
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, aggregation induced emission and through space conjugation of triphenylvinylphenyl substituted [2.2]paracyclophane-1,9-diene†
Chin-Yang Yu * and
Yu-Chun Lai
* and
Yu-Chun Lai
Department of Materials Science and Engineering, National Taiwan University of Science and Technology, 43, Section 4, Keelung Road, Taipei, 10607, Taiwan. E-mail: cyyu@mail.ntust.edu.tw
First published on 25th May 2018
Abstract
4-Bromo substituted [2.2]paracyclophane-1,9-diene was synthesized from the corresponding dithia[3.3]paracyclophane in three steps through benzyne Steven rearrangement, oxidation, and a thermal elimination reaction. 4-Triphenylvinylphenyl substituted [2.2]paracyclophane-1,9-diene was successfully prepared by the Suzuki–Miyaura cross-coupling reaction of 4-bromo substituted [2.2]paracyclophane-1,9-diene and 4,4,5,5-tetramethyl-2-(4-(1,2,2-triphenylvinyl)phenyl)-1,3,2-dioxaborolane using Pd(OAc)2 as a catalyst, S-Phos as a ligand and K3PO4 as a base. The structures of bromo substituted [2.2] paracyclophane-1,9-diene and triphenylvinylphenyl substituted [2.2]paracyclophane-1,9-diene were fully characterized by 1H NMR spectroscopy and X-ray crystallography. 4-Triphenylvinylphenyl substituted [2.2]paracyclophane-1,9-diene exhibited aggregation-induced emission characteristics when the water fraction was higher than 80% in the THF/water mixtures. 4-Triphenylvinylphenyl substituted [2.2]paracyclophane-1,9-diene displays much higher fluorescence when the water fraction is 90% compared to that of model compounds due to both through bond and through space conjugation. To the best for our knowledge, we are the first to synthesize triphenylvinylphenyl substituted [2.2]paracyclophane-1,9-diene with aggregation-induced emission characteristics.
Introduction
[2.2]Paracyclophanes have received tremendous attention over the past six decades.1–5 The structures of [2.2]paracyclophanes which comprise intramolecular face-to-face π-conjugated phenyl rings give rise to unique electronic and optical properties.3,6 The introduction of substituents onto the individual phenyl rings of [2.2]paracyclophanes makes them become chiral molecules that were widely used as the building blocks for organic reactions and catalyst applications.7–9 Recently, a number of conjugated molecules bonded to the two individual phenyl rings of the [2.2]paracyclophanes have been intensively prepared and investigated.10–12 These types of substituted [2.2]paracyclophanes exhibited remarkable intramolecular through space conjugation and charge transfer features which lead to potential applications in optoelectronics.[2.2]Paracyclophane-1,9-dienes were first synthesized by Dewhirst and Cram in the 1960s.13 [2.2]Paracyclophanedienes are one of the paracyclophene families in which the two bridges are replaced by cis-vinylenes that increase the angles between the bridgehead carbons and the other central carbons of the phenyl rings. Therefore, the highly twisted, ring strained nature with cis-vinylene bridges of [2.2]paracyclophanedienes makes them enable to be ring-opened by carbene initiator to form phenylenevinylenes.14–17 The alkoxy or alkyl substituted paracyclophanedienes and their soluble phenylenevinylene homopolymers and block copolymers prepared by direct ring-opening metathesis polymerization (ROMP) have been reported.18–22 In addition, a variety of phanedienes, phanetrienes or phanetetraenes have been synthesized and investigated.23–25 One of the advantages for soluble phenylenevinylene polymers by ROMP of alkoxy or alkyl substituted paracyclophanedienes is that the precise control of the molecular weights, polydispersity index (PDI) as well as the functional end groups as this give low defect structures of the polymers.
In general, most of the conjugated small molecules showed significant fluorescence quenching in the aggregate state due to strong intermolecular interaction. The aggregation-caused quenching (ACQ) effect therefore limits the uses of conjugated molecules in aqueous solution or in solid state. This issue can be solved after the aggregation induced emission (AIE) phenomenon was first discovered by Tang and co-workers.26 After that, the AIE active molecules have been intensively prepared. In particular, tetraphenylethenes (TPE) with propeller-like structures have attracted much attention possibly due to the facile preparation and purification. The TPE and its derivatives have been intensively investigated and widely used as sensors for the detection of a variety of analysts27–29 owing to high luminescence efficiency and good photostability.
Herein, we reported the synthesis of 4-triphenylvinylphenyl substituted [2.2]paracyclophane-1,9-diene (TPE-PCPDE) by Suzuki–Miyaura cross-coupling reaction of 4-bromo substituted [2.2]paracyclophane-1,9-diene and 4,4,5,5-tetramethyl-2-(4-(1,2,2-triphenylvinyl)phenyl)-1,3,2-dioxaborolane using Pd(OAc)2 as a catalyst, S-Phos as a ligand and K3PO4 as a base. The structural characterization, optical properties as well as the aggregation-induced emission characteristics of TPE-PCPDE compared to the model compounds such as paracyclophanediene (PCPDE), tetraphenylethene (TPE) and phenyl substituted tetraphenylethene (P-TPE) were investigated. The structure of triphenylvinylphenyl substituted paracyclophanediene TPE-PCPDE and the model compounds such as PCPDE, TPE and P-TPE is shown in Fig. 1.
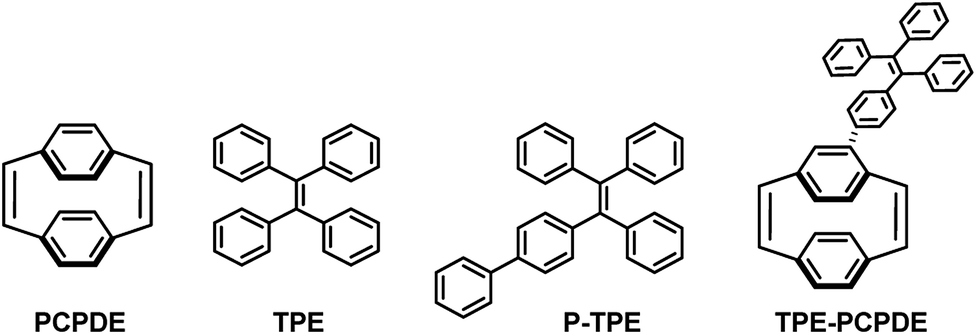 | ||
| Fig. 1 The structure of PCPDE, TPE, P-PCPDE and TPE-PCPDE. | ||
Experimental
Materials and instrumentation
All reagents were from commercial sources and were used without further purification. Solvents used for spectroscopic measurements were spectrograde. Most of the reactions were monitored by thin-layer chromatography carried out on silica gel plates. Preparative separations were performed by column chromatography on silica gel grade 60 (0.040–0.063 mm) from Merck. The starting material, dithiaparacyclophane 1, was synthesized by the modification of previously established synthetic methods (more details see ESI†). Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker ultrashield spectrometer operating at 600 MHz for 1H nuclei and 150 MHz for 13C nuclei. Chemical shifts are reported in ppm. All coupling constants are reported in hertz (Hz). The abbreviations are used to indicate multiplicity: s = singlet, d = doublet, dd = doublet of doublets, t = triplet and m = multiplet. UV-vis absorption and photoluminescence spectra were recorded on the Jasco (V −670) UV-Vis-NIR spectrophotometer and on the Jasco (FP-8500) fluorescence spectrophotometer, respectively. X-ray crystallographic data for single crystals were determined using a Bruker Kappa APEX II diffractometer equipped with an Oxford Cryosystems 700 series cryostream cooler using graphite monochromated Mo-Kα radiation. Electron impact mass spectra (EI-MS) were recorded on a JEOL JMS-700 spectrometer. The melting point of compounds were recorded on a MEL-TEMP 1001D capillary melting point apparatus without further calibration. In order to model the electron affinity and ionization potential, the HOMO and LUMO of PCPDE, TPE, P-TPE and TPE-PCPDE were computed using density functional theory. Becke's three parameter exchange and the Lee–Young–Parry correlation functionals (B3LYP) were used with added polarization basis functions (6-31G*).Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane (3:7) as eluents. The main fraction was collected to give bis(sulfide) compounds as the yellow oils in a yield of 74%. Bis(sulfide) compounds were very difficult to characterize by NMR spectroscopy due to the large numbers of overlapping signals. HRMS (EI, M+): calculated for [C28H23BrS2]+: m/z 502.0425, found m/z 502.0425. Hydrogen peroxide (1.07 mL, 35 wt%) was added dropwise to the solution of bis(sulfide) compounds (3.1 g, 6.16 mmol) in toluene (145 mL) and acetic acid (48 mL) over a period of 30 minutes at 0 °C. The solution was allowed to warm up to room temperature and kept stirring for another 12 hours. The resulting solution was extracted with DCM and brine, dried with anhydrous MgSO4 and concentrated to give compound 2 as yellow oils in a yield of 93% without further purification. Again, the 1H NMR spectrum of compound 2 exhibited a complex range of signals and characterization was not possible. HRMS (EI, M+): calculated for [C28H23BrO2S2]+: m/z 534.0323, found m/z 534.0320 (see Fig. S21 in ESI†).
:hexane (3:7) as eluents. The main fraction was collected to give bis(sulfide) compounds as the yellow oils in a yield of 74%. Bis(sulfide) compounds were very difficult to characterize by NMR spectroscopy due to the large numbers of overlapping signals. HRMS (EI, M+): calculated for [C28H23BrS2]+: m/z 502.0425, found m/z 502.0425. Hydrogen peroxide (1.07 mL, 35 wt%) was added dropwise to the solution of bis(sulfide) compounds (3.1 g, 6.16 mmol) in toluene (145 mL) and acetic acid (48 mL) over a period of 30 minutes at 0 °C. The solution was allowed to warm up to room temperature and kept stirring for another 12 hours. The resulting solution was extracted with DCM and brine, dried with anhydrous MgSO4 and concentrated to give compound 2 as yellow oils in a yield of 93% without further purification. Again, the 1H NMR spectrum of compound 2 exhibited a complex range of signals and characterization was not possible. HRMS (EI, M+): calculated for [C28H23BrO2S2]+: m/z 534.0323, found m/z 534.0320 (see Fig. S21 in ESI†).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 7.19 (d, J = 10.2 Hz, 1H, ArCHCH–), 7.10 (d, J = 10.2 Hz, 1H, ArCHCH–), 7.07 (d, J = 7.8 Hz, 1H, ArH), 6.93 (d, J = 10.2 Hz, 1H, ArCHCH–), 6.66 (s, 1H, ArH), 6.64 (d, J = 7.8 Hz, 1H, ArH), 6.56 (d, J = 7.8 Hz, 1H, ArH), 6.47 (d, J = 7.8 Hz, 2H, ArH), 6.41 (d, J = 7.8 Hz, 1H, ArH). 13C NMR (150 MHz, CDCl3, δ): δ 139.99, 138.08, 138.05, 137.94, 137.59, 136.48, 136.42, 135.75, 135.69, 133.09, 131.67, 130.75, 130.47, 130.40, 126.71, 124.44. HRMS (EI, M+): calculated for [C16H11Br]+: m/z 282.0044, found m/z 282.0044 (see Fig. S22 in ESI†). Mp: 135 °C.:hexane, 1:9 (v/v)) to give compound TPE-PCPDE as white solids in a yield of 81%. 1H NMR (600 MHz, CDCl3, δ): 7.25 (d, J = 10.5 Hz, 1H, ArCHCH–), 7.22 (d (AA′BB′), J = 8.4 Hz, 2H, ArH), 7.20 (d, J = 10.5 Hz, 1H, ArCHCH–), 7.14 (d, J = 10.5 Hz, 1H, ArCHCH–), 7.18–7.04 (m, 16H, ArH), 6.90 (d, J = 10.5 Hz, 1H, ArCHCH–), 6.72 (s, 1H, ArH), 6.65 (d, J = 8.1 Hz, 1H, ArH), 6.56 (d, J = 8.1 Hz, 1H, ArH), 6.51 (d, J = 7.8 Hz, 1H, ArH), 6.47 (d (A2), J = 7.8 Hz, 2H, ArH), 6.36 (d, J = 7.8 Hz, 1H, ArH). 13C NMR (150 MHz, CDCl3, δ): 143.92, 143.81, 143.62, 142.63, 141.39, 141.26, 140.88, 138.62, 138.48, 138.22, 137.81, 137.29, 137.00, 136.07, 134.57, 133.31, 131.60, 131.55, 131.51, 131.50, 131.12, 130.74, 130.54, 130.51, 129.33, 128.66, 127.84, 127.79, 127.76, 127.32, 126.63, 126.58, 126.56, 138.24. HRMS (EI, M+): calculated for [C42H30]+: m/z 534.2348, found m/z 534.2345 (see Fig. S23 in ESI†). Mp: 143 °C.
CH–), 7.19 (d, J = 10.2 Hz, 1H, ArCHCH–), 7.10 (d, J = 10.2 Hz, 1H, ArCHCH–), 7.07 (d, J = 7.8 Hz, 1H, ArH), 6.93 (d, J = 10.2 Hz, 1H, ArCHCH–), 6.66 (s, 1H, ArH), 6.64 (d, J = 7.8 Hz, 1H, ArH), 6.56 (d, J = 7.8 Hz, 1H, ArH), 6.47 (d, J = 7.8 Hz, 2H, ArH), 6.41 (d, J = 7.8 Hz, 1H, ArH). 13C NMR (150 MHz, CDCl3, δ): δ 139.99, 138.08, 138.05, 137.94, 137.59, 136.48, 136.42, 135.75, 135.69, 133.09, 131.67, 130.75, 130.47, 130.40, 126.71, 124.44. HRMS (EI, M+): calculated for [C16H11Br]+: m/z 282.0044, found m/z 282.0044 (see Fig. S22 in ESI†). Mp: 135 °C.:hexane, 1:9 (v/v)) to give compound TPE-PCPDE as white solids in a yield of 81%. 1H NMR (600 MHz, CDCl3, δ): 7.25 (d, J = 10.5 Hz, 1H, ArCHCH–), 7.22 (d (AA′BB′), J = 8.4 Hz, 2H, ArH), 7.20 (d, J = 10.5 Hz, 1H, ArCHCH–), 7.14 (d, J = 10.5 Hz, 1H, ArCHCH–), 7.18–7.04 (m, 16H, ArH), 6.90 (d, J = 10.5 Hz, 1H, ArCHCH–), 6.72 (s, 1H, ArH), 6.65 (d, J = 8.1 Hz, 1H, ArH), 6.56 (d, J = 8.1 Hz, 1H, ArH), 6.51 (d, J = 7.8 Hz, 1H, ArH), 6.47 (d (A2), J = 7.8 Hz, 2H, ArH), 6.36 (d, J = 7.8 Hz, 1H, ArH). 13C NMR (150 MHz, CDCl3, δ): 143.92, 143.81, 143.62, 142.63, 141.39, 141.26, 140.88, 138.62, 138.48, 138.22, 137.81, 137.29, 137.00, 136.07, 134.57, 133.31, 131.60, 131.55, 131.51, 131.50, 131.12, 130.74, 130.54, 130.51, 129.33, 128.66, 127.84, 127.79, 127.76, 127.32, 126.63, 126.58, 126.56, 138.24. HRMS (EI, M+): calculated for [C42H30]+: m/z 534.2348, found m/z 534.2345 (see Fig. S23 in ESI†). Mp: 143 °C.Results and discussion
The routes to synthesize 4-triphenylvinylphenyl-[2.2]paracyclophane-1,9-diene are shown in Scheme 1. The starting material, dithiaparacyclophane 1 (see ESI†), was prepared by the cyclization of an equimolar amount of 2-bromo-1,4-bis(bromomethyl)benzene S2 and 1,4-phenylenedimethanethiol S4 under a high dilution in the presence of aqueous potassium hydroxide for at least 3 days to avoid the formation of oligomers and polymers.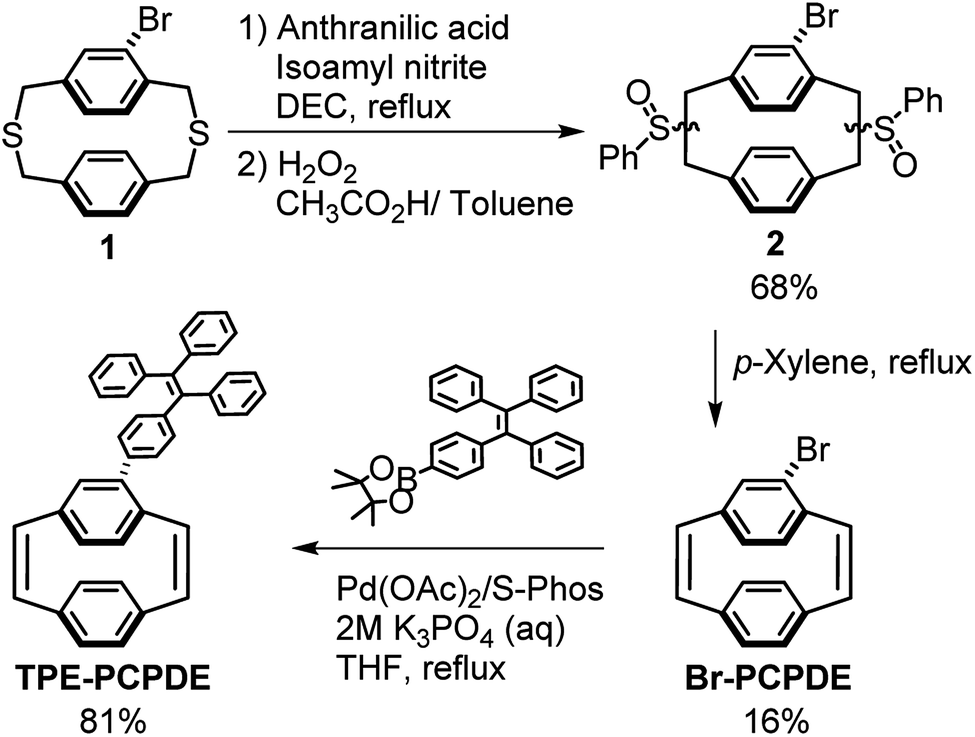 | ||
| Scheme 1 Synthetic route to triphenylvinylphenyl substituted [2.2]paracyclophane-1,9-diene (TPE-PCPDE). | ||
Compound 2 was prepared by the benzyne induced Stevens rearrangement using anthranilic acid and isoamyl nitrite following by oxidation using H2O2 (35 wt%).30,31 Compound 2 was then obtained as semi-solids in an overall yield of 68%. Compound 2 was then dissolved in p-xylene at reflux under an argon atmosphere for 20 hours. The 4-bromo-[2.2]paracyclophane-1,9-diene (Br-PCPDE) was obtained in a yield of 16% after purification by column chromatography and recrystallization. The 4-triphenylvinylphenyl substituted [2.2]paracyclophane-1,9-diene (TPE-PCPDE) was synthesized by the reaction of Br-PCPDE and 4,4,5,5-tetramethyl-2-(4-(1,2,2-triphenylvinyl)phenyl)-1,3,2-dioxaborolane in the presence of Pd(OAc)2 and 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (S-Phos) in THF and aqueous K3PO4 at reflux for 72 hours. After purified by column chromatography, the TPE-PCPDE was obtained in a yield of 81%.
The 1H NMR spectrum of Br-PCPDE in CDCl3 is shown in Fig. 2a. The four doublets (7.24, 7.19, 7.10 and 6.93 ppm, each J = 10.2 Hz, 1H) were assigned to the cis-vinylic hydrogens. Two doublets at 7.07 and 6.41 ppm with J = 7.8 Hz were associated with the hydrogens of non-substituted phenyl ring. In addition, the doublet at 6.47 ppm (J = 7.8 Hz) integrating to two hydrogens corresponds to the hydrogens to the non-substituted phenyl ring. A singlet at 6.66 ppm corresponds to the aromatic hydrogen at the ortho-position to the bromine atom. The two doublets (6.64 and 6.56 ppm, each J = 7.8 Hz, 1H) were assigned to the hydrogens at the para- and meta-positions to the bromine atom, respectively. The 1H NMR spectrum of TPE-PCPDE in CDCl3 is shown in Fig. 2b. The two doublets (7.25 and 7.20 ppm, each J = 10.5 Hz, 1H) were assigned to the cis-vinylic hydrogens. In addition, a doublet at 6.90 ppm (J = 10.5 Hz, 1H) was assigned to the cis-vinylic hydrogen. It should be noted that a doublet at around 7.14 ppm (J = 10.5 Hz, 1H) can be assigned to the remaining cis-vinylic hydrogen which is overlapping with the hydrogens of TPE moiety. The doublet at 7.22 ppm (AA′BB′ system, J = 8.4 Hz, 2H) corresponds to the hydrogens to the ortho position bonded to the carbon of the PCPDE. The multiple between 7.18 and 7.04 ppm represent for the rest of the hydrogens of TPE moiety. A singlet peak at 6.72 ppm and two doublet peaks (6.65 and 6.56 ppm, each J = 8.1 Hz, 1H) were assigned to the aromatic hydrogens at the ortho-, para- and meta-positions to the bromine atom, respectively. The two doublets (6.51 and 6.36 ppm, each J = 7.8 Hz, 1H) were assigned to hydrogens of the non-substituted phenyl ring. In addition, the two doublet peaks at 6.47 ppm (an A2 system) with J = 7.8 Hz are two hydrogens belong to the non-substituted phenyl ring. 2D 1H–1H COSY can confirm the peak assignment and can be found in ESI.†
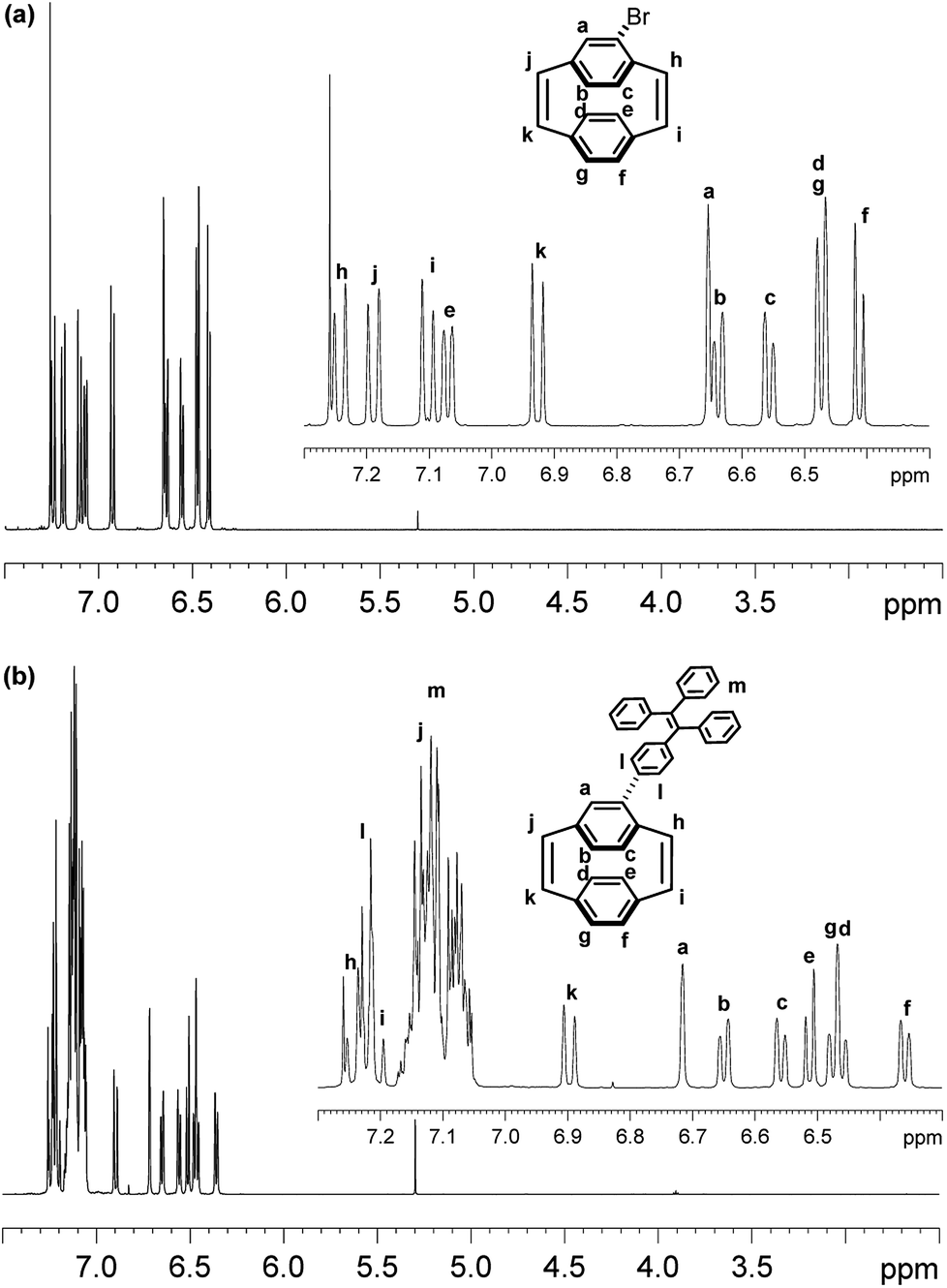 | ||
| Fig. 2 1H NMR spectra of (a) 4-bromo-[2.2]paracyclophane-1,9-diene Br-PCPDE and (b) 4-triphenylvinylphenyl-[2.2]paracyclophane-1,9-diene TPE-PCPDE in CDCl3. Signals associated with alkenic and aromatic hydrogens are shown in the insets. | ||
The single crystal of Br-PCPDE and TPE-PCPDE can be obtained by slow evaporation from a dilute hexane solution at room temperature and the solid-state structure of Br-PCPDE and TPE-PCPDE was determined by X-ray crystallography. For solid-state structure of Br-PCPDE (Fig. 3a), the average vinylene bond length is 1.34 Å which is in agreement with a standard cis-vinylene bond (1.32 Å). The average intramolecular distance between the two carbons of the phenyl rings bonded to carbons of vinylene is 2.78 Å which is the same value with that of the unsubstituted PCPDE reported previously. The average intramolecular separation between the central carbon atoms of the phenyl rings is 3.11 Å. The carbons of the phenyl rings bonded to the carbons of vinylene are significantly out of plane of the four central carbons and the average deviation is of around 13.1°. It should be noted that the largest value (13.43°) of out of plane for the carbon which is in the ortho-position relative to the carbon bonded to bromine atom was observed. The average C(aryl)–C(methine) bond length is 1.50 Å. The solid-state structure of TPE-PCPDE determined by X-ray crystallography is shown in Fig. 3b. The average bond length of vinylene is 1.32 Å which is the same value compared to a standard cis-vinylene bond (1.32 Å). The average intramolecular separation between the central carbon atoms of the phenyl rings is 3.12 Å. The average intramolecular distance between the carbon atoms of the phenyl rings linked to carbons of vinylene is significantly shortened to 2.79 Å. The dihedral angle between the phenyl rings of PCPDE and TPE moieties is around 39°. The carbons of the phenyl rings connected to the vinylene in PCPDE moieties are significantly out of plane of the four central carbons and the average deviation is of around 13.4°. A large distortion in the phenyl rings can be seen which indicates the highly ring strained nature of this molecule. The average C(aryl)–C(methine) bond length is 1.50 Å. Both of the crystal packing structures of compounds Br-PCPDE and TPE-PCPDE (space group: P21/c, Z = 4) showed four molecules in one unit cells and two pairs of them are racemic mixtures due to the chirality center of substituted phenyl rings of paracyclophanedienes.
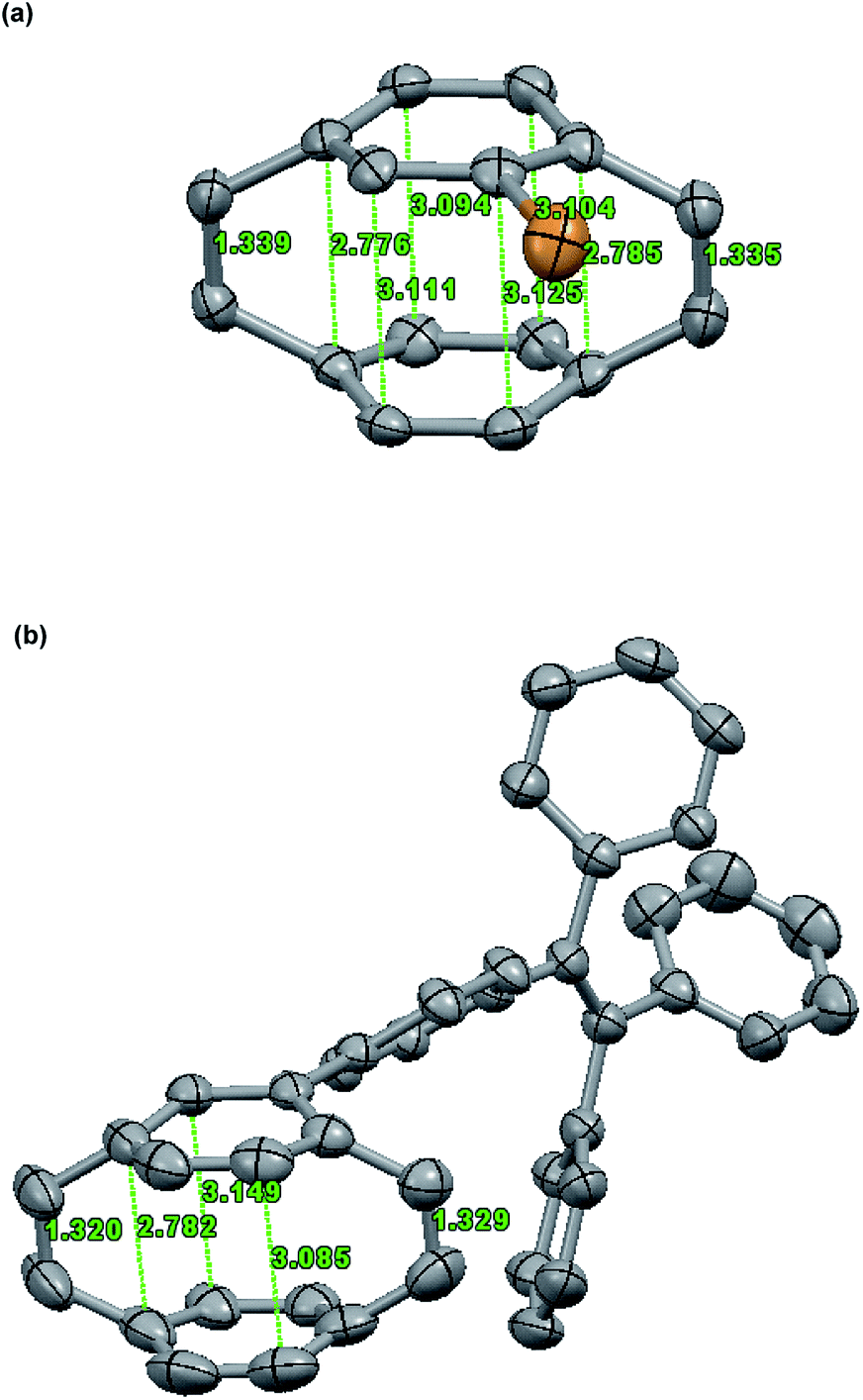 | ||
| Fig. 3 Solid-state structure of (a) Br-PCPDE and (b) TPE-PCPDE. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at 50% probability. | ||
The model compounds PCPDE, TPE and P-TPE were prepared by the modification of established procedures reported previously.32–34 The initial concentration of compounds PCPDE, TPE, P-TPE and TPE-PCPDE is 2 × 10−6 M in dilute THF solution in order to eliminate the potential interference of intermolecular interactions. The UV-vis absorption peak of PCPDE, TPE, P-TPE and TPE-PCPDE in dilute THF solution exhibited at 242, 309, 321 and 325 nm, respectively (Fig. 4a). The notably bathochromic shift from PCPDE, TPE, P-TPE to TPE-PCPDE can be attributed to the extension of the conjugation. It should be noted that the longest absorption wavelength of TPE-PCPDE is red-shifted by 4 nm compared to that of P-TPE possibly due to the through space conjugation of TPE-PCPDE.
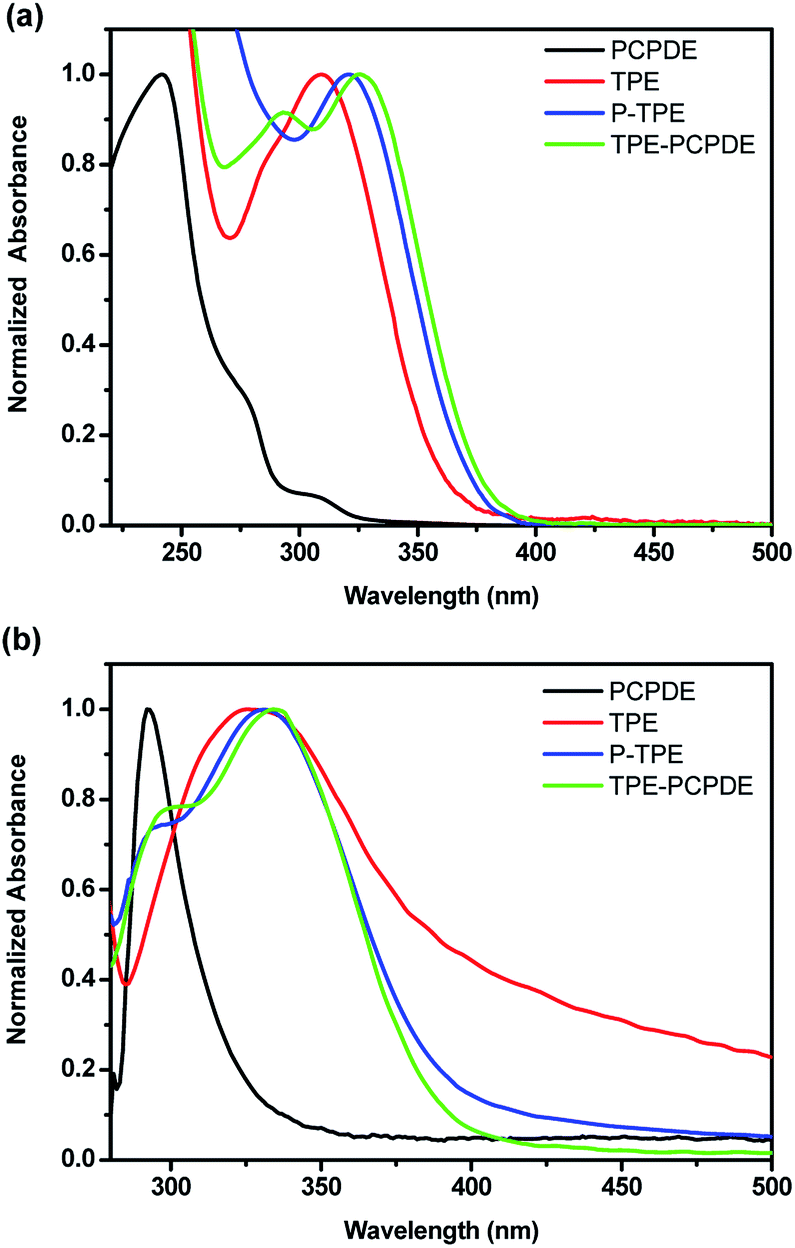 | ||
| Fig. 4 Normalized (a) solution and (b) solid state UV-vis absorption spectra of PCPDE, TPE, P-TPE and TPE-PCPDE. (The initial concentration for solution is 2 × 10−6 M). | ||
The thin films were prepared by spin-casting a solution of the compounds (3 mg mL−1 in toluene) onto glass slides (2.0 cm × 2.0 cm) at a rotation speed of 1500 rpm for 20 seconds and then 3000 rpm for 20 seconds. The normalized solid state UV-vis absorption spectra of PCPDE, TPE, P-TPE and TPE-PCPDE were shown in Fig. 4b. The solid state UV-vis absorption maximum of PCPDE, TPE, P-TPE and TPE-PCPDE is at 292, 326, 331 and 335 nm, respectively which shows bathochromic shifts compared to that of solution due to the relatively strong intermolecular interaction in solid state. The details of optical properties of PCPDE, TPE, P-TPE and TPE-PCPDE are summarized in Table 1.
Again, an absorption maximum of TPE-PCPDE in solid state exhibited a bathochromic shift compared to the model compounds due to the through bond and through space conjugation. It should be noted that the solid state UV-vis absorption spectrum of TPE exhibited a very long tail extending through the whole visible region possibly due to the scattering effect as the quality of films of TPE is poor. In addition, the comparative HOMO and LUMO structures and levels of PCPDE, TPE, P-TPE and TPE-PCPDE using density functional theory calculation (B3LYP functional, 6-31G* basis set) were shown in ESI (Fig. S27 and Table S1†). The HOMO levels and LUMO levels became upper and lower, respectively on going from PCPDE, TPE, P-TPE to TPE-PCPDE. Based on the experimental measurements and theoretical calculations, the through bond and through space conjugation make significant contributions to the absorption wavelength and bandgap in TPE-PCPDE when compared with PCPDE, TPE and P-TPE.
The emission spectra of TPE-PCPDE with different water fractions (fw) in THF are shown in Fig. 5a. The maximum emission intensity of TPE-PCPDE is very low with little change when the water fraction is lower than 80%. However, the maximum emission intensity of TPE-PCPDE shows significant increase when the water fraction is higher than 80%. This indicates that the TPE-PCPDE exhibited aggregation-induced emission characteristics in THF/water mixtures when the water fraction is higher than 80%. The maximum emission wavelength of TPE-PCPDE is at 475 nm in its most aggregated state. The pictures of TPE-PCPDE in THF with different water contents under the UV lamp irradiation with a wavelength of 365 nm are shown in Fig. 5b. Clearly, the TPE-PCPDE in pure THF solution and lower water contents (<80% of water) showed very weak fluorescence. In addition, the fluorescence can be seen by naked eyes for TPE-PCPDE when the water fraction is higher than 80% in the THF/water mixture.
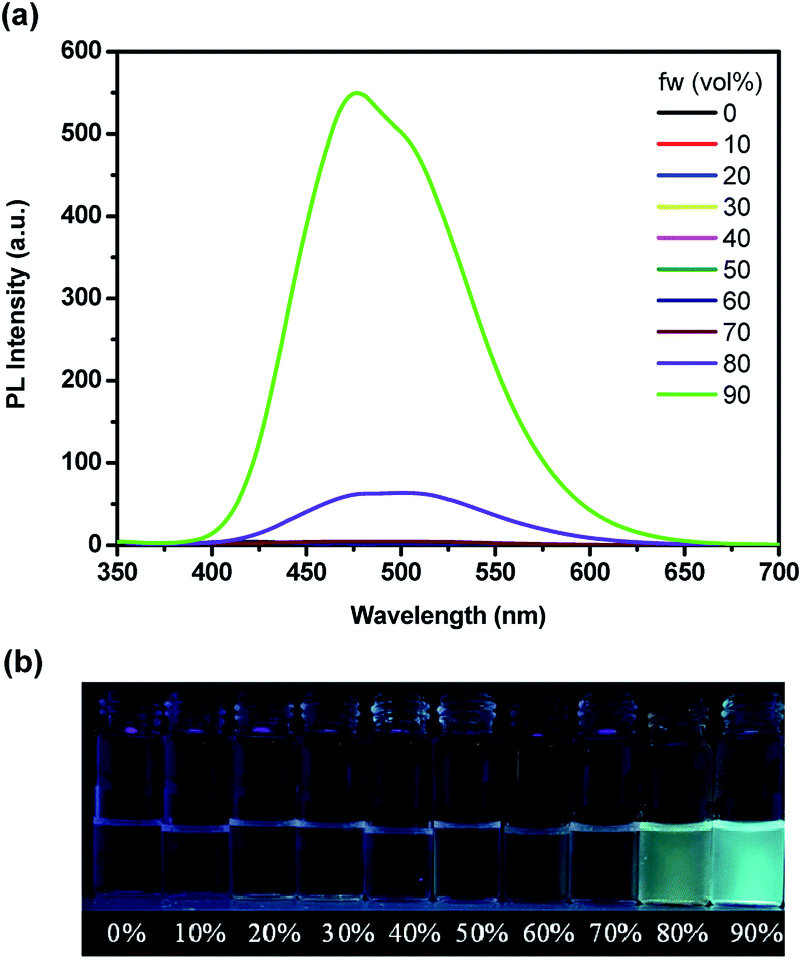 | ||
| Fig. 5 (a) Photoluminescence spectra of TPE-PCPDE in THF/water mixtures with different water fractions (initial concentration: 2 × 10−6 M) and (b) the pictures of TPE-PCPDE in THF with different water contents under the UV lamp at a wavelength of 365 nm. | ||
The photoluminescence quantum yields of TPE, P-TPE to TPE-PCPDE with different water fractions are shown in Fig. 6a. The maximum photoluminescence quantum yield of TPE, P-TPE and TPE-PCPDE is 0.049, 0.140 and 0.266, respectively when the water fraction is 90% compared to quinine sulfate (0.546) in 0.1 M H2SO4. TPE is a typical AIE molecule. However, the quantum yield of TPE in 90% water content reported here was only about 5% compared to 14–18% reported previously. This is due to the low initial concentration used in this experiment (2 × 10−6 M) which is more than 5 times diluted than reported in previous literatures.35,36 Clearly, compound TPE-PCPDE displays much higher fluorescence when the water fraction is 90% compared to that of TPE and P-TPE molecules. This indicates that through bond and through space conjugation of TPE-PCPDE which leads to a higher fluorescence than that of TPE and P-TPE. The pictures of PCPDE, TPE, P-TPE and TPE-PCPDE in THF and in 90% water fraction of the THF/water mixture under the UV lamp at a wavelength at 365 nm are shown in Fig. 6b. The notable fluorescence can be seen by naked eyes for P-TPE and TPE-PCPDE when the water fraction is 90% in the THF/water mixture. No fluorescence for PCPDE and very little fluorescence for TPE can be seen by naked eyes.
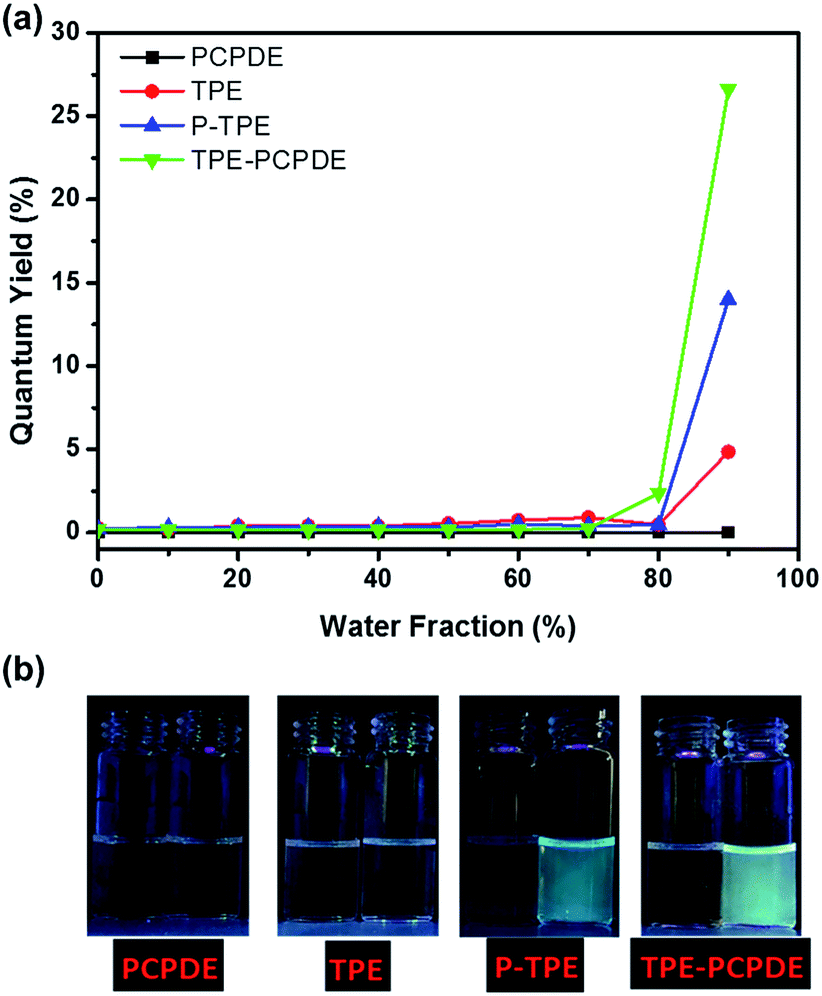 | ||
| Fig. 6 (a) Plots of photoluminescence quantum yields of PCPDE, TPE, P-TPE and TPE-PCPDE versus water fraction and (b) the pictures of PCPDE, TPE, P-TPE and TPE-PCPDE in THF and in 90% water/THF mixture under the UV lamp at a wavelength at 365 nm. | ||
Conclusions
Br-PCPDE was synthesized from the corresponding dithia[3.3]paracyclophane in three steps through benzyne Steven rearrangement, oxidation and thermal elimination reaction. TPE-PCPDE was successfully synthesized by the reaction of Br-PCPDE and monoboronic ester substituted tetraphenylethene using Pd(OAc)2 as a catalyst, S-Phos as a ligand and K3PO4 as a base. Functionalized paracyclophanedienes were fully characterized by nuclear magnetic resonance spectroscopy, high resolution mass spectrometry and X-ray crystallography. The longest absorption wavelength of TPE-PCPDE showed bathochromic shift compared to that of P-TPE due to the through space conjugation. TPE-PCPDE exhibited aggregation-induced emission characteristics when the water fraction was higher than 80% in the THF/water mixtures. TPE-PCPDE exhibited high fluorescence when the water fraction is 90% compared to that of TPE and P-TPE due to the through bond and through space conjugation. The ROMP of TPE-PCPDE by ruthenium carbene initiators is currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Ministry of Science and Technology and the National Taiwan University of Science and Technology – Taiwan is gratefully acknowledged.Notes and references
- C. J. Brown and A. C. Farthing, Nature, 1949, 164, 915–916 CrossRef.
- S. E. Gibson and J. D. Knight, Org. Biomol. Chem., 2003, 1, 1256–1269 Search PubMed.
- H. Hopf, Angew. Chem., Int. Ed., 2008, 47, 9808–9812 CrossRef PubMed.
- Y. Morisaki and Y. Chujo, Prog. Polym. Sci., 2008, 33, 346–364 CrossRef.
- P. G. Ghasemabadi, T. Yao and G. J. Bodwell, Chem. Soc. Rev., 2015, 44, 6494–6518 RSC.
- E. Elacqua and L. R. MacGillivray, Eur. J. Org. Chem., 2010, 6883–6894 CrossRef.
- Y. Morisaki, M. Gon, T. Sasamori, N. Tokitoh and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 3350–3353 CrossRef PubMed.
- B. Jiang, Y. Lei and X.-L. Zhao, J. Org. Chem., 2008, 73, 7833–7836 CrossRef PubMed.
- H. Hinrichs, A. J. Boydston, P. G. Jones, K. Hess, R. Herges, M. M. Haley and H. Hopf, Chem.–Eur. J., 2006, 12, 7103–7115 CrossRef PubMed.
- S. P. Jagtap, S. Mukhopadhyay, V. Coropceanu, G. L. Brizius, J. L. Brédas and D. M. Collard, J. Am. Chem. Soc., 2012, 134, 7176–7185 CrossRef PubMed.
- H. Y. Woo, J. W. Hong, B. Liu, A. Mikhailovsky, D. Korystov and G. C. Bazan, J. Am. Chem. Soc., 2005, 127, 820–821 CrossRef PubMed.
- J. L. Zafra, A. M. Ontoria, P. M. Burrezo, M. Peña-Alvarez, M. Samoc, J. Szeremeta, F. J. Ramírez, M. D. Lovander, C. J. Droske, T. M. Pappenfus, L. Echegoyen, J. T. López Navarrete, N. Martin and J. Casado, J. Am. Chem. Soc., 2017, 139, 3095–3105 CrossRef PubMed.
- K. C. Dewhirst and D. J. Cram, J. Am. Chem. Soc., 1958, 80, 3115–3125 CrossRef.
- E. Thorn-Csányi, P. Kraxner and J. Hammer, J. Mol. Catal., 1994, 90, 15–20 CrossRef.
- V. P. Conticello, D. L. Gin and R. H. Grubbs, J. Am. Chem. Soc., 1992, 114, 9708–9710 CrossRef.
- Y. J. Miao and G. C. Bazan, J. Am. Chem. Soc., 1994, 116, 9379–9380 CrossRef.
- Y. J. Miao and G. C. Bazan, Macromolecules, 1994, 27, 1063–1064 CrossRef.
- C.-Y. Yu and M. L. Turner, Angew. Chem., Int. Ed., 2006, 45, 7797–7800 CrossRef PubMed.
- C.-Y. Yu, M. Horie, A. M. Spring, K. Tremel and M. L. Turner, Macromolecules, 2009, 43, 222–232 CrossRef.
- C.-Y. Yu, J. W. Kingsley, D. G. Lidzey and M. L. Turner, Macromol. Rapid Commun., 2009, 30, 1889–1892 CrossRef PubMed.
- A. M. Spring, C.-Y. Yu, M. Horie and M. L. Turner, Chem. Commun., 2009, 2676–2678 RSC.
- B. J. Lidster, D. R. Kumar, A. M. Spring, C.-Y. Yu and M. L. Turner, Polym. Chem., 2016, 7, 5544–5551 RSC.
- C.-Y. Yu, S.-H. Yu, S.-H. Wen and C.-C. Wang, Tetrahedron Lett., 2017, 58, 3854–3858 CrossRef.
- D. Mäker, C. Maier, K. Brödner and U. H. F. Bunz, ACS Macro Lett., 2014, 3, 415–418 CrossRef.
- C.-Y. Yu, Y.-C. Chen and C.-C. Wang, New J. Chem., 2017, 41, 14116–14121 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 381, 1740–1741 RSC.
- Z. Luo, B. Liu, S. Si, Y. Lin, C. S. Luo, C. Pan, C. Zhao and L. Wang, Dyes Pigm., 2017, 143, 463–469 CrossRef.
- H.-T. Feng, J.-H. Wang and Y.-S. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 20067–20074 Search PubMed.
- C.-Y. Yu and C.-C. Hsu, Polymer, 2018, 137, 30–37 CrossRef.
- B. J. Lidster, D. R. Kumar, A. M. Spring, C.-Y. Yu, M. Helliwell, J. Raftery and M. L. Turner, Org. Biomol. Chem., 2016, 14, 6079–6087 Search PubMed.
- C.-Y. Yu, C.-H. Sie and C.-Y. Yang, New J. Chem., 2014, 38, 5003–5008 RSC.
- C.-Y. Yu, M. Helliwell, J. Raftery and M. L. Turner, Chem.–Eur. J., 2011, 17, 6991–6997 CrossRef PubMed.
- J. L. Muzyka and M. A. Fox, J. Org. Chem., 1991, 56, 4549–4552 CrossRef.
- C. Braun, E. Spuling, N. B. Heine, M. Cakici, M. Nieger and S. Bräse, Adv. Synth. Catal., 2016, 358, 1664–1670 CrossRef.
- Z. Zhao, S. Chen, X. Shen, F. Mahtab, Y. Yu, P. Lu, J. W. Y. Lam, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 686–688 RSC.
- H. Li, J. Wang, J. Z. Sun, R. Hu, A. Qin and B. Z. Tang, Polym. Chem., 2012, 3, 1075–1083 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: the synthesis of starting materials for compound 1 and X-ray crystallographic data. CCDC 1830878 (TPE-PCPDE) and 1833417 (Br-PCPDE). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra03025a |
| This journal is © The Royal Society of Chemistry 2018 |