
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed tandem allylic substitution/cyclization and cascade hydrosilylated reduction: the influence of reaction parameters and hydrosilanes on the stereoselectivity†
Peng-Wei Longa,
Jian-Xing Xua,
Xing-Feng Baiab,
Zheng Xu*a,
Zhan-Jiang Zhenga,
Ke-Fang Yanga,
Li Lia and
Li-Wen Xu *ab
*ab
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou 311121, P. R. China. E-mail: liwenxu@hznu.edu.cn; Fax: +86 2886 5135; Tel: +86 2886 5135
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, P. R. China. E-mail: licpxulw@yahoo.com
First published on 22nd June 2018
Abstract
To shed light on the influence of reaction parameters on palladium-catalyzed tandem allylic alkylation in the presence of Fei-Phos (a chiral trans-1,2-diaminocyclohexane-derived phosphine ligand), the effect of different phosphine ligands, inorganic or organic bases, Brønsted acids, and other additives on the asymmetric palladium-catalysed alkylation of catechol with allylic diacetate was investigated. In this reaction, 2-vinyl-2,3-dihydro-benzo[1,4]dioxin products with promising enantioselectivity were achieved in good yields. In addition, a novel palladium-catalyzed three-component and one-pot allylic substitution/cyclization/reduction reaction assisted by methylphenylsilane was reported with good selectivity.
Introduction
Transition-metal-catalyzed allylic substitution reactions are an extremely useful class of organic transformation, revealed several decades ago, and offer simple and practical approaches for the synthesis of alkene-substituted molecular frameworks.1 Notably, the allylic substitution reaction has been proven to be one of the most important and fundamental C–C bond-forming processes in organic synthesis.2 In this regard, the catalytic asymmetric version of allylic alkylation has also become a powerful carbon–carbon or carbon–heteroatom bond-forming approach to a range of structurally diverse allyl-substituted compounds. These are prevalent functional molecules and building blocks for the construction of natural products and biologically important complexes in synthetic chemistry.3 In this approach, it is well-known that catalytic asymmetric allylic substitution largely relies on the capability for stereoselective induction of the chiral ligands.4 In fact, numerous phosphine ligands have been utilized as they are extremely effective and are key factors in this allylic substitution reaction, including allylic cyclization.1–5 However, it is still highly desirable to search for a highly efficient ligand, which is needed to accommodate low-reactivity substrates for catalytic asymmetric allylic substitution reactions with improved activity and stereoselectivity. Despite the recent success in allylic alkylation of various carbon- or heteroatom-based nucleophiles, including cyanoacetate, alcohols, and heterocycles,6 transition-metal-catalyzed asymmetric allylic substitutions of functional molecules with structurally varied allylic acetates continue to receive much attention from synthetic chemists. Thus the development of new reaction systems with good yields and stereoselectivity is a valuable topic and is still highly desirable.Among allylic substitution reactions, the stereoselective allylic alkylation of oxygen-nucleophiles, such as alcohols, is one of the most important C–O bond-forming transformations for the construction of chiral ether-containing compounds.7 Notably, in this context, palladium complexes with bidentate phosphine or phosphite ligands have been proven to be effective catalysts for the construction of different C–O bonds and chiral ethers with good stereoselectivity.7 For example, we developed the chiral trans-1,2-diaminocyclohexane-derived multiple stereogenic and multifunctional CycloN2P2-Phos (also called Fei-Phos) for catalytic asymmetric allylic substitutions of benzyl alcohols and silanols with allylic acetates.8 Good to excellent yields and high enantioselectivities (up to 99% ee) were achieved in this reaction, which motivated us to expand the synthetic functions of Fei-Phos in palladium catalysis.9 Although asymmetric palladium-catalyzed allylic alkylation of catechol with (Z)-2-butene-1,4-diylbis(methylcarbonate) gave the desired 2-vinyl-1,4-benzodioxane with up to 45% ee in the presence of chiral ligands such as BINAP was realized by Sinou in 1994,10 the use of simple allylic acetates as the electrophiles in this reaction has not been described yet. In addition, it is surprising that limited progress has been made so far on the tandem allylic alkylation of catechol with allylic reagents for the synthesis of chiral 2-vinyl-2,3-dihydro-benzo[1,4]dioxanes.11 Thus in this work, we continue to carry out further investigations on the catalytic asymmetric allylic substitution of catechol with allylic diacetates in the presence of a palladium catalyst with Fei-Phos or other P-ligands. This approach could be useful for the construction of 1,4-benzodioxane ring systems that are found in various natural products and are key skeletons in pharmaceutical candidates possessing important biological activities.12 In addition, the effect of various phosphine ligands, inorganic or organic bases, Brønsted acids, and other additives, on the asymmetric palladium-catalysed alkylation of catechol with allylic diacetate was investigated in this work.
Results and discussion
Palladium-catalyzed tandem allylic alkylation of catechol with allylic compounds
Firstly, the allylic substitution/cyclization reaction of benzene-1,2-diol 1a with the (Z)-but-2-ene-1,4-diol-derived allylic acetate 2a was used as a model reaction at room temperature in the presence of a palladium complex which was generated in situ by mixing Pd2(dba)3 [tris(benzylideneacetone)dipalladium] with various phosphine ligands (Scheme 1). It should be noted that there is no effective phosphine ligand previously reported for this reaction.10,11 To evaluate the feasibility of the tandem allylic alkylation reactions, we wanted to explore the potential of commercially available phosphine ligands and our P-ligands, which we have reported in the past few years,13 including Fei-Phos. Results from the optimization studies employing the phosphine ligands are summarized in Table 1. Notably, most of the phosphine ligands gave poor or no yield of the corresponding product 3a. Only L3, L8, and L12 (Fei-Phos) were proven to be effective in this reaction (entries 3, 8, and 12, respectively), and L3 and L8 gave low to moderate yields (30–50%) as well as low enantioselectivity (25–39% ee). Interestingly, our Fei-Phos is still the best choice in this reaction in comparison with the other P-ligands evaluated in this work (entry 12, 60% yield and 39% ee). Notably, when the reaction temperature was decreased to 0 or −20 °C, both the yield and enantioselectivity decreased obviously (entries 17 and 18). With the optimized ligand in THF, the solvent effect was also evaluated in the catalytic asymmetric allylic substitution/cyclization reaction (Table 1, entries 19–22). Although this tandem allylic substitution reaction occurred smoothly to deliver 2-vinyl-2,3-dihydro-benzo[1,4]dioxane (3a) with good yields (70%) in 1,4-dioxane, the solvents evaluated in this work gave inferior performance in comparison with THF. | ||
| Scheme 1 The various phosphine ligands evaluated in the palladium-catalyzed tandem allylic alkylation of benzene-1,2-diol 1a with the (Z)-but-2-ene-1,4-diol derived allylic acetate 2a. | ||
|
||||
|---|---|---|---|---|
| Entrya | Ligand | Solvent | Yieldd (%) | eee,f (%) |
| a Reaction conditions: 1a (0.25 mmol), 2a (1.4 eq.), Pd2(dba)3 (2.5 mol%) and Fei-Phos (5 mol%), N2, rt, 24 h.b The temperature was 0 °C.c The temperature was −20 °C.d Isolated yields after silica gel chromatography.e Determined using chiral HPLC. n.r is no reaction; a trace is represented by <5% yield; n.d is not determined.f Determined by the sign of the specific rotation.14 | ||||
| 1 | L1 | THF | n.r | — |
| 2 | L2 | THF | <5 | — |
| 3 | L3 | THF | 50 | 39 |
| 4 | L4 | THF | n.r | — |
| 5 | L5 | THF | n.r | — |
| 6 | L6 | THF | n.r | — |
| 7 | L7 | THF | n.r | — |
| 8 | L8 | THF | 30 | 25 |
| 9 | L9 | THF | n.r | — |
| 10 | L10 | THF | n.r | — |
| 11 | L11 | THF | n.r | — |
| 12 | L12 | THF | 60 | 39 |
| 13 | L13 | THF | n.r | — |
| 14 | L14 | THF | n.r | — |
| 15 | L15 | THF | n.r | — |
| 16 | L16 | THF | n.r | — |
| 17b | L12 | THF | 24 | 31 |
| 18c | L12 | THF | 5 | 33 |
| 19 | L12 | 1,4-Dioxane | 72 | 35 |
| 20 | L12 | DCM | <5 | — |
| 21 | L12 | DCE | 12 | n.d |
| 22 | L12 | DMSO | n.r | — |
Then, we continued to investigate the effect of the palladium catalyst precursors or other transition metal catalysts on the catalytic asymmetric allylic alkylation. As shown in Table 2, most of the palladium salts were not suitable for this tandem allylic alkylation reaction, in which only [Pd(η-C3H5)Cl]2 and Pd(PPh3)4 had the ability to promote tandem allylic substitution to give the corresponding 2-vinyl-2,3-dihydro-benzo[1,4]dioxane (3a) in moderate yield (35–58%). Interestingly, other palladium salts or transition metal salts, such as PdCl2(CH3CN)2, Pd(OAc)2, [RhCl(cod)]2, [Ir(cod)OMe]2, or RuCl(PPh)3, were not suitable catalyst precursors in this reaction. These experimental results showed that the enhancement of enantioselectivity in the palladium-catalyzed allylic substitution/cyclization reaction of benzene-1,2-diol 1a with the (Z)-but-2-ene-1,4-diol derived allylic acetate 2a is not an easy task. The major reason possibly originated in the acidity of catechol because we have found that the equilibrium acidity (pKa) is an important factor in the reactivity of structurally diverse alcohols.9
|
|||
|---|---|---|---|
| Entrya | Catalyst | Yieldb (%) | eec (%) |
| a Reaction conditions: the reaction of 1a (0.25 mmol) and 2a (1.4 eq.) was carried out under N2 at room temperature for 24 h by mixing with [Pd] (5 mol%), [Rh] (5 mol%), [Ru] (5 mol%), or [Ir] (5 mol%) and the chiral ligand (5 mol%).b Isolated yields after silica gel chromatography.c Determined using chiral HPLC. | |||
| 1 | [Pd(C3H5)Cl]2 | 58 | 35 |
| 2 | (Ph3P)4Pd | 35 | 19 |
| 3 | PdCl2(CH3CN)2 | n.r | — |
| 4 | PdCl2(PhCN)2 | <5 | — |
| 5 | Pd(OAc)2 | n.r | — |
| 6 | RhCl(PPh3) | n.r | — |
| 7 | [RhCl(coe)]2 | n.r | — |
| 8 | [RhCl(cod)]2 | n.r | |
| 9 | [RhCl2(pmcpd)]2 | n.r | |
| 10 | RuCl(PPh)3 | n.r | |
| 11 | [Ir(cod)OMe]2 | n.r | |
Therefore, on the basis of the above findings, the modification of the reaction parameters by the addition of acidic or basic additive to tune the acidity is not trivial in this work. Initially, the Pd/Fei-Phos-catalyzed tandem allylic substitution of benzene-1,2-diol 1a with the (Z)-but-2-ene-1,4-diol derived allylic acetate 2a was carried out in the presence of various inorganic bases or organic bases at room temperature. As shown in Fig. 1, the effect of the bases on the enantioselectivity in the palladium/Fei-Phos catalyzed allylic substitution/cyclization reaction provided direct information that this reaction largely relied on the acidity of the reaction system (Table S6 of ESI†). For example, when KF or Na2CO3 was used as the base, almost no enantioselectivity was observed and almost all the bases decreased the stereoselectivity (3–37% ee).
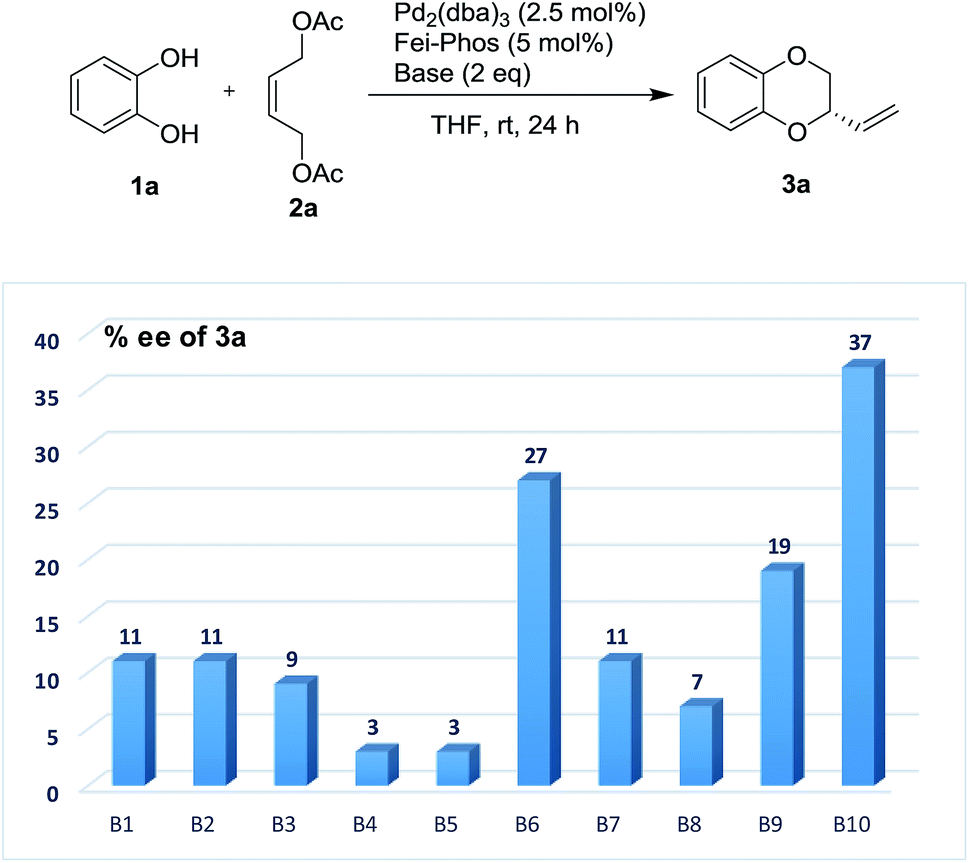 | ||
| Fig. 1 The effect of base (B1–B10) on the enantioselectivity in the Pd-catalyzed tandem allylic substitution/cyclization: B1 (K2CO3), B2 (KHCO3), B3 (K3PO4), B4 (KF), B5 (Na2CO3), B6 (NaHCO3), B7 (Cs2CO3), B8 (DBU), B9 (Et3N) and B10 (DIPEA). | ||
Similarly, the effect of organic acids on the enantioselective Pd-catalyzed allylic alkylation was also performed under the optimized reaction conditions. As shown in Fig. 2, it was found that all the chiral organic acids, such as mandelic acid and proline, gave the desired product 3a with a low enantioselectivity (5–25% ee). Interestingly, the absolute configuration of the chiral organic acid played important role in the influence of the additive, for example, (S)-(+)-mandelic acid or L-(−)-proline gave higher enantioselectivity than (R)-(−)-mandelic acid or D-(−)-proline. Although most of the organic acids had no ability to improve the enantioselectivity, the experimental results clearly supported the theory that the organic acids could affect the catalytic efficiency of the palladium catalyst in this reaction. Gratifyingly, among the different organic acids used as additives in this reaction, phenylboronic acid was determined to be an excellent additive for the enhancement of enantioselectivity.
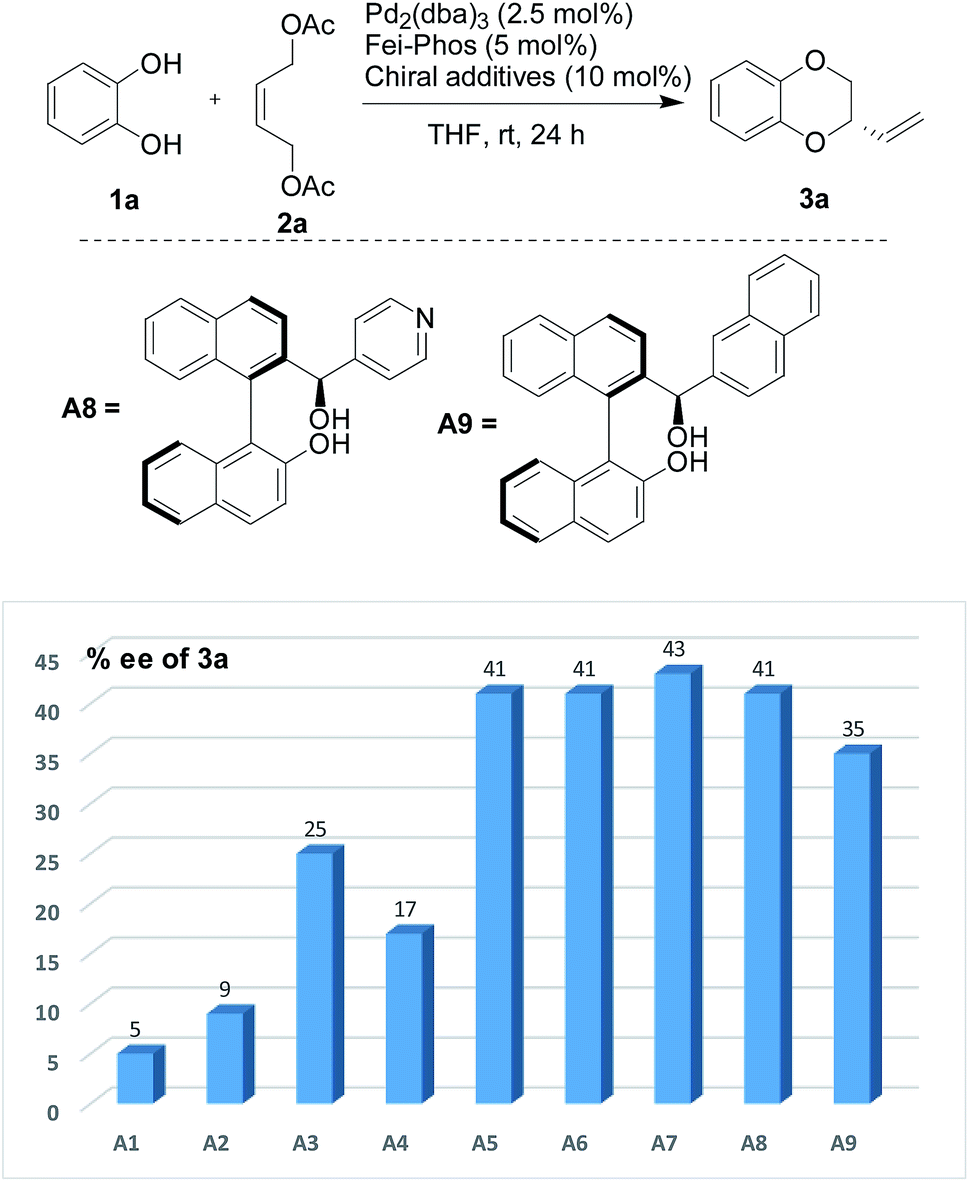 | ||
| Fig. 2 The effect of chiral organic acids on the enantioselectivity in the Pd-catalyzed tandem allylic substitution/cyclization: A1 [(S)-(+)-mandelic acid], A2 [(R)-(+)-mandelic acid], A3 [L-(−)-proline], A4 [D-(+)-proline], A5 [(S)-BNIOL], A6 [(R)-BINOL], A7 (race-BINOL), A8, and A9.15 | ||
As shown in Fig. 3, phenylboronic acid improved the enantioselectivity from 39% ee to 51% ee when 10 mol% of PhB(OH)2 was used in this reaction. However, the yield was quite low in this case (10% isolated yield). Inspired by this finding, the role of various arylboronic acids in the palladium-catalyzed tandem allylic substitution of benzene-1,2-diol 1a with the (Z)-but-2-ene-1,4-diol derived allylic acetate 2a was then investigated using a series of experimental studies. As shown in Table S7 (see the ESI†) about 20 examples revealed that phenylboronic acid was still the best additive in this reaction.
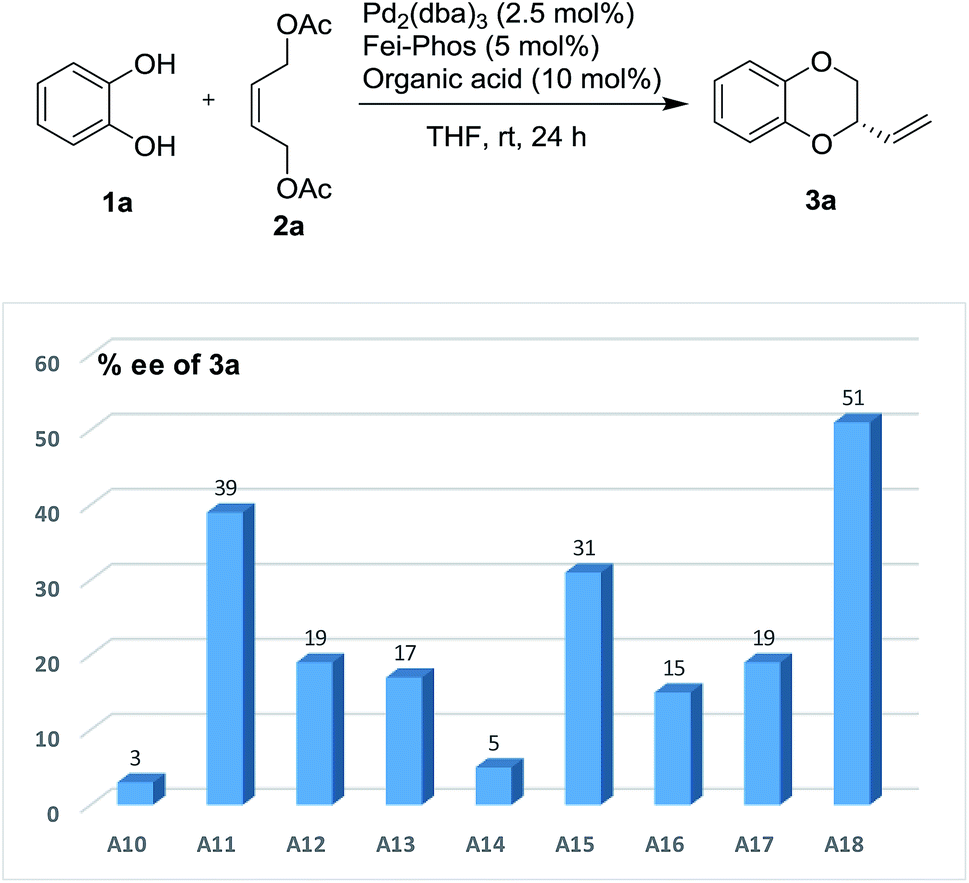 | ||
| Fig. 3 The effect of organic acids on the enantioselectivity in the Pd-catalyzed tandem allylic substitution/cyclization: A10 (benzoic acid), A11 (2-methylbenzoic acid), A12 (abietic acid), A13 (ascorbic acid), A14 (pivalic acid), A15 (boric acid), A16 (cyclopropylboronic acid), A17 (2-naphthaleneacetic acid) and A18 (phenylboronic acid). | ||
Then the effect of the amount of phenylboronic acid on the enantioselectivity was further investigated. Unfortunately, it was difficult to exceed 51% ee when larger amounts of phenylboronic acid were used as the additive in the palladium-catalyzed allylic substitution/cyclization reaction (Fig. 4).
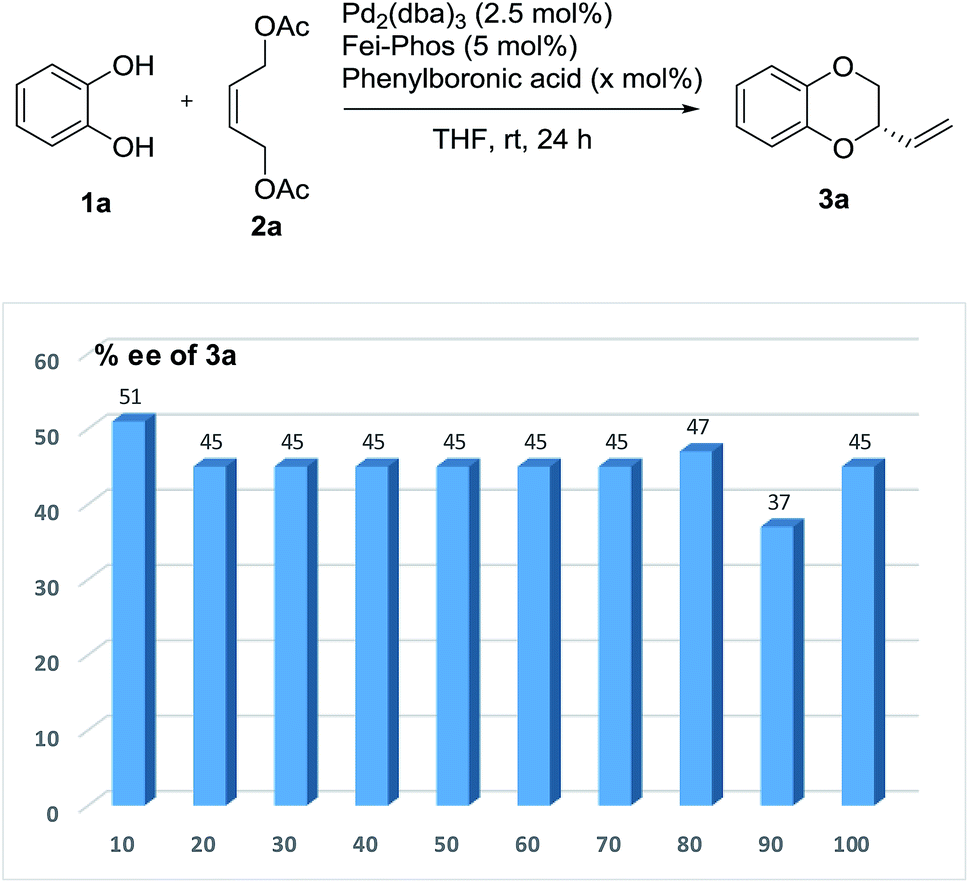 | ||
| Fig. 4 The effect of the amount of phenylboronic acid on the enantioselectivity in the Pd-catalyzed tandem allylic substitution/cyclization. | ||
It is well-known that the leaving group is also an important factor in the palladium-catalyzed allylic alkylation reaction.11 Thus its effect on the enantioselectivity of the allylic substitution/cyclization reaction is shown in Fig. 5. The results indicate that the benzoyl group as the leaving group has the best enantioselectivity (45% ee). In addition, the enantioselectivity due to the leaving group was influenced largely by whether it was an electron-donating group, for example, with a p-methoxybenzyl group as the leaving group, the enantioselectivity of the reaction system reduced to 23% ee. However, other fatty acids and unsaturated fatty acids other than acetic acid esters had no significant enhancement on the enantioselectivity of the tandem allylic alkylation. Moreover, under the optimized reaction conditions, all these leaving groups decreased the yields to lower than that with the acetyl group.
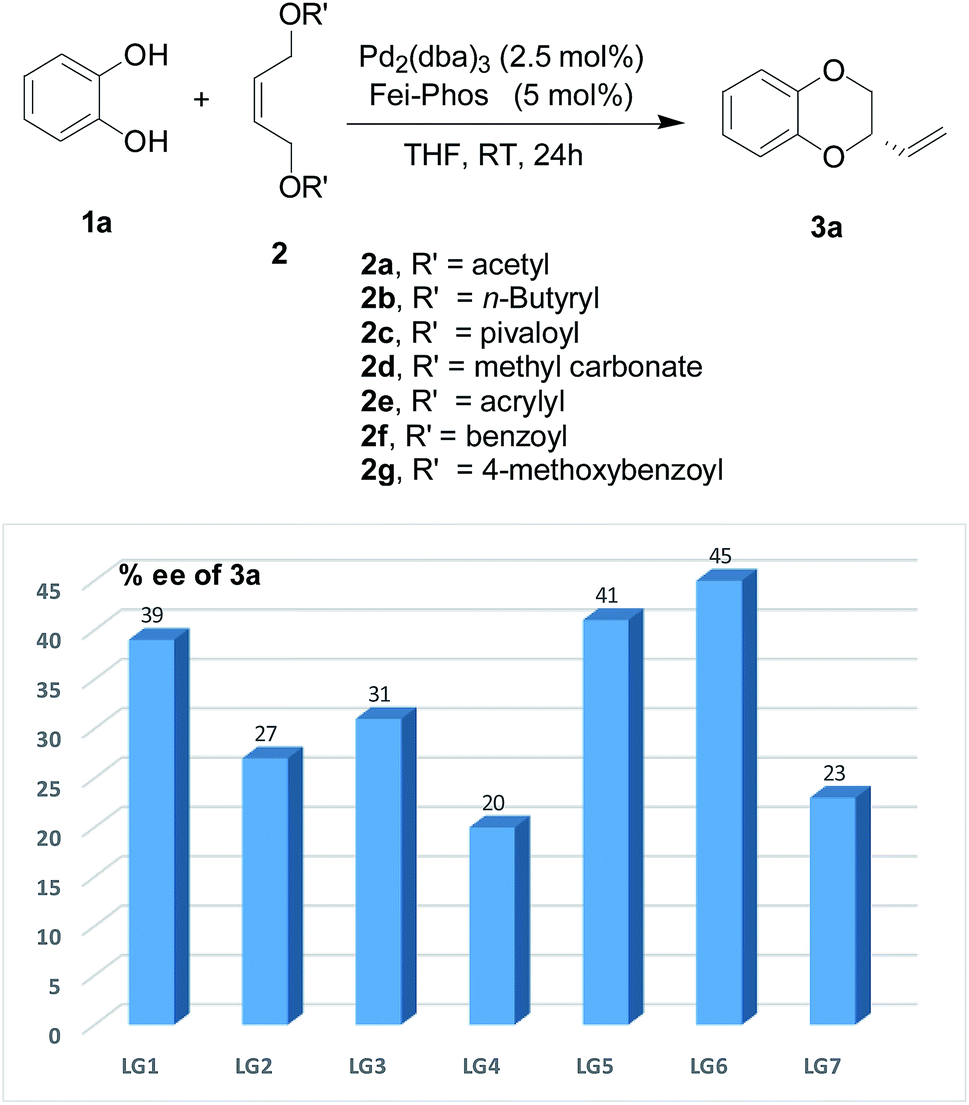 | ||
| Fig. 5 The effect of the leaving group of substrate 2 on the enantioselectivity in the Pd-catalyzed tandem allylic substitution/cyclization: LG1 (acetyl), LG2 (n-butyryl), LG3 (pivaloyl), LG4 (methyl carbonate), LG5 (acrylyl), LG6 (benzoyl) and LG7 (4-methoxybenzoyl). | ||
Therefore, under the optimized reaction conditions shown in Table 1, we further investigated the substrate scope of the catalytic asymmetric tandem allylic substitution/cyclization reaction of different 1,2-bifunctional nucleophiles 1 with the (Z)-but-2-ene-1,4-diol derived allylic acetate 2a. As shown in Scheme 2, good yields and promising ee values were obtained for various substituted 2-heterocycles 3 (up to 7–39% ee). Although the enantioselectivity is not good enough, the present method is quite simple and features good yields. In addition, the reaction information described above provides a springboard to modify the reaction conditions of the palladium-catalyzed tandem allylic alkylation reactions in the near future.
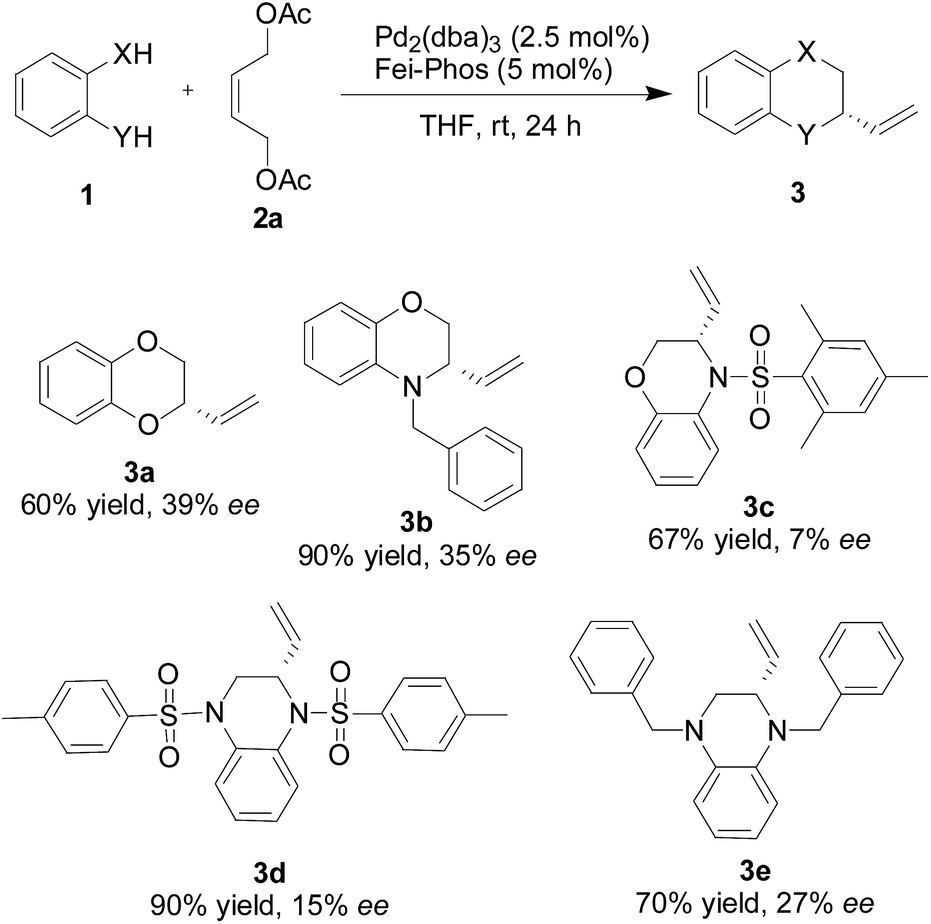 | ||
| Scheme 2 Fei-Phos ligand-controlled palladium-catalyzed tandem allylic alkylation of different 1,2-bifunctional nucleophiles 1 with the (Z)-but-2-ene-1,4-diol derived allylic acetate 2a: a representative substrate scope. | ||
Palladium-catalyzed three-component tandem allylic substitution/cyclization and cascade hydrosilylated reduction
Encouraged by the experimental results for the palladium-catalyzed tandem allylic alkylation of different 1,2-bifunctional nucleophiles 1 with the (Z)-but-2-ene-1,4-diol-derived allylic acetate 2a shown in Scheme 2, we turned our attention to the design of a novel one-pot multicomponent asymmetric allylic alkylation and cascade hydrosilylated reduction. In fact, we found that 2-vinyl-2,3-dihydro-benzo[1,4]dioxane (3a) and the other products 3 were not stable enough in the presence of the palladium catalyst. We proposed that the palladium-promoted isomerisation of the alkene group resulted in the deceased enantioselectivity.16 Therefore, the reduction of the vinyl group by hydrosilane would be an interesting topic because the palladium complex associated with Fei-Phos would be effective in this reaction. However, to the best of our knowledge, despite the previous success of Pd-catalyzed tandem reactions,17 there are no past reports on a palladium-catalyzed cascade allylic alkylation/hydrosilylated reduction. In addition, the different activities of hydrosilanes makes this reaction quite difficult and unpredictable. Herein, with commercially available hydrosilanes to hand, including PMHS (polymethylhydrosiloxane), Ph2SiH2, MePhSiH2, PhSiH3, and MePh2SiH, a one-pot palladium-catalyzed asymmetric allylic alkylation and cascade hydrosilylated reduction reaction was attempted under optimized reaction conditions (see Table 3 and Table S10 in the ESI;† 2.5 mol% of palladium catalyst, Pd2(dba)3, 5 mol% of Fei-Phos, and 3 equiv. hydrosilane in THF were determined as the optimal reaction conditions).
|
|||
|---|---|---|---|
| Entrya | Silane | Yieldb (%) | 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4ac :4ac |
| a Reaction conditions: 1a (0.25 mmol), 2a (1.4 eq.), Pd2(dba)3 (2.5 mol%), hydrosilane (3 eq.), Fei-Phos (5 mol%), N2, rt, 48 h.b Isolated yields after silica gel chromatography.c Determined from GC-MS analysis. | |||
| 1 | PMHS | 99% | 1:0.48 |
| 2 | Ph2SiH2 | 99% | 1:0.59 |
| 3 | MePhSiH2 | 99% | 0:1 |
| 4 | PhSiH3 | n.r | — |
| 5 | MePh2SiH | 87% | 1:0 |
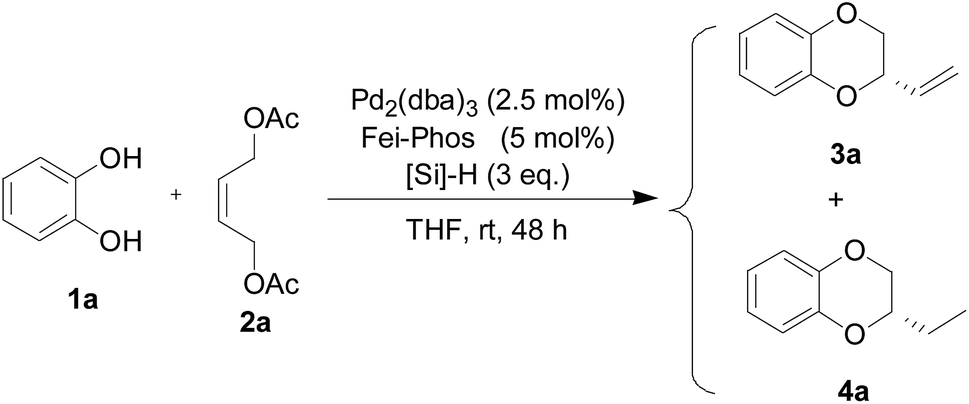
To our delight, the palladium catalyst with Fei-Phos was also highly effective in the asymmetric tandem allylic alkylation of benzene-1,2-diol 1a with the (Z)-but-2-ene-1,4-diol derived allylic acetate 2a. The results presented in Table 3 clearly demonstrate the efficiency of Fei-Phos in this multicomponent allylic alkylation transformation. The corresponding product 4a could be obtained with varied yields with better enantioselectivity (45% ee), which supported our hypothesis that the direct reduction of 2-vinyl-2,3-dihydro-benzo[1,4]dioxane (3a) could inhibit the isomerization of the double carbon–carbon bond in this reaction. In particular, when MePhSiH2 was used as a reductive hydrosilane, only the desired product 4a was obtained, while other hydrosilanes gave poor chemoselectivity or had no activity for the cascade hydrosilylated reduction. Therefore, we have successfully developed a novel strategy for the asymmetric palladium-catalyzed multicomponent allylic alkylation and one-pot reduction of an alkene product.
Finally, under the optimized reaction conditions that were established, shown in Table 3, the substrate scope of this palladium-catalyzed multicomponent allylic substitution/cyclization and cascade reduction with hydrosilane (MePhSiH2) was examined with the representative 1,2-bifunctional nucleophiles 1. As summarised in Scheme 3, all the corresponding heterocycle products 4 were obtained in excellent yields (80–99% isolated yields) with moderate to low enantioselectivities (up to 46% ee).
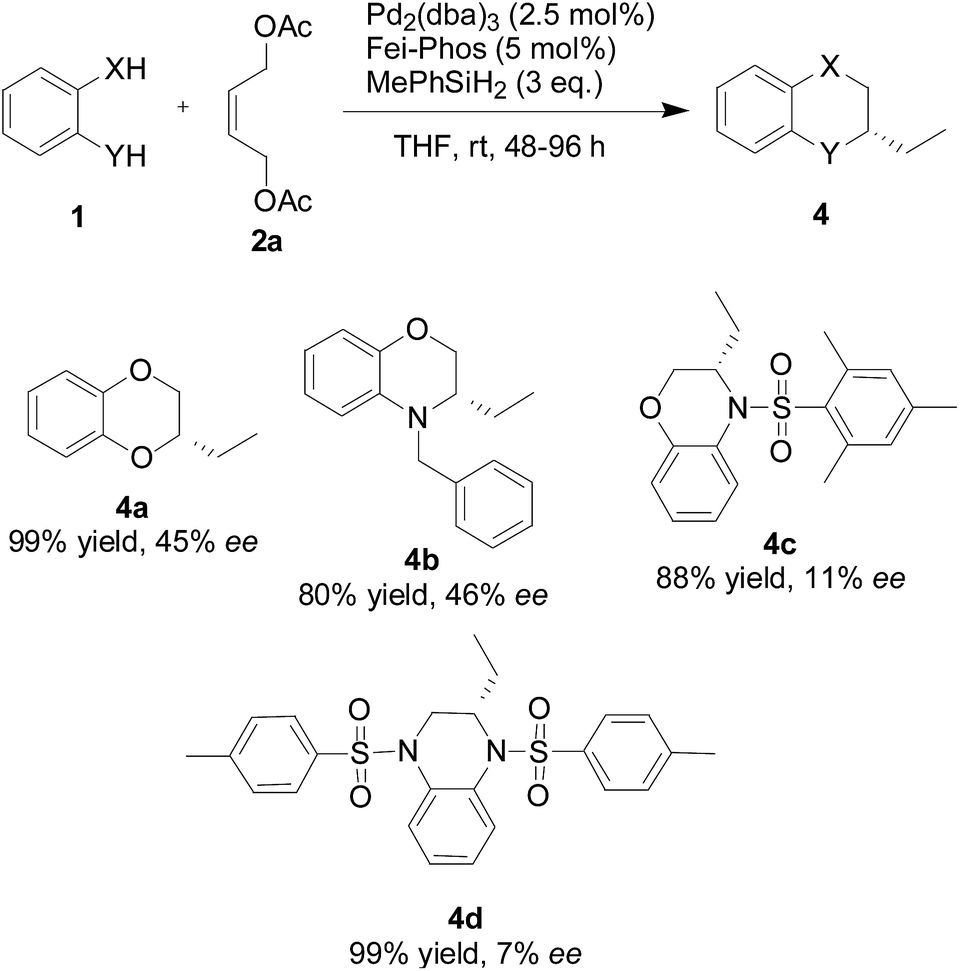 | ||
| Scheme 3 Palladium-catalyzed multicomponent allylic substitution/cyclization and cascade reduction with hydrosilane (MePhSiH2). | ||
On the basis of the experimental results and the generally accepted mechanism for palladium-catalyzed allylic alkylation,18 we proposed a mechanistic procedure with the corresponding transition-state model, as shown in Scheme 4. In the first step, there was an initial oxidative addition of the low valent palladium species (Pd(0)L2), which formed in situ from Pd2(dba)3 and Fei-Phos, to the (Z)-but-2-ene-1,4-diol derived allylic acetate 2a (electrophile) and then nucleophilic attack on the corresponding allylic Pd(II) cation. Subsequently, the reductive elimination of the palladium intermediate II that had coordinated with the ligand resulted in the formation of the single alkylation intermediate III, and then intramolecular allylic alkylation led to the regeneration of the Pd(0) complex when intermediate IV liberated the product.
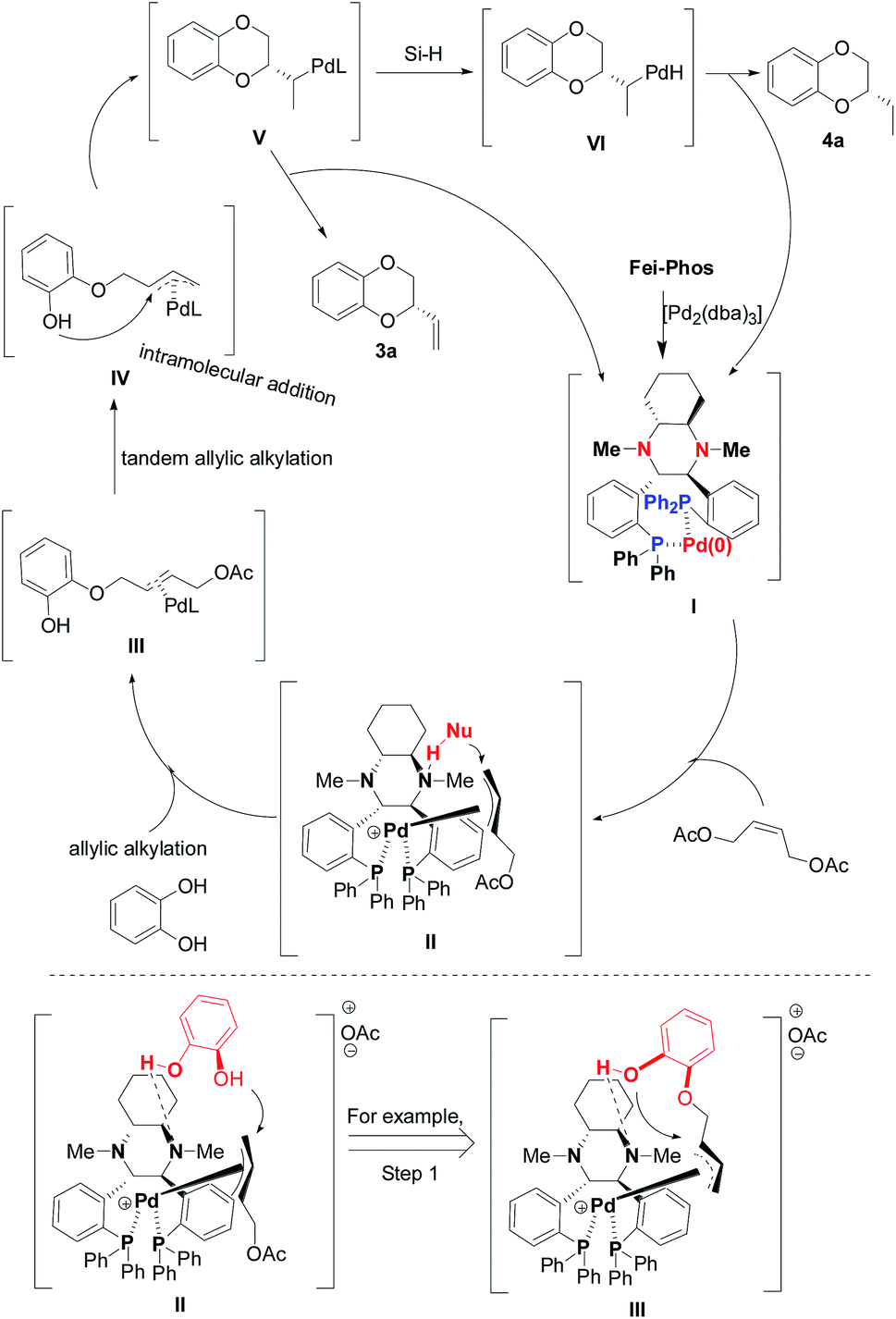 | ||
| Scheme 4 The proposed mechanism for asymmetric palladium-catalyzed allylic substitution/cyclization and cascade hydrosilylated reduction in the presence of hydrosilane. | ||
Conclusions
In summary, we have determined the stereoselective activity of Fei-Phos in the asymmetric palladium-catalyzed tandem allylic alkylation of 1,2-bifunctional nucleophiles 1 with the (Z)-but-2-ene-1,4-diol derived allylic acetate 2a. In addition, the influence of the reaction parameters on palladium-catalyzed tandem allylic alkylation in the presence of Fei-Phos, including the effect of various phosphine ligands, inorganic or organic bases, Brønsted acids, and other additives on the asymmetric palladium-catalysed alkylation of catechol with allylic diacetate, was investigated in detail. Under optimized reaction conditions, the corresponding 2-vinyl-2,3-dihydro-benzo[1,4]dioxin products were achieved with promising enantioselectivity in good yields. In addition, a novel one-pot allylic substitution/cyclization and cascade reduction with the aid of methylphenylsilane was further developed in this work. It provides an interesting example of the concept of silicon-mediated organic synthesis (SiMOS) because the three-component reaction is precisely controlled by certain hydrosilanes. Finally, on the basis of the present work, we believe that the modification of Fei-Phos would be an effective strategy for the improvement of asymmetric palladium-catalyzed transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thank the National Natural Science Foundation of China (No. 21472031, 21703051, 21702211, and 21773051), and the Zhejiang Provincial Natural Science Foundation of China (LZ18B020001, LY16E030009, LY17B030005, and LY17E030003) for financial support, which is appreciated. The authors also thank Dr J. Cao, Dr Y. M. Cui, Dr F. Ye, Dr K. Z. Jiang, Dr C. Q. Sheng, and Dr W. G. Yin (all at HZNU) for their technical and analytical support.Notes and references
- (a) B. M. Trost, Acc. Chem. Res., 1980, 13, 385–393 CrossRef; (b) J. Tsuji, I. Minami and I. Shimizu, Chem. Lett., 1983, 1325–1326 CrossRef , and references cited therein.
- For representative reviews, see: (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef PubMed; (b) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef PubMed; For representative book, see: (c) D. Caine, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 3, pp. 1–63 Search PubMed.
- For recent examples, see: (a) S. Chang, L. Wang and X. Lin, Org. Biomol. Chem., 2018, 16, 2239–2247 RSC; (b) I. Szulc, R. Kolodziuk and A. Zawisza, Tetrahedron, 2018, 74, 1476–1485 CrossRef; (c) Z. P. Yang, R. Jiang, C. Zheng and S. L. You, J. Am. Chem. Soc., 2018, 140, 3114–3119 CrossRef PubMed; (d) Y. J. Yang, G. P. Zhang, J. Q. Huang, D. Chen, C. H. Ding and X. L. Hou, Org. Lett., 2017, 19, 5932–5935 CrossRef PubMed; (e) E. J. Alexy, S. C. Virgil, M. D. Bartberger and B. M. Stoltz, Org. Lett., 2017, 19, 5007–5009 CrossRef PubMed; (f) L. Wei, S. M. Xu, Q. Zhu, C. Che and C. J. Wang, Angew. Chem., Int. Ed., 2017, 56, 12312–12316 CrossRef PubMed; (g) J. Wang, P. X. Wang, L. Q. Wang, D. Li, K. Z. Wang, Y. Wang, H. Y. Zhu, D. X. Yang and R. Wang, Org. Lett., 2017, 19, 4826–4829 CrossRef PubMed; (h) S. Kayal and S. Mukherjee, Org. Lett., 2017, 19, 4944–4947 CrossRef PubMed; (i) S. E. Shockley, J. C. Hethcox and B. M. Stoltz, Angew. Chem., Int. Ed., 2017, 56, 11545–11548 CrossRef PubMed; (j) P. Starkov, J. T. Moore, D. C. Duquette, B. M. Stoltz and I. Marek, J. Am. Chem. Soc., 2017, 139, 9615–9620 CrossRef PubMed.
- For recent reviews or books: (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3069 CrossRef PubMed; (b) Privileged Chiral Ligands and Catalysts, ed. Q. L. Zhou, Wiley-VCH, Singapore, 2011 Search PubMed; (c) M. P. Carroll and P. J. Guiry, Chem. Soc. Rev., 2014, 43, 819–833 RSC.
- For recent reviews, see: (a) J. P. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539–2555 CrossRef PubMed; (b) I. G. Rios, A. Rosas-Hernandez and E. Martin, Molecules, 2011, 16, 970–1010 CrossRef PubMed; (c) H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119–2176 CrossRef PubMed. For recent examples, see: (d) J. Q. Huang, W. Liu, B. H. Zheng, X. Y. Liu, Z. Yang, C. H. Ding, H. Li, Q. Peng and X. L. Hou, ACS Catal., 2018, 8, 1964–1972 CrossRef; (e) B. P. Pritchett, E. J. Donckele and B. M. Stoltz, Angew. Chem., Int. Ed., 2017, 56, 12624–12627 CrossRef PubMed; (f) M. A. Schafroth, S. M. Rummelt, D. Sarlah and E. M. Carreira, Org. Lett., 2017, 19, 3235–3238 CrossRef PubMed; (g) T. Y. Lin, H. H. Wu, J. J. Feng and J. L. Zhang, Org. Lett., 2017, 19, 2897–2900 CrossRef PubMed; (h) A. Saito, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2017, 56, 5551–5555 CrossRef PubMed; (i) Y. Suzuki, N. Vatmurge, S. Tanaka and M. Kitamura, Chem.–Asian J., 2017, 12, 633–637 CrossRef PubMed; (j) J. D. Sieber, V. V. Angeles, D. Chennamadhavuni, D. R. Fandrick, N. Haddad, N. Grinberg, D. Kurouski, H. Lee, J. H. J. Song, N. K. Yee, A. E. Mattson and C. H. Senanayake, Adv. Synth. Catal., 2016, 358, 3062–3068 CrossRef; (k) Q. L. Liu, W. Chen, Q. Y. Jiang, X. F. Bai, Z. Li, Z. Xu and L. W. Xu, ChemCatChem, 2016, 8, 1495–1499 CrossRef.
- For a representative review on catalytic asymmetric allylic alkylation employing heteroatom nucleophiles, see: (a) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427–440 RSC; (b) J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539–2555 CrossRef PubMed; For recent examples, see: (c) X. Caldentey and M. A. Pericàs, J. Org. Chem., 2010, 75, 2628–2644 CrossRef PubMed; (d) M. Austeri, D. Linder and J. Lacour, Adv. Synth. Catal., 2010, 352, 3339–3347 CrossRef; (e) L. Dai, X. Li, H. Yuan, X. Li, Z. Li, D. Xu, F. Lei, Y. Liu, J. Zhang and Z. Zhou, Tetrahedron: Asymmetry, 2011, 22, 1379–1389 CrossRef; (f) M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5568–5571 CrossRef PubMed; (g) K. N. Gavrilov, S. V. Zheglov, A. A. Shiryaev, N. N. Groshkin, E. A. Rastorguev, E. B. Benetskiy and V. A. Davankov, Tetrahedron Lett., 2011, 52, 964–968 CrossRef; (h) W. H. Deng, F. Ye, X. F. Bai, Z. J. Zheng, Y. M. Cui and L. W. Xu, ChemCatChem, 2015, 7, 75–79 CrossRef; (i) C. Xia, J. Shen, D. Liu and W. Zhang, Org. Lett., 2017, 19, 4251–4254 CrossRef PubMed; (j) P. Paz and A. Baeza, Adv. Synth. Catal., 2017, 359, 1735–1741 CrossRef; (k) Y. Suzuki, N. Vatmurge, S. Tanaka and M. Kitamura, Chem.–Asian J., 2017, 12, 633–637 CrossRef PubMed; (l) Z. Wu, X. Fang, Y. Leng, H. Yao and A. Lin, Adv. Synth. Catal., 2018, 360, 1289–1295 CrossRef.
- (a) F. L. Lam, T. T. L. Au-Yeung, F. Y. Kwong, Z. Zhou, K. Y. Wong and A. S. C. Chan, Angew. Chem., Int. Ed., 2008, 47, 1280–1283 CrossRef PubMed; (b) M. Kato, T. Nakamura, K. Ogata and S.-I. Fukuzawa, Eur. J. Org. Chem., 2009, 5232–5238 CrossRef; (c) P. Fang, C. H. Ding, X. L. Hou and L. X. Dai, Tetrahedron: Asymmetry, 2010, 21, 1176–1178 CrossRef; (d) Z. Liu and H. Du, Org. Lett., 2010, 12, 3054–3057 CrossRef PubMed; (e) J. Mazuela, O. Pàmies and M. Diéguez, Chem.–Eur. J., 2013, 19, 2416–2432 CrossRef PubMed; (f) B. Feng, H. G. Cheng, J. R. Chen, Q. H. Deng, L. Q. Lu and W. J. Xiao, Chem. Commun., 2014, 50, 9550–9553 RSC; (g) M. Coll, O. Pàmies and M. Diéguez, Org. Lett., 2014, 16, 1892–1895 CrossRef PubMed; (h) P. Trillo and I. M. Pastor, Adv. Synth. Catal., 2016, 358, 2929–2939 CrossRef; (i) A. Khan, S. Khan, I. Khan, C. Zhao, Y. Mao, Y. Chen and Y. J. Zhang, J. Am. Chem. Soc., 2017, 139, 10733–10741 CrossRef PubMed.
- F. Ye, Z. J. Zheng, L. Li, K. F. Yang, C. G. Xia and L. W. Xu, Chem.–Eur. J., 2013, 19, 15452–15457 CrossRef PubMed.
- (a) J. X. Xu, F. Ye, X. F. Bai, Y. M. Cui, Z. Xu, Z. J. Zheng and L. W. Xu, RSC Adv., 2016, 6, 70624–70631 RSC; (b) J. X. Xu, F. Ye, X. F. Bai, J. Zhang, Z. Xu, Z. J. Zheng and L. W. Xu, RSC Adv., 2016, 6, 45495–45502 RSC.
- (a) M. Massacret, C. Goux, P. Lhoste and D. Sinou, Tetrahedron Lett., 1994, 35, 6093–6096 CrossRef; (b) C. Goux, M. Massacret, P. Lhoste and D. Sinou, Organometallics, 1995, 14, 4585–4593 CrossRef.
- (a) S. Tanimori, Y. Kato and M. Kirihata, Synthesis, 2004, 2103–2106 CrossRef; (b) K. Ito, Y. Imahayashi, T. Kuroda, S. Eno, B. Saito and T. Katsuki, Tetrahedron Lett., 2004, 45, 7277–7281 CrossRef; (c) M. Massacret, R. Lakhmiri, P. Lhoste, C. Nguefack, A. Ben, B. Fouad, R. Fadel and D. Sinou, Tetrahedron: Asymmetry, 2000, 11, 3561–3568 CrossRef; (d) M. Massacret, P. Lhoste, R. Lakhmiri, T. Parella and D. Sinou, Eur. J. Org. Chem., 1999, 1999, 2665–2673 CrossRef.
- (a) M. R. Stillings, C. B. Chapleo, R. C. M. Butler, J. A. Davis, C. D. England, M. Myers, P. L. Myers, N. Tweddle, A. P. Welbourn, J. C. Doxey and C. F. C. Smith, J. Med. Chem., 1985, 28, 1054–1062 CrossRef PubMed; (b) D. Giardina, R. Bertini, E. Brancia, L. Brasili and C. Melchiorre, J. Med. Chem., 1985, 28, 1354–1357 CrossRef PubMed; (c) G. P. Fagan, C. B. Chapleo, A. C. Lane, M. Myers, A. G. LarRoach, C. F. C. Smith, M. R. Stillings and A. P. Welbourn, J. Med. Chem., 1988, 31, 944–948 CrossRef PubMed; (d) G. Marciniak, A. Delgado, G. Leclerc, J. Velly, N. Decker and J. Schwartz, J. Med. Chem., 1989, 32, 1402–1407 CrossRef PubMed; (e) R. R. Ruffolo Jr, W. Bondinell and J. P. Hieble, J. Med. Chem., 1995, 38, 3681–3716 CrossRef; (f) N. R. Guz and F. R. Stermitz, J. Nat. Prod., 2000, 63, 1140–1145 CrossRef PubMed; (g) R. Gazak, K. Valentova, K. Fuksova, P. Marhol, M. Marek, A. Miguel, I. Oborna, J. Jitka and V. Kren, J. Med. Chem., 2011, 54, 7397–7407 CrossRef PubMed.
- For HZNU-Phos (L14): (a) F. Ye, Z. J. Zheng, W. H. Deng, L. S. Zheng, Y. Deng, C. G. Xia and L. W. Xu, Chem.–Asian J., 2013, 8, 2242–2253 CrossRef PubMed; For Ar-NNP (L9): (b) L. S. Zheng, L. Li, K. F. Yang, Z. J. Zheng, X. Q. Xiao and L. W. Xu, Tetrahedron, 2013, 69, 8777–8784 CrossRef; For Tao-Phos (L10): (c) T. Song, L. S. Zheng, F. Ye, W. H. Deng, Y. L. Wei, K. Z. Jiang and L. W. Xu, Adv. Synth. Catal., 2014, 356, 1708–1718 CrossRef; (d) T. Song, L. Li, W. Zhou, Z. J. Zheng, Y. Deng, Z. Xu and L. W. Xu, Chem.–Eur. J., 2015, 21, 554–558 CrossRef PubMed; For Xing-Phos (L13): (e) X. F. Bai, T. Song, Z. Xu, C. G. Xia, W. S. Huang and L. W. Xu, Angew. Chem., Int. Ed., 2015, 54, 5255–5259 CrossRef PubMed; For L4: (f) L. Chen, J. B. Huang, Z. Xu, Z. J. Zheng, K. F. Yang, Y. M. Cui, J. Cao and L. W. Xu, RSC Adv., 2016, 6, 67113–67117 RSC; For L11: (g) J. X. Xu, M. Y. Chen, Z. J. Zheng, J. Cao, Z. Xu, Y. M. Cui and L. W. Xu, ChemCatChem, 2017, 9, 3111–3116 CrossRef , and references cited therein.
- J. S. Cannon, A. C. Olson, L. E. Overman and N. S. Solomon, J. Org. Chem., 2012, 77, 1961–1973 CrossRef PubMed.
- The synthesis of A8 and A9, see: (a) G. Gao, F. L. Gu, J. X. Jiang, K. Jiang, C. Q. Sheng, G. Q. Lai and L. W. Xu, Chem.–Eur. J., 2011, 17, 2698–2703 CrossRef PubMed; (b) C. Dong, S. Tao, X. F. Bai, Y. M. Cui, Z. Xu and L. W. Xu, Catal. Sci. Technol., 2015, 5, 4755–4759 RSC.
- (a) X. F. Bai, T. Song, W. H. Deng, Y. L. Wei, L. Li, C. G. Xia and L. W. Xu, Synlett, 2014, 25, 417–422 Search PubMed; (b) X. F. Bai, L. W. Xu, L. S. Zheng, J. X. Jiang, G. Q. Lai and J. Y. Shang, Chem.–Eur. J., 2012, 18, 8174–8179 CrossRef PubMed.
- For recent examples, see: (a) S. S. M. Spoehrle, T. H. West, J. E. Taylor, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 11895–11902 CrossRef PubMed; (b) K. Kim, Y. Jung, S. Lee, M. Kim, D. Shin and H. Byun, Angew. Chem., Int. Ed., 2017, 56, 6952–6956 CrossRef PubMed; (c) S. J. Cho, H. Song, H. Kim, L. Qi, K. Hu, S. Yu, J. Zhu, T. Cheng, X. Wang, J. Chen and H. Wu, Org. Lett., 2017, 19, 218–221 CrossRef PubMed; (d) Q. S. Gu and D. Yang, Angew. Chem., Int. Ed., 2017, 56, 5886–5889 CrossRef PubMed; (e) K. Hu, L. Qi, S. Yu, T. Cheng, X. Wang, Z. Li, Y. Xia, J. Chen and H. Wu, Green Chem., 2017, 19, 1740–1750 RSC; (f) K. Hu, Q. Zhen, J. Gong, T. Cheng, L. Qi, Y. Shao and J. Chen, Org. Lett., 2018, 20, 3083–3087 CrossRef PubMed; (g) X. Huang, W. Wu, S. Song, C. Fu and S. Ma, Adv. Synth. Catal., 2016, 358, 2791–2805 CrossRef.
- (a) L. A. Evans, N. Fey, J. N. Harvey, D. Hose, G. C. Lloyd-Jones, P. Murray, A. G. Orpen, R. Osborne, G. J. J. Owen-Smith and M. Purdie, J. Am. Chem. Soc., 2008, 130, 14471–14473 CrossRef PubMed; (b) C. P. Butts, E. Filali, G. C. Lloyd-Jones, P.-O. Norrby, D. A. Sale and Y. Schramm, J. Am. Chem. Soc., 2009, 131, 9945–9957 CrossRef PubMed; (c) M. H. Katcher, P.-O. Norrby and A. G. Doyle, Organometallics, 2014, 33, 2121–2133 CrossRef; (d) M. Magre, M. Biosca, P. O. Norrby, O. Pamies and M. Dieguez, ChemCatChem, 2015, 7, 4091–4107 CrossRef; (e) A. J. DeAngelis, P. G. Gildner, R. Chow and T. J. Colacot, J. Org. Chem., 2015, 80, 6794–6813 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02995d |
| This journal is © The Royal Society of Chemistry 2018 |