
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improving the photocatalytic reduction of CO2 to CO for TiO2 hollow spheres through hybridization with a cobalt complex†
Jinliang Lin *a,
Xiaoxiang Sunb,
Biao Qina and
Ting Yua
*a,
Xiaoxiang Sunb,
Biao Qina and
Ting Yua
aDepartment of Chemical and Engineering, Zunyi Normal College, 563000, Zunyi, P. R. China. E-mail: jinliang_lin@163.com; Fax: +86-851-28927159; Tel: +86-851-28927159
bDepartment of Chemical and Engineering, Qiannan Normal University for Nationalities, 558000, DuYun, P. R. China
First published on 5th June 2018
Abstract
A chemical system with enhanced efficiency for electron generation and transfer was constructed by the integration of TiO2 hollow spheres with [Co(bipy)3]2+. The introduction of [Co(bipy)3]2+ remarkably enhances the photocatalytic activity of pristine semiconductor photocatalysts for heterogeneous CO2 conversion, which is attributable to the acceleration of charge separation. Of particular interest is that the excellent photocatalytic activity of the heterogeneous catalysts can be utilised for a universal photocatalytic CO2 reduction system. Yields of 16.8 μmol CO and 6.6 μmol H2 can be obtained after 2 h of the photoredox reaction, and the apparent overall quantum yield was estimated to be 0.66% under irradiation at λ = 365 nm. The present findings clearly demonstrate that the integration of electron mediators with semiconductors is a feasible process for the design and development of efficient photochemical systems for CO2 conversion.
1 Introduction
The photochemical reduction of CO2, as a model reaction of artificial photosynthesis, has obtained much attention because it may provide solutions for overcoming both the problem of global warming and the shortage of fossil resources.1 Of particular interest is the conversion of CO2 into the energy carrier CO, which is an important chemical feedstock used to form syngas in combination with H2, and can be used to replace the steam reforming of fossil fuels in the petrochemical industry.2Photoelectrocatalytic CO2 reduction has been achieved on various kinds of material, including inorganic semiconductors, carbon based semiconductors, metal complexes, supermolecules, and their derivatives.3 Of these, TiO2 is one of the most well-known and widely used materials due to its prominent properties, namely that it is non-toxic, low cost, chemically stable, and exhibits good radiosity.4 In order to improve optical performance, it has been combined with various metals or other semiconductors to form hybridized composites in an attempt to reduce the band gap or suppress the recombination of photogenerated charge carriers.5 Another way to achieve desirable photocatalytic behavior is by preparing catalytic materials that possess special structures. Hierarchical structured TiO2 with high surface areas such as hollow spheres has been designed for the purpose of increasing active site exposure. The beneficial effects of particle morphology on photochemistry have also been proven, with previous examples investigating geometric effects and electronic nature.6 Additionally, previous works have proven that spherical morphology endowed Ti-base materials exhibit improved photocatalytic performances.7
Furthermore, hollow sphere structured TiO2 can be further modified through hybridization or ion-doping strategies, and exhibits an enhanced photocatalytic performance for CO2 activation. Fang et al. has presented novel Ti-based spherical photocatalysts with a large mesoporous volume and hierarchical porosity.8 These exhibited an improvement in photocatalytic activity and an enhancement in the efficiency for the solar reduction of CO2. Additionally, a one-pot template-free method has been reported for the preparation of Cu(II) incorporated TiO2 hollow microspheres for photo-driven CO2 reduction. The effect of metal vacancies on the physicochemical properties has been proven to be critically important for metal oxides.9 Hollow spheres consisting of alternating titania nanosheets (Ti0.91O2) and graphene nanosheets have been prepared by Zou’s group.10 There was a nine-fold enhancement of the photocatalytic activity on such hollow spherical materials compared to commercial P25 for photocatalytic CO2 conversion. They have also reported a Au–TiO2 nanocomposite with bifunctional linker molecules which was applied for the photocatalytic reduction of CO2 into hydrocarbon fuels.11 Several carbon fuels were produced over the Au–TiO2 nanocomposite, including CO, CH4, CH3OH and CH3CH2OH, under different forms of light irradiation and in different reaction systems. A Cu2O/TiO2 composite exhibited higher efficiency in the photocatalytic reduction of CO2 into CH4 under visible-light irradiation (λ ≥ 420 nm).12 This is because of the formation of a p–n heterojunction in the composites, resulting in the efficient suppression of the recombination of the photogenerated electrons and holes as well as an improved stability of the catalyst and, accordingly, an improved visible-light photocatalytic activity. Additionally, Jiang et al. found that a significantly improved photocatalytic activity obtained by hybrid carbon@TiO2 hollow spheres was mainly due to the increased specific surface area, CO2 uptake, local photothermal effect, enhancement of light absorption and charge transfer efficiency.5b A comprehensive review on surface and interface design in cocatalysts concerning CO2 reduction has been presented by Bai et al.13 As discussed, the rational material design of cocatalysts would be an effective route for pursuing their maximum contribution to the performance of photocatalysts.
The strategy of hybrid photocatalysts is a promising approach for the rational design of efficient and stable artificial photosynthetic systems. This is because homogeneous processes exhibit high quantum efficiencies, while heterogeneous processes possess desirable stabilities. Z-scheme water splitting systems, redox couples (e.g., Fe3+/Fe2+, IO3−/I−, [Co(bipy)3]3+/2+ and [Co(phen)3]3+/2+) and reduced grapheme oxides have been revealed to act as electron mediators to deliver photogenerated electrons from the O2-evolving catalyst to the H2-evolving catalyst in the two-photon type photocatalytic system.14 Also, various metal complexes, which mostly can facilitate charge separation and lower the energy barrier, are usually employed as co-catalysts for CO2 conversion. In earlier years, a high-efficiency photocatalytic CO2 reduction system consisting of semiconductor powders and a Ru-based complex was reported by Bhatt and coworkers.15 Xu et al. have developed an efficient hybrid CO2 photoreduction system by mixing CdS nanoparticles and CoCl2/bipy in an acetonitrile solution, which affords a high apparent quantum yield of 1.0% for CO formation under monochromatic irradiation (λ = 470 nm).16 Our previous work revealed that a similar reaction mechanism can be further extended to both heterogeneous and homogenous photocatalytic CO2 reduction systems in acetonitrile solution.17 More recently, Kuriki et al. has successfully developed several types of hybrid photocatalyst consisting of semiconductors and metal complexes, which have both the efficient CO2 reduction abilities supplied by the metal-complex unit and the strong oxidation power of semiconductors.18
Complex/semiconductor hybrid photocatalysts represent a feasible approach to achieving high efficiency semiconductors for solar fuel production. However, the research in this direction is still in an early stage and therefore leaves much room for further development. In this paper we present the synthesis of spherical TiO2 (denoted as “sTiO2”), and then employ it as photocatalyst for CO2 reduction under UV-light irradiation. Based on the analysis, it is reasonable to think that the hybrid of TiO2 hollow sphere and homogeneous methods may be an ideal pathway towards high efficiency. The spherical TiO2 with higher surface area exhibits a superior photocatalytic performance to bulk TiO2 (denoted as “bTiO2”). More importantly, this study also shows that the combination of the sTiO2 catalyzed photosystem with [Co(bipy)3]2+ as a cobalt complex redox shuttle induces a highly efficient and selective electron transfer for the reduction of CO2. The reaction activity in the presence/absence of cobalt complex shows that this is a feasible way to obtain high efficiency photocatalytic CO2 conversion.
2 Experimental
2.1 Chemicals
All reagents were commercially provided and used without further purification. Diethylenetriamine (DETA, 97%), titanium(IV) isopropoxide (TIP, 97%) and 2,2-bipyridine (bipy, 98%) were purchased from Alfa. Isopropyl alcohol (IPA, 98%), acetonitrile (MeCN, AR), triethanolamine (TEOA, AR) and cobalt chloride hexahydrate (CoCl2·6H2O, 99.9%) were purchased from China Sinopharm Chemical Reagent Co. Ltd.2.2 Preparation of TiO2 samples
The TiO2 spheres were prepared using a hydrothermal method as described in previous reports:19 0.05 mL of DETA was added to 75 mL of isopropyl alcohol, and the mixture was stirred for 5 minutes. Then, 2.5 mL of titanium(IV) isopropoxide was added. The reaction solution was then transferred to a 100 mL Teflon-lined stainless steel autoclave and the temperature was maintained at 180 °C for 24 h. The autoclave was then taken out of the oven and left to cool naturally to room temperature. The puce products were isolated via centrifugation, cleaned with four cycles of centrifugation and washing in ethanol and water respectively, and dried at 60 °C overnight. All of the products were calcined at 450 °C for 2 h with a heating rate of 1 °C min−1 to obtain a highly crystalline anatase phase. The bulk TiO2 was directly obtained through hydrolysis: 5 mL of titanium(IV) isopropoxide was added to 95 mL of water, and the mixture was stirred for 10 minutes. The following processes, including isolation and calcination, were the same as those for the TiO2 sphere.2.3 Characterization
X-ray diffraction (XRD) patterns were collected using a Bruker D8 Advance X-ray diffractometer (Cu Kα irradiation, λ = 1.5406 Å), irradiating with a scanning rate of 0.02 deg s−1. A transmission electron microscope (TEM, JEOL-200CX) was used to observe the morphology of the particles. The scanning electron microscope (SEM, FEI-Philips CM-20) images were obtained with an electron microscope coupled to an energy-dispersive X-ray spectrometer (EDS) with an accelerating voltage of 200 kV. The Brunauer–Emmett–Teller (BET) specific surface areas of initially treated samples at 453 K for 8 h were calculated from the nitrogen adsorption–desorption isotherms obtained at 77 K with the Micromeritics ASAP 2020 system. Ultraviolet and visible absorption spectra were recorded at room temperature with a UV-vis (Varian Cary 5000) spectrophotometer. The detection of the CoI transition was conducted in a sealed container adapted to accommodate the recording equipment, and the sample was exposed to light irradiation for 10 min before the test. Photoluminescence (PL) spectra were observed with a spectrophotometer (Hitachi, F-4600), with an excitation wavelength of 400 nm. The electrochemical behavior was tested on a CHI660E workstation (Shanghai Chenhua Instruments, China) using a three-electrode electrochemical cell with a working electrode, a platinum counter electrode and a saturated calomel electrode (SCE) as a reference electrode in a 0.1 M KCl aqueous solution. The working electrode was made of indium-tin oxide (ITO) glass.2.4 Photocatalysis test
All experiments were performed in a Schlenk flask (80 mL) under an atmospheric pressure of CO2 (1 atm).20 In the Schlenk flask, TiO2 (50 mg), bipy (15 mg) and CoCl2 (50 μmol) were added to a mixture of solvent (5 mL) and TEOA (1 mL). The system was subjected to vacuum degassing and backfilling with pure CO2 gas. This process was repeated three times and after the last cycle the flask was back filled with CO2. Then the system was irradiated with five non-focused 6 W UV lights (Hitachi F6T5, 365 nm) with vigorous stirring at 20 °C controlled by a water-cooling system. The produced gases (CO, H2) were detected by a gas chromatograph (Agilent 7890B, Agilent Technologies) equipped with a packed column (TDX-1 mesh 42/10). Ar was used as the carrier gas. The apparent quantum yield (AQY) for CO/H2 generation was measured using the same photochemical experimental setup. The intensity of light irradiation was measured as 2.6 mW cm−2 (CEAulight, AULTT-P4000) and the irradiated area was 1.0 cm2.3 Results and discussion
The high crystallinity and phase purity of the spherical resultant material (sTiO2) were confirmed via XRD (Fig. 1). The diffraction peaks of TiO2 were observed at 2θ = 25.1° (101), 37.6° (004), 47.9° (200), 53.7° (105), 54.5° (211) and 62.7° (204), which agrees with the corresponding crystal planes of anatase TiO2 (JCPDS file no. 21-1272). Additionally, the TiO2 directly obtained via hydrolysis and then calcination at 450 °C that was used as a reference sample (bTiO2) exhibited a pattern corresponding to the anatase phase, which is the same as the spherical sample.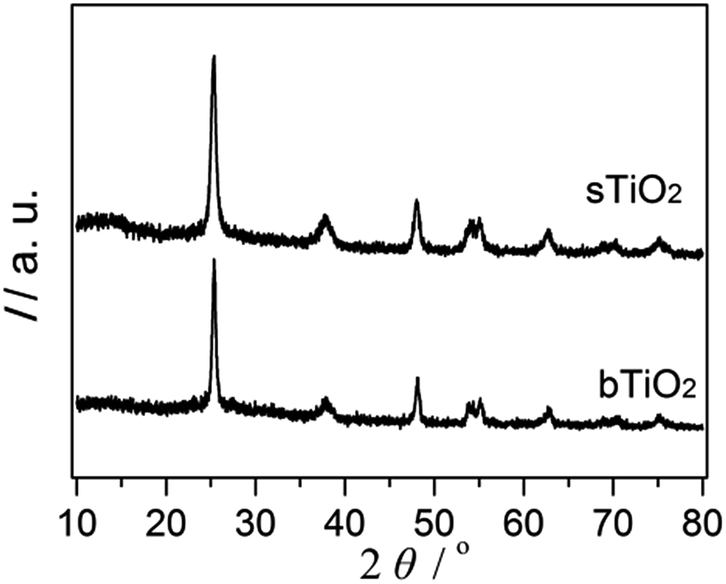 | ||
| Fig. 1 XRD spectra of the as-prepared sTiO2 and bTiO2. | ||
The morphology of the TiO2 sphere was verified via SEM and TEM. As shown in Fig. 2a, the SEM images display that the as-synthesized TiO2 structures are typically spherical in shape. Fig. 2b indicates that the units of the spheres are constructed with nanosheets. These plates with interlaced structure are also revealed in the SEM image. The superstructure was further confirmed via TEM (Fig. 2c), and is in accord with previous reports.19 These results indicate that the three-dimensional structure with pieces supporting each other greatly contributes to the high surface area and the stability of the catalytic material during the photochemical reaction.
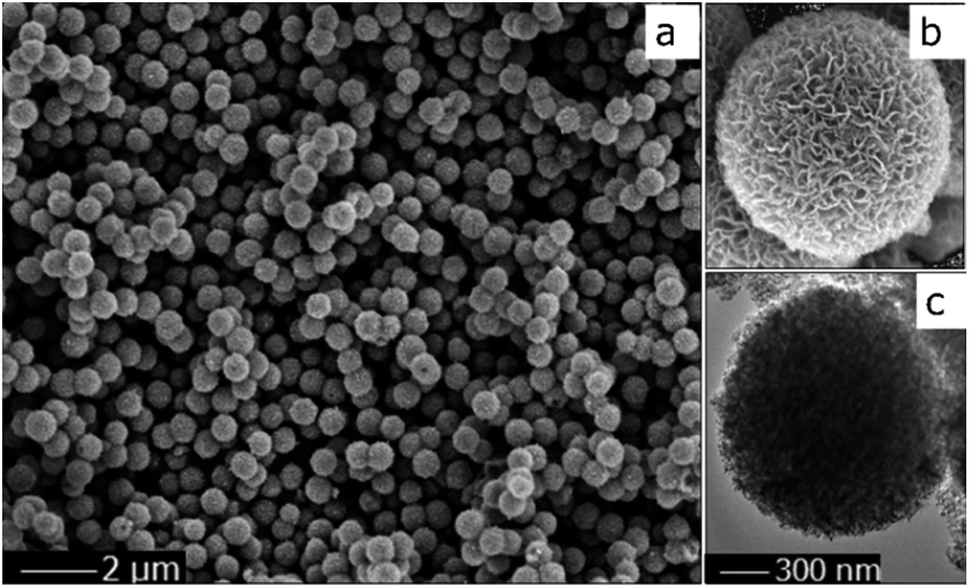 | ||
| Fig. 2 SEM (a and b) and TEM (c) images of the TiO2 spheres. | ||
The self-assembly of the hollow structured TiO2 produces a nanoporous structure, as confirmed by the N2 adsorption/desorption isotherm shown in Fig. 3. It gives a type-IV isotherm with a type H3 hysteresis loop, which is indicative of a mesoporous structure.21 The relatively narrow pore size distribution of sTiO2 (Fig. 3, inset) calculated using the Barrett–Joyner–Halenda (BJH) method from the two branches of the isotherm signifies that most of the pores have sizes in the range 5–15 nm. Such a porous structure gives rise to a surface area of 136 m2 g−1, as calculated via the Brunauer–Emmett–Teller (BET) method. Obviously, as demonstrated in Fig. 3, the surface area of sTiO2 is much higher than that of bTiO2 (ca. 14 m2 g−1).
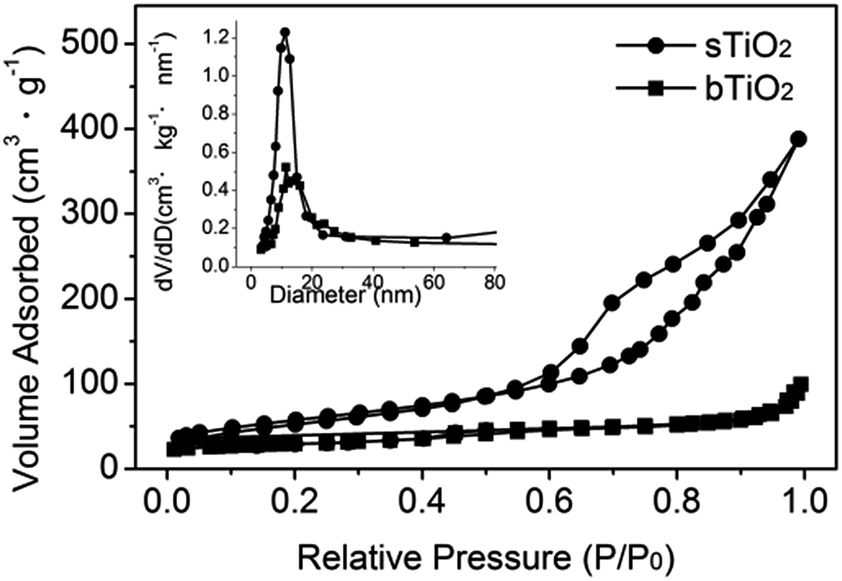 | ||
| Fig. 3 N2 adsorption/desorption isotherm of the as-prepared spherical TiO2 (●) and bulk TiO2 (■). The inset shows the pore size distributions calculated using the BJH method. | ||
As shown in Fig. 4, only a trace amount of CO (<1 μmol) was detected in the current system, and the yield of H2 was also moderate (3 μmol). When Co(bipy)32+ was present as an effective electron transport carrier, as expected, the reaction was promoted dramatically, yielding nearly a 20-fold increase in CO (16.8 μmol) and twice the amount of H2 (6.6 μmol). The apparent overall quantum yield of this CO2 photoreduction system was estimated to be 0.66% under irradiation at λ = 365 nm. The production of CO and H2 on bTiO2 was 9.4 μmol and 3.2 μmol respectively, in spite of this system also containing [Co(bipy)3]Cl2. We have also tested the photocatalytic performance on P25 (Degussa) as a standard reference, which leads to the formation of CO (19.3 μmol) and H2 (8.9 μmol) in the current system. This is mainly attributable to its intrinsic structure being a 70% rutile phase and 30% anatase phase titanium mixture. CO production on sTiO2 is 2.5 μmol less than that on P25. When CO2 was replaced by N2 in the system, evidence was observed that confirmed the participation of CO2 in the reaction, because only H2 gas (3.9 μmol) was detected under these reaction conditions, and no CO was detected. This result confirmed that the carbon source of CO production is CO2, and not other organic ingredients. Additionally, control experiments showed that there was no CO generation in the absence of light or TiO2. In addition, energy-dispersive X-ray spectroscopy (EDS) analysis and elemental mapping are shown in Fig. S1 to S6†, and were used to determine the Co content in bTiO2, sTiO2 and sTiO2 samples after reaction. There was no change detected in the Co content in sTiO2 after the reaction, compared with the samples before the reaction. The results indicate the existence of the Co-complex in this system throughout the studied process.
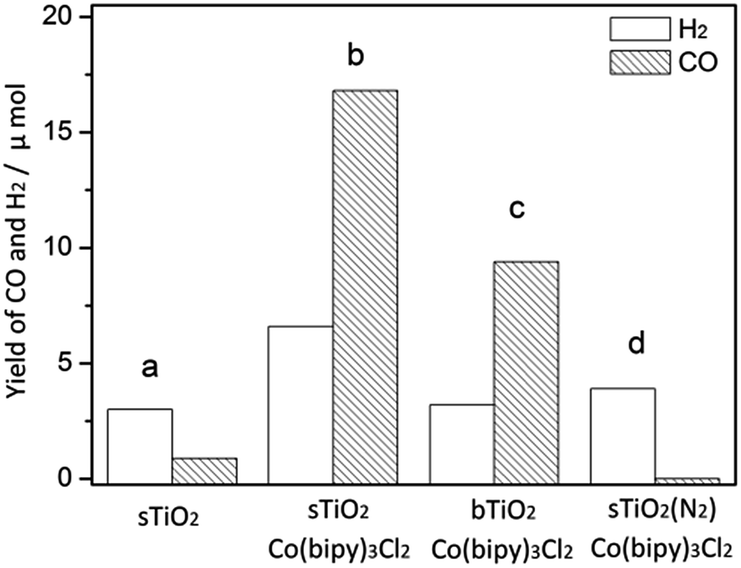 | ||
| Fig. 4 The photocatalytic performance of (a) sTiO2, (b) sTiO2 + Co(bipy)3Cl2, (c) bTiO2 + Co(bipy)3Cl2 and (d) sTiO2 + Co(bipy)3Cl2 under an N2 atmosphere. | ||
To identify the formation of cobalt species, the optical absorption spectra of several solutions including Co2+, [CoII(bipy)3]Cl2, and [CoI(bipy)3]Cl are presented in Fig. 5. The solution containing Co2+ exhibited an absorption band positioned at 550–720 nm (red line), which is due to the d–d transition in CoL4 tetrahedral complexes.22 After the introduction of bipy, a new absorption peak at 400–570 nm was observed in the UV-vis spectrum (blue line), which was in accord with results for Co(bipy)3Cl2 in previous literature.23 These results are due to the formation of a new photo-responsive compound [Co(bipy)3]2+ when Co2+ and bipy were combined. To investigate the effect upon CoII when catalyzed by sTiO2, the reaction solution was measured via UV-Vis absorption spectroscopy after 10 min of light irradiation (green line). The formation of a new absorption band (ca. 450–720 nm) was observed in the UV-Vis absorption spectrum. The broad absorption band is attributed to the intramolecular charge-transfer from the metal center to the pyridinium moiety. This change is mainly due to the generation of a reducing CoI-complex during the photocatalytic CO2 reduction reaction.
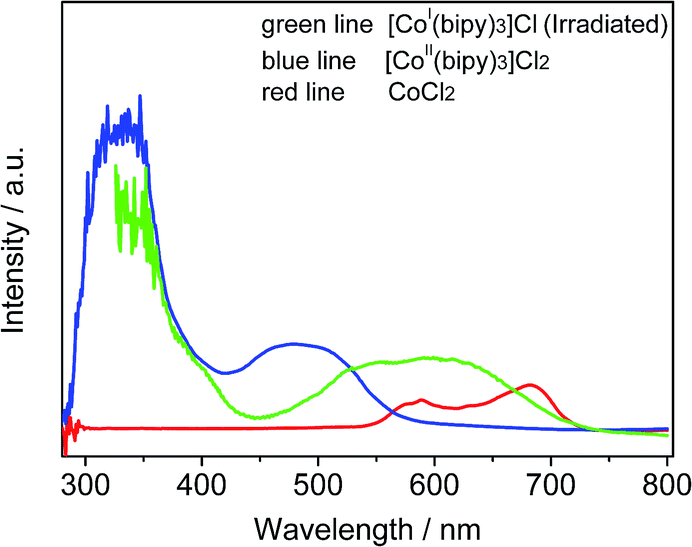 | ||
| Fig. 5 Absorption spectra of acetonitrile solutions in the forms of [CoI(bipy)3]Cl (green line, containing TiO2 spheres), [CoII(bipy)3]Cl2 (blue line) and CoCl2 (red line). | ||
To determine whether the cobalt complex accelerates charge carrier transfer, we used a PL technique to investigate the fluorescence, and the results are shown in Fig. 6. The sTiO2 exhibited a strong PL band that was assigned to the inherent recombination of photogenerated electrons and holes.24 In the presence of [Co(bipy)3]2+, there is a significant decrease in the PL intensity. A weaker intensity of the PL peak represents a lower recombination probability of photo-generated charge carriers. Therefore, the integration of cobalt redox mediators with sTiO2 could effectively inhibit the recombination of photogenerated charge carriers and accelerate electron migration.
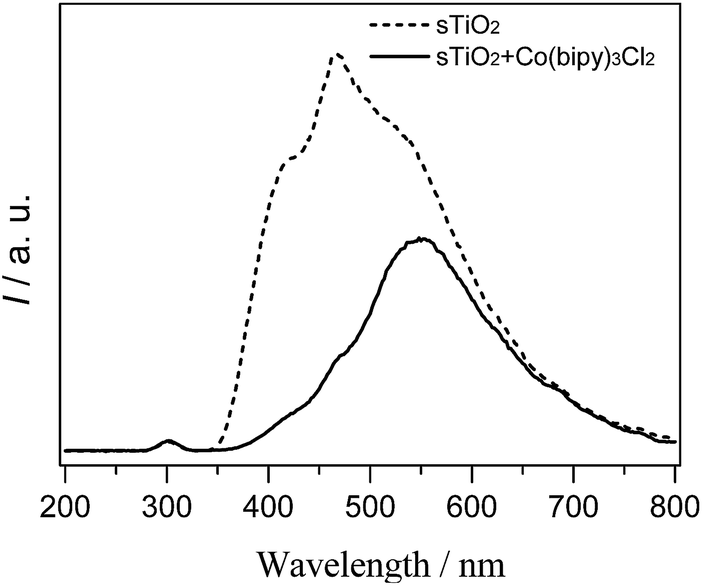 | ||
| Fig. 6 Photoluminescence spectra of semiconductor photocatalysts without (dashed line) and with (solid line) [Co(bipy)3]2+ . | ||
The process of cobalt complex transformation during CO2 reduction can be inspected via cyclic voltammetry (CV). In electrochemical tests, the transition state of the cobalt species can be monitored indirectly, which involves the initial reduction of CoII complexes and their subsequent reaction with CO2 to form Co–CO2 intermediates.25 In the absence of CO2, the obtained redox potential (E0 = −0.8 V) in MeCN solution is attributed to the conversion of the CoII complex to CoI species.26 When CO2 is introduced (Fig. 7), the intensity of the peak observed in N2 is enhanced considerably. The markedly increased current densities reveal that the reaction system undergoes the electrocatalytic CO2 reduction reaction with high efficiency.
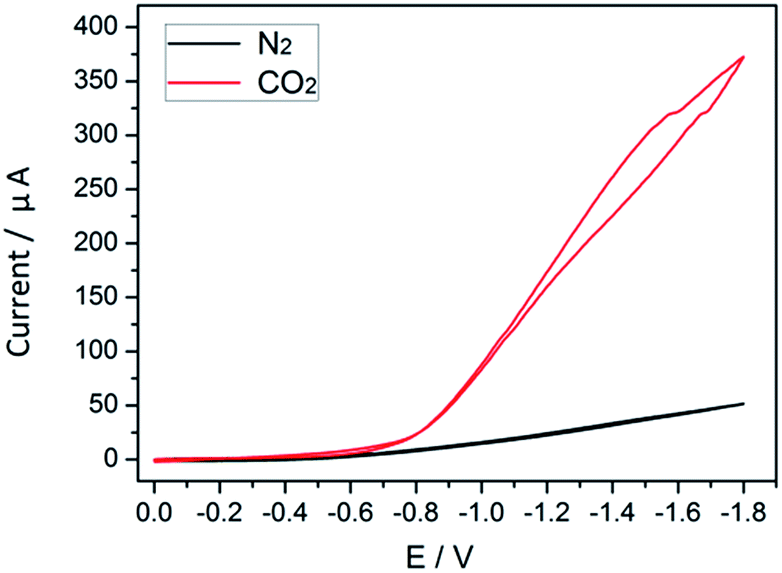 | ||
| Fig. 7 Cyclic voltammograms in N2 (black) and CO2 (red) saturated solutions. | ||
The advantage of hollow spherical TiO2 has been demonstrated in the photocatalytic performance of CO2 reduction (Fig. 4). We suggest that the hierarchical structure plays a important role in the current reaction. As revealed in Fig. 2, the ultrathin nature of the TiO2 nanosheets allows charge carriers to move rapidly onto the surface. In addition, the sufficient separation of the ultrathin TiO2 nanosheets enables photogenerated electrons to quickly transfer from the TiO2 nanosheets to the cobalt complex to enhance utilization of the charge carriers. With respect to the photoresponse, the hollow structure potentially acts as a photon trap well to allow the multiscattering of incident light for the enhancement of light absorption.
Upon irradiation, the excited TiO2 liberates the electrons from the valence band to the conduction band.27 CO2 activation happens in the absence of [Co(bipy)3]2+ through charge transfer from the semiconductor to the CO2 molecule adsorbed on the TiO2 surface. While the presence of massive photogenerated electron–hole pairs is in vain because of the fast recombination, especially in the heterogeneous photocatalytic system, as expected, there is a dominant pathway that is designed for electron transfer towards the CO2 molecule via the cobalt complex.28 After CoII gains an electron, the low valent Co intermediates are stabilized by pyridine ligands through weak coordination,29 promoting the activation of the CO2 molecules via nucleophilic attack. The thus formed CoI complexes are important redox active catalysts.16 In our previous work, evidence of the generation of CoI through light excitation was observed via UV-vis spectroscopy.30 Such intermediates may also result in the protonation of the CoI–H complex when hydron is present, subsequently yielding H2.31 It should be noted that the activity of the CO2 conversion reaction is determined not only by the rate of formation of the photogenerated charge carrier, but also the energy barrier to formation of the relevant CO2 intermediates. The activation energy of molecular CO2 is lowered after the combination of CO2 with cobalt species. Thus, CO2 reduction exhibits superior competitiveness towards H2 generation. As discussed, the following processes are revealed by the electrochemical performance, and are described in eqn (1)–(7).
| TiO2 + hν = e− + h+ | (1) |
In N2-saturated solution
| Co(II) + e− → Co(I) | (2) |
| Co(I) − e− → Co(II) | (3) |
In CO2-saturated solution
| Co(II) + e− → Co(I) | (4) |
| Co(I) + CO2 ↔ CO2Co(I) | (5) |
| CO2Co(I) + H+ + e− → (CO2–)(H–)Co(I) | (6) |
| (CO2)(H)Co(I) + H+ → Co(II) + CO + H2O | (7) |
4 Conclusions
In summary, we have explored improving the efficiency of photocatalytic CO2 reduction with TiO2 hollow spheres as photocatalysts in synergetic combination with [Co(bipy)3]Cl2. This hybrid system exhibits high photocatalytic reactivity towards CO2-to-CO conversion due to the addition of [Co(bipy)3]2+ as a promoter for charge-carrier separation, transfer kinetics, and interface interaction. This result provides insight to the construction of efficient heterogeneous photochemical CO2 reduction systems under mild conditions. This finding has the potential to open up new opportunities in artificial photosynthesis, catalytic chemistry, and energy conversion with excellent morphology catalytic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored by the Natural Science Foundation of Guizhou Province ([2017]1198 & [2017]7089 & [2016]7010) and Construction Projects of Undergraduate Course Teaching of the Guizhou Province Education Department ([2015]337 & [2017]352).Notes and references
- (a) X. Z. Zheng, Y. Yang, S. F. Chen and L. W. Zhang, Chin. J. Catal., 2018, 39, 379–389 CrossRef; (b) T. Gasser, C. Guivarch, K. Tachiiri, C. D. Jones and P. Ciais, Nat. Commun., 2015, 6, 7958 CrossRef PubMed.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018 10.1039/c8ee00097b.
- (a) B. Wang, C. Li, H. Cui, J. Zhang, J. P. Zhai and Q. Li, J. Mater. Sci., 2014, 49, 1336–1344 CrossRef; (b) Z. M. Li, P. Y. Zhang, T. Shao and X. Y. Li, Appl. Catal., B, 2012, 125, 350–357 CrossRef; (c) J. Zhu and X. F. Qian, J. Solid State Chem., 2010, 183, 1632–1639 CrossRef; (d) C. Q. Zhu, B. G. Lu, Q. Su, E. Q. Xie and W. Lan, Nanoscale, 2012, 4, 3060–3064 RSC; (e) G. F. Lin, J. W. Zheng and R. Xu, J. Phys. Chem. C, 2008, 112, 7363–7370 CrossRef; (f) A. Nikokavoura and C. Trapalis, Appl. Surf. Sci., 2017, 391, 149–174 CrossRef; (g) J. M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701–704 CrossRef PubMed.
- (a) M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef; (b) D. R. Fattakhova, A. Zaleska and T. Bein, Chem. Rev., 2014, 114, 9487–9558 CrossRef PubMed.
- (a) L. Pan, S. Wang, J. Xie, L. Wang, X. Zhang and J. J. Zou, Nano Energy, 2016, 28, 296–303 CrossRef; (b) W. K. Wang, D. Xu, B. Cheng, J. G. Yu and C. J. Jiang, J. Mater. Chem. A, 2017, 5, 5020–5029 RSC.
- (a) G. Liu, J. C. Yu, G. Q. Lu and H. M. Cheng, Chem. Commun., 2011, 47, 6763–6783 RSC; (b) M. A. Butler and D. S. Ginley, J. Electrochem. Soc., 1978, 125, 228–232 CrossRef.
- D. Zhang, J. Y. Zhu, N. Zhang, T. Liu, L. M. Chen, X. H. Liu, R. Z. Ma, H. T. Zhang and G. Z. Qiu, Sci. Rep., 2015, 5, 8737 CrossRef PubMed.
- B. Fang, A. Bonakdarpour, K. Reilly, Y. Xing, F. Taghipour and D. P. Wilkinson, ACS Appl. Mater. Interfaces, 2014, 6, 15488–15498 Search PubMed.
- S. B. Wang, L. Pan, J. J. Song, W. B. Mi, J. J. Zou, L. Wang and X. W. Zhang, J. Am. Chem. Soc., 2015, 137, 2975–2983 CrossRef PubMed.
- W. Tu, Z. Tian, J. Gao, Y. Zhou, Q. Liu, X. Y. Chen, H. T. Zhang, J. G. Liu and Z. G. Zou, Adv. Funct. Mater., 2012, 22, 1215–1221 CrossRef.
- M. Wang, Q. T. Han, Y. Zhou, P. Li, W. G. Tu, L. Q. Tang and Z. G. Zou, RSC Adv., 2016, 6, 81510–81516 RSC.
- F. Bi, M. N. Ashiq, M. F. Ehsan, W. Liu and T. He, Chin. J. Chem., 2015, 33, 112–118 CrossRef.
- S. Bai, W. J. Yin, L. L. Wang, Z. Q. Li and Y. J. Xiong, RSC Adv., 2016, 6, 57446–57463 RSC.
- (a) Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441–5449 CrossRef PubMed; (b) A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef PubMed; (c) R. Abe, T. Takata, H. Sugihara and K. Domen, Chem. Commun., 2005, 3829–3831 RSC.
- M. M. T. Khan, D. Chatterjee and J. Bhatt, J. Chem. Sci., 1992, 104, 747–752 Search PubMed.
- Z. G. Chai, Q. Li and D. S. Xu, RSC Adv., 2014, 4, 44991–44995 RSC.
- J. L. Lin, Y. D. Hou, Y. Zheng and X. C. Wang, Chem.–Asian J., 2014, 9, 2468–2474 CrossRef PubMed.
- R. Kuriki, H. Kumagai, K. Maeda and O. Ishitani, Hyomen Kagaku, 2017, 38, 291–296 CrossRef.
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah and D. Luan, J. Am. Chem. Soc., 2010, 132, 6124–6130 CrossRef PubMed.
- J. L. Lin, Z. X. Ding, Y. D. Hou and X. C. Wang, Sci. Rep., 2013, 3, 1056 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouqerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef.
- K. Sivaraj and K. P. Elango, J. Solution Chem., 2010, 39, 1681–1697 CrossRef.
- H. A. Schwarz, C. Creutz and N. Sutin, Inorg. Chem., 1985, 24, 433–439 CrossRef.
- (a) R. R. Prabhu and M. A. Khadar, J. Phys. D: Appl. Phys., 2005, 65, 801–807 Search PubMed; (b) J. Zhou, M. Takeuchi, A. K. Ray, M. Anpo and X. S. Zhao, J. Colloid Interface Sci., 2007, 311, 497–501 CrossRef PubMed; (c) X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef PubMed.
- J. D. Froehlich and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 3565–3573 CrossRef PubMed.
- H. Fillon, E. L. Gall, C. Gosmini and J. Périchon, Tetrahedron Lett., 2002, 43, 5941–5944 CrossRef.
- F. Wenand and C. Li, Acc. Chem. Res., 2013, 46, 2355–2364 CrossRef PubMed.
- A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef.
- Y. G. Budnikova, A. Kafiyatullina, Y. M. Kargin and O. Sinyashin, Russ. Chem. Bull., 2003, 52, 1504–1511 CrossRef.
- J. L. Lin, B. Qin and G. L. Zhao, J. Photochem. Photobiol., A, 2018, 354, 181–186 CrossRef.
- S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127–15131 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of element analysis are listed. See DOI: 10.1039/c8ra03211d |
| This journal is © The Royal Society of Chemistry 2018 |