only in small amounts for catalytic purposes.10,11,30,31Accordingly, the present work aims the study of the effect of the addition of an electrochemically active oxide such as CoO (25 mol%) on the cycling performance of MgH2 anode, a mixture having a theoretical capacity around ∼1350 mA h g−1. The final goal is to promote the cycling reversibility and inhibit the formation of Li–Mg alloys for low voltage operation in the presence of an oxide with large voltage window and better cycling response upon discharge/charge. Considering the effect of the cell assembly (additives, heat treatment, mechanical pressing) which may influence the cycling performance,5,7,8,10,32 the optimization of the design of the battery cells was performed first with MgH2 electrode without CoO addition. The prepared and optimized composite electrodes were characterized using powder X-ray diffraction (PXD), X-ray photoelectron spectroscopy (XPS) and scanning transmission electron microscopy (STEM) before and after the electrochemical tests toward Li metal. The mechanistic and synergetic behaviours of the hydride–oxide paired anode are addressed for the first time in this contribution.
Experimental
MgH2 (98% purity), CoO (99.9% purity) and Co (99.9% purity) were purchased from Alpha-Aesar. The MgH2 powders were ball-milled (24 h) under 50 bar H2 using a Fritsch Pulverisette 6 planetary ball-mill with stainless steel vials and balls (ball-to-powder ratio 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, rotation speed 400 rpm) to hydrogenate any metallic Mg impurities. The LiBH4 (95%) chemical was purchased from Sigma-Aldrich and stored in its delivery flask in an argon filled glove box. The LiBH4 was ball-milled for 3 h using the Fritsch Pulverisette 6 planetary ball-mill and dried overnight at 120 °C under dynamic vacuum. The carbon black Super P® (CSP, Timcal) was dried in the same way. Synchrotron radiation powder X-ray diffraction (SR-PXD) patterns were obtained at the Swiss-Norwegian Beamlines (SNBL, BM01), ESRF, Grenoble with a Pilatus 2M 2-dimensional detector and a wavelength of 0.78956 Å. The samples were contained in 0.5 mm borosilicate capillaries that were rotated 90° during 30 seconds exposure. The sample – detector distance was 345.97 mm. 1D data were obtained by integration of the 2D diffraction patterns using the program Bubble.33
:1, rotation speed 400 rpm) to hydrogenate any metallic Mg impurities. The LiBH4 (95%) chemical was purchased from Sigma-Aldrich and stored in its delivery flask in an argon filled glove box. The LiBH4 was ball-milled for 3 h using the Fritsch Pulverisette 6 planetary ball-mill and dried overnight at 120 °C under dynamic vacuum. The carbon black Super P® (CSP, Timcal) was dried in the same way. Synchrotron radiation powder X-ray diffraction (SR-PXD) patterns were obtained at the Swiss-Norwegian Beamlines (SNBL, BM01), ESRF, Grenoble with a Pilatus 2M 2-dimensional detector and a wavelength of 0.78956 Å. The samples were contained in 0.5 mm borosilicate capillaries that were rotated 90° during 30 seconds exposure. The sample – detector distance was 345.97 mm. 1D data were obtained by integration of the 2D diffraction patterns using the program Bubble.33
MgH2 and CoO were mixed using both an agate mortar/pestle and Spex mill. For battery tests, the MgH2/CoO composite electrode and Li foil were used as active anode material and counter-reference electrode, respectively. The active material (MgH2 or MgH2/CoO), CSP and SE powders were weighed in a 40:30:30 mass ratio, and then hand mixed by an agate mortar/pestle to obtain a composite mixture. Around 50 mg of the SE was introduced first in a 15 mm die-set and pre-pressed uniaxially at 100 MPa, and followed by 5 mg of the composite mixture and pressed together with Cu foil on top at 180 MPa. A Li foil was placed on the opposite side of the material composite. The resulted multi-layer pellet was inserted in a CR2032 coin cell fitted with PTFE gasket for cycling at 120 °C.
The following cells were assembled:
Battery cell (1): MgH2/CSP/LiBH4 | LiBH4 | Li.
Battery cell (2): MgH2/CoO/CSP/LiBH4 | LiBH4 | Li.
All sample handling was performed in a MBraun® glove box under argon atmosphere (H2O and O2 < 1 ppm). The cells were cycled at different rates between 0.3–1 V, using a battery cycler (Hioki®, Japan). In this study, a voltage cut-off at 0.3 V was applied to avoid formation of Mg–Li solid solutions and thus focus on the main conversion reaction occurring around 0.5 V.4,6
XPS technique was carried out using a Thermo Fisher Scientific ESCALAB 250Xi unit with an Al-Kα (1486.6 eV) X-ray source. A dedicated sample holder was employed to transfer the powder samples from the glovebox to the vacuum chamber of the XPS without air exposure.
STEM was performed with a JEOL JEM-2100F microscope operated at 200 kV, with an Oxford 80 mm2 EDS detector. Focused ion beam-scanning electron microscopy (FIB-SEM) was carried out with a FEI Helios G4 dualBeam FIB. The Ga+ ion beam was used to cut down through the electrode for cross-section imaging, EDS analysis and STEM sample preparation. Owing to their high reactivity after cycling, the electrode samples for STEM/SEM were prepared with the addition of the polyvinylidene fluoride (PVDF) binder using the slurry method to protect the particles surface as short air exposure cannot be avoided.
Results and discussion
Fig. 1 shows the SR-PXD patterns of the as received CoO, mixed MgH2/CoO before and after addition of the LiBH4 electrolyte and CSP carbon black. The as-received CoO material is well crystalline without any impurity phases. After hand mixing of MgH2/CoO, the corresponding pattern shows both tetragonal and orthorhombic MgH2 with major presence of the former modification in addition to CoO.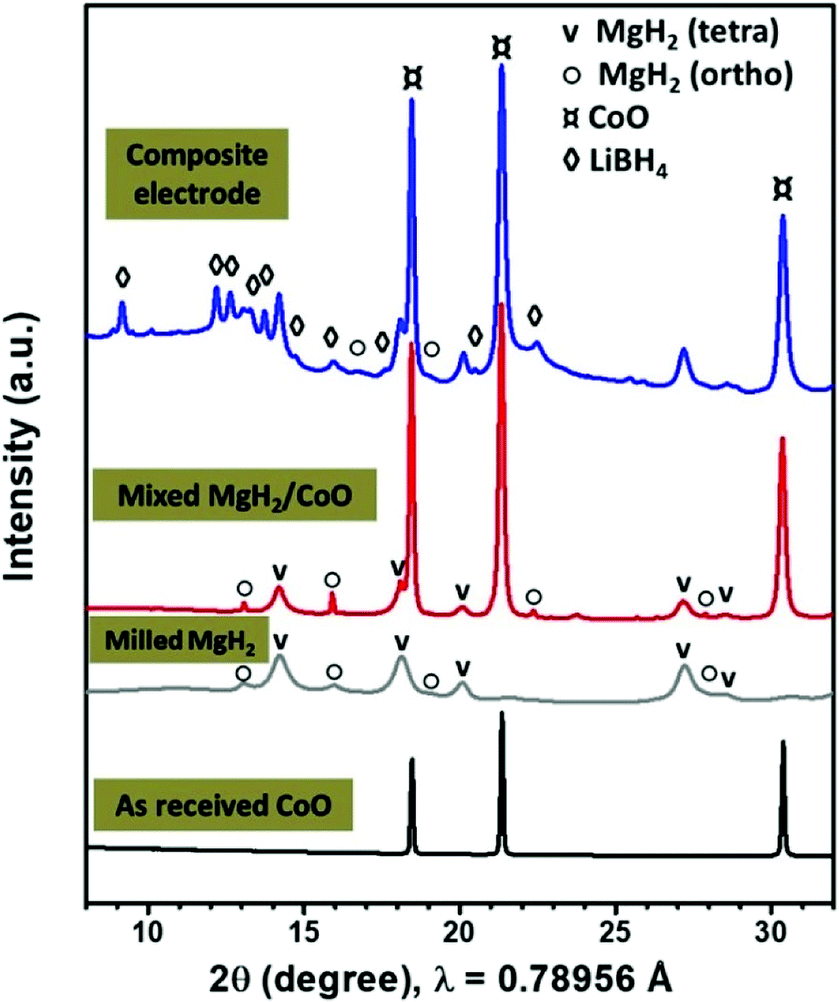 | ||
| Fig. 1 SR-PXD patterns of the as-received CoO, 24 h milled MgH2 and mixed MgH2/CoO, and after addition of the LiBH4 solid electrolyte and CSP carbon black. | ||
Ball milling of MgH2 and metallic Co is reported to result in formation of Mg2CoH5.34,35 We find that ball-milling MgH2/CoO using Spex for 3 h is enough to form significant amounts of this phase. In this case, the analyzed patterns (Fig. S1 in ESI section†) indicate that MgH2 is almost disappeared, while CoO still remains. During this solid-state reaction, the formation of water molecules may occur (hydrated or gaseous phase) which is beyond the scope of the present study. As the electrochemical performance of Mg2CoH5 compound is poor, its formation during the milling should be avoided.36,37 Thus, the preparation was then limited to hand mixing and Spex-milling with LiBH4/CSP for 1 h, which ensure good dispersion without formation of Mg2CoH5 according to SR-PXD. TEM analysis indicates that the 24 h milled MgH2 powders have an average particle size of about 150 nm.8,9 The starting hand-mixed CoO powders are in the micron range. However, after Spex-milling, Fig. S2 (ESI section†) shows well-dispersed CoO particles with sizes of 20–100 nm in the MgH2/C matrix.
Preliminary battery tests were carried out with the MgH2 single anode without CoO. Fig. 2a shows the notable reversible cyclability of the MgH2-based anode with flat discharge/charge plateaus, mirroring merely the solid-state diffusion process. This behaviour agrees with the fact that the conversion reaction undergoes a stable separation in the three-phase system (MgH2–Mg–LiH) during the thermodynamic non-equilibrium state at constant current.38–40 The presence of solid solution reactions and how the different phases are formed at the early stage (nucleation/growth) of the conversion mechanism can be a subject of debate which are not approached in this study.
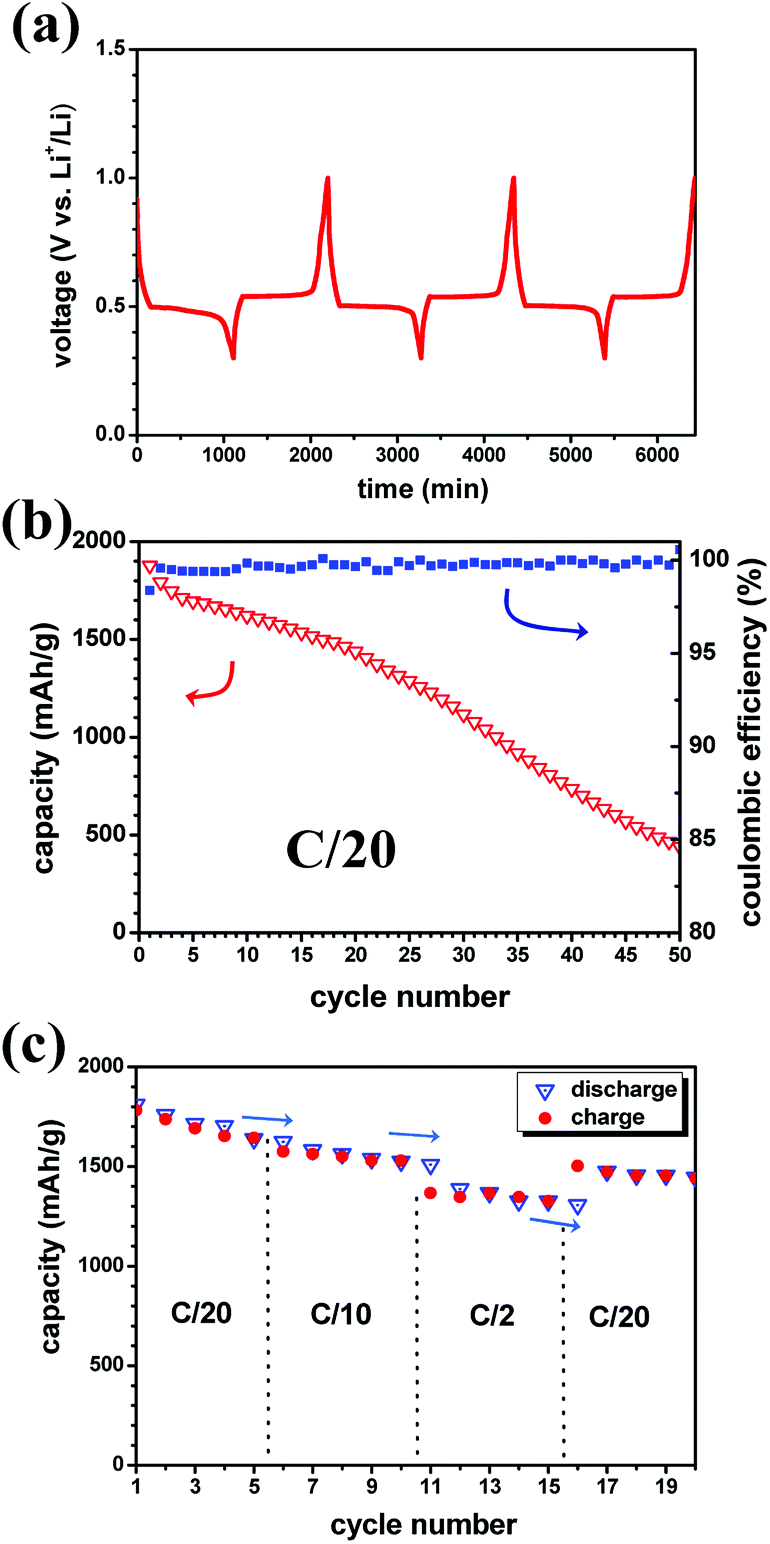 | ||
| Fig. 2 (a) Display of the notable cyclability (voltage vs. time) of MgH2 single anode using LiBH4 SE toward Li (C-rate C/20, 120 °C). (b) Cycling performance as function of cycle number with the corresponding coulombic efficiency. (c) Rate capability tests of the same electrode. | ||
Furthermore, the cycling curves show no unwanted effects at extreme compositions. The sudden change in the slope after the charge-transfer process during lithiation may be explained by the poor conductivity of the native LiH formed at the MgH2 surface. The resulting discharge/charge hysteresis is around 42 mV with a working potential of ∼0.5 V; values close to the obtained ones in liquid electrolyte.8 A small perturbation around 0.7 V can be discerned, and it needs to be clarified if it is related to any interaction with the electrolyte as reported at higher voltage.11 Further cycling (Fig. 2b) shows the enhanced capacity retention compared to previous studies during the first 20 cycles even though no catalyst is employed here. Afterwards up to 50 cycles, the MgH2 anode follows a progressive decrease of the capacity. Remarkably, owing to the high coulombic efficiency, the loss of the capacity may occur during discharge as observed in previous studies.11 Fig. 2c shows the variation of the capacity as function of the C-rate and cycle number for the MgH2 battery cell. From C/20 to C/10, the capacity retention is unaffected by the C-rate during discharge and charge. At the highest employed C-rate (C/2), a kinetic effect can be observed and the capacity is somewhat lower than at C/10 and C/20 rates, but it does not decrease further over 5 cycles. It can be noticed that the kinetic rate seems to have more significant effect on the charge (LiH/Mg → MgH2) than the discharge reaction (MgH2 → LiH/Mg).
After assessing the performance of the MgH2 anode, the electrochemical behaviour of the MgH2–CoO composite electrode assembled in the same way as for the single anode needs to be elucidated. Fig. 3 shows the obtained discharge–charge galvanostatic profiles of the 75MgH2·25CoO anode in which two reversible processes at 0.89 V and 0.5 V (inset of Fig. 3) can be distinguished during the first cycle. At discharge, the observed two plateaus are related to the successive reduction of CoO and MgH2, respectively. It presents flat discharge–charge plateaus and low polarization-hysteresis; a remarkable electrochemical asset with high conversion yield. Higher capacity is observed for the first discharge which can be attributed to an activation process of the electrolyte/electrode interface as reported previously.11,12
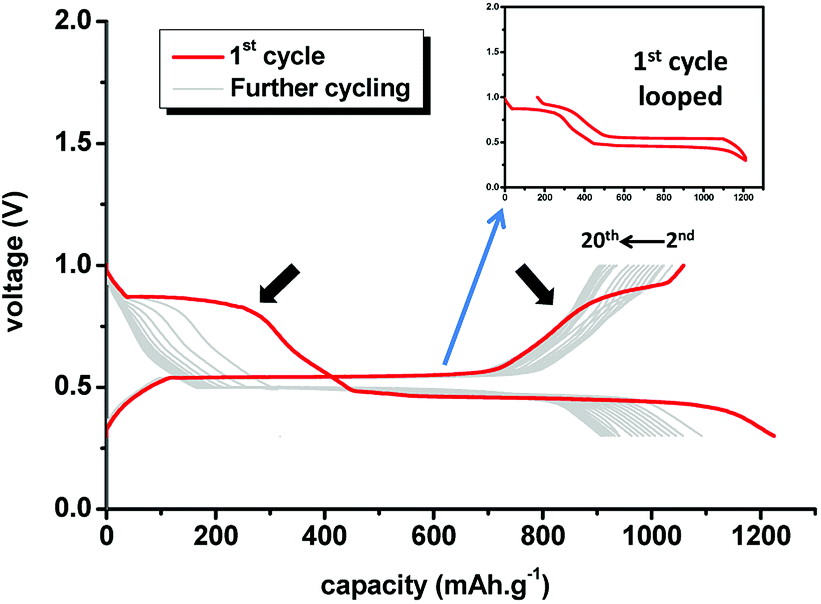 | ||
| Fig. 3 Discharge–charge galvanostatic profiles of the 75MgH2·25CoO-based anode using LiBH4 SE toward Li (0.05C rate, 120 °C). Inset: looped discharge/charge curve for clarity. | ||
Further cycling leads to the gradual decline of the 1st plateau in both discharge and charge, and its disappearance after 5 cycles.
This point needs to be clarified if the nanocrystalline nature of CoO/Co can be connected to the decline of the plateau and resulting to a pseudocapacitor behaviour. Interestingly despite the observed structural changes during the (de)lithiation processes, the capacity retention remains less affected. During the first 5 cycles, about 94% of the capacity is retained compared to only 90% for MgH2 anode. Such improvement could raise the question if the CoO/Co particles are incorporated in the MgH2 matrix leading to a nano-composite with high reversibility yield and thus overcoming the issues of the electronic conductivity and particles aggregation. According to literature, CoO has been reported to undergo reversible cycling at constant current density with formation of nano-sized particles in the range of 1–2 nm.28 Such an eventual nanoscale interaction can make the Li ions and electrons available in close vicinity at the surface of MgH2/Mg via the highly dispersive CoO/Co nanoparticles. At first glance, the interaction of Li2O with Mg and LiH is not conceivable. These findings may suggest preferred reaction pathways for the composite-electrode system in a synergetic-like behaviour, and with a compromise between reversibility and kinetically-induced hysteresis offered by CoO and MgH2 at different degrees.
Fig. 4a displays the rate capability of the MgH2/CoO anode with good capacity retention and a coulombic efficiency close to 100%, except the 1st cycle after the rate is changed. Only slight decrease of the capacity can be detected at high rate (C/2). Contrary to MgH2, in the MgH2/CoO anode both discharge and charge states are sensitive to the C-rate. However, the composite-electrode exhibits a stable capacity and a good rate capability. A priori it seems like there is a beneficial kinetic effect from the presence of CoO. Fig. 4b shows the good cyclability of the MgH2/CoO composite-electrode at high rate (C/2) with a moderate charge/discharge hysteresis of 75 mV compared to 32 mV obtained at C/20. This latter value is even lower than the one found for the MgH2 anode (42 mV) at the same rate. At this stage it is difficult to conclude if this is related to mass effect or due to the presence of CoO, though the same total active masses (∼2 mg) were used. However, the presence of CoO is clearly contributing to the improvement of the cyclability and reversibility while remaining active and delivering an extra capacity. Reduced diffusion pathways and less polarized electrodes are believed to be at the origin of such beneficial properties.
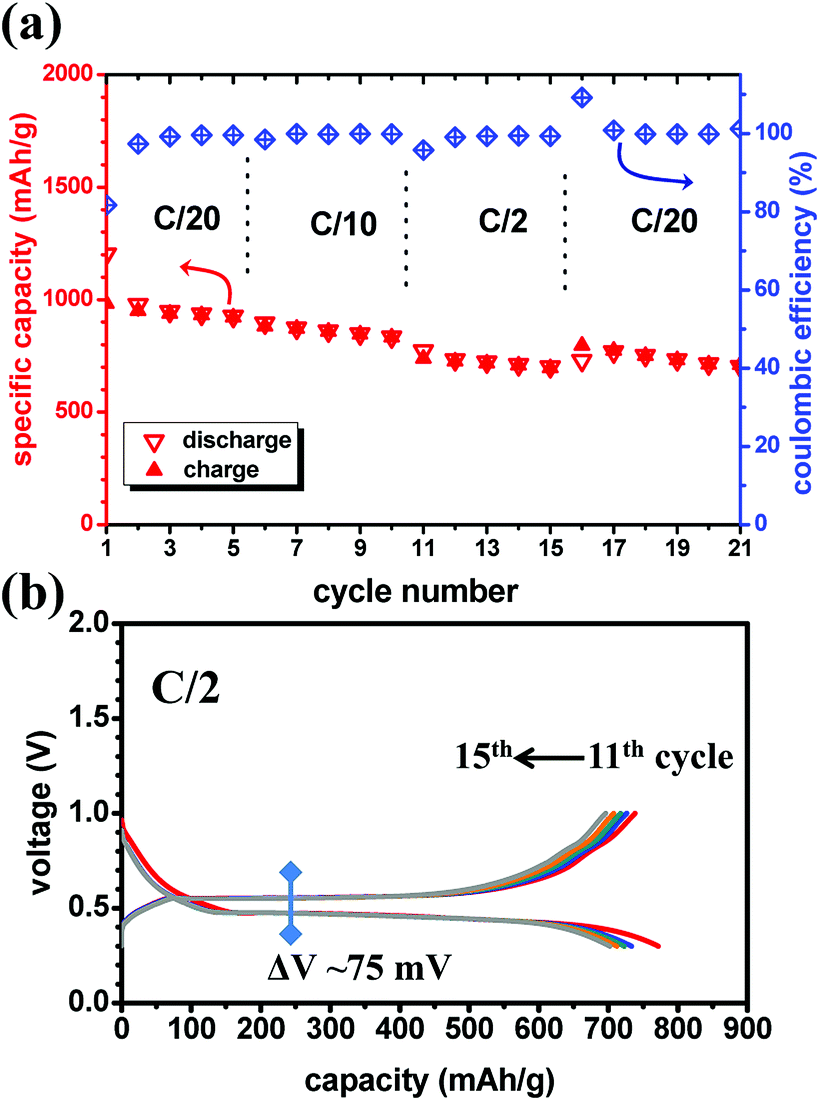 | ||
| Fig. 4 (a) Rate capability tests of the 75MgH2·25CoO-based anode using LiBH4 SE toward Li (different C-rates, 120 °C). (b) Display of the galvanostatic discharge–charge profiles from 11th to 15th cycle (C/2 rate). | ||
High angle annular dark field (HAADF) STEM imaging combined with EDS were used to map the different elements before and after battery cycling. Fig. 5 shows the STEM observations of the electrode composite before cycling. The prepared sample section (Fig. 5a) reveals a homogenous electrode with the presence of small micro-cracks. EDS mapping (Fig. 5b and c) shows the Mg-based matrix with uniform distribution of CoO particles and with some C-rich areas. The CoO particles have an average size of about 50–100 nm in the uncycled electrode, in agreement with the first TEM analysis (Fig. S2†) of the as-prepared composite powders. The presence of O throughout the sample comes from the inevitable short exposure to air between FIB preparation and STEM analysis.
 | ||
| Fig. 5 STEM images of the as-assembled 75MgH2·25CoO-based cell before cycling: (a) cross-section of the electrode side during thinning with FIB, (b) HAADF STEM image and the corresponding (c) EDS mapping. | ||
A SEM-EDS map of the cross section of the interface between the electrode and the SE after 2 cycles is shown in Fig. 6. The electrolyte side can be distinguished by the B-rich region. The electrode part shows an even distribution of Mg and Co expected in the nano-range of few nm. According to cycling curves, MgH2 is formed with possible presence of residual metallic Mg. Small amounts of O can be seen on the SE side, which may be due to possible surface contamination. Furthermore, O is depleted around Co (also seen in other maps), implying that some Co particles are metallic.
 | ||
| Fig. 6 Electron micrographs (FIB-SEM) of 75MgH2·25CoO-based cell after 2 cycles at the electrode/electrolyte interface and the corresponding EDS mapping of the selected area. | ||
PXD analysis of the cycled electrodes yielded limited information, and the data were dominated by Bragg peaks from the SE LiBH4. This indicates that MgH2 and CoO take a nanocrystalline or amorphous state as reported previously for CoO anode.28,29 Samples of the first discharged and charged states were therefore analysed by XPS and compared to the as-received CoO and MgH2. The peaks deconvolution in the Co 2p XPS spectra was not possible owing to dominant thin oxide layer on the surface of Co particles, in addition to the multiplicity of the 2p peaks.41 Fig. 7a shows the O 1s XPS core spectra obtained for the analysed samples. The starting CoO (sample a) presents a peak at 530.2 eV, characteristic of O2− anions in the crystalline network,42 and slightly shifted from the expected position (529.9 eV) toward higher energies which may suggest the presence of mixed valence. A second peak at 531.6 eV is observed which agree with weakly adsorbed species at the surface, but also due to oxygen “O22−” anions of CoO with particular coordination.41,42 After discharge down to 0.3 V (sample b), an additional peak appears at 528.2 eV, indicating the formation of Li2O. The broad peak at 531.7 eV can be attributed to many oxygen-containing species, particularly dissociation products of possible interaction with the electrolyte at the surface of the Co particles. Note that XPS is surface specific with an analysis depth of about 5 nm. This abovementioned layer (531.7 eV) is thick enough to make the Co atoms undetectable.28 These results are in agreement with the mechanism of formation of Co nanoparticles dispersed in the Li2O matrix.28 After recharge at 1 V (sample c), the spectrum shows the disappearance of Li2O and consists then of one main broad peak at 531.6 eV. The restitution of CoO can not be clearly evidenced because of possible layer on the surface of the nanoparticles owing to intimate interaction with the electrolyte after cycling. A shoulder at 533.0 eV is also observed and its assignment here is not possible; as many inorganic/organic species can be attributed to this signal.
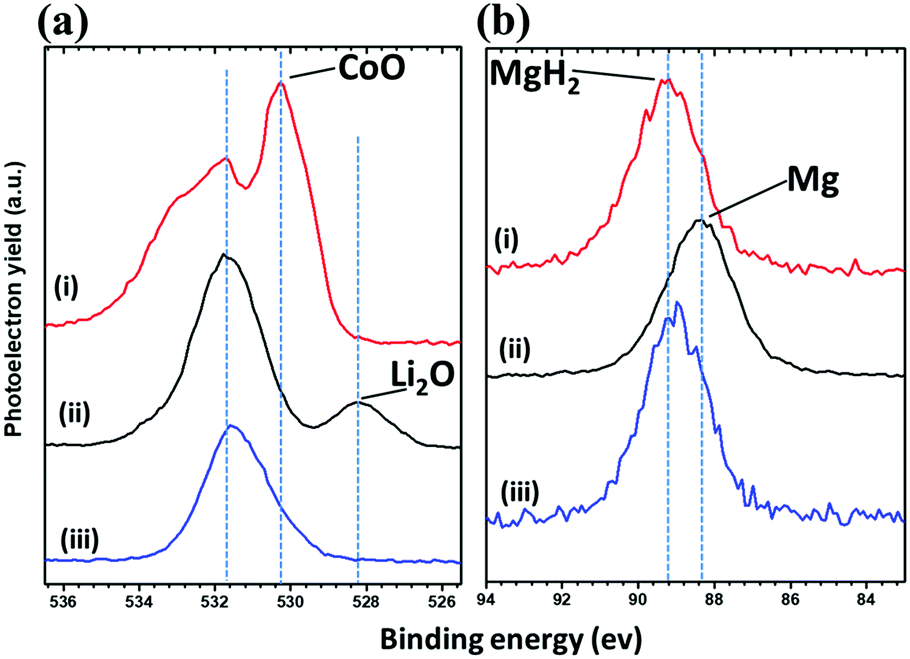 | ||
| Fig. 7 (a) O 1s and (b) Mg 2s photoelectrons spectra regions of (i) the starting CoO/MgH2 powders, (ii) discharged 75MgH2·25CoO-based cell at 0.3 V and (iii) charged at 1 V. | ||
The Mg 2s core spectra of the same samples are shown in Fig. 7b. Before cycling (sample a), given the peak position at 89.2 eV, it shows the presence of MgH2. After full discharge (sample b), the Mg 2s single peak shifts toward lower binding energies (88.4 eV) in agreement with elemental Mg. After charge at 1 V (sample c), the spectrum is less resolved and the Mg 2s peak is centred at ∼89.0 eV, implying the attenuation of Mg and formation of MgH2 as expected from cycling profiles. In other words, this analysis based on the study of O 1s and Mg 2s photoelectrons spectra regions allows us to confirm the independent reaction mechanism of MgH2 and CoO and their reversibility during cycling. These parallel reactional processes seem to make possible the formation/decomposition of Li2O and LiH without chemical interference, despite their supposed high surface energy at the nanometric scale.28
Conclusions
New insights are reported for the enhancement of the reversible capacity of nano-composite electrodes combining two different materials with complementary electrochemical properties. A novel strategy is followed to improve the cycling performance of MgH2 by adding an electrochemically active oxide, CoO. Battery tests demonstrate possible co-existence of both MgH2/CoO anodes in a synergetic way, compensating mutually each other mainly regarding the mediocre reversibility of MgH2 and presence of highly dispersive CoO/Co nanoparticles. After prolonged cycling, the CoO plateau declines gradually, presumably owing to the nanocrystallinity of the involved entities in agreement with FIB-SEM analysis and XPS characterizations. The reversibility of MgH2 is confirmed by XPS while the CoO/Co entities are probably partially involved after cycling which may require the adjustment of the voltage window. Such electrochemical incorporation of CoO/Co nanoparticles in the MgH2 matrix is believed to contribute to the improvement of the cycling performance at high rates. This work may open up for a new composite-electrode concept where the electrochemical properties can be tuned for better cycling performance of electrode materials for application in all-solid-state batteries which will undoubtedly become a hot topic.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by Research Council of Norway under the program EnergiX, Project no. 244054, LiMBAT-“Metal hydrides for Li-ion battery anodes”. AE is indebted to Prof. D. Larcher for introducing him to conversion reactions during the postdoctoral stay at LRCS-Amiens. The authors thank Prof. M. Latroche, F. Cuevas and J. Zhang for preliminary X-ray diffraction analysis and discussions. We acknowledge the skilful assistance from the staff of SNBL at ESRF, Grenoble, France.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef PubMed.
- S.-K. Jung, H. Kim, M. G. Cho, S.-P. Cho, B. Lee, H. Kim, Y.-U. Park, J. Hong, K.-Y. Park, G. Yoon, W. M. Seong, Y. Cho, M. H. Oh, H. Kim, H. Gwon, I. Hwang, T. Hyeon, W.-S. Yoon and K. Kang, Nat. Energy, 2017, 2, 16208 CrossRef.
- A. Grimaud, Nat. Energy, 2017, 2, 17003 CrossRef.
- Y. Oumellal, A. Rougier, G. A. Nazri, J. M. Tarascon and L. Aymard, Nat. Mater., 2008, 7, 916–921 CrossRef PubMed.
- Y. Oumellal, C. Zlotea, S. Bastide, C. Cachet-Vivier, E. Leonel, S. Sengmany, E. Leroy, L. Aymard, J.-P. Bonnet and M. Latroche, Nanoscale, 2014, 6, 14459–14466 RSC.
- L. Aymard, Y. Oumellal and J.-P. Bonnet, Beilstein J. Nanotechnol., 2015, 6, 1821–1839 CrossRef PubMed.
- S. Brutti, G. Mulas, E. Piciollo, S. Panero and P. Reale, J. Mater. Chem., 2012, 22, 14531–14537 RSC.
- A. El Kharbachi, H. F. Andersen, M. H. Sørby, P. E. Vullum, J. P. Mæhlen and B. C. Hauback, Int. J. Hydrogen Energy, 2017, 42, 22551–22556 CrossRef.
- A. El Kharbachi, Y. Hu, M. H. Sørby, P. E. Vullum, J. P. Mæhlen, H. Fjellvåg and B. C. Hauback, J. Phys. Chem. C, 2018, 122, 8750–8759 CrossRef.
- S. Ikeda, T. Ichikawa, K. Kawahito, K. Hirabayashi, H. Miyaoka and Y. Kojima, Chem. Commun., 2013, 49, 7174–7176 RSC.
- L. Zeng, K. Kawahito, S. Ikeda, T. Ichikawa, H. Miyaoka and Y. Kojima, Chem. Commun., 2015, 51, 9773–9776 RSC.
- L. Zeng, T. Ichikawa, K. Kawahito, H. Miyaoka and Y. Kojima, ACS Appl. Mater. Interfaces, 2017, 9, 2261–2266 CrossRef PubMed.
- A. El Kharbachi, I. Nuta, F. Hodaj and M. Baricco, Thermochim. Acta, 2011, 520, 75–79 CrossRef.
- A. Unemoto, T. Ikeshoji, S. Yasaku, M. Matsuo, V. Stavila, T. J. Udovic and S.-i. Orimo, Chem. Mater., 2015, 27, 5407–5416 CrossRef.
- A. Unemoto, M. Matsuo and S.-i. Orimo, Adv. Funct. Mater., 2014, 24, 2267–2279 CrossRef.
- M. Matsuo, Y. Nakamori, S.-i. Orimo, H. Maekawa and H. Takamura, Appl. Phys. Lett., 2007, 91, 224103 CrossRef.
- M. Matsuo and S.-i. Orimo, Adv. Energy Mater., 2011, 1, 161–172 CrossRef.
- K. Kawahito, L. Zeng, T. Ichikawa, H. Miyaoka and Y. Kojima, Mater. Trans., 2016, 57, 755–757 CrossRef.
- J. Weeks, S. Tinkey, P. Ward, R. Lascola, R. Zidan and J. Teprovich, Inorganics, 2017, 5, 83 CrossRef.
- P. López-Aranguren, N. Berti, A. H. Dao, J. Zhang, F. Cuevas, M. Latroche and C. Jordy, J. Power Sources, 2017, 357, 56–60 CrossRef.
- P. Huen and D. B. Ravnsbæk, Electrochem. Commun., 2018, 87, 81–85 CrossRef.
- P. Vajeeston, P. Ravindran, A. Kjekshus and H. Fjellvåg, J. Alloys Compd., 2005, 387, 97–104 CrossRef.
- Y. Bouhadda, S. Djellab, M. Bououdina, N. Fenineche and Y. Boudouma, J. Alloys Compd., 2012, 534, 20–24 CrossRef.
- H. Benzidi, M. Lakhal, A. Benyoussef, M. Hamedoun, M. Loulidi, A. El kenz and O. Mounkachi, Int. J. Hydrogen Energy, 2017, 42, 19481–19486 CrossRef.
- U. Bösenberg, S. Doppiu, L. Mosegaard, G. Barkhordarian, N. Eigen, A. Borgschulte, T. R. Jensen, Y. Cerenius, O. Gutfleisch, T. Klassen, M. Dornheim and R. Bormann, Acta Mater., 2007, 55, 3951–3958 CrossRef.
- L. Zeng, H. Miyaoka, T. Ichikawa and Y. Kojima, J. Phys. Chem. C, 2010, 114, 13132–13135 CrossRef.
- A. El Kharbachi, Y. Hu, M. H. Sørby, J. P. Mæhlen, P. E. Vullum, H. Fjellvåg and B. C. Hauback, Solid State Ionics, 2018, 317, 263–267 CrossRef.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. c. Dupont, B. Beaudoin and J.-M. Tarascon, C. R. Acad. Sci., Ser. IIc: Chim., 2000, 3, 681–691 CrossRef.
- S. Ikeda, T. Ichikawa, S. Yamaguchi, H. Miyaoka and Y. Kojima, J. Jpn. Inst. Energy, 2014, 93, 926–930 CrossRef.
- S. Ikeda, T. Ichikawa, K. Goshome, S. Yamaguchi, H. Miyaoka and Y. Kojima, J. Solid State Electrochem., 2015, 19, 3639–3644 CrossRef.
- W. Zaïdi, Y. Oumellal, J. P. Bonnet, J. Zhang, F. Cuevas, M. Latroche, J. L. Bobet and L. Aymard, J. Power Sources, 2011, 196, 2854–2857 CrossRef.
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, J. Synchrotron Radiat., 2016, 23, 825–829 CrossRef PubMed.
- M. Norek, T. K. Nielsen, M. Polanski, I. Kunce, T. Płociński, L. R. Jaroszewicz, Y. Cerenius, T. R. Jensen and J. Bystrzycki, Int. J. Hydrogen Energy, 2011, 36, 10760–10770 CrossRef.
- M. G. Verón, A. M. Condó and F. C. Gennari, Int. J. Hydrogen Energy, 2013, 38, 973–981 CrossRef.
- W. Zaïdi, J. P. Bonnet, J. Zhang, F. Cuevas, M. Latroche, S. Couillaud, J. L. Bobet, M. T. Sougrati, J. C. Jumas and L. Aymard, Int. J. Hydrogen Energy, 2013, 38, 4798–4808 CrossRef.
- K. Provost, J. Zhang, W. Zaïdi, V. Paul-Boncour, J. P. Bonnet, F. Cuevas, S. Belin, L. Aymard and M. Latroche, J. Phys. Chem. C, 2014, 118, 29554–29567 CrossRef.
- M. Hess, T. Sasaki, C. Villevieille and P. Novak, Nat. Commun., 2015, 6 Search PubMed.
- V. Srinivasan and J. Newman, Electrochem. Solid-State Lett., 2006, 9, A110–A114 CrossRef.
- T. R. Ferguson and M. Z. Bazant, Electrochim. Acta, 2014, 146, 89–97 CrossRef.
- R. Dedryvère, S. Laruelle, S. Grugeon, P. Poizot, D. Gonbeau and J. M. Tarascon, Chem. Mater., 2004, 16, 1056–1061 CrossRef.
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed SR-PXD and TEM of milled MgH2–CoO composites. See DOI: 10.1039/c8ra03340d |
| This journal is © The Royal Society of Chemistry 2018 |