
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of Helicobacter pylori in dental plaque using a DNA biosensor for noninvasive diagnosis†
Li-Li Chena,
Hui-Fang Cui *a,
Shuang-Fei Fana,
Zong-Yi Lia,
Shuang-Yin Hanb,
Xin Mac,
Shu-Wen Luoc,
Xiaojie Songa and
Qi-Yan Lva
*a,
Shuang-Fei Fana,
Zong-Yi Lia,
Shuang-Yin Hanb,
Xin Mac,
Shu-Wen Luoc,
Xiaojie Songa and
Qi-Yan Lva
aDepartment of Bioengineering, School of Life Sciences, Zhengzhou University, Zhengzhou, 450001, P. R. China. E-mail: hfcui@zzu.edu.cn; Fax: +86-371-67783235; Tel: +86-371-67781325
bDivision of Gastroenterology, Henan Provincial People's Hospital, #7 Weiwu Road, Zhengzhou, 450000, P. R. China
cDivision of Stomatology, Henan Provincial People's Hospital, #7 Weiwu Road, Zhengzhou, 450000, P. R. China
First published on 8th June 2018
Abstract
Noninvasive diagnosis of Helicobacter pylori (H. pylori) infection is very attractive. This study investigated the single strand DNA (ssDNA) acquisition method from H. pylori in dental plaque, and the integration of our previously developed 43-mer H. pylori DNA biosensor with the obtained target ssDNA (tDNA). Dental plaque samples were collected from 34 patients/volunteers, whose gastric H. pylori infection statuses were tested with the 13C urea breath test (UBT). The samples were treated with colony polymerase chain reaction (PCR) to obtain double strand DNA (dsDNA) of 104 basepairs (bp) long. A blocker ssDNA was designed and used in thermal treatment of the dsDNA to release the 104-mer tDNA, which contains the 43-mer DNA sequence in the middle. PCR primers were designed, and the tDNA releasing and detection conditions with the biosensor were optimized. The limit of detection with the biosensor was 12 fM dsDNA. The dental plaque detection results correlated quite well with the UBT results, with a sensitivity of 100%, and specificity of 97%. These results indicate that the residence of H. pylori in dental plaque is highly associated with gastric H. pylori infection, and detection of dental plaque samples with our DNA biosensor is promisingly applicable in noninvasive diagnosis of H. pylori infection.
1. Introduction
Helicobacter pylori (H. pylori) is a crucial etiological agent in the pathogenesis of gastroduodenal diseases including peptic ulcers, mucosa-associated lymphoid tissue lymphoma, and gastric adenocarcinoma.1,2 About 15% of the global population has been infected by H. pylori.3 Reliable and fast diagnosis of H. pylori infection before and after eradication therapy is of great clinical importance for monitoring and management of gastroduodenal diseases. The diagnostic methods for H. pylori infection are generally grouped as “invasive”, requiring endoscopy to sample gastric tissue or mucus, or “noninvasive”, requiring only blood, breath, urine, saliva, or stool samples.4,5 As the invasive methods can cause pain and psychological burden to patients, the noninvasive methods are the tests of choice for many patients. Among the noninvasive methods developed, 13C-urea breath test (UBT) is one of the most reliable tests for diagnosing H. pylori infection, providing accuracy for both the initial diagnosis and the confirmation of eradication.6,7 However, although UBT is safe, noninvasive, and usable for children older than 6 years old, it has shown heterogeneous accuracy in the pediatric population, especially in young children.8 In addition, for some specific clinic settings, such as peptic ulcer bleeding, or partial gastrectomy patients, UBT showed relatively low sensitivity and specificity.9 In contrast, the noninvasive method of serology (i.e. antibody-based test), although highly sensitive and specific, fails to distinguish between past and current H. pylori infection.10 Improvement of the current diagnosis approaches and development of novel diagnosis methods are necessary, especially for specific age groups and clinical conditions.Sequence-specific DNA detection has been of great importance for clinical diagnostics, molecular biology, agriculture, forensic science, and pathogen detection.11,12 Polymerase chain reaction (PCR), with primer-mediated enzymatic amplification of DNA segments, has presented to be a highly sensitive, fast, and accurate method for H. pylori detection, especially for detection of antibiotic resistance, virulence determinants, and bacterial quantification.13–18 Nested PCR19 and real-time PCR20 have been demonstrated to be sensitive and specific in H. pylori detection in dental plaque samples, and the results have a good association with gastric H. pylori infection. In contrast, single-step PCR did not detect H. pylori as sensitive as nested PCR.19 It could not detect H. pylori in dental plaque samples.19 However, nested PCR and real-time PCR method suffers from the disadvantages of complex and expensive.21 Furthermore, nonspecific amplification is a major drawback of PCR with high-number cycles.22
The efforts in recent years have been focused on the development of DNA biosensors or other DNA sensing techniques possessing the detection limits of ≤femtomolar (fM), to meet the requirement of DNA detection in very minute physiological and other biological samples.23–27 Among different sensitivity enhancing strategies in DNA biosensors, nanostructure-based labelling method combined with sandwich DNA sensing has emerged as an effective way to approach the PCR detection sensitivity.25–29 Au nanoparticles (AuNPs) have become one of the most-employed labelling nanomaterials for fabricating sensitive DNA biosensors, thanks to their large surface to volume ratio, excellent biocompatibility, terrific electrocatalytic activity, dramatic electrical conductivity, and excellent chemical and physical stability.25,29–32 In contrast, electrochemical DNA biosensors, with the advantages of simple and fast, sensitive, accurate, specific, free of interference from color, and cheap instrumentation, have emerged as a promisingly practical strategy for DNA detection in physiological/biological samples. We recently reported a novel and sensitive electrochemical sandwich DNA biosensing approach using hairpin DNA (hpDNA) as a novel biobarcode modified on AuNPs, and using [Ru(NH3)5L]2+, where L is referred to 3-(2-phenanthren-9-ylvinyl)-pyridine, as an indicator of DNA hybridization.29 Electroactive indicators of DNA hybridization can interact with dsDNA through intercalation, e.g. the [Ru(NH3)5L]2+ and [Os(bpy)2(phe-dione)]3+/2+ (bpy = 2,2′-bipyridyl),33 or through groove binding, e.g. Oracet Blue.34 [Ru(NH3)5L]2+, has been demonstrated being an electro-active intercalator of double strand DNA (dsDNA).35,36 Under high ionic strength, the [Ru(NH3)5L]2+ complex binds with DNA mainly through intercalation mode, thus minimizing background noise and interferences.29 The novel biosensor was demonstrated being able to detect a 43-mer single strand DNA (ssDNA) specific in the UreB gene sequence of H. pylori, with a detection limit down to 1 fM.29 Although big efforts have been focused on developing various DNA biosensors, seldom was found on H. pylori detection in physiological samples, in which the DNA sequence is in double stranded form in the H. pylori genome, and is in the trace concentration of fM or even attomolar (aM) levels.
To develop a noninvasive H. pylori diagnostic method, this study investigated a short ssDNA acquisition method from H. pylori in dental plaque, and the integration of our 43-mer H. pylori DNA biosensor with the obtained target ssDNA (tDNA). Firstly, a dsDNA sequence of 104 basepairs (bp) containing the 43 bp oligonucleotide sequence specific in the UreB gene of H. pylori was obtained by colony PCR with the assistance of two primers of 19–20 nucleotides (nt) (Table 1), which were specially designed based on the UreB gene of H. pylori. The tDNA of 104 nt containing the 43 nt target sequence (the underlined and the italic sequence in tDNA in Table 1) for DNA biosensor detection was then released by heat denaturation of the dsDNA in the presence of a blocker ssDNA. The blocker is complementary to a small portion of tDNAcm, an ssDNA completely complementary to tDNA (Table 1 and Scheme 1). The released tDNA was then captured and detected by our DNA biosensor. The biosensor method was evaluated by comparing the detection results with those from UBT. This study could not only provide a novel and noninvasive method for diagnosis of H. pylori infection, but also provide information on association of dental residence of H. pylori with gastric H. pylori infection.
| Oligonucleotides | Sequences (from left to right: 5′ to 3′) |
|---|---|
| a The cpDNA sequence is complementary to the 3′ underlined segment in tDNA.b The underlined sequences in tDNA are the portions that are complementary to the capture DNA or the reporter DNA.c The underlined italic sequence in tDNAcm is the portion that is complementary to the blocker.d The underlined sequence in rpDNA is complementary to the 5′ underlined segment in tDNA.e The two underlined segments in hpDNA are complementary with each other. | |
| Primer F | CCT AGC AAA GAA GAA CTG G |
| Primer R | CCA GCG ATT TTG CCA TCT TT |
| Capture probe (cpDNA)a |  |
| Blocker | CGT GGA TTA CAC CGG |
| Target DNA (tDNA)b | 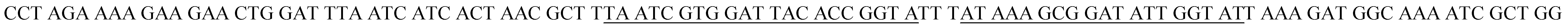 |
| Complementary DNA of tDNA (tDNAcm)c |  |
| Reporter probe (rpDNA)d | 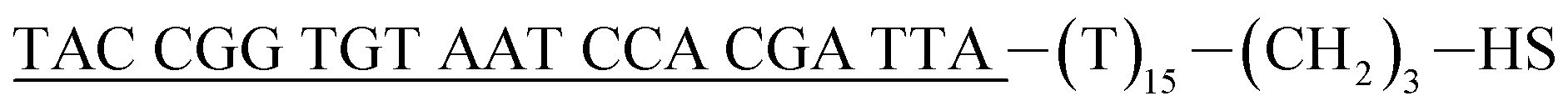 |
| Hairpin DNA (hpDNA)e | 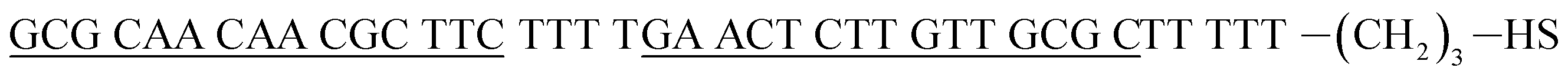 |
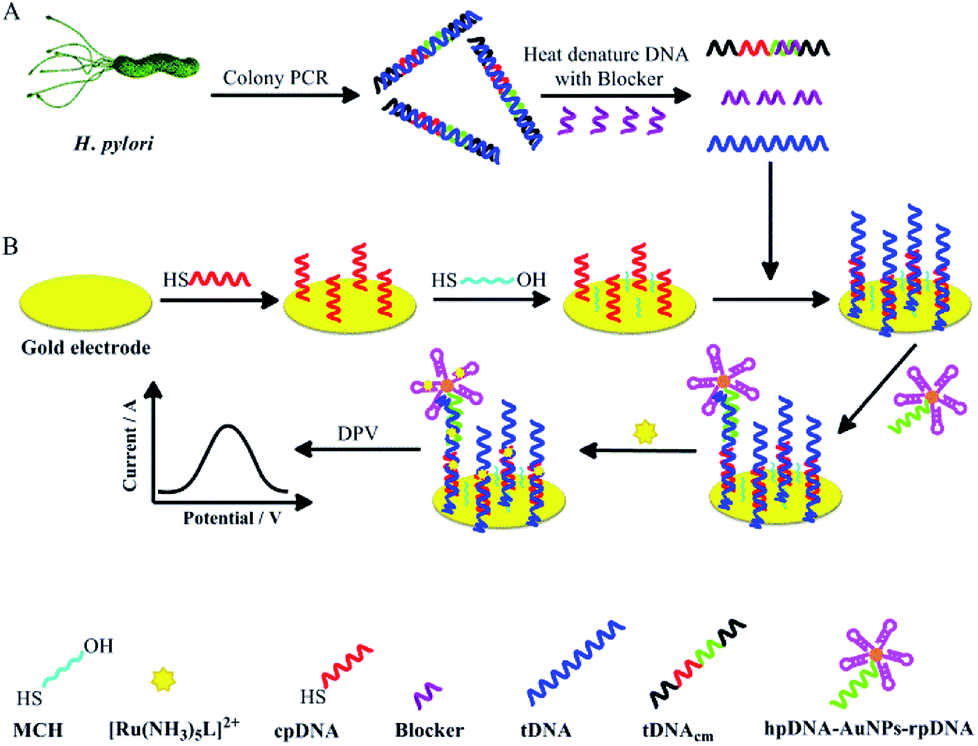 | ||
| Scheme 1 Schematic illustrations of (A) the amplification and acquirement of target dsDNA from H. pylori and releasing of the single strand tDNA from the dsDNA, and (B) the construction of the electrochemical sandwich DNA biosensor and the tDNA detection with the biosensor. | ||
2. Experimental
2.1. Materials and chemicals
Hydrogen tetrachloroaurate (III) hydrate, 9-bromophenanthrene, 4-vinylpyridine, palladium acetate, palladium triphenylphosphine, silver trifluoroacetate, ammonium hexafluorophosphate, tris-(2-carboxyethyl)phosphine hydrochloride (TCEP), N,N,N′,N′-tetramethylethylenediamine, hexaamineruthenium(III) chloride and N,N-dimethylformamide (DMF) were purchased from Alfa Aesar (MA, U.S.A.). KNO3 (99.99%) was obtained from Aladdin Industrial (Shanghai, China). H. pylori strain (11637WT) was generally provided by The 5th Affiliated Hospital of Zhengzhou University. TransTaq-T DNA Polymerase, deoxynucleotide solution mixture (dNTPs), 10× Trans Taq-T buffer, and 6× loading buffer were obtained from TransGen Biotech (Beijing, China). DNA ladder markers (20–500 bp) were purchased from TaKaRa Bio Inc (Shiga Japan). GoldView I was purchased from Solarbio Bioscience & Technology (Beijing, China). All other chemicals were of analytical grade and obtained from Sinopharm Chemical Reagent (Shanghai, China). H. pylori strain (11637WT) was generally provided by The 5th Affiliated Hospital of Zhengzhou University.Tris–HCl buffer (10 mM, pH: 7.0) was prepared from tris(hydroxymethyl)aminomethane and 0.1 M HCl. Annealing buffer (10×, pH: 7.4) contains 100 mM Tris–HCl, 10 mM ethylenediaminetetraacetic acid (EDTA), and 1 M NaCl. Phosphate buffer solution (PBS, 10 mM, pH: 7.4) consists of 1.9 mM NaH2PO4 and 8.1 mM Na2HPO4. Assay buffer is the 10 mM PBS containing 0.15 M NaCl and 0.1% sodium dodecyl sulfate (SDS). TE buffer (pH 8.0) consists of 10 mM Tris–HCl and 1 mM EDTA. The PBS/KNO3 buffer is the 10 mM PBS with 100 mM KNO3. Immobilization buffer is the 10 mM PBS containing 1.0 mM EDTA and 0.6 M NaCl. The 6-mercapto-1-hexanol (MCH) solution (1 mM) was prepared by diluting the stock solution (100 mM in ethanol) with the immobilization buffer. Deionized water obtained from a Millipore water system was used throughout the experiment. Oligonucleotide sequences were custom-made by Invitrogen Biotech (Shanghai, China) and listed in Table 1.
2.2. Instrumentation
Electrochemical measurements were carried out using an electrochemical analyzer (CHI660E, Shanghai Chenhua Instrument, China) consisting of a three-electrode system: a working gold electrode, a platinum counter electrode, and a 3 M KCl–Ag/AgCl reference electrode, at room temperature (∼25 °C). The gold and the platinum electrodes are disk electrodes with a diameter of 2 mm. The agarose gel electrophoresis was realized by using an electrophoresis apparatus (DYY-6D, Beijing Liuyi Instrument Factory, China), and the gels were imaged on a G:BOX Chemi XT4 imaging system (Syngene, UK). UV-vis spectroscopy was performed using a Shimadzu UV-2600 UV-vis spectrophotometer (Shimadzu Scientific Instrument, Japan). The PCR amplification was realized with Professional Standard Gradient 96 (Analytik Jena, Germany).2.3. Dental plaque sample collection
Dental plaque samples were collected from 34 patients/volunteers (14 men, 20 women; from 23 to 71 years old) by dentists complying with strict sterilization conditions in Henan Provincial People's Hospital. All the patients/volunteers did UBT measurement. The sample collection was approved by the Life Science Ethics Committee of Zhengzhou University and the Medial Science Ethics Committee of Henan Provincial People's Hospital. According to the UBT results, 28 patients/volunteers were H. pylori positive and the rest were H. pylori negative. There was no any treatment such as antibiotics, proton pump inhibitors, H2 blockers, antacids, or bismuth compounds to the people in the last three months to avoid false negative results. The dental plaque samples were collected from molars, and then immediately put into 0.5 mL physiological saline solution in Eppendorf tubes. Afterwards, 0.5 mL physiological saline solution and glycerol mixture (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, volume ratio) was added immediately to the Eppendorf tube. The dental plaque samples were stored at −80 °C until use.
:2, volume ratio) was added immediately to the Eppendorf tube. The dental plaque samples were stored at −80 °C until use.
2.4. H. pylori cultivation
The H. pylori strain (11637WT) was cultured on Brucella broth agar medium supplemented with 10% goat blood. The culture plates were incubated under a microaerophilic atmosphere (85% N2, 5% O2, 10% CO2) at 37 °C for 72–96 h. The H. pylori colonies on the plates were collected, and then suspended in sterile water, ready for the following PCR amplification. For verification of the PCR specificity, Escherichia coli (E. coli) DH5α as a negative control was cultured in Luria–Bertani broth at 37 °C with vigorous shaking, followed by the same treatment procedures with the H. pylori suspension.2.5. H. pylori dsDNA fragment acquisition and verification
To detect microbe with DNA biosensors or other DNA sensing techniques, a short DNA fragment must be obtained from the microbe genome first. In order to obtain the H. pylori dsDNA fragment, 330 μL dental plaque suspension (containing about 2 mg sample) or 100 μL H. pylori culture suspension (2 × 104 CFU mL−1) was centrifuged at 8000 rpm for 10 min first. The E. coli culture was processed in the same way with the H. pylori culture for comparison. The precipitates were suspended in 100 μL sterile water followed by incubation in boiled water bath for 15 min. After cooled to room temperature, the suspensions were centrifuged again, resulting in the supernatants as the colony PCR templates. The PCR reaction solution was prepared on ice by mixing 8 μL of the colony PCR template solution, 28.5 μL sterile water, 5.0 μL 10× Trans Taq-T Buffer, 4.0 μL dNTP (2.5 mM for each kind of nucleotide), 2.0 μL 10 μM primer F, 2.0 μL 10 μM primer R, and 0.5 μL 5 U mL−1 TransTaq-T DNA polymerase. The PCR was realized through initial denaturation at 95 °C for 2 min; 10 or 25 cycles of 95 °C for 30 s, 53 °C for 30 s, 72 °C for 30 s; and an additional extension step at 72 °C for 10 min. The H. pylori and the E. coli culture were subject to 25 cycles of PCR for verification of the PCR product. In contrast, the dental plaque supernatant was subject to 10 PCR cycles for detection of the obtained tDNA with the DNA biosensor. The PCR products from the microbe cultures were analyzed by using agarose gel electrophoresis under 120 V for 1 h. The gels after electrophoresis were stained with GoldView I.2.6. Releasing tDNA from H. pylori dsDNA fragment
Almost all the reported DNA biosensors have been developed for detecting ssDNA. The target DNA of our DNA biosensor27 was a 43-mer ssDNA specific in the UreB gene of H. pylori. Releasing the single strand tDNA from the dsDNA obtained in the colony PCR is prerequisite for detecting the physiological samples with the biosensor. To release the tDNA, 100 μL PCR products or standard target dsDNA samples of various concentrations (in TE buffer) were firstly mixed with 100 μL 10−6 M blocker, and the mixture was then diluted to 1 mL with the immobilization buffer. The standard target dsDNA was a duplex form of tDNA and tDNAcm with concentrations calibrated with UV-vis technique. The tDNA sequence was released by thermal denaturation at 95 °C for 10 min, and then renaturation at 30 °C for 15 min. In the renaturation process, the blocker with a shorter sequence and in a higher concentration was expected to hybridize more rapidly than tDNA with tDNAcm, limiting the rehybridization between tDNA and tDNAcm, thus releasing free tDNA strand.2.7. Fabrication of the DNA biosensor and detection of H. pylori
The DNA biosensor in detection of H. pylori dsDNA was fabricated similarly with our biosensor29 in ssDNA detection (Scheme 1). Briefly, capture DNA (denoted as cpDNA) (Table 1) was firstly immobilized on cleaned gold electrode from TCEP treated cpDNA solution (100 nM in immobilization buffer) by applying a constant potential of +0.4 V at the electrode for 500 s. The electrode was then passivated with MCH (1 mM) for 1 h. The single strand tDNA released from the H. pylori PCR products or standard dsDNA samples was captured on the electrode by hybridization with the immobilized cpDNA at 40 °C under gentle shaking for 3 h. The electrode was then washed with the assay buffer and the immobilization buffer successively. It should be noted here that an additional washing step with the assay buffer was introduced compared with our ssDNA biosensor fabrication processes, to remove the nonspecific adsorbed DNA. Afterwards, 200 μL hpDNA-AuNPs-rpDNA nanoparticle solution was dropped onto the electrode surface. The term ‘rpDNA’ is referred to reporter DNA (Table 1), which is a DNA sequence complementary to the 5′ underlined segment in tDNA. The electrode was then kept at 47 °C under gentle shaking for 3 h for rpDNA/tDNA hybridization. The as-fabricated electrode was washed and submerged in the immobilization buffer, ready for [Ru(NH3)5L]2+ intercalation and signal detection. The electro-oxidation current signal from the intercalated [Ru(NH3)5L]2+ was recorded with differential pulse voltammetry (DPV) technique (from −0.6 V to +0.1 V; amplitude, 0.05 V; pulse width, 0.01 s; pulse period, 0.02 s) in the PBS/KNO3 buffer.3. Results and discussion
3.1. H. pylori dsDNA acquisition
The colony PCR products from the H. pylori culture were tested on agarose gel electrophoresis to evaluate the selectivity of our designed PCR primers and the PCR process (Fig. 1). Before the PCR process, the H. pylori culture was verified through morphological observation, and various biochemistry tests (Fig. S1, ESI†). Fig. 1A and B shows the electrophoresis analytical results of the two PCR products from the H. pylori culture, and the E. coli culture, respectively. For the H. pylori culture, a band at ∼100 bp can be obviously observed. In contrast, the PCR products from the E. coli culture did not exhibit obvious bands. These results indicate that the primers and the PCR process can specifically amplify the designated 104 bp product from H. pylori.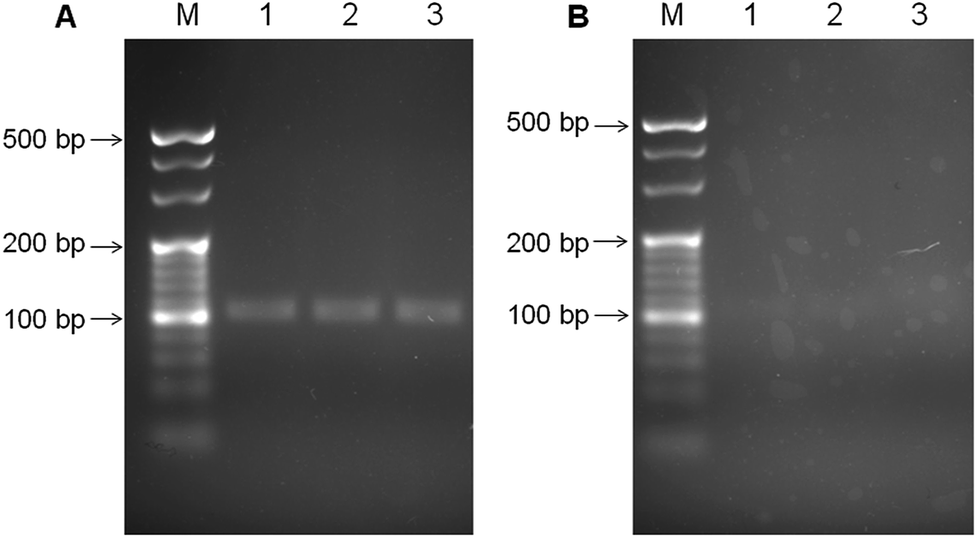 | ||
| Fig. 1 The agarose (3.5%) gel electrophoresis results of the colony PCR products from (A) the H. pylori culture, and (B) the E. coli culture. M is denoted for the DNA marker. Lane 1, lane 2 and lane 3 were the repetitive loadings from the PCR products. The gels were stained with GoldView I for observation. | ||
3.2. Releasing single strand tDNA and detecting the dsDNA with the biosensor
It is challenging to keep the denatured DNA duplex strands apart long time enough for hybridization with the cpDNA.37 In this study, the 15 nt blocker designed to target a small portion of the tDNAcm sequence was added into the tDNA/tDNAcm solution in the denaturation/renaturation process to hybridize with the tDNAcm, for limiting the rehybridization of tDNAcm with tDNA, and thus releasing the tDNA sequence. To testify the feasibility of the tDNA releasing and detection strategies with our biosensor, the standard target dsDNA (104 bp) of 1 pM was detected with the biosensor first by using the DPV measurement. Compared to linear sweep voltammetry, DPV has better sensitivity and the electrode reactions can be analyzed more precisely, because in DPV measurements, the effect of the charging current can be minimized, and only faradaic current is extracted. Fig. 2 illustrates the DPV detection curves for the 1 pM dsDNA treated with 0.1 μM blocker (curve a), the dsDNA treated without the presence of blocker (curve b), and the blocker only (curve c). With the presence of the dsDNA and blocker, a very strong oxidation peak with a peak current (Ip) of ∼0.37 μA, coming from the intercalated [Ru(NH3)5L]2+ signal molecules appeared (curve a). In contrast, the control groups only exhibited very weak oxidation peaks (Ip: 0.087 μA and 0.025 μA, respectively for curve b and c), significantly smaller than that of the ‘dsDNA + blocker’ group (inset in Fig. 2). This result indicates that introduction of the blocker can drastically release the tDNA, and the tDNA can be captured and detected efficiently with the biosensor. During the renaturation process in the dsDNA treatment, the short blocker DNA at a much high concentration (blocker:dsDNA = 105:1, molar ratio) could rapidly hybridize with the tDNAcm sequence produced in the preceding denaturation step. The hybridization speed between blocker and tDNAcm is very fast, surpassing that between tDNA and tDNAcm, thus releasing the single strand tDNA. It should be noted that the tDNA sequence is 104 nt, longer than the target DNA sequence in our reported DNA biosensor (43 nt) (the underlined sequence and the italic sequence in tDNA in Table 1). The biosensor detection results indicate that the 104 nt tDNA can hybridize with the cpDNA and the rpDNA for fabrication of the biosensor.
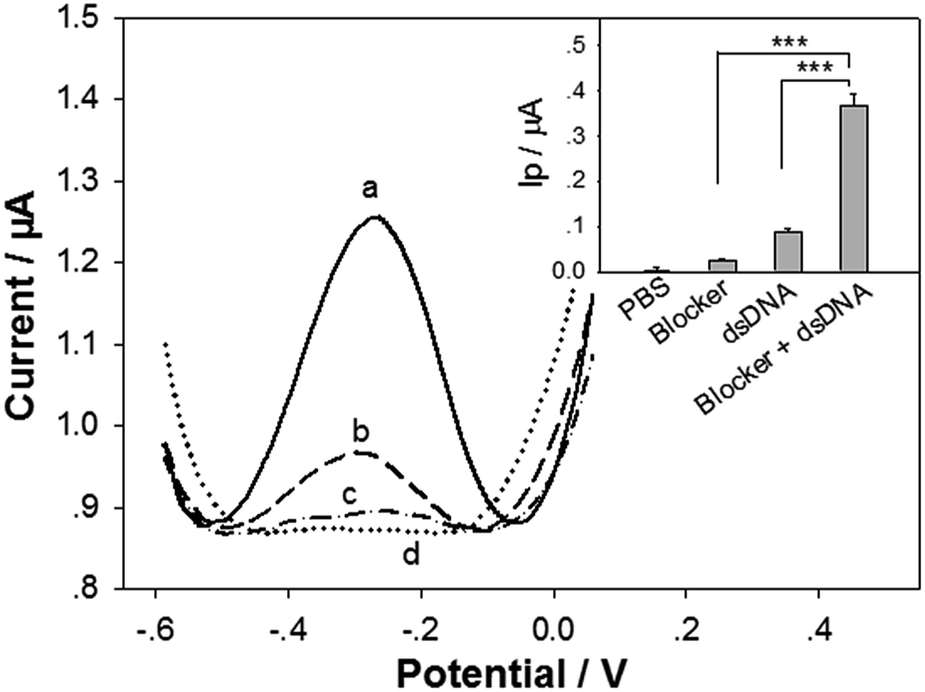 | ||
| Fig. 2 DPV curves of the DNA biosensors in detection of (a, dash line) 1 pM target dsDNA treated with 100 nM blocker, (b, dash line) 1 pM target dsDNA treated without the presence of blocker, (c, dash-dot line) 100 nM blocker, and (d, dotted line) blank PBS. Inset: the statistic peak current (Ip) of the corresponding DPV curves (n = 3 repetitive biosensor fabrications and detections). *** represents that the p value in the t test is less than 0.001. | ||
3.3. Optimization of the tDNA releasing conditions
Experimental conditions for releasing the tDNA from the target dsDNA were then optimized. Firstly the blocker concentration was varied, when other conditions were kept at constant. The DPV Ip values of the biosensor in detection of the standard dsDNA (Ip1) and the blank samples (Ip0) are shown in Fig. 3A. The Ip value increment (ΔIp = Ip1 − Ip0) for 1 pM target dsDNA is shown in the inset in Fig. 3A. The Ip1 value increased monotonically with the blocker concentration from 0.001 μM to 10 μM. In contrast, the ΔIp value increased obviously with the blocker concentration from 0.001 μM to 0.1 μM, but reached a plateau at 0.1 μM. The blocker of higher concentration could bind more rapidly with tDNAcm, resulting in higher releasing efficiency for the single strand tDNA. However, too much blocker could also result in more nonspecific DNA adsorption onto the electrode surface, leading to a high background noise. The blocker concentration was therefore selected as 0.1 μM.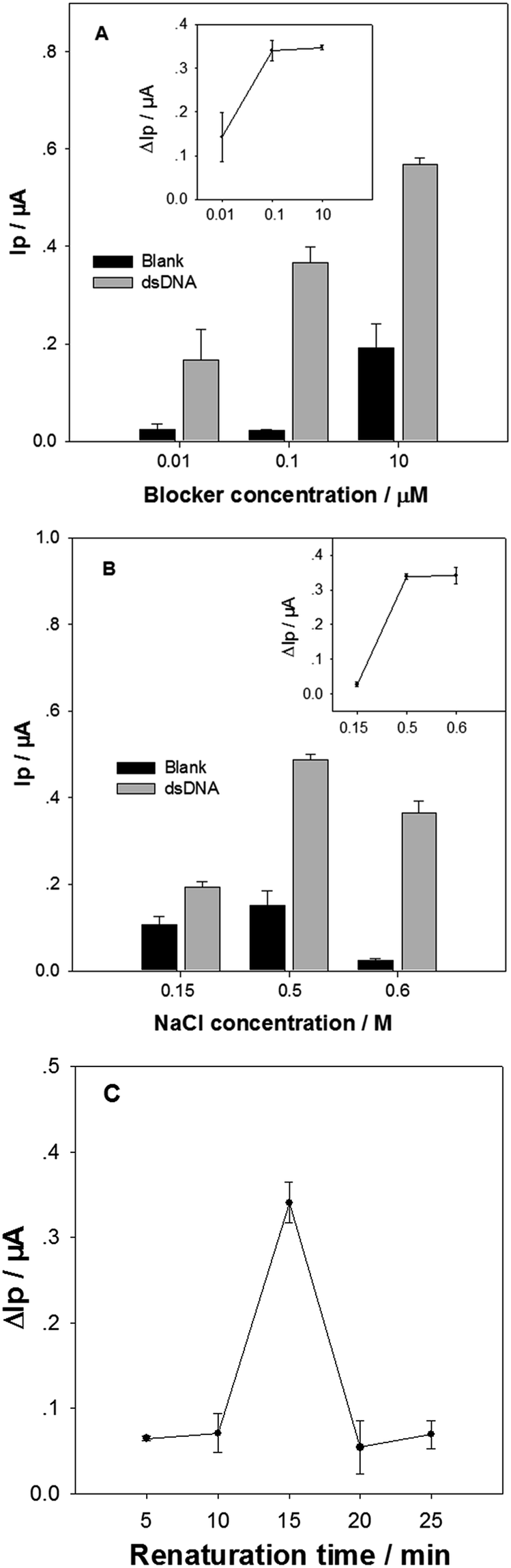 | ||
| Fig. 3 The DPV Ip values of the biosensor in detection of 1 pM target dsDNA, and blank samples, treated and captured at various conditions. (A) The blocker concentration was varied in the immobilization buffer containing 0.6 M NaCl, and the renaturation was at 30 °C for 15 min. (B) The NaCl concentration was varied in the immobilization buffer containing 0.1 μM blocker, and the renaturation was at 30 °C for 15 min. (C) The renaturation time at 30 °C was varied, and the immobilization buffer contained 0.1 μM blocker and 0.6 M NaCl. Insets: the corresponding ΔIp values. Error bars represent the standard deviation for 3 repetitive biosensor fabrications and detections. | ||
The NaCl concentration in the immobilization buffer in the dsDNA sample treatment was then optimized (Fig. 3B). When the NaCl concentration was increased from 0.15 M to 0.5 M, the Ip1 value in response to the 1 pM target dsDNA increased obviously, accompanied with a slight increase of background noise (i.e. Ip0). In contrast, increasing the NaCl concentration further from 0.5 M to 0.6 M resulted in an obvious Ip1 value decrease. However, it is very good to be noticed that the background noise became very small at 0.6 M NaCl. As a consequence, the ΔIp value at 0.6 M NaCl was similar with that at 0.5 M NaCl (inset in Fig. 3B). It is well known that NaCl concentration greatly affects the hybridization efficiency and speed between DNA strands: higher NaCl concentration, easier hybridization. In addition, the nonspecific adsorption of DNA and [Ru(NH3)5L]2+ molecules on electrode surface are also affected by ionic strength and cationic ion concentration: higher ionic strength and cationic ion concentration, less nonspecific adsorption. In the tDNA releasing and capturing processes, the hybridizations of the three DNA pairs (tDNA/tDNAcm, tDNAcm/blocker, and cpDNA/tDNA) would compete with each other. The results suggest that the tDNA amount specifically captured by the cpDNA at 0.6 M NaCl may be similar to that at 0.5 M NaCl, and the nonspecific adsorption of blocker and [Ru(NH3)5L]2+ on the electrode surface is the smallest at 0.6 M NaCl. In the following experiments, 0.6 M NaCl in the immobilization solution was used.
Finally, the renaturation time at 30 °C in the tDNA releasing treatment was optimized (Fig. 3C). The ΔIp value in response to 1 pM target dsDNA increased significantly from 10 min to 15 min, and then decreased dramatically when the renaturation time further increased to 20 min. In the renaturation process, the tDNAcm could hybridize with the blocker, or the tDNA. Due to the short size and the high concentration of blocker, the tDNAcm/blocker pair would hybridize faster than the tDNA/tDNAcm, dominating at the short period of 15 min. When the renaturation period is long enough, the ratio of the tDNA/tDNAcm duplex would increase due to its higher melting temperature. Thus, the renaturation time was chosen as 15 min.
3.4. The biosensor performances in detecting dsDNA
Under the optimum conditions, the detection sensitivity and linearity of the biosensor in detecting the target dsDNA was evaluated. Fig. 4 illustrates the DPV curves of the biosensor in detecting different concentrations of the dsDNA. The Ip value increased monotonically with the dsDNA concentration (CdsDNA) from 3.28 × 10−14 M to 6.55 × 10−12 M, with a semi-log linear calibration equation of Ip (μA) = 3.27 + 0.24 log[CdsDNA] (M) (inset in Fig. 4). The linear regression coefficient was 0.9977, with a limit of detection (LOD) of 1.2 × 10−14 M (i.e. 12 fM), estimated based on 3σ rule. This LOD value is one order of magnitude higher than our published LOD value (1 fM) for detecting the 43-mer ssDNA.27 The releasing efficiency for the tDNA could not be 100%. In addition, the hybridization efficiency of cpDNA/tDNA may also be lower than that of the cpDNA/(43-mer ssDNA), due to steric hindrances from the overhangs at the two ends of tDNA (Scheme 1). However, the only one order of magnitude higher LOD value indicates that the tDNA releasing efficiency and the cpDNA/tDNA hybridization efficiency are good. In addition, the LOD value is still comparable or even better than most of the reported ssDNA biosensors (Table S1, ESI†).38–42 More importantly, it should be noted that detecting dsDNA with biosensors, which has been seldom reported, is much more difficult and challenging than detecting ssDNA. In addition, the biosensor has very good reproducibility. As shown in inset in Fig. 4, the relative standard deviations of the detection signals for the detected DNA levels were within 10%.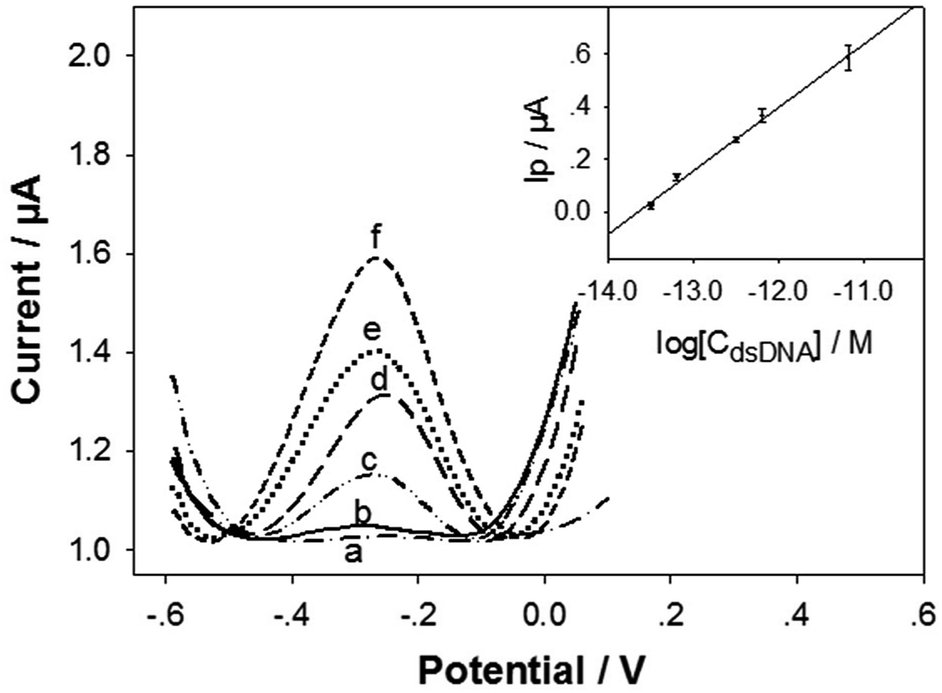 | ||
| Fig. 4 The DPV response curves of the biosensor to the target dsDNA with the concentrations of (a) 0, (b) 3.28 × 10−14, (c) 6.55 × 10−14, (d) 3.28 × 10−13, (e) 6.55 × 10−13, and (f) 6.55 × 10−12 M. Inset: the plot of the DPV Ip values versus the logarithm of the dsDNA concentration (CdsDNA). Error bars represent the standard deviation for 3 repetitive and independent biosensors. | ||
3.5. Detection of H. pylori DNA in dental plaque
The successful tDNA releasing and detecting with the biosensor enabled us to investigating the H. pylori detection in dental plaque samples. The colony PCR products (10 PCR cycles) of the dental plaque samples were treated under the optimum tDNA releasing conditions and detected with the electrochemical biosensor. It should be noted that the 10 cycles PCR product was not able to be detected by the agarose gel electrophoresis. Although the product of ≥25 PCR cycles could be detected with the gel electrophoresis (Fig. S2†), the detection specificity (∼47%) and sensitivity (∼35%) are very low (taking the UBT results as standards), indicating nonspecific amplification with high PCR cycles and insufficient amplification with low PCR cycles. This result is consistent with the report published by Ismail et al.,19 showing that single-step PCR could not detect H. pylori in dental plaque samples. Nonspecific amplification is a major drawback of PCR with high PCR cycles.21The typical DPV curves in detecting the dental plaque samples from the H. pylori negative and positive patients/volunteers (classified based on the UBT results) are illustrated in Fig. 5A. The Ip value from the H. pylori negative patient/volunteer (UBT value: 0.5) was 0.011 μA (curve a), obviously smaller than those (curve b and curve c, 0.056 μA and 0.59 μA respectively) from the H. pylori positive counterparts (UBT value: 5.4 and 53.2, respectively). Fig. 5B shows the average Ip values and the target dsDNA concentrations detected (calculated from the above semi-log linear calibration equation) for all the dental plaque samples. It has been generally accepted that the H. pylori positive UBT threshold is ≥4.0 DOB (indicated with the vertical line in Fig. 5B). Based on our biosensor detection results and the UBT data, the threshold for H. pylori positive residence in dental plaque was arbitrarily determined to be Ip ≥ 0.05 μA in our biosensor DPV test, corresponding to 3.98 × 10−14 M target dsDNA (indicated with the horizontal line in Fig. 5B). The biosensor detection results correlated very well with the UBT results, with a high sensitivity of 100%, and specificity of 97%. These results indicate that the developed sample treatment and biosensing method can sensitively and specifically detect dental plaque physiological samples, and the residence of H. pylori in dental plaque highly associates with gastric H. pylori infection. The association of dental H. pylori residence and gastric H. pylori infection was also found by other researchers.19,20 Dental plaque can be one of the main causes of H. pylori re-infection and also be the cause of oral–oral transmission.43 The colony PCR of only 10 cycles from the dental plaque sample produced not only enough but also specific H. pylori dsDNA fragment for the biosensors. The sensitive and specifical detection of dental plaque H. pylori with the DNA biosensor could provide an H. pylori infection diagnosis method for patients unfit for endoscopic examination, or unfit for UBT.
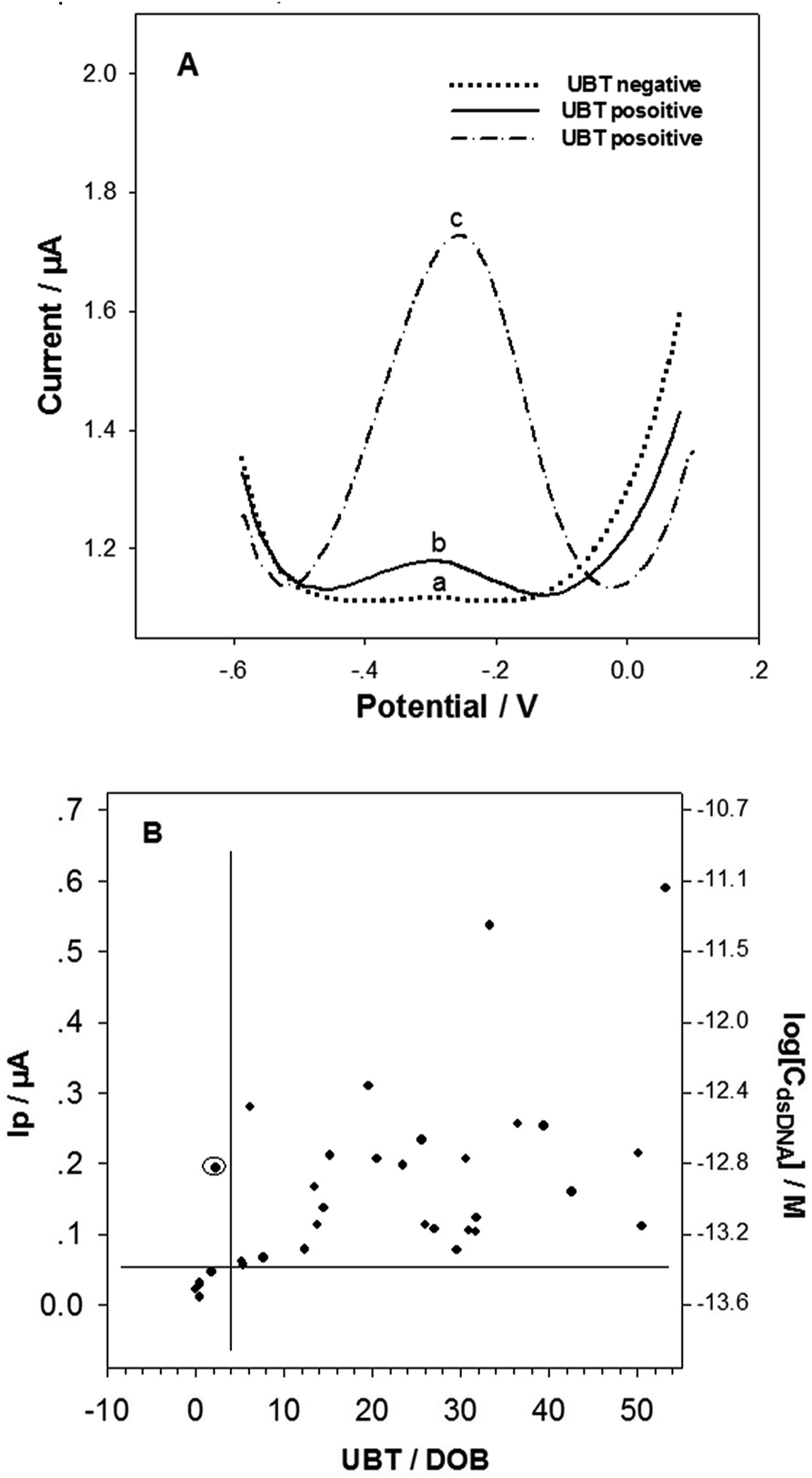 | ||
| Fig. 5 (A) Representative DPV curves of the developed biosensors in detecting H. pylori DNA in dental plaque samples collected from (a) gastric H. pylori negative, and (b and c) gastric H. pylori positive patients/volunteers (classified based on the UBT results). (B) Correlations between the detection results using the developed biosensing method in this work, and the UBT measurement in hospital. The lines represent the H. pylori infection thresholds based on the tests. The circled spot represents the inconsistent testing result between the two methods. | ||
4. Conclusions
A sensitive and specific detection method for H. pylori in dental plaque has been developed. With the specially designed primers, specific dsDNA segments have been obtained from H. pylori in dental plaque by colony PCR. Single strand tDNA was efficiently released under the help of the blocker, and then sensitively detected with the DNA biosensor. The dental plaque H. pylori detection results using the biosensor correlate well with the UBT results. The residence of H. pylori in dental plaque highly associates with gastric H. pylori infection. Detection of dental plaque physiological samples with the DNA biosensor is promisingly applicable in noninvasive diagnosis of H. pylori infection, and could provide an H. pylori infection diagnosis method for patients unfit for endoscopic examination, or unfit for UBT.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Plan for Scientific Innovation Talent of Henan Province to H. F. Cui (Grand number 154200510007), the National Natural Science Foundation of China (Grand number NSFC 21345007), and the Henan Open-up and Collaboration Program of Science and Technology (Grand number 132106000070). We thank Ke Wang for UBT measurement, and Dr Huang for providing the H. pylori strain.Notes and references
- B. Marshall and J. R. Warren, Lancet, 1984, 323, 1311–1315 CrossRef.
- F. Bazzoli, L. Olivieri, L. De Luca, P. Pozzato, P. Lehours and F. Megraud, Dig. Liver Dis., 2000, 32, 207–210 CrossRef.
- P. B. Ernst and B. D. Gold, J. Pediatr. Gastroenterol. Nutr., 1999, 28, 462–473 CrossRef PubMed.
- R. Nosrati, B. Golichenari, A. Nezami, S. M. Taghdisi, B. Karimi, M. Ramezani, K. Abnous and S. A. M. Shaegh, Trends Anal. Chem., 2017, 97, 428–444 CrossRef.
- R. S. P. Malon, S. Sadir, M. Balakrishnan and E. P. Córcoles, BioMed Res. Int., 2014, 2014, 962903 Search PubMed.
- A. I. Lopes, F. F. Vale and M. Oleastro, World J. Gastroenterol., 2014, 20, 9299–9313 Search PubMed.
- P. Malfertheiner, F. Megraud, C. A. Morain, J. Atherton, A. T. R. Axon, F. Bazzoli, G. F. Gensini, J. P. Gisbert, D. Y. Graham, T. Rokkas, E. M. El-Omar and E. J. Kuipers, Gut, 2012, 61, 646 CrossRef PubMed.
- J. Guarner, N. Kalach, Y. Elitsur and S. Koletzko, Eur. J. Pediatr., 2010, 169, 15–25 CrossRef PubMed.
- B. Velayos, L. Fernández-Salazar, F. Pons-Renedo, M. F. Muñoz, A. Almaraz, R. Aller, L. Ruíz, L. Del Olmo, J. P. Gisbert and J. M. González-Hernández, Dig. Dis. Sci., 2012, 57, 1880–1886 CrossRef PubMed.
- P. Malfertheiner, F. Megraud, C. A. O'Morain, J. P. Gisbert, E. J. Kuipers, A. T. Axon, F. Bazzoli, A. Gasbarrini, J. Atherton, D. Y. Graham, R. Hunt, P. Moayyedi, T. Rokkas, M. Rugge, M. Selgrad, S. Suerbaum, K. Sugano and E. M. El-Omar, Gut, 2017, 66, 6–30 CrossRef PubMed.
- R. K. Saiki, S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich and N. Arnheim, Science, 1985, 230, 1350–1354 Search PubMed.
- G. M. Makrigiorgos, S. Chakrabarti, Y.-Z. Zhang, M. Kaur and B. D. Price, Nat. Biotechnol., 2002, 20, 936–939 CrossRef PubMed.
- M. Oleastro, A. Ménard, A. Santos, H. Lamouliatte, L. Monteiro, P. Barthélémy and F. Mégraud, J. Clin. Microbiol., 2003, 41, 397–402 CrossRef PubMed.
- I. C. A. Scaletsky, K. R. S. Aranda, G. T. Garcia, M. E. P. Gonçalves, S. R. Cardoso, K. Iriya and N. P. Silva, Helicobacter, 2011, 16, 311–315 CrossRef PubMed.
- A. Vécsei, A. Innerhofer, U. Graf, C. Binder, H. Giczi, K. Hammer, A. Bruckdorfer, A. M. Hirschl and A. Makristathis, J. Pediatr. Gastroenterol. Nutr., 2011, 53, 65–70 CrossRef PubMed.
- P. Lehours, E. Siffré and F. Mégraud, BMC Gastroenterol., 2011, 11, 112–116 CrossRef PubMed.
- R. M. Ferreira, J. C. Machado, D. Letley, J. C. Atherton, M. L. Pardo, C. A. Gonzalez, F. Carneiro and C. Figueiredo, J. Clin. Microbiol., 2012, 50, 3983–3989 CrossRef PubMed.
- J. Saez, S. Belda, M. Santibáñez, J. C. Rodríguez, J. Sola-Vera, A. Galiana, M. Ruiz-García, A. Brotons, E. López-Girona, E. Girona, C. Sillero and G. Royo, J. Clin. Microbiol., 2012, 50, 3233–3237 CrossRef PubMed.
- H. Ismail, C. Morgan, P. Griffiths, J. Williams and G. Jenkins, J. Clin. Gastroenterol., 2016, 50, 17–22 CrossRef PubMed.
- D. Aksit Bıcak, S. Akyuz, B. Kıratlı, M. Usta, N. Urganci, B. Alev, A. Yarat and F. Sahin, BMC Oral Health, 2017, 17, 67 CrossRef PubMed.
- B. Schweitzer and S. Kingsmore, Curr. Opin. Biotechnol., 2001, 12, 21–27 CrossRef PubMed.
- D. B. Zhu, Y. B. Tang, D. Xing and W. R. Chen, Anal. Chem., 2008, 80, 3566–3571 CrossRef PubMed.
- J. G. Hacia, L. C. Brody, M. S. Chee, S. P. Fodor and F. S. Collins, Nat. Genet., 1996, 14, 441–447 CrossRef PubMed.
- F. S. Santiago, A. V. Todd, N. J. Hawkins and R. L. Ward, Mol. Cell. Probes, 1997, 11, 33–38 CrossRef PubMed.
- J. M. Nam, S. I. Stoeva and C. A. Mirkin, J. Am. Chem. Soc., 2004, 126, 5932–5933 CrossRef PubMed.
- X. X. Yu, Y. Chai, J. Jiang and H. Cui, J. Photochem. Photobiol., A, 2012, 241, 45–51 CrossRef.
- X. Y. Dong, X. N. Mi, L. Zhang, T. M. Liang, J. J. Xu and H. Y. Chen, Biosens. Bioelectron., 2012, 38, 337–341 CrossRef PubMed.
- C. Thiruppathiraja, S. Kamatchiammal, P. Adaikkappan, D. J. Santhosh and M. Alagar, Anal. Biochem., 2011, 417, 73–79 CrossRef PubMed.
- H. F. Cui, T. B. Xu, Y. L. Sun, A. W. Zhou, Y. H. Cui, W. Liu and J. H. T. Luong, Anal. Chem., 2015, 87, 1358–1365 CrossRef PubMed.
- D.-B. Zhu, Y. B. Tang, D. Xing and W. R. Chen, Anal. Chem., 2008, 80, 3566–3571 CrossRef PubMed.
- G. J. Li, X. L. Li, J. Wan and S. S. Zhang, Biosens. Bioelectron., 2009, 24, 3281–3287 CrossRef PubMed.
- A. Q. Shi, J. Wang, X. W. Han, X. Fang and Y. Z. Zhang, Sens. Actuators, B, 2014, 200, 206–212 CrossRef.
- M. V. del Pozo, C. Alonso, F. Pariente and E. Lorenzo, Anal. Chem., 2005, 77, 2550–2557 CrossRef PubMed.
- S. Hajihosseini, N. Nasirizadeh, M. S. Hejazi and P. Yaghmaei, Mater. Sci. Eng., C, 2016, 61, 506–515 CrossRef PubMed.
- T. García, M. Revenga-Parra, H. D. Abruña, F. Pariente and E. Lorenzo, Anal. Chem., 2008, 80, 77–84 CrossRef PubMed.
- T. García, E. Casero, M. Revenga-Parra, J. Martín-Benito, F. Pariente, L. Vázquez and E. Lorenzo, Biosens. Bioelectron., 2008, 24, 184–190 CrossRef PubMed.
- M. Minunni, S. Tombelli, J. Fonti, M. M. Spiriti, M. Mascini, P. Bogani and M. Buiatti, J. Am. Chem. Soc., 2005, 127, 7966–7967 CrossRef PubMed.
- D. S. Campos-Ferreira, G. A. Nascimento, E. V. M. Souza, M. A. Souto-Maior, M. S. Arruda, D. M. L. Zanforlin, M. H. F. Ekert, D. Bruneska and J. L. Lima-Filho, Anal. Chim. Acta, 2013, 804, 258–263 CrossRef PubMed.
- S. Liu, J. Liu, L. Wang and F. Zhao, Bioelectrochemistry, 2010, 79, 37–42 CrossRef PubMed.
- H. P. Peng, Y. Hu, P. Liu, Y. N. Deng, P. Wang, W. Chen, A. L. Liu, Y. Z. Chen and X. H. Lin, Sens. Actuators, B, 2015, 207, 269–276 CrossRef.
- Z. Shakoori, S. Salimian, S. Kharrazi, M. Adabi and R. Saber, Anal. Bioanal. Chem., 2015, 407, 455–461 CrossRef PubMed.
- L. Zhu, L. Luo and Z. Wang, Biosens. Bioelectron., 2012, 35, 507–511 CrossRef PubMed.
- N. Amiri, R. Abiri, M. Eyvazi, M. R. Zolfaghari and A. Alvandi, Arch. Oral Biol., 2015, 60, 782–788 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information and figures. See DOI: 10.1039/c8ra03134g |
| This journal is © The Royal Society of Chemistry 2018 |