
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe trans/cis photoisomerization in hydrogen bonded complexes with stability controlled by substituent effects: 3-(6-aminopyridin-3-yl)acrylate case study†
Adam Kwiatkowskia,
Beata Jędrzejewskaa,
Marek Józefowiczb,
Izabela Grelaa and
Borys Ośmiałowski *c
*c
aFaculty of Chemical Technology and Engineering, UTP University of Science and Technology, Seminaryjna 3, PL-85326 Bydgoszcz, Poland
bFaculty of Mathematics, Physics and Informatics, University of Gdańsk, Wita Stwosza 57, 80-308 Gdańsk, Poland
cFaculty of Chemistry, Nicolaus Copernicus University in Toruń, 7 Gagarin Street, 87-100 Torun, Poland. E-mail: borys.osmialowski@umk.pl
First published on 29th June 2018
Abstract
The association of aminopyridine-based acrylic acid and its salt was studied by NMR titration experiments. The AA (acceptor, acceptor) hydrogen-bonding pattern present in the salt forms a complex readily with a DD (donor, donor) hydrogen-bonding pattern of the substituted ureas even in polar and competitive environment. The double carbon–carbon bond in the acrylic acid derivative is subjected to photoisomerization. This is dependent on the association with substituted urea derivatives. The substituent in ureas influences the trans/cis isomerization kinetics and position of the photostationary state. Two mechanisms that influence the photoisomerization were proposed. To the best of our knowledge, the trans/cis photoisomerization influenced by the substituent in such a hydrogen-bonding pattern has not observed previously. It was shown that interaction with urea derivatives causes lowering of the trans-to-cis photoreaction rates.
Introduction
The urea moiety is one of the most popular supramolecular synthons.1–3 It forms double hydrogen-bonding but this feature is just a starting point in research on its properties. These molecules may be used in crystal engineering, as a selective sensor for nitro-compounds4 and are able to form intramolecular hydrogen bond that is broken upon association.5–7 Their main popularity in organic chemistry lies in organocatalytic properties of sulfur derivatives (thioureas).8 The studies on complexation of ureas are quite popular especially in light of interactions of these molecules with oxoanions or carboxylates.9–11 Regarding other simple supramolecular synthons able to form two intermolecular hydrogen bonds carboxylic acids are good co-crystallizing agents that interact with 2-acylaminopyridines forming co-crystals while the same acids form salts with aminopyridine in the solid state.12–14 These examples of doubly-hydrogen bonded associates show a number of possibilities of inter-synthon interactions, which is further extended due to the proton transfer reaction (probably also in crystals15). Taking the proton transfer process into account some general remarks should be made. First, it is worth mentioning that carboxylic acids are self-complementary (Fig. 1a) and the same is realized in 2-aminopyridine (Fig. 1b) or its N-acylated (Fig. 1c) derivative.16 Secondly, the carboxylic acid is complementary to the 2-acylaminopyridine (Fig. 1d). On the other hand, its deprotonated form (carboxylate) may interact with ureas (Fig. 1e) readily but not with 2-acylaminopyridine. Thus, the dissociation of the carboxylic acid causes that its hydrogen-bonding pattern does not fit the interaction regime in 2-acylaminopyridine.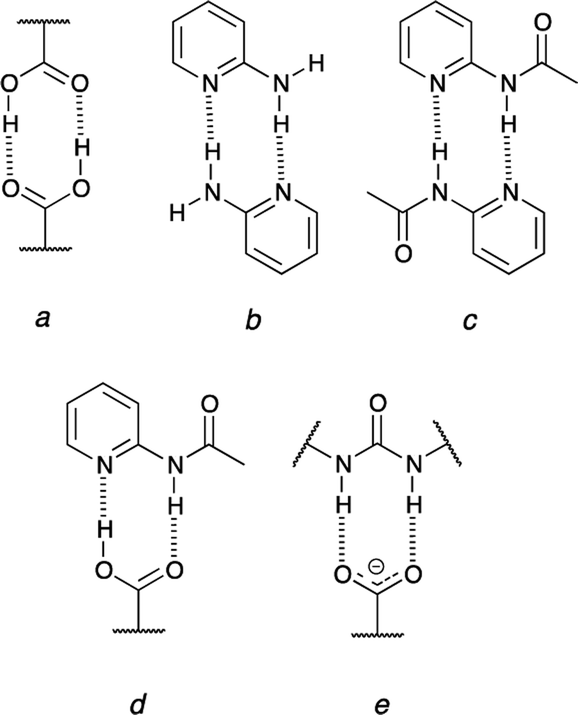 | ||
| Fig. 1 Inter-synthon interaction by double hydrogen bonding. | ||
Over the plethora of possible interactions the most challenging ones are those that may be present in the investigated system simultaneously. One of examples may be the bis(urea) that is, in theory, able to break two intramolecular hydrogen bonds upon association.5,7 Thus, it is crucial to know the interaction of the simple supramolecular synthons. This, for sure, move the research towards more complex systems as it was realized for ternary ones.17 This is important to keep in mind that non-covalent interactions are geometry-dependent. This includes the rotational movements of one part of molecule with respect to another one. These rotations play a crucial role in fitting molecule to various guests as, for example, in 2,4-bis(acylamino)pyrimidines when no intramolecular hydrogen bond is broken18 but also when rotamerism must be present to form complexes6 and the dissociation of the intramolecular hydrogen bond is a condicio sine qua non.
The isomerism of the double bonds is one of the most well investigated elementary reaction in photochemistry.19 Although, the geometrical isomerization of alkenes can be effected thermally, catalytically and photochemically, the light induced process is crucial due to variety of applications.20 In particular, the process was investigated from the theoretical, mechanistic and synthetic point of view. It proceeds in about 200 fs with quantum efficiency reaching up to 67%.21–24 The photoisomerization of alkenes is believed to take place by the excited state (singlet or triplet) in which the two sp2 carbons are twisted by about 90° with respect to one another leading to an intermediate in a form of zwitterionic or biradical. The basic features of the process is that the ground state thermodynamics does not influence the trans/cis ratio in the photostationary state but is, instead, controlled by the excited state potential surfaces.19
A numerous studies on the application of photoisomerization have been conducted. For instance, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond conversion plays a role in many molecules25 that are called molecular machines.26,27 The photoisomerization is responsible for reversible processes in crystals28–30 and in solution.31–33 It can be used in photoswitches34 or photoswitchable polymers.35
C double bond conversion plays a role in many molecules25 that are called molecular machines.26,27 The photoisomerization is responsible for reversible processes in crystals28–30 and in solution.31–33 It can be used in photoswitches34 or photoswitchable polymers.35
The trans/cis isomerism changes the photophysical properties of dyes. Thus, the stabilization of one form over another might be crucial to material chemistry and molecular design. Joining the photoisomerization with intermolecular interactions may deliver interesting results. This is even more interesting and challenging in the light of mentioned proton transfer reaction that may take place between hydrogen-bonded counterparts. It is worth noting that excited state proton transfer is the mechanism that causes non-radiative energy loss. Our interests in this topic came from the idea that association and energy loss within the supramolecular complex may have an impact on the stabilization of the double bond. Our intention was to use the hydrogen bonding in studies of stability of double CC bonds in compounds that are prone to photoisomerization. The aim of this work is to (a) study the effect of irradiation on isomerization of 1 and 2, (b) study the isomerization together with interaction of 2 with substituted ureas, (c) check if the photoisomerization or thermal back-reaction might be influenced by hydrogen bonding. The said thermal reaction usually proceeds in cis-to-trans direction. It may also be guided by irradiation of the cis isomer by light of higher energy than in case of trans-to-cis photoisomerization. The initial idea to study this particular structure and its photoisomerization was inspired by the curiosity if quadruple hydrogen bonding in the dimer of 1 (vide infra) is able to hinder cis-to-trans thermal isomerization. For comparison purposes two simpler compounds were used (3 and 4). Fig. 2 shows structures used in the current study.
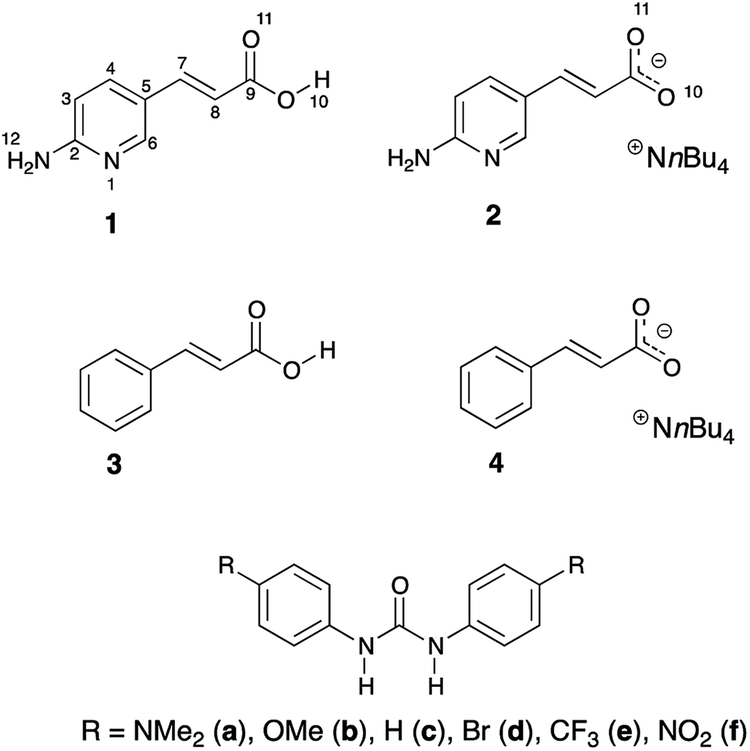 | ||
| Fig. 2 Structures of compounds used in photoisomerization study. | ||
Results and discussion
Photoisomerization of 3-(6-aminopyridin-3-yl)acrylate derivatives
The NMR data confirm that aminopyridine-based acrylic acid (1) and its salt (2) exist as trans isomer in solution. This isomer may be partially transformed into a cis-form upon irradiation. To do so the solution of compounds 1 and 2 in DMSO-d6 were irradiated with xenon lamp. The trans-to-cis conversion was monitored by 1H NMR spectrometry. The spectra were recorded at each step of irradiation (time intervals, see ESI, Fig. S1 and S2†).The obtained data revealed that both compounds (1 and 2) isomerize upon irradiation but their photostationary states are different. Because the proton spectra of the compounds show considerable changes in chemical shifts, the kinetics of the photoisomerization was studied. This was monitored by the changes in the area under corresponding trans and cis peaks as a function of irradiation time (t). The observed rate constant (k) was determined based on the analysis of the experimental data applying the following equation:
| ln[% trans] = ln[% trans]0 − kt |
In Fig. 3 the comparison of the kinetic behavior for aminopyridine-based acrylic acid (1) and its salt (2) are shown. The corresponding rate constants36 are first-order reaction:37 11.2 × 10−4 s−1 (1) and 29.1 × 10−4 s−1 (2). It is reasonable to tell that the proton transfer in 1 is responsible for lower k value than that in 2 (lack of OH proton, Fig. 4). That supports the general conclusion on the proton-transfer assisted mechanism of the stabilization (non-radiative energy loss) of trans form of 2 by hydrogen bonded urea counterparts discussed in next sections. Except the proton transfer within dimers it is reasonable to tell the –COOH group dissociation in polar environment is important.
 | ||
| Fig. 3 Top: The isomerization of 1 and 2 as a function of irradiation time (H8 chemical shift based data, in DMSO-d6, c = 10.6 mmol dm−3); Bottom: Kinetics of the photoisomerization process induced by irradiation. | ||
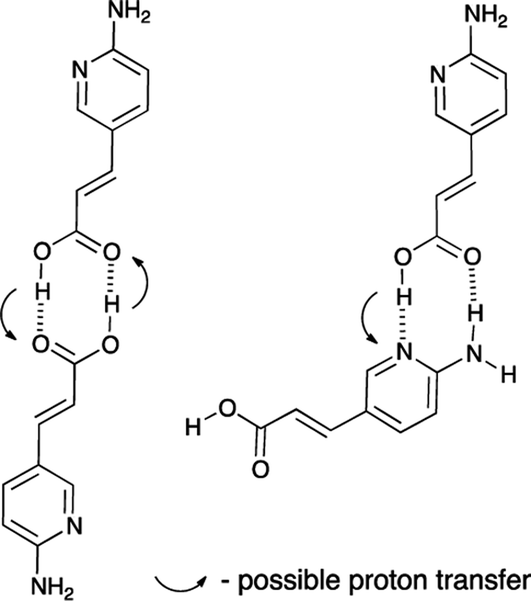 | ||
| Fig. 4 Two dimers of 1 and possible proton transfer that could lead to the non-radiative energy loss. | ||
For comparison the behaviour of the cinnamic acid (3) and its tetrabutylammonium salt (4) upon irradiation was also determined (see ESI, Fig. S3 and S4†). The respective rate constants of trans-to-cis isomerization are 2.9 × 10−4 s−1 (3) and 6.8 × 10−4 s−1 (4). The higher values for 1 and 2 than that for 3 and 4 suggest the aminopyridine moiety plays a role in this process. Most probably the charge transfer by electron donating NH2 group causes photoisomerization to proceed faster. The charge transfer in 1 and 2 was studied by spectroscopic methods and further described in ESI.†
Except NMR the photoisomerization process was also studied by the UV-vis spectroscopy. Fig. 5 shows the absorption spectra before and after (time intervals) irradiation at the 330 nm with the xenon lamp equipped with monochromator.
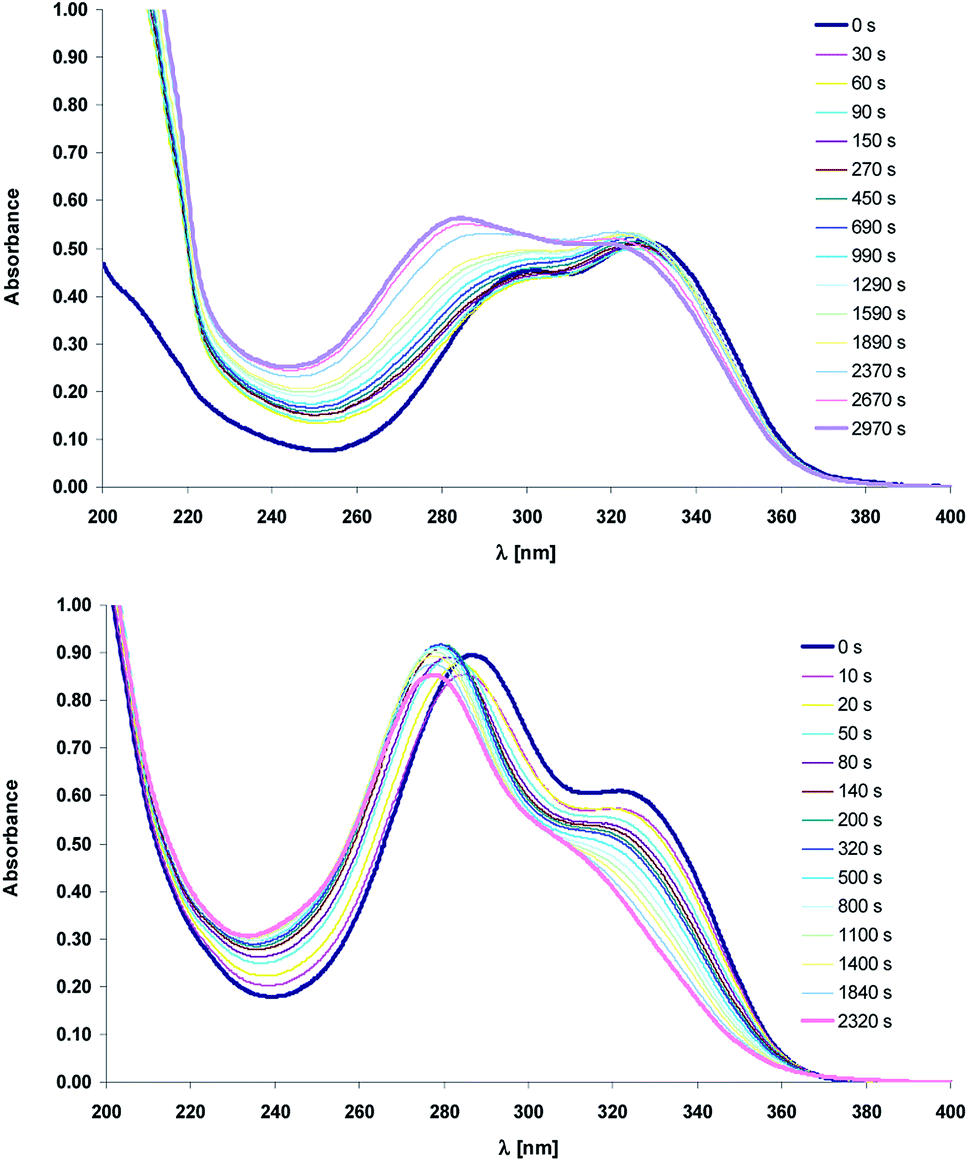 | ||
| Fig. 5 Electronic absorption spectra of compounds 1 (top) and 2 (bottom) recorded before and after irradiation ([s], 330 nm) in acetonitrile. | ||
Comparison of the structure–isomerization relationships for 1 and 2 suggests that upon UV irradiation (330 nm) the absorption band position of the acid and its salt moves to the higher energy level, which is accompanied by non-monotonic changes in their absorbance (Fig. 5). The absorption spectra in the near ultraviolet of 1 and 2 consist of two intense absorption bands with maxima at ca. 290 nm and 340 nm. The trans-1 absorbs more light at 340 nm, whereas for its salt (trans-2), the more intense is the short-wavelength absorption band. Upon irradiation the inversion in the intensity of the main bands of the acid 1 is observed. The short-wavelength band is more intense like in the trans form of the salt 2 whereas the intensity of the absorption band with maximum at 340 nm does not change significantly. This effect may be due to the different molecular configuration of trans and cis isomers. The short-wavelength band would be assigned to the electron transition in 2-aminopyridine group whereas the peak at ca. 340 nm should belong to the intramolecular charge transfer (ICT). Assuming that ethylene group of cis-1 adopts more twisted conformation, which hinders the ICT process, the incidence of charge transition is reduced, so the main absorption band is localized at the short-wavelength region.
The transformation between trans and cis forms is also reflected in the occurrence of the sharp isosbestic point at ca. 370 nm (ESI, Fig. S5†). Due to the overlapping of the electronic absorption spectra of the two isomers, the isomerization quantum yields were not studied quantitatively.
Association studies
The compounds 1 and 2 were studied for their self-association in their trans and cis forms. It is worth mentioning that the trans and cis forms of 1 have much different self-interacting scheme, id est. the trans form should be able to form cyclic hexamers or oligomers, while the cis form should be able to form dimers stabilized by four hydrogen bonds (Fig. 6).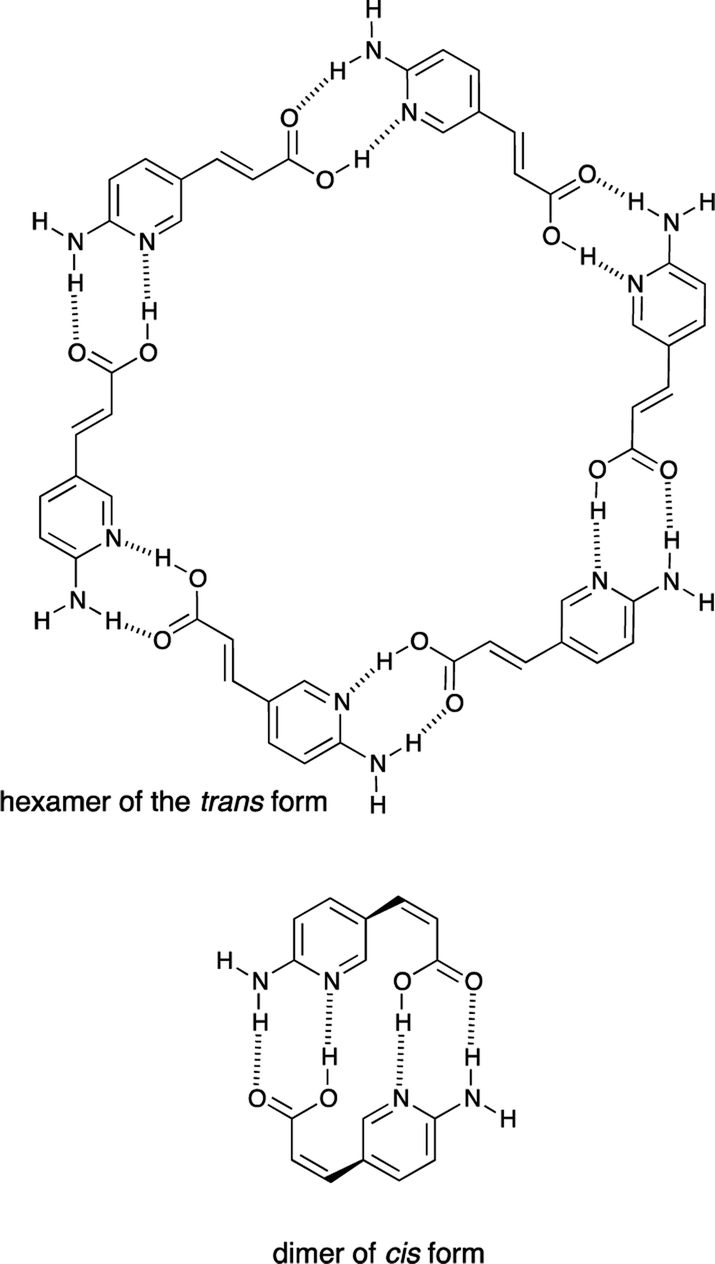 | ||
| Fig. 6 Probable arrangement of self-interacting trans and cis forms of 1. | ||
However, the results show that 1 does not form any associate (trans or cis) in DMSO solution due to highly polar character of this solvent and its solvation abilities towards compounds carrying NH/OH groups. Unfortunately 1 is not soluble in CDCl3. The fact that double hydrogen bonded neutral molecules barely form dimers in polar solvent is in agreement with the studies of 2-acylamino-1,8-naphthyridin-7-ones that are stabilized in its dimeric form by quadruple hydrogen bonding (in CDCl3).38 The same hydrogen-bonding pattern is present in the cis form of 1, but both, the mentioned naphthyridine derivative and cis-1, do not dimerize in DMSO. Since anions39–43 are able to interact with urea moiety in polar and highly competitive solvents,7 the tetrabutylammonium salt 2 was used in association (vide infra) experiments.
As before for anionic species7 the anion of salt 2 interacts readily with ureas in polar solution. The association was monitored by the change in chemical shift of several protons in 2 (host molecule). Table 1 shows the complexation induced shift (CIS) values and association constants (Kassoc) determined, as before,7 with the use of various protons. The CIS value is the difference between chemical shift of the chosen proton in a free molecule and the chemical shift of the same proton with the concentration of the guest molecule extrapolated to infinity. Not all data are collected in Table 1 because, most probably, proton transfer accompanies association. This is further discussed in next sections. Thus the ureas substituted with electron accepting groups are not included in correlation (R in Table 1). In Fig. 7 two titration curves are shown. The comparison is made between ureas carrying extreme substituents (NMe2 and NO2).
| Guest | H4 | H6 | H7 | H8 | H12 |
|---|---|---|---|---|---|
| a Correlation coefficient for the trans form. | |||||
| a (NMe2) | 200, 0.112 | 200, 0.108 | 150, 0.153 | 130, 0.270 | 170, 0.097 |
| b (OMe) | 240, 0.124 | 300, 0.120 | 220, 0.156 | 170, 0.289 | 280, 0.112 |
| c (H) | 430, 0.139 | 480, 0.133 | 300, 0.200 | 190, 0.359 | 400, 0.126 |
| c (H, cis) | 300, 0.187 | 260, 0.135 | 300, 0.369 | 250, 0.156 | 250, 0.169 |
| Ra | 0.97, 0.96 | 0.93, 0.97 | 0.97, 0.78 | 1.00, 0.87 | 0.97, 0.98 |
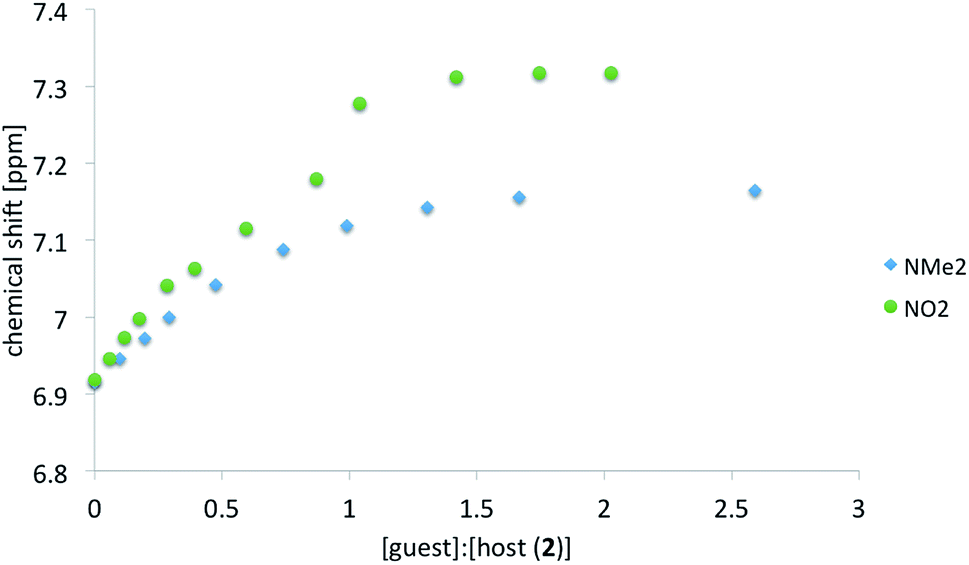 | ||
| Fig. 7 Example of the titration curve based on H8 chemical shift of 2+a (NMe2) and 2+f (NO2). | ||
It is seen that for ureas carrying electron-donating substituents the association constants based on various protons are in line with the character of the substituent.5,6 The mentioned substituent effect in ureas is manifested by association but also by the chemical shift of the NH protons. For δ[ppm] = f(σ) where σ is a substituent constant the correlation coefficient R = 0.97 for pure a–f recorded in DMSO-d6. Thus it is safe to tell the substituent effect is transmitted in a classical way.
For most of complexes the course of the titration curves are regular. A serious deviation of the titration curve (Fig. 7) from the ideal course of the Benesi–Hildebrand equation is observed for NO2 substituted urea (compare also other electron-acceptor substituted compounds used in titrations in ESI†). Previously the sigmoidal shape of the titration curve was caused by the coexistence of at least two forms in solution.5 Currently the proton transfer may be observed in the ground state of the complex.
Since the acidity of NH groups in ureas is tuned by substituent the binding of those should also influence the change in electronic structure of the anion of 2 in complex in a regular way. The same association-induced charge reorganization influences the photoisomerization. It is worth remembering that trans and cis forms differ in their geometry and thus their intermolecular interactions might be different to some extent even if the hydrogen-bonding pattern of the carboxylate group is preserved. Thus, to study these differences the cis form of 2 was also titrated by c but the curve obtained by fitting the data had larger residuals than that for trans form (see ESI†). For the cis form the data fit linear function at the beginning of titration as in case of triuret studied by us recently.5 That suggest two geometrically different forms of cis-2 co-exist in solution. These were further studied with the use of DFT methods (vide infra).
Photoisomerization of the complexes tested
The next step in experiments was to probe the photoisomerization (as before for 1–4 including the same concentrations) with the assistance of ureas as a hydrogen bonding counterparts. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of 2 and a–f was used. Samples were irradiated as before and the procedure of the trans:cis ratio determination was repeated (see Experimental section). In Fig. 8 the substituent effect on the position of photostationary state was shown as a percentage of the cis form dependent from the substituent constant. Two sets of data represent all ureas (a–f, in blue) and “all except NO2 derivative” (in red).
:2 ratio of 2 and a–f was used. Samples were irradiated as before and the procedure of the trans:cis ratio determination was repeated (see Experimental section). In Fig. 8 the substituent effect on the position of photostationary state was shown as a percentage of the cis form dependent from the substituent constant. Two sets of data represent all ureas (a–f, in blue) and “all except NO2 derivative” (in red).
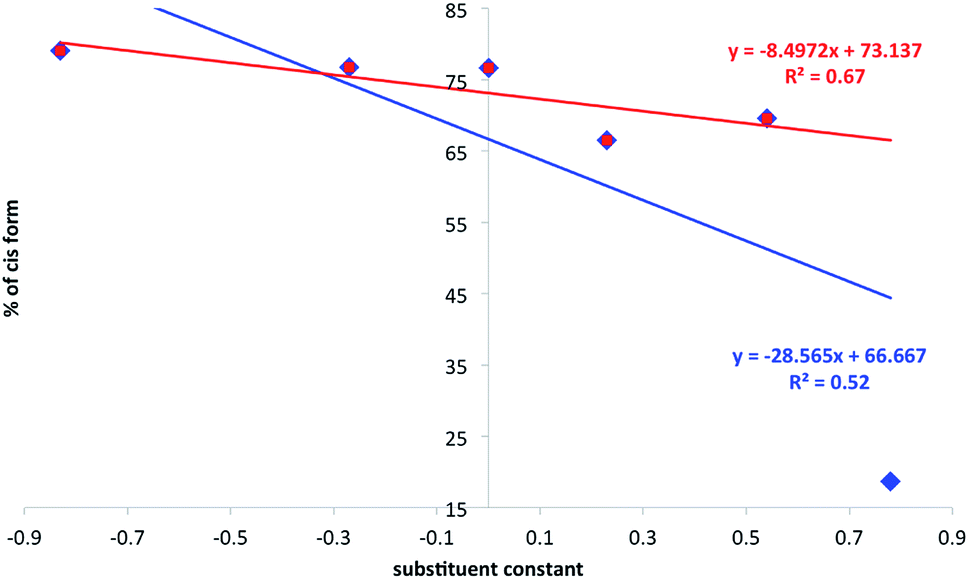 | ||
| Fig. 8 The relation between substituent constant and the percentage of the cis form (photostationary state) in 2. | ||
It is easy to see that the data presented do not fit well to the linear function as defined by classical Hammett relation. The reasons for that may be the mentioned proton transfer or complicated association/dissociation equilibrium present in the studied system. The proton transfer, for sure, causes additional loss of excitation energy so it decreases the amount of the cis form in the mixture. This is especially important when one realize that, most probably, in 2+f mixture most of the 2 is transformed into 1 due to the proton transfer in the ground state in DMSO. The data obtained suggest the association causes the decrease of photoisomerization rate constant (compare irregular changes in 2+f titration curve). We have recently shown that stronger electron donating group leading to the intramolecular charge transfer causes easier photoisomerization of the double CC bond.44 Currently the data presented in Fig. 8 shows donor–acceptor properties of substituent in ureas influences charge transfer in 2 and thus its photoisomerization. That leads to the association-assisted stabilization of the CC double bond. It is worth to note that for 2+f (NO2) complex the amount of the cis form in photostationary state after irradiation (18.7%) is even lower than that in 1 (42.8%). Thus the position of the photostationary state in 1 is different from that for 1+f−/2+f complex and may be interpreted as the loss of excitation energy by intermolecular proton transfer. The proton transfer may also be present in other ureas but the most efficient reaction of this type is observed for NO2-derivative due to the highest acidic character of NH protons. Thus, lowering acidity of NHs and lowering the association constants weakens the proton transfer in the ground state giving relatively lowered stabilization of the trans CC bond. That, in turn, causes the photoisomerization proceeds faster. The photoisomerization rate constants and half-life of reaction are collected in Table 2.
| Compound/complex | k | t1/2 | % cis |
|---|---|---|---|
| 2+a | 14.0 | 495 | 79.1 |
| 2+b | 21.3 | 326 | 76.7 |
| 2+c | 23.7 | 293 | 76.6 |
| 2+d | 16.7 | 414 | 66.5 |
| 2+e | 12.2 | 566 | 68.3 |
| 2+f/1+f− | 10.6 | 6540 | 18.7 |
| 1 | 11.2 | 620 | 42.8 |
| 2 | 29.1 | 238 | 82.2 |
| 3 | 2.9 | 2402 | 42.8 |
| 4 | 6.8 | 1023 | 64.5 |
Based on the data from Table 2 it is clear that k for 2 is ca. three times higher than that in 2+f (R = NO2) complex and only ca. twice higher than that in 2+a (R = NMe2). Still the double hydrogen bonding of the anion carrying CC double bond causes photoisomerization to slow down. It is worth noting that only in case of 2+c the value of k is comparable to that of 2 (Fig. 9). Substitution of urea with strong electron donor (a) or strong electron acceptor (f) causes that the photoisomerization rate constant of the 2 in these complex is similar to the 1. Thus, the photoisomerization rate constants do not follow the substituent effect in the whole series. Instead, a good correlation was found separately for donors + unsubstituted derivatives and the same for acceptors + unsubstituted derivatives.
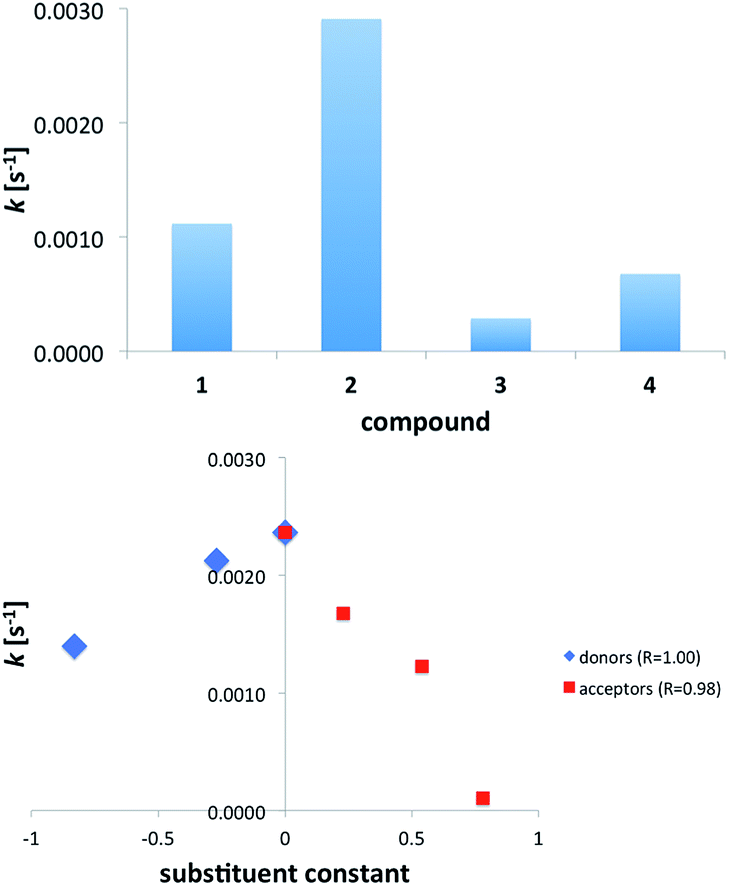 | ||
| Fig. 9 The rate constant of trans/cis photoisomerization in acids and salts (top) and complexes of 2 with substituted ureas (bottom). | ||
The unsubstituted derivative is a common point for both series since c is a reference molecule (no substituent present). Both correlations are very good (Rs are given in Fig. 9b) albeit it is fair to note that the number of points is three and four for donors and acceptors, respectively.
Fig. 9b shows that any electronic effect by substitution of ureas causes change in value of k. Since the slopes of the fitted linear functions have opposite signs we assumed two different mechanisms of photoisomerization reaction. The similar non-linearity in the larger series have been observed for binding barbital with triple hydrogen-bonding.45 It is worth noting that, except for 2+f, photoisomerization rate constant for complexes fall between values of k for 1 and 2. This shows the hydrogen bonding with ureas slows down the photoisomerization of the anion 2 and the changes in k are, most probably, related to the existence of equilibrium between –COO− and –COOH upon irradiation. As already said, for comparison purposes, the cinnamic acid (3) and its anion (4, salt) was used. These experiments confirm that anions (2, 4) undergo faster isomerization than acids (1, 3). Again, the loss of excitation energy by proton transfer, acid dissociation or vibrations of OH groups may cause the slower photoisomerization in acids.
The photoisomerization of 2 with the assistance of ureas as a hydrogen bonding counterparts was also studied by UV-vis spectroscopy (Fig. 10). Upon irradiation a decrease of the main absorption band with a clear two isosbestic points (270 nm and 350 nm) are observed. The existence of the isosbestic points confirms that two parallel processes occur. The first isosbestic point indicates a transition between two species, trans and cis form of the salt. The second one may be connected with formation of the complex between 2 and urea that affects the probability and potential of the transformation. Upon prolonged irradiation, the changes become smaller and a photostationary state is reached. The absorption thus consists of two contributions, from the original trans-2 and from the newly formed isomer cis-2 influenced by association with ureas.
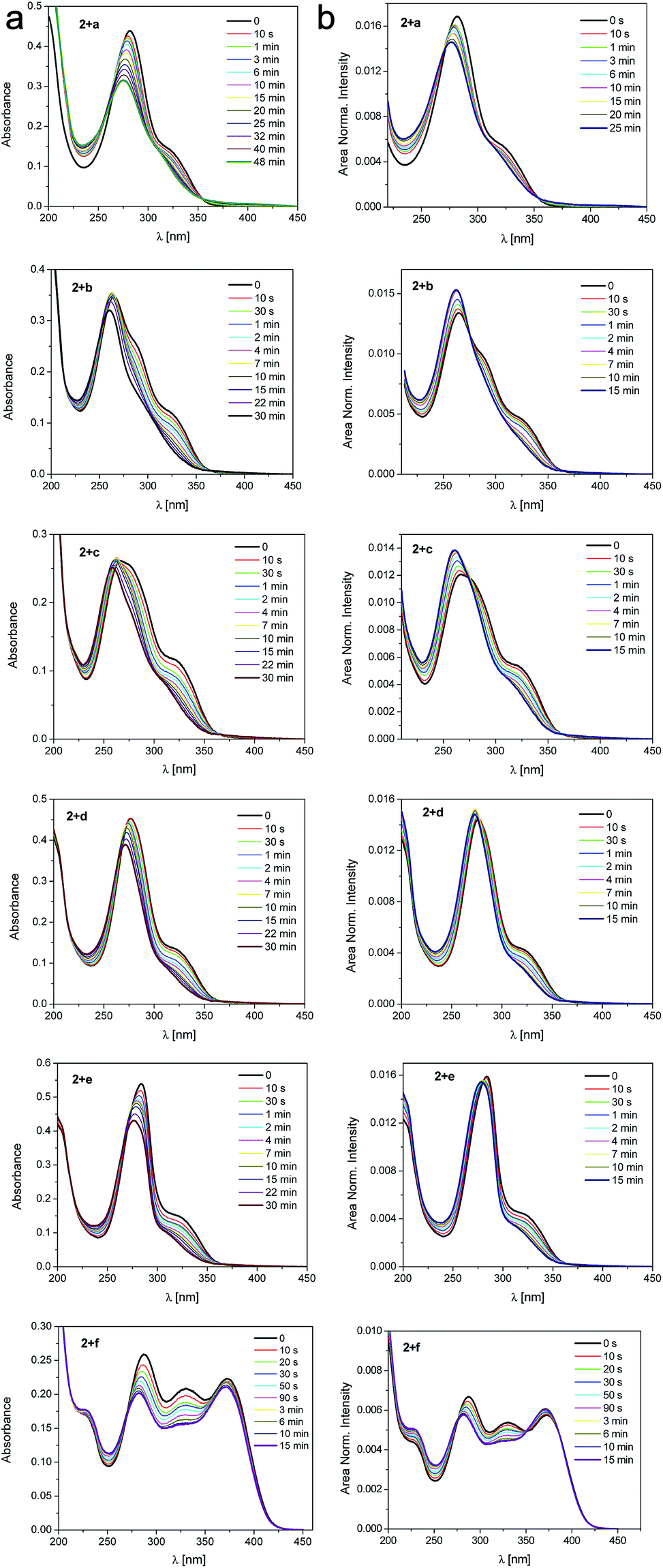 | ||
| Fig. 10 (a) Changes in UV-vis absorption spectra of complexes upon irradiation with xenon lamp equipped with monochromator (330 nm), (b) area normalized absorption spectra of the complexes upon irradiation (time given). | ||
To sum up, the rate constant of photoisomerization in the studied series is dependent on the following factors: (a) the association constant of counterparts and (b) the proton transfer that can accompany the association in ground or excited state.46 The proton transfer takes place in case of NO2 substituted urea similarly as during interaction with fluoride anion.47 However, as opposite to previous works, it was impossible to study proton transfer by UV-vis absorption spectroscopy because the absorption spectra of aromatic, interacting molecules overlap.
As mentioned above the association of 2 with substituted ureas causes the stabilization of the trans form. The lowest k value was obtained for the complex with nitro-carrying urea. In this case, the intermolecular proton transfer that is present in the ground state causes the excited state relaxation so efficiently that most of energy is consumed for this reaction. This conclusion is supported by a well-known observation telling that the excited state proton transfer quenches fluorescence and/or shifts its maximum towards red part of the spectrum.48,49
Proposed mechanism of the reaction in tested complexes
We propose two mechanisms that influence photoisomerization in studied complexes. However, before some already observed conclusions are given. The position of the photostationary state follows the general substituent effect giving the lowest value of the percentage of the cis form for CF3 substituted urea (Fig. 8). In the a–c part of data (Fig. 9b) the more electron-donating substituent is the lower k value is obtained. The decrease of the k in 2+a and 2+b with respect to 2+c is, most probably, attributed to the intramolecular charge transfer in a and b (NMe2 and OMe substituted urea derivatives). Thus, the negative charge transferred from electron donating group (for example NMe2) influences the complex stability, which dissociate (Fig. 11) upon excitation – mechanism no. 1. The said association/charge transfer/dissociation might be the source of the energy loss by thermal vibrations (geometry relaxation).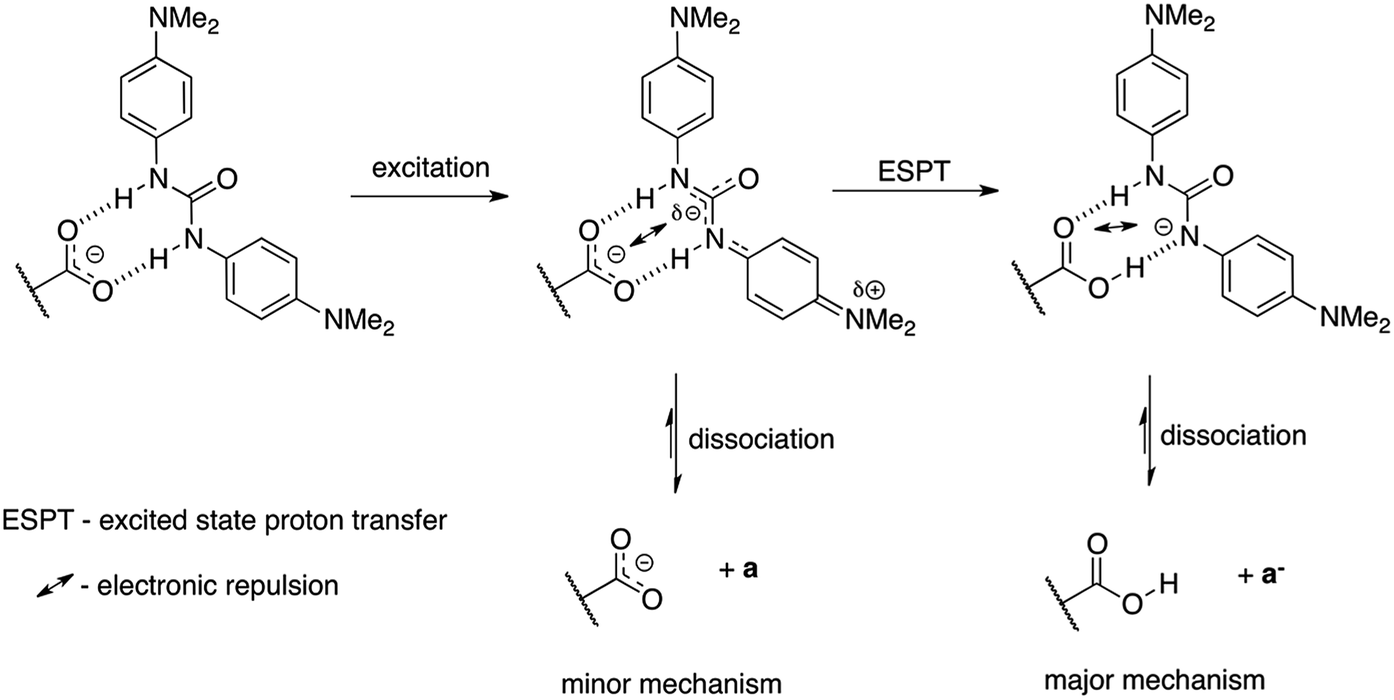 | ||
| Fig. 11 The charge transfer in a and its influence on complex dissociation. | ||
Moreover, the said dissociation of the complex caused by electronic repulsion between counterparts, most probably, leads to the photoisomerization of the acid 1 (major part of mechanism) and hydrogen-bonded 2 (minor part of mechanism) simultaneously. The effectiveness of acid formation affects the rate of photoisomerisation and causes that the k value tends the photoisomerization rate constant close to k value for pure 1. The dissociation of two complexes in the same solution might also be responsible for lowered correlation coefficient between the position of the photostationary state and substituent constant as mentioned above (see Fig. 8). Since the photoisomerization rate constant for 2+a mixture is closer by its value to k for 1 the photoisomerization in dissociated state was proposed as a major sub-mechanism (Fig. 11).
The second mechanism is proposed for complexes containing an electron acceptor substituent in series d–f. In this case, most probably, the tendency to intermolecular proton transfer that is dependent from the substituent takes place. The NO2 group is an exception since it transfers proton in the ground state. This reaction was already noticed for similar urea derivatives.50,51 The proton transfer in the current complexes takes place in much more complicated system than simple anion:urea associate. This is due to the fact that if the proton was transferred from urea derivative to the carboxylate group the obtained acid 1 might be subjected to other reactions. For that to happen, the dissociation of the complex must take place. Taking into account the strongest acid in urea series (f) two complexes of compounds 2 and f should be considered. These are 2+f and 1+f−(a) (complex after proton transfer reaction, Fig. 12) and 1+f−(b) (rotameric form of 1+f−(a)).
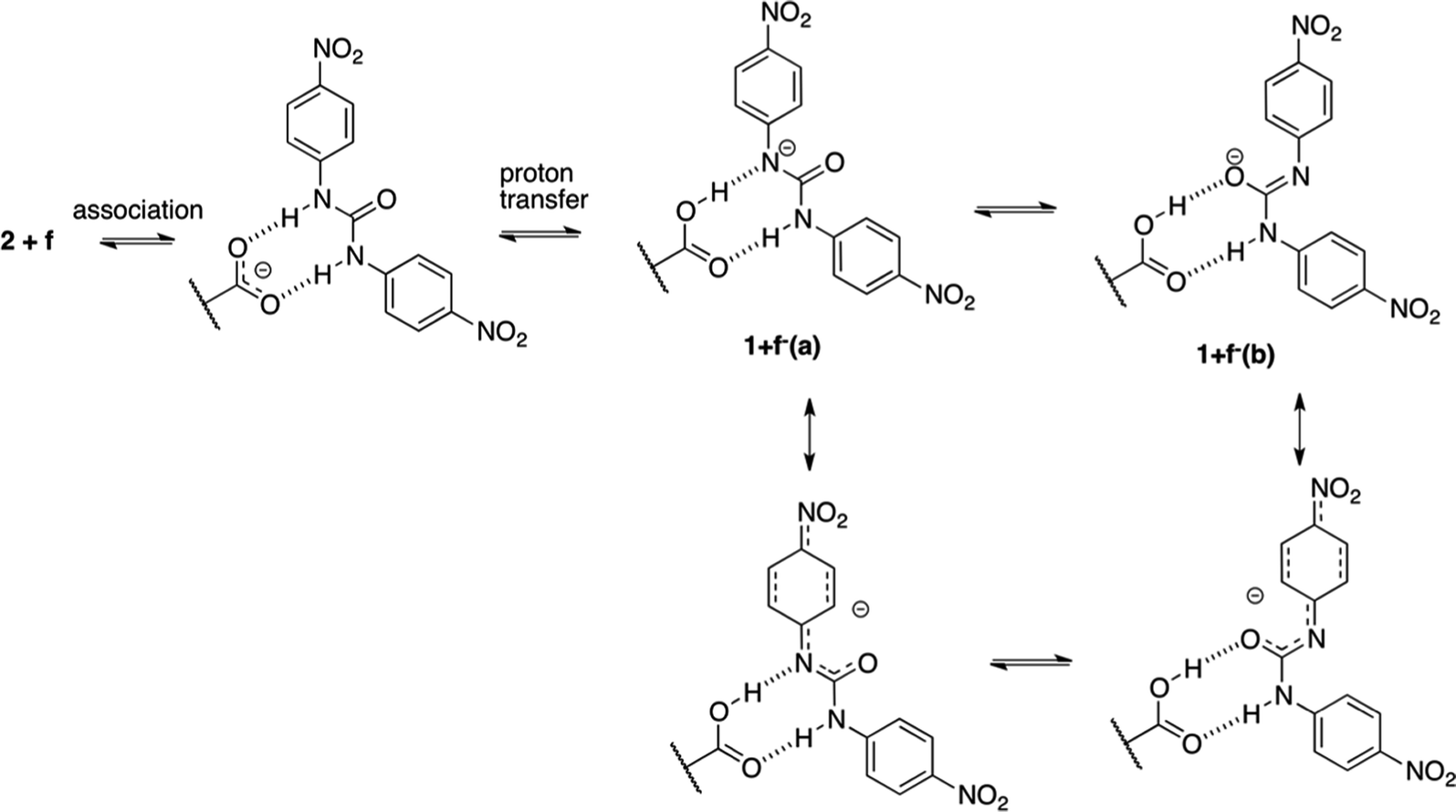 | ||
| Fig. 12 Structure of 2+f complex exhibiting intermolecular proton transfer. | ||
In the above discussion on proposed mechanisms it is important to keep in mind that stability of complexes in the ground state is variable thus some effects may vanish. Unfortunately compounds used here do not exhibit efficient fluorescence that could be used during additional experiments in very systematic way. Still, it was possible to record fluorescence spectra and to conclude the origins of the variable photoisomerization reactions. Thus, the respective spectra were recorded in chosen solvents to study the ICT within 1 and 2. Due to the space limitations those results were placed in ESI† and more detailed studies are planned with the use of highly fluorescent compounds.
The change in properties of the substituent is able to tune the photophysical properties of molecules as, for example, association constants but also the molar attenuation coefficient (ε). Table 3 contains the ε and λ for 1–4 and a–f.
| Compound | ε [dm3 mol−1 cm−1], λ [nm] | Compound | ε [dm3 mol−1 cm−1], λ [nm] |
|---|---|---|---|
| 1 | 19200, 326 |
b | 14200, 256 |
| 2 | 22700, 280 |
c | 12700, 252 |
| 3 | 19800, 271 |
d | 14100, 262 |
| 4 | 22500, 258 |
e | 11700, 266 |
| a | 16300, 276 |
f | 19700, 340 |
It is worth reminding here that the association in the ground state is substituent-dependent and the amount of the cis form in the photostationary state also follows the substituent effect (Fig. 8). A correlation for k = f(ε) for the acceptor part of data is equal to R = 0.79 while for donors R = 0.98. The similar applies for k = f (Hammett substituent constant) where for acceptors R = 0.98 and donors R = 1.00 (n = 4 and n = 3, respectively). No such tendency was observed for the whole set of substituents. This suggests the light-initiated proton transfer in the complexes takes place in case of acceptors and another (vide supra) mechanism is present in donor part of series a–f. This type of correlations (separate for acceptors and donors) has been already described in a series of azobenzenes suggesting two different mechanisms of photoreaction.37,52,53
Computations
In order to have a deeper insight into the non-covalent interactions a series of computations were performed. These are: (a) geometry optimization of 1, 2 and a–f and their complexes, (b) determination of intermolecular interaction energy, (c) QTAIM-based54,55 calculation of the hydrogen bond energy,56,57 (d) calculation of the barrier of proton transfer in the ground state for NO2 derivative. The results are discussed below while geometries of the optimized structures are collected in ESI.† The QTAIM data consist of Laplacian value calculated for hydrogen bond critical point (H-BCP) and the energy of the hydrogen bond (EHB) calculated using Espinosa's approach (Table 4).56,57| Complex | trans | cis | ||
|---|---|---|---|---|
| H⋯O10 | H⋯O11 | H⋯O10 | H⋯O11 | |
| a N⋯HO hydrogen bond. | ||||
| 2+a | 1.865 | 1.866 | 1.880 | 1.886 |
| 0.0937 | 0.0933 | 0.0907 | 0.0897 | |
| −31.7 | −31.5 | −30.2 | −29.8 | |
| 2+b | 1.869 | 1.855 | 1.879 | 1.867 |
| 0.0926 | 0.0955 | 0.0914 | 0.0937 | |
| −31.4 | −32.7 | −30.7 | −31.6 | |
| 2+c | 1.852 | 1.847 | 1.870 | 1.860 |
| 0.0956 | 0.0967 | 0.0925 | 0.0946 | |
| −32.9 | −33.3 | −31.4 | −32.1 | |
| 2+d | 1.833 | 1.841 | 1.847 | 1.854 |
| 0.0992 | 0.0976 | 0.0962 | 0.0970 | |
| −34.8 | −33.9 | −33.2 | −33.3 | |
| 2+e | 1.830 | 1.817 | 1.836 | 1.832 |
| 0.0994 | 0.1022 | 0.0980 | 0.0997 | |
| −35.1 | −36.4 | −34.2 | −34.9 | |
| 2+f | 1.791 | 1.800 | 1.806 | 1.801 |
| 0.1058 | 0.1064 | 0.1035 | 0.1050 | |
| −38.8 | −38.9 | −37.4 | −37.9 | |
| 1+f− | 1.721a | 1.922 | ||
| 0.0881 | 0.0853 | |||
| −56.5 | −26.8 | |||
Based on the data in Table 4 it is easy to note that the hydrogen bond length follows the character the substituent expressed as the Hammett sigma constant58 (σ) giving the correlation coefficients (R) for NH⋯O = f(σ) linear function from 0.88 to 0.95 (NH⋯O10 in trans form and NH⋯O11 in the cis form, respectively). The same is realized for values of Laplacian (R = 0.87–097) and the EHB (R = 0.88–0.95) in complexes. That shows the effect of the substituent on association is regular. The last row in Table 4 refers to 1+f− complex and shows the hydrogen bonding in the ground state after proton transfer is stronger than before such reaction (complex 2+f). Also, the BSSE (Basis Set Superposition Error) corrected energies of intermolecular interaction (Eint) collected in Table 5 show the character of the substituent in urea derivatives influences linearly the Eint values. As expected the strongest interaction was found for NO2 substituted derivative.
| trans form | cis form | ||
|---|---|---|---|
| Complex | Eint | Complex | Eint |
| 2+a | −47.1 | 2+a | −45.7 |
| 2+b | −50.0 | 2+b | −47.5 |
| 2+c | −53.6 | 2+c | −52.8 |
| 2+d | −57.1 | 2+d | −55.9 |
| 2+e | −62.0 | 2+e | −59.3 |
| 2+f | −69.7 | 2+f | −67.7 |
| 1+f− | −43.5 | 1+f− | −37.4 |
The computationally obtained data related to the energy of intermolecular interaction, in general, follow the effect of the substituent in anion binding (both in trans and cis forms) as for previously studied derivatives.5,6 Thus, it is reasonable to tell the same effects drive the association in the currently studied compounds. Moreover, the proton transfer reaction suggested by the experimental data is supported by the strength of the hydrogen bonds in 1+f− complex versus that in 2+f. In these complexes the sum of the energy of hydrogen bonds is higher in 1+f− than that in 2+f. Unfortunately the studied complexes and processes are too large to consider computations of reactions taking place in their excited state. Instead, the calculations were performed to inspect the proton transfer reaction in ground state. The DFT calculations show the proton transfer in 2+f complex is a barrier-less process (or the real barrier is very low), while the energy of the 1+f− complex is 26.9 kJ mol−1 higher than that for 2+f (relatively low comparing to Eint for both complexes).
It was mentioned that during the titration of the trans-2/cis-2 mixture the data suggest two individuals are present in solution for cis isomer. The only possibility is that two cis forms in various rotameric forms are present. This might be a bit confusing since rotation around single bonds should be easy in solution and, for sure, it is so in the trans form (no steric intramolecular interaction is present in this isomer). However, for cis-2 form it is easy to recognize the competition between two opposite mechanisms responsible for rotamerism. These are (a) steric hindrance that acts as a destabilizing force and (b) the π-electron conjugation that stabilize the conjugated structure. The said π-electron conjugation in cis form is not as efficient as in trans conformation because steric interaction prohibits the co-planarity of two parts of molecule. We have studied rotamerism in cis form by DFT methods. Indeed, two rotamers of the cis-2 (a and b, Fig. 13) are different in energy by ca. 12 kJ mol−1, but what is more important the transition state for such process (cis(TS)) is located at the energy level higher than 30 kJ mol−1 with respect to the most stable form (cis(a)).
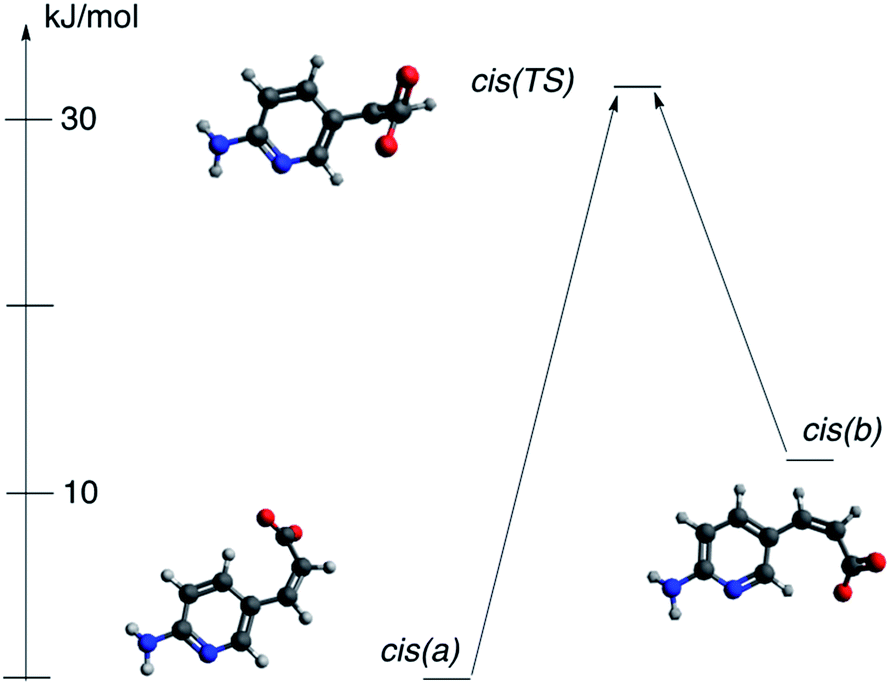 | ||
| Fig. 13 Two rotameric forms of anion of 2 in cis form and transition state between those forms. | ||
The variable stability of forms cis(a) and cis(b) may be attributed to the relatively weak CH⋯O interaction. This kind of interaction, although, weak, might be responsible for molecular packing in crystal as shown by us59 and others.60,61 If so, the CH moiety being in close proximity to the negatively charged oxygen may stabilize considered rotamers (both H6 and H4 protons interact with oxygen carrying negative charge). The respective H⋯O distances are: 2.051 and 2.125 Å for cis(a) and cis(b), respectively. As shown in Fig. 13 the energy of cis(b) form with respect to the cis(a) is ca. 12 kJ mol−1. The QTAIM-based data for CH⋯O interaction energy calculated by Espinosa's approach gives values of −20.6 kJ mol−1 and −17.2 kJ mol−1 for cis(a) and cis(b) forms, respectively. The Laplacian of electron density is equal to 0.0740 and 0.0668 confirming the interaction is of hydrogen bond nature according to the recent definition of hydrogen bond.62 Thus, in a middle-complicated system studied here the computations shed additional light on properties of molecules.
Experimental
To keep the conditions of experiments constant procedures related to intermolecular interactions were the same as in our recent publication on bisurea and association in polar solvent.7 The synthesis of the main compound (1) was performed by the reaction of 2-amino-5-bromopyridine with acrylic acid under the Heck coupling conditions, while substituted ureas were obtained by reaction of the respective anilines with 1,1′-carbonyldiimidazole.63 The salt 2 was obtained by reaction of 1 with tetrabutylammonium hydroxide, as before.6 All NMR spectra were recorded on Bruker Avance 3HD 400 MHz spectrometer working at 400 MHz and 100 MHz for 1H and 13C, respectively. Irradiation was performed with the use of xenon lamp with water filter to remove the heat flow from the lamp to studied solution. The samples were kept in dark after irradiation. Degree of isomerization was calculated by the comparison of the integrals of respective signals in 1H NMR spectra of the irradiated sample. The signal at ca. 7.22 ppm (3JH,H = ca. 16 Hz) was used to identify trans isomer and the one at ca. 6.15 ppm (3JH,H = ca. 12 Hz) for cis isomer. The back reaction (cis → trans) was not detected after keeping solution at 50 °C for 4 h. The photoisomerization rate constants were determined with the use of integrals of three protons (H4, H6 and H7) in trans and cis forms of 2. The irradiation for UV-vis measurements of the trans–cis isomerization was performed with xenon lamp equipped with monochromator, λEM = 330 nm in a quartz cuvette. Acetonitrile was used as a solvent. The samples were stirred during irradiation. The steady-state absorption and emission spectra were recorded at room temperature using a Shimadzu UV-vis Multispec-1501 spectrophotometer and a Hitachi F-7100 spectrofluorimeter, respectively. The fluorescence quantum yields for the acid (1), its salt (2) and complexes (2+a, 2+c, 2+f) in acetonitrile were calculated using following equation:where: Is and Iref are the integrated emission intensity, ns and nref are the refractive indexes of the solvents used for the sample and the reference, respectively.
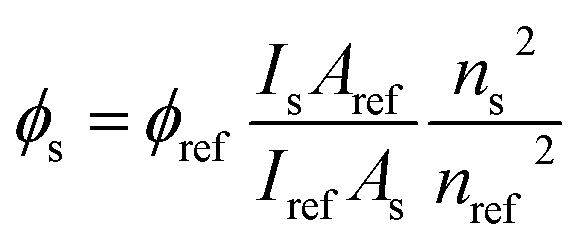
The absorbances (A) of both the sample tested and reference solution at an excitation wavelength (325 nm) was ca. 0.1. 9,10-Diphenylanthracene in cyclohexane (ϕref = 0.90 (ref. 64)) was used as reference. Computations were carried out with Gaussian program65 (M05/6-311++G(2d,2p) level of computation) with the PCM66 model of solvation and the same solvent as in experiments. The frequency calculations (the same level of theory) showed all structures were optimized (zero imaginary frequencies) except for the transition state investigated (one imaginary frequency). The interaction energy (Eint.) was corrected to the basis set superposition error (BSSE) in vacuum with the use of counterpoise methods as implemented in Gaussian with default settings. The zero-point energy correction was applied.
3-(6-Aminopyridin-3-yl)acrylic acid (1)67
Acrylic acid (2.38 g, 0.033 mol) was added carefully to a solution of 2-amino-5-bromopyridine (2.59 g 0.015 mol) and Na2CO3, (5.56 g 0.053 mol) in H2O (50 cm3). Next, PdCl2 (0.053 g, 0.0003 mol) was added and initiated the Heck reaction. Mixture was heated (100 °C) at reflux for 24 h. After, the reaction was cooled to room temperature and filtered. Filtrate was adjusted to pH 6 with aqueous HCl. Additional H2O (20 cm3) was added to improve mixing, and the mixture was stirred for 1 h. Then the solid was collected by filtration. The filter pad was washed sequentially with H2O (20 cm3), cold absolute EtOH (20 cm3). Drying under vacuum gave 1.53 g (62%) as pale yellow solid, mp 224.6–228.2 °C. 1H NMR (TMS, DMSO) δ: 12.08 (bs, 1H), 8.12 (d, 3JHH = 2.3 Hz, 1H), 7.76 (dd, 3JHH = 8.7 Hz, 1H), 7.43 (d, 3JHH = 15.9 Hz, 1H), 6.53 (bs, 1H), 6.45 (d, 3JHH = 8.7 Hz, 1H), 6.22 (d, 3JHH = 15.9 Hz, 1H). 13C NMR (TMS, DMSO) δ: 168.54, 161.41, 151.02, 142.47, 135.59, 118.97, 114.09, 108.77. Anal. calc. for C8H8N2O2: C 58.53, H 4.91, N 17.06. Found C 58.46, H 5.02, N 16.98.Tetra-n-butylammonium 3-(6-aminopyridin-3-yl)acrylate (2)
3-(6-Aminopyridin-3-yl)acrylic acid (1 g, 0.006 mol) was dissolved in MeOH (30 cm3). Solution was adjusted to pH = 7 using tetrabutylammonium hydroxide. Then, MeOH was evaporated. Residue was washed with cold MeOH. Drying under vacuum gave 1.84 g (74%) as brown yellow solid, mp 114.6–117.2 °C. 1H NMR (TMS, DMSO) δ: 7.91 (d, 3JHH = 2.2 Hz, 1H), 7.54 (d, 3JHH = 8.6 Hz, 1H), 6.90 (d, 3JHH = 16.1 Hz, 1H), 6.41 (d, 3JHH = 8.6 Hz, 1H), 6.11 (d, 3JHH = 16.1 Hz, 1H), 6.09 (bs, 1H), 3.17 (m, 8H), 1.57 (m, 8H), 1.3 (m, 8H), 0.94 (m, 12H). 13C NMR (TMS, DMSO) δ: 169.81, 160.03, 148.26, 134.67, 133.06, 126.67, 121.34, 108.64, 57.97, 23.54, 19.67, 13.97. Anal. calc. for C24H43N3O2: C 71.07, H 10.69, N 10.36. Found C 70.98, H 10.83, N 10.27. trans-3-Phenylacrylic acid (3) was available from commercial sources.Tetra-n-butylammonium (2E)-3-phenylprop-2-enoate (4)
Cinnamic acid was (1 g, 0.007 mol) dissolved in MeOH (30 cm3) and the solution was adjusted to pH = 7 using tetrabutylammonium hydroxide. Then, MeOH was evaporated and residue was washed with small portions of cold MeOH and dried under vacuum. That 1.89 g (72.00%) of 4 as light-yellow oil. 1H NMR (TMS, DMSO) δ: 7.46 (d, 3JHH = 7.6 Hz, 2H), 7.32 (t, 2H), 7.25 (t, 1H), 7.11 (d, 3JHH = 16 Hz, 1H), 6.42 (d, 3JHH = 16 Hz, 1H), 3.17 (m, 8H), 1.54 (m, 8H), 1.3 (m, 8H), 0.94 (m, 12H). 13C NMR (TMS, DMSO) δ: 169.70, 137.24, 135.68, 130.72, 129.04, 128.32, 127.27, 57.98, 23.56, 19.65, 13.94. Anal. calc. for C25H43NO2: C 77.07, H 11.12, N 3.60. Found C 76.97, H 11.30, N 3.53.General procedure for synthesis of substituted diphenylureas (a–f)
In anhydrous THF (50 cm3) was dissolved 4-substituted aniline (0.01 mol) and 1,1′-carbonyldiimidazole (0.81 g, 0.005 mol). Mixture was heated at reflux during 24 h. Then, solvent was evaporated, residue was recrystallized from MeOH and dried under vacuum.N,N′-Bis[4-(dimethylamino)phenyl]urea (a)
N,N-Dimethylbenzene-1,4-diamine (1.36 g) gave product (1.26 g, 85%) as brown solid, mp 252.3–257.8 °C (lit.68 250 °C). 1H NMR (TMS, DMSO) δ: 8.13 (bs, 2H), 7.23 (d, 3JHH = 9.2 Hz, 4H), 6.68 (d, 3JHH = 9.0 Hz, 4H), 2.82 (s, 12H). 13C NMR (TMS, DMSO) δ: 153.50, 146.72, 130.40, 120.41, 113.67, 41.23. Anal. calc. for C17H22N4O: C 68.43, H 7.43, N 18.78. Found C 68.52, H 7.51, N 18.63.N,N′-Bis(4-methoxyphenyl)urea (b)
4-Methoxyaniline (1.23 g) gave product (1.19 g, 87%) as white-grey powder, mp 239.5–241.6 °C (lit.69 239–240 °C). 1H NMR (TMS, DMSO) δ: 8.37 (bs, 2H), 7.33 (d, 3JHH = 9.2 Hz, 4H), 6.85 (d, 3JHH = 9.2 Hz, 4H), 3.71 (s, 6H). 13C NMR (TMS, DMSO) δ: 154.7, 153.4, 133.39, 120.35, 114.41, 55.62. Anal. calc. for C15H16N2O3: C 66.16, H 5.92, N 10.29. Found C 66.02, H 5.99, N 10.15.N,N′-Diphenylurea (c)
Aniline (0.93 g) gave product (0.81 g, 76%) as a white powder, mp 239–241 °C (lit.70 235–237 °C). 1H NMR (TMS, DMSO) δ: 8.65 (bs, 2H), 7.44 (d, 3JHH = 8.4 Hz, 4H), 7.28 (t, 4H), 6.97 (t, 2H). 13C NMR (TMS, DMSO) δ: 153.00, 140.17, 129.26, 122.27, 118.85. Anal. calc. for C13H12N2O: C 73.56, H 5.70, N 13.20. Found C 73.49, H 5.79, N 13.01.N,N′-Bis(4-bromophenyl)urea (d)
4-Bromoaniline (1.7 g) gave product (1.29 g, 70%) as white powder, mp 291–294 °C (lit.71 292–293 °C). 1H NMR (TMS, DMSO) δ: 8.85 (bs, 2H), 7.44 (m, 8H). 13C NMR (TMS, DMSO) δ: 152.73, 139.42, 131.99, 120.99, 113.85. Anal. calc. for C13H10Br2N2O: C 42.20, H 2.72, N 7.57. Found C 42.09, H 2.80, N 7.43.N,N′-Bis[4-(trifluoromethyl)phenyl]urea (e)
4-(Trifluoromethyl)aniline (1.61 g) gave product (1.31 g, 75%) as a pale yellow needles, mp 231.2–234.5 °C (lit.72 228–230 °C). 1H NMR (TMS, DMSO) δ: 9.25 (bs, 2H), 7.67 (m, 8H). 13C NMR (TMS, DMSO) δ: 152.58, 143.58, 126.65 (q, JC,F = 3.6 Hz), 123.63 (q, JC,F = 270.6 Hz), 122.10 (q, JC,F = 31.7 Hz), 118.59. Anal. calc. for C15H10F6N2O: C 51.73, H 2.89, N 8.04. Found C 51.80, H 2.94, N 7.98.N,N′-Bis(4-nitrophenyl)urea (f)
4-Nitroaniline (1.38 g) gave product (0.94 g, 62%) as yellow solid, mp 299–305 °C (lit.73 299–305 °C). 1H NMR (TMS, DMSO) δ: 9.71 (bs, 2H), 8.22 (d, 3JHH = 12 Hz, 4H), 7.73 (d, 3JHH = 8.8 Hz, 4H). 13C NMR (TMS, DMSO) δ: 152.12, 146.17, 141.97, 125.62, 118.45. Anal. calc. for C13H10N4O5: C 51.66, H 3.33, N 18.54. Found C 51.55, H 3.40, N 18.45.Conclusions
The synthesized acid isomerizes but it does not form dimers in polar solvent. However, we observed that the trans/cis photoisomerization of its salt is influenced by non-covalent interactions with ureas. It was shown, in contrast to usual role of ureas (organocatalysis), that these compounds might act as organoinhibitor in photoisomerization reaction. In this sense the supramolecular counterpart carrying acidic group with acidity tuned by the substituent character is able to influence the photochemical processes within the complexes. The said tuned acidity results in variable association but also influences the tendency of intermolecular, excited state proton transfer. The substituent effect might be used as a method for studying photochemical behavior of supramolecular complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre, Poland under Grant 2013/09/B/ST4/02308. PL-Grid Infrastructure supported this research.Notes and references
- G. R. Desiraju, Angew. Chem., Int. Ed., 1995, 34, 2311–2327 CrossRef.
- V. R. Hathwar, T. S. Thakur, T. N. G. Row and G. R. Desiraju, Cryst. Growth Des., 2011, 11, 616–623 CrossRef.
- J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2012, 12, 5873–5877 CrossRef.
- A. Azhdari Tehrani, L. Esrafili, S. Abedi, A. Morsali, L. Carlucci, D. M. Proserpio, J. Wang, P. C. Junk and T. Liu, Inorg. Chem., 2017, 56, 1446–1454 CrossRef PubMed.
- K. Mroczyńska, M. Kaczorowska, E. Kolehmainen, I. Grubecki, M. Pietrzak and B. Ośmiałowski, Beilstein J. Org. Chem., 2015, 11, 2105–2116 CrossRef PubMed.
- B. Ośmiałowski, K. Mroczyńska, E. Kolehmainen, M. Kowalska, A. Valkonen, M. Pietrzak and K. Rissanen, J. Org. Chem., 2013, 78, 7582–7593 CrossRef PubMed.
- A. Kwiatkowski, I. Grela and B. Ośmiałowski, New J. Chem., 2017, 41, 1073–1081 RSC.
- P. R. Schreiner and A. Wittkopp, Org. Lett., 2002, 4, 217–220 CrossRef PubMed.
- A. Zafar, S. J. Geib, Y. Hamuro and A. D. Hamilton, New J. Chem., 1998, 22, 137–141 RSC.
- A. E. Hooper, S. R. Kennedy, C. D. Jones and J. W. Steed, Chem. Commun., 2016, 52, 198–201 RSC.
- P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289–296 RSC.
- S. Mohamed, D. A. Tocher, M. Vickers, P. G. Karamertzanis and S. L. Price, Cryst. Growth Des., 2009, 9, 2881–2889 CrossRef.
- C. B. Aakeröy, M. E. Fasulo and J. Desper, Mol. Pharm., 2007, 4, 317–322 CrossRef PubMed.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharm., 2007, 4, 323–338 CrossRef PubMed.
- A. Parkin, C. C. Seaton, N. Blagden and C. C. Wilson, Cryst. Growth Des., 2007, 7, 531–534 CrossRef.
- C. B. Aakeröy, I. Hussain and J. Desper, Cryst. Growth Des., 2005, 6, 474–480 CrossRef.
- D. A. Adsmond, A. S. Sinha, U. B. R. Khandavilli, A. R. Maguire and S. E. Lawrence, Cryst. Growth Des., 2016, 16, 59–69 CrossRef.
- B. Ośmiałowski, E. Kolehmainen, S. Ikonen, A. Valkonen, A. Kwiatkowski, I. Grela and E. Haapaniemi, J. Org. Chem., 2012, 77, 9609–9619 CrossRef PubMed.
- T. Mori and Y. Inoue, in Molecular and supramolecular photochemistry, ed. A. G. Griesbeck and J. Mattay, CRC Press Taylor & Francis Group, 2004, ch. CC Photoinduced Isomerization Reactions, p. 417 Search PubMed.
- W. M. Horspool, in Photochemistry, ed. I. Dunkin, RCS Publishing, 2007, vol. 36, pp. 55–90 Search PubMed.
- J. Herzfeld and J. C. Lansing, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 73–95 CrossRef PubMed.
- H. Kandori, Y. Shichida and T. Yoshizawa, Biochemistry, 2001, 66, 1197–1209 Search PubMed.
- T. Okada and K. Palczewski, Curr. Opin. Struct. Biol., 2001, 11, 420–426 CrossRef PubMed.
- J. L. Spudich, Science, 2000, 288, 1358–1359 CrossRef PubMed.
- A. S. Lubbe, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2017, 46, 1052–1079 RSC.
- M. Asakawa, P. R. Ashton, V. Balzani, C. L. Brown, A. Credi, O. A. Matthews, S. P. Newton, F. M. Raymo, A. N. Shipway, N. Spencer, A. Quick, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem.–Eur. J., 1999, 5, 860–875 CrossRef.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- V. V. Avdeeva, M. I. Buzin, E. A. Malinina, N. T. Kuznetsov and A. V. Vologzhanina, CrystEngComm, 2015, 17, 8870–8875 RSC.
- G. Kaupp and J. Schmeyers, J. Photochem. Photobiol., B, 2000, 59, 15–19 CrossRef.
- Y. Hasegawa, K. Takahashi, S. Kume and H. Nishihara, Chem. Commun., 2011, 47, 6846–6848 RSC.
- Y. Norikane, K. Kitamoto and N. Tamaoki, J. Org. Chem., 2003, 68, 8291–8304 CrossRef PubMed.
- D. Aronzon, E. P. Levy, P. J. Collings, A. Chanishvili, G. Chilaya and G. Petriashvili, Liq. Cryst., 2007, 34, 707–718 CrossRef.
- E. Merino and M. Ribagorda, Beilstein J. Org. Chem., 2012, 8, 1071–1090 CrossRef PubMed.
- T.-T. Yin, Z.-X. Zhao and H.-X. Zhang, New J. Chem., 2017, 41, 1659–1669 RSC.
- P. Weis, D. Wang and S. Wu, Macromolecules, 2016, 49, 6368–6373 CrossRef.
- E. Bernard, P. Britz-McKibbin and N. Gernigon, J. Chem. Educ., 2007, 84, 1159–1161 CrossRef.
- G. Angelini, N. Canilho, M. Emo, M. Kingsley and C. Gasbarri, J. Org. Chem., 2015, 80, 7430–7434 CrossRef PubMed.
- B. Ośmiałowski, E. Kolehmainen, E. Kalenius, B. Behera, R. Kauppinen and E. Sievänen, Struct. Chem., 2011, 22, 1143 CrossRef.
- L. Cao, R. Jiang, Y. Zhu, X. Wang, Y. Li and Y. Li, Eur. J. Org. Chem., 2014, 2687–2693 CrossRef.
- C. R. Bondy and S. J. Loeb, Coord. Chem. Rev., 2003, 240, 77–99 CrossRef.
- N. A. Esipenko, P. Koutnik, T. Minami, L. Mosca, V. M. Lynch, G. V. Zyryanov and P. Anzenbacher, Chem. Sci., 2013, 4, 3617–3623 RSC.
- A. J. Evans and P. D. Beer, J. Chem. Soc., Dalton Trans., 2003, 4451–4456, 10.1039/b308541d.
- X. Huang, B. Wu, C. Jia, B. P. Hay, M. Li and X.-J. Yang, Chem.–Eur. J., 2013, 19, 9034–9041 CrossRef PubMed.
- B. Jędrzejewska, B. Ośmiałowski and R. Zaleśny, Photochem. Photobiol. Sci., 2016, 15, 117–128 RSC.
- J. M. McGrath and M. D. Pluth, J. Org. Chem., 2014, 79, 11797–11801 CrossRef PubMed.
- L. G. Arnaut and S. J. Formosinho, J. Photochem. Photobiol., A, 1993, 75, 1–20 CrossRef.
- D. Esteban-Gómez, L. Fabbrizzi and M. Licchelli, J. Org. Chem., 2005, 70, 5717–5720 CrossRef PubMed.
- Y.-A. Chen, F.-Y. Meng, Y.-H. Hsu, C.-H. Hung, C.-L. Chen, K.-Y. Chung, W.-F. Tang, W.-Y. Hung and P.-T. Chou, Chem.–Eur. J., 2016, 22, 14688–14695 CrossRef PubMed.
- S. Tasaki, A. Momotake, Y. Kanna, T. Sato, Y. Nishimura and T. Arai, Photochem. Photobiol. Sci., 2015, 14, 1864–1871 RSC.
- M. Boiocchi, L. Del Boca, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, Chem.–Eur. J., 2005, 11, 3097–3104 CrossRef PubMed.
- M. Boiocchi, L. Del Boca, D. E. Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507–16514 CrossRef PubMed.
- R. Cimiraglia and H.-J. Hofmann, Chem. Phys. Lett., 1994, 217, 430–435 CrossRef.
- H. Rau and E. Lueddecke, J. Am. Chem. Soc., 1982, 104, 1616–1620 CrossRef.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef.
- E. Espinosa, M. Souhassou, H. Lachekar and C. Lecomte, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 563–572 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef.
- B. Ośmiałowski, E. Kolehmainen, A. Valkonen, M. Kowalska and S. Ikonen, Struct. Chem., 2013, 24, 2203–2209 CrossRef.
- S. Scheiner, Phys. Chem. Chem. Phys., 2011, 13, 13860–13872 RSC.
- J. L. Dragelj, G. V. Janjic, D. Z. Veljkovic and S. D. Zaric, CrystEngComm, 2013, 15, 10481–10489 RSC.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 Search PubMed.
- W. C. Drewe, R. Nanjunda, M. Gunaratnam, M. Beltran, G. N. Parkinson, A. P. Reszka, W. D. Wilson and S. Neidle, J. Med. Chem., 2008, 51, 7751–7767 CrossRef PubMed.
- M. Brouwer Albert, Pure Appl. Chem., 2011, 83, 2213–2228 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Pittsburgh PA, 2009 Search PubMed.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef.
- W. H. Miller, M. A. Seefeld, K. A. Newlander, I. N. Uzinskas, W. J. Burgess, D. A. Heerding, C. C. K. Yuan, M. S. Head, D. J. Payne, S. F. Rittenhouse, T. D. Moore, S. C. Pearson, V. Berry, W. E. DeWolf, P. M. Keller, B. J. Polizzi, X. Qiu, C. A. Janson and W. F. Huffman, J. Med. Chem., 2002, 45, 3246–3256 CrossRef PubMed.
- D. Asthana, R. Pandey and P. Mukhopadhyay, Chem. Commun., 2013, 49, 451–453 RSC.
- S. Gavade, R. Balaskar, M. Mane, P. N. Pabrekar and D. Mane, Synth. Commun., 2012, 42, 1704–1714 CrossRef.
- A. Krogul and G. Litwinienko, J. Mol. Catal. A: Chem., 2015, 407, 204–211 CrossRef.
- R. Chutia and G. Das, Dalton Trans., 2014, 43, 15628–15637 RSC.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef PubMed.
- R. P. McGeary, A. J. Bennett, Q. B. Tran, J. Prins and B. P. Ross, Tetrahedron, 2009, 65, 3990–3997 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, titration curves, absorption spectra, Cartesian of structures. See DOI: 10.1039/c8ra03042a |
| This journal is © The Royal Society of Chemistry 2018 |