
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrazone-based cobalt complexes toward multielectron redox and spin crossover†
Wei Huang,
Yujie Li,
Juan Yong,
Yang Liu and
Dayu Wu *
*
Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology, Collaborative Innovation Center of Advanced Catalysis & Green Manufacturing, School of Petrochemical Engineering, Changzhou University, Changzhou, Jiangsu 213164, China. E-mail: wudy@cczu.edu.cn
First published on 9th May 2018
Abstract
Hydrazone-based derivatives modified by substitution at different positions were utilized to prepare a series of bis-homoleptic cobalt complexes. One species, [CoIII(L1)2]+ (1), which incorporated deprotonated ligands, adopted a Co(III) diamagnetic ground state. However, the substituent of a hydrogen atom with a methyl group precluded the possibility of deprotonation upon metal coordination, which led to two species, [CoII(L2Me)2]2+ (2) and [CoII(L3NO2)2]2+, (3) which underwent a gradual spin crossover with an adjustable substituent effect and a mixed character of low-spin (doublet) and high-spin (quartet) populations in wide temperature ranges. Depending on the electronic effects of the substituents on the ligand, the multielectron redox behavior of the cobalt center was systematically modulated as well. This result demonstrates redox-switchable spin crossover in a new hydrazone-based Co(II) system, in which the deprotonation of the coordination pocket and substituent groups in aromatic ligands can have a profound effect on the redox potential and spin state of the metal center.
Introduction
The study of molecular complexes based on a redox-controllable metal center has been a hot topic in research because of the potential application in the field of redox shuttles for dye-sensitized solar cells (DSSC),1 anolytes for redox flow batteries, and as a redox mediator for water splitting and CO2 reduction.2,3 The function of such complexes has been proved to be strongly dependent on the electronic property of the N-donor ligands.4 Among these subjects, polypyridyl complexes containing iron and ruthenium metal centers have been widely investigated as photosensitizers during the catalytic cycle.5 Although great achievements have been made concerning terpyridine ligands, the main shortcoming lie in the lack of efficient tools to investigate the new structure–function relationship, in particular for those with electronically tunable substituents, which still deserve to be currently studied.6 Another interesting point for cobalt complexes is that their spin state, electronic properties, and redox chemistry can be tailored by optimizing the ligand environment.7–10 For the purpose of potential application, it is of paramount importance to tune the donor type and ligand field strength of the ligands available to form the cobalt coordination sphere.11–13 Just recently, Yang et al. reported the redox tuning and electronic effects of proximal nonredox active cations in cobalt Schiff-base complexes. The tunability in the reversible redox potential was ascribed to the control of the absolute energy of the molecular orbital participating in electron transfer.14 In the previous work, we investigated how the electronic effect of the ligand influences the spin crossover of Co(II) ion within a metallogrid complex.15 Based on the electrochemical results, modification of the ligand field strength would give rise to a variation in the electronic structure (or molecular orbitals) of the redox active cation. However, spin crossover in a multinuclear system is not easy to control due to the synthetic difficulty of modifying the ligand field of the bis-multidendate ligand. In contrast, the mononuclear system could provide a convenient tool to tune the electrostatic effect, which would uniformly shift the molecular orbitals on the redox active metal.16–19 To further elucidate the source of the change in redox potential and to optimize the spin crossover of the Co(II) ion starting from Schiff-base pyrazine ligands (Scheme 1, R1 = R2 = H: HL1, R1 = Me, R2 = H: L2Me, R1 = Me, R2 = NO2: L3NO2), we synthesized a series of mononuclear cobalt complexes, aiming at achieving control of the local spin state as well as the redox chemistry. The ligand environments with the N4O2 donor permitted facile comparisons of the electronic structure. In compound 1, ligand HL1 was deprotonated upon coordination and Co(II) was oxidized to Co(III). In other cases, L2Me and L3NO2 were not deprotonated, whereas HL4 in compound 4 was deprotonated, and divalent cobalt ion was stable. The ligand provided a coordination pocket of a transition-metal center, and thus could have a minimal effect on the coordination geometry of the cobalt center. Through this framework, a multielectron redox couple, such as Co(III/II) or Co(II/I), and ligand-based reduction were reversible, conveniently allowing systematic tuning of the electronic effect on the redox potential.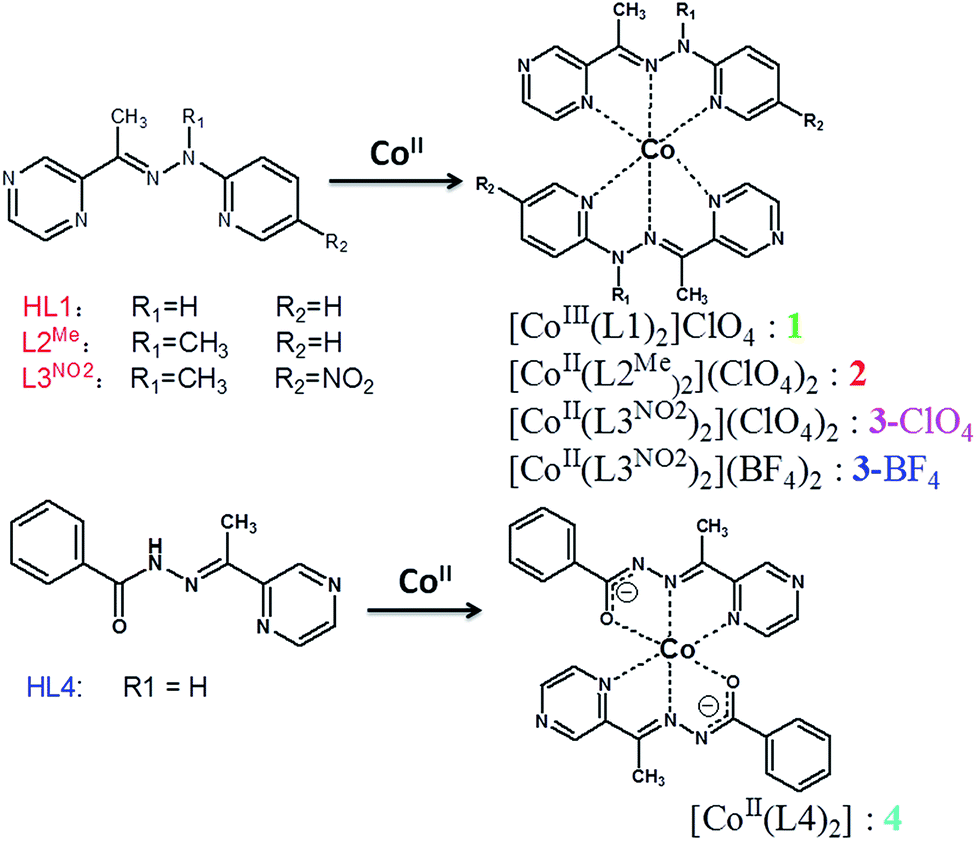 | ||
| Scheme 1 General structures of the ligands and complexes in this work. | ||
Although a handful of mononuclear 2,2′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6′,2′′-terpyridine-derivative Co(II)-center spin-crossover compounds have been previously investigated,20–25 other systems have been rarely reported and should be further developed.26–28 In the previous work, we investigated the deprotonation effects at the coordination pocket, such as how LH1 forms diamagnetic [Co(LH1)2]X complexes irrespective of the counterion X and how the introduction of substituents at the imine position has an effect on the variation of the spin state.15 As a result, the cobalt(II) complex of 2,5-bis[1-[2-methyl-2-(pyridin-2-yl)-hydrazono]ethyl]pyrazine (LMe), in which the methyl substituent was adopted to prevent deprotonation on the imine group, showed an incomplete spin crossover at higher temperature. In this paper we continue to expand the new ligand system and describe the synthesis of a number of ligands with modified substituents, aiming at achieving an enhanced spin crossover property of their Co(II) complexes, and also we discuss the effect of the substituents on the spin state of the cobalt(II) center.29
:6′,2′′-terpyridine-derivative Co(II)-center spin-crossover compounds have been previously investigated,20–25 other systems have been rarely reported and should be further developed.26–28 In the previous work, we investigated the deprotonation effects at the coordination pocket, such as how LH1 forms diamagnetic [Co(LH1)2]X complexes irrespective of the counterion X and how the introduction of substituents at the imine position has an effect on the variation of the spin state.15 As a result, the cobalt(II) complex of 2,5-bis[1-[2-methyl-2-(pyridin-2-yl)-hydrazono]ethyl]pyrazine (LMe), in which the methyl substituent was adopted to prevent deprotonation on the imine group, showed an incomplete spin crossover at higher temperature. In this paper we continue to expand the new ligand system and describe the synthesis of a number of ligands with modified substituents, aiming at achieving an enhanced spin crossover property of their Co(II) complexes, and also we discuss the effect of the substituents on the spin state of the cobalt(II) center.29
Experimental section
Materials and general procedures
All the reagents employed were commercially available and used without further purification. Methanol and acetonitrile were dried using standard procedures. Predeuterated dimethyl sulfoxide (DMSO-d6) was purchased from Alfa Aesar Co. Ltd., while 2-hydrazinylpyridine, benzohydrazide, 2-Br-5-nitropyridyl, and 2-acetylpyrazine were obtained from a commercial source and used directly without any purification. 1-Methyl-1-pyridylhydrazine was prepared according to reported procedures.30UV-visible studies were performed on a Perkin-Elmer Lambda 950 UV-vis instrument. Elemental analyses (C, H, and N) were conducted with a Perkin-Elmer 2400 analyzer. Micro-IR spectroscopy studies were performed on a Nicolet Magna-IR 750 spectrophotometer in the 4000–400 cm−1 region (w, weak; b, broad; m, medium; s, strong) with a KBr disc. 1H NMR spectra were obtained from a solution in deuterated dimethyl sulfoxide using a Bruker-400 spectrometer. (s, singlet; d, doublet; t, triplet; m, multiplet; dd, double doublet). Magnetic susceptibility measurements were carried out using a superconducting quantum interference device (SQUID) magnetometer (Quantum Design MPMS-XL) under applied magnetic fields of 2500 Oe. Corrections for diamagnetism were applied using Pascal's constants.31 Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) experiments were undertaken in acetonitrile (0.05 M [Bu4N](PF6)) at 293 ± 2 K using a CHI620E computer-controlled electrochemical workstation and a standard three-electrode cell employing a glassy carbon microelectrode as the working electrode, a Pt wire counter electrode, and a Ag wire in a tip separated from the bulk solution, which was used as the quasi-reference electrode. All the potentials given in this paper are referred to the ferrocene/ferrocenium ([FeCp2]0/+) reference couple under the same conditions as an internal reference.
Data collection and refinement
Single-crystal X-ray data for complexes 1–4 were collected on a Bruker APEX-2 CCD using Mo-Kα radiation (λ = 0.71073 Å). Data collection, data reduction, and cell refinement were performed by using the Bruker Instrument Service v4.2.2 and SAINT V8.34A software, respectively.32,33 The structure was solved by direct methods using the SHELXS program, and refinement was performed using SHELXL based on F2 through a full-matrix least squares routine.34 Empirical multi-scan absorption corrections using equivalent reflections were performed with the SADABS program.35 All the non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were set in the calculated positions and refined as a riding model.36,37Synthetic procedures
Caution! Perchlorate salts are potentially explosive and should be treated with great caution. Only small amounts were used in the present work.
:MeOH (v/v = 1:1) solvent was carefully layered on the top of this solution as a buffer layer. Finally, a 2 mL solution of LH1 (1.9 mg, 9 μmol in MeOH) was layered on top of the buffer layer. The tiny tube was kept in the dark and left undisturbed without protection from air. Over the course of two weeks, dark-red block-shaped crystals suitable for X-ray diffraction studies were obtained at the buffer layer. Yield: 1.1 mg, 37%. IR (KBr pallet, cm−1): 628(m), 780(m), 1092(s), 1316(m), 1472(m), 1621(m). Elemental analysis (%) calculated for C22H20ClCoN10O4: C, 45.33; H, 3.46; N, 24.03. Found: C, 45.44; H, 3.35; N, 24.23.:MeOH (v/v = 1:1) solvent was carefully added to the top of this solution as a buffer layer. Finally, a 2 mL solution of Co(ClO4)2·6H2O (0.5 mg, 1.5 μmol in MeOH) was layered on top of the buffer layer. The tiny tube was kept in the dark and left undisturbed without protection from air. Over the course of two weeks, dark-red block-shaped crystals unsuitable for X-ray diffraction studies were obtained at the interface. Yield: 0.17 mg, 16%. IR (KBr pallet, cm−1): 625(m), 773(m), 850(w), 943(w), 1091(s), 1316(m), 1469(m), 1607(m). Elemental analysis (%) calculated for C24H26Cl2CoN10O8: C, 40.46; H, 3.68; N, 19.66. Found: C, 40.72; H, 3.82; N, 19.44.Complex [CoII(L3NO2)2](BF4)2·0.5CH3OH (3BF4) was prepared in a similar manner as for 3ClO4 except that Co(BF4)2·6H2O was used instead of Co(ClO4)2·6H2O. Yield: 27.12 mg, 68%. Elemental analysis (%) calculated for C24.5H26B2CoF8N12O4.5: C, 37.10; H, 3.30; N, 21.19. Found: C, 36.92; H, 3.12; N, 21.76.
:MeOH (v/v = 1:1) solvent was carefully added to the top of this solution as a buffer layer. Finally, a 2 mL solution of HL4 (4.8 mg, 20 μmol in MeOH) was layered on top of the buffer layer. The tiny tube was kept in the dark and left undisturbed without protection from air. Over the course of two weeks, dark-red block-shaped crystals suitable for X-ray diffraction studies were obtained at the interface. Yield: 2.0 mg, 38%. IR (KBr pallet, cm−1): 626(w), 709(m), 1068(m), 1405(s), 1621(m). Elemental analysis (%) calculated for C26H22CoN8O2: C, 58.10; H, 4.13; N, 20.85. Found: C, 58.11; H, 4.21; N, 21.04.Results and discussion
The non-symmetric ligands used in this work were prepared through reacting 1-pyridylhydrazine and commercially available 2-acetylpyrazine. It should be noted that the reaction to produce L3NO2 was difficult to occur, possibly due to the relatively low electron density on the carbohydrazide nitrogen, which was unfavorable to nucleophilic attack, and so forcing conditions were required, namely 120 °C in toluene for 1 day, to complete the reaction. This method, previously used to generate the symmetric pyrazine-bridged bis-tridentate ligand (LR),15 should open up access to a wide variety of non-symmetric pyrazine-based ligands. HL1 and HL4 were readily deprotonated so the complexes [CoIII(L1)2]ClO4 (1) and [CoII(L4)2](ClO4)2 (4) were initially prepared by simply combining a 2:1 ratio of the ligand and [CoII(H2O)6](ClO4)2 salt in methanol. However the oxidation of cobalt(II) in complex 1 cannot be avoided even by using a Schlenk flask in an inert atmosphere of nitrogen. So all the synthetic operations were undertaken in air. Single crystals were grown by a layering method of a CH3OH solution of ligand HL1 and HL4 and, on one occasion, of an ethyl acetate–MeOH solution for L2Me. However, the reaction between electron-deficient ligand L3NO2 and cobalt salt was not easy to occur at room temperature due to the dynamic inertia. Thus the syntheses of compounds 3ClO4 and 3BF4 were further optimized by holding the reaction suspension containing L3NO2 and cobalt salt in acetonitrile at 55 °C for 30 min, which was then diffused with ether vapor to produce higher-quality, more uniform single crystals suitable for X-ray diffraction analysis. It is necessary to note that the reaction to produce 1, 2, and 4 should be operated in a low concentration of reactants. The higher concentration was also attempted, but the diffusion reaction was so rapid that the precipitate with high impurity rather than crystals was obtained. Hence, to obtain spectroscopic, magnetic, and electrochemical data, we had to set up many reactions in parallel to accumulate enough sample.
The UV-vis absorption spectra of all the ligands and their cobalt complexes are shown in Fig. 1, where the features closely match those of the previously investigated [Co4] complexes.15 For the complexes 1 and 4, two transitions appear at ∼280 nm and ∼320 nm that can also be observed in the free ligand spectra (see Fig. 1(a)). These could be expected to be ligand-based π → π* transition in nature. A similar transition was also observed in complexes 2 and 3. The transitions can be observed at ∼390 nm for 1 and 415 nm for 4, and are red-shifted relative to the free ligand, and could possibly be assigned as a deprotonated ligand-based transition. The most striking point that can be observed in [CoIII(L1)2]+ (1) is a strong absorption that spans from 450 to 650 nm, reminiscent of a similar feature in the [CoIII4]4+ spectrum.15 This observation in solution at room temperature distinguishes these species into three categories: 1, 2 and 3, and 4 in view of the ground state electronic absorption spectra, which is consistent with the later analysis of the valence state.
 | ||
| Fig. 1 UV-vis absorption spectra for all the ligands (a) and the corresponding cobalt complexes (b) in acetonitrile at room temperature. | ||
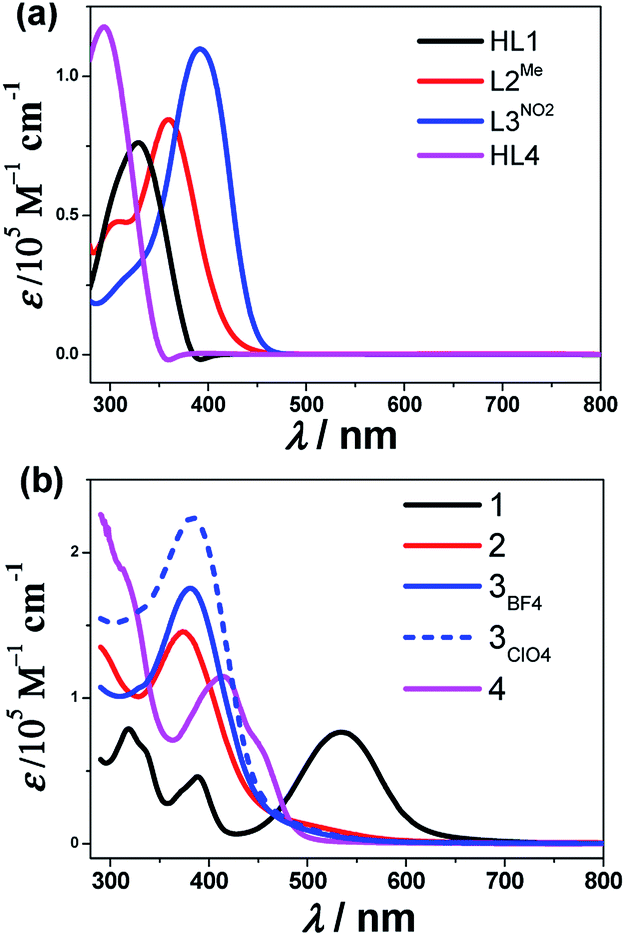
X-ray diffraction data were collected for single crystals of the cobalt complexes at 298 K. In addition, the data at 150 K were collected for complex 3ClO4 considering the potential spin-state transition-induced structure changes. The structure refinement parameters are collected in Table 1. The cobalt(III) complex 1 crystallized in the monoclinic space group P21/c. Here, the central cobalt atom is coordinated by two deprotonated L1 ligands binding via chelating in a mer fashion the pyridyl nitrogens (N5 and N10), imide N8 and N3, and pyrazine nitrogens (N1 and N6) (Fig. 2(A)). Furthermore, the bond lengths of C7–N4 (1.312 Å) and N4–N3 (1.332 Å), as well as another pair of bond lengths (1.330 Å for C18–N9 and 1.310 Å for N9–N8) on the ligand, are intermediate between a normal single bond and a double one, suggesting that the protons on the N4 and N9 sites are lost upon metal binding. The lengths of the Co–N bonds are quite characteristic of the respective spin and oxidation states of the coordinated metal ion and, therefore, can be used as diagnostic tools for determining the electronic state of the metal ion.38 The Co–N bond distances range from 1.817(2) to 1.877(2) Å, typical of LS CoIII,39 and the angular distortion parameter (Σ, defined as the sum of deviation from 90° of 12 cis-N–Co–N angles about the Co atom) is 76.68°. The resultant structural models of Co(II) complexes display geometries that are consistent with a singlet ground state for complex 1 at room temperature, while a quartet at the same temperature for complexes 3 and 4, highlighting the chemical modification of the electronic configuration of the metal-center orbitals.
| Complex 1 | Complex 3BF4 | Complex 3ClO4 | Complex 4 | ||
|---|---|---|---|---|---|
| 150 K | 296 K | 296 K | 150 K | 296 K | |
| a R1 = ∑(|Fo| − |Fc|)/∑|Fo|.b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2 | |||||
| Formula | C22H20ClCoN10O4 | C24.5H26B2CoF8N12O4.5 | C25H24Cl2CoN12O13 | C25H28Cl2CoN12O13 | C26H22CoN8O2 |
| F.W. | 582.86 | 793.12 | 830.39 | 834.42 | 537.45 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P21/n | P21/n | P21/n | P21/n | Aba2 |
| a [Å] | 8.6063(5) | 14.713(8) | 14.826(4) | 14.286(8) | 12.408(2) |
| b [Å] | 9.7092(8) | 13.235(7) | 13.366(4) | 13.218(7) | 19.368(3) |
| c [Å] | 28.092(2) | 18.309(9) | 18.433(5) | 18.043(10) | 9.8080(16) |
| α [°] | 90 | 90 | 90 | 90 | 90 |
| β [°] | 98.083(2) | 102.214(12) | 102.714(7) | 102.175(11) | 90 |
| γ [°] | 90 | 90 | 90 | 90 | 90 |
| Volume [Å3] | 2324.1(3) | 3485 (3) | 3563.1(18) | 3331(3) | 2357.0(7) |
| Z | 4 | 4 | 4 | 4 | 4 |
| Cal. Density (g cm−3) | 1.666 | 1.512 | 1.548 | 1.664 | 1.515 |
| μ/mm−1 | 0.909 | 0.587 | 0.710 | 0.760 | 0.771 |
| F(000) | 1192 | 1608 | 1636 | 1724 | 1108 |
| θ range [°] | 2.93–25.36 | 0.25–30.87 | 0.23–30.32 | 1.04–30.54 | 0.33–30.12 |
| Goodness of fit on F2 | 1.132 | 1.035 | 1.055 | 1.032 | 1.028 |
| Final R indices | R1a = 0.0466 | R1 = 0.0903 | R1 = 0.1001 | 0.1061 | R1 = 0.0427 |
| [I > 2σ(I)] | wR2 = 0.0828 | wR2 = 0.2357 | wR2 = 0.2484 | 0.2512 | wR2 = 0.1522 |
| R Indices (all data) | R1 = 0. 0801 | R1 = 0.1508 | R1 = 0.2189 | 0.2142 | R1 = 0.0435 |
| wR2b = 0.0905 | wR2 = 0.2621 | wR2 = 0.3089 | 0.2945 | wR2 = 0.1530 | |
 | ||
| Fig. 2 Crystal structure of the Co-containing complexes in 1 (A), 3ClO4 (B), 4 (C) and crystal packing mode in 3ClO4 (D) with the selected atom labels with ball-and-stick representation. Co = pink ball, C = gray, N = blue, O = red. Hydrogen atoms have been removed for clarity. | ||
The cobalt(II) complexes 3ClO4 and 3BF4 crystallized in the monoclinic space group P21/c and had similar structures except for the anions (Fig. 2(B) and Fig. S9, ESI†). Here, the binding mode of ligand L3NO2 to CoII is identical between complexes 3ClO4 and 3BF4 and so only the structure of 3ClO4 will be described herein. Single-crystal X-ray structures were determined at 296 K (3ClO4RT) and 150 K (3ClO4LT), respectively (Table 1). As in the case of 1, the coordination sphere of the CoII ion adopts a distorted octahedron with a CoN6 chromophore. Four equatorial N atoms, including N2, N3, N5, N9 with the average Co⋯N distance of 2.034(6) Å and two axial N atoms of N7 and N11 with Co⋯N distance of 2.114(4) and 2.122(3) Å that come from the pyrazine and pyridyl group of the individual ligand consist of an elongated octahedral environment, which is characteristic of CoII-HS systems. In the crystal packing mode, some significant intermolecular weak interactions were found, such as π⋯π stacking between two neighboring pyrazine rings with the shortest atom⋯atom distance of 3.317 Å (Fig. 2(D)). In addition, the intermolecular contacts through the nitro-group also support the framework, such as O⋯O contact of 2.952 Å and O⋯N contact of 3.001 Å.
In view of the spin-crossover behavior, we continued to determine the structure of 3ClO4 at low temperature (150 K). The coordination environment of the metal center was analogous to that observed in 3ClO4RT. However, the average Co⋯N bond lengths (Table 2) within the equatorial plane now shrink to 1.942(5) Å, and here the striking point is that the axial coordination bonds of 2.086(8) and 2.119(7) Å were not significantly changed, giving rise to more elongated octahedron as expected for a Jahn-Teller distorted low-spin octahedral d7 system. The observed distortion for the low-spin cobalt(II) complex was reminiscent of other low-spin mononuclear cobalt(II) complexes of 2,2′:6′,2′′-terpyridine (terpy) and other N-donor complexes.20–24 The distortion situation is also consistent with our recently reported SCO Co4II metallogrid [Co4II(LMe)4](ClO4)8·4MeCN·H2O,15 but contrasts with that recently observed in the LS Co4II grid [Co4II(HL3)4](ClO4)4·8H2O, where a compressed octahedron was observed.40 Alternatively, the distortion parameter, Σ, defined as the sum of the deviation from 12 cis-N–Co–N angles, is sensitive to the changes of the spin state of the center cobalt atom.41 In general, HS Co(II) compounds have a larger value of Σ than that in LS ones because HS metal compounds allow for more distorted coordination spheres. In the reported Co-terpy SCO compound,20–24 the values of Σ ranged from 92° corresponding to the LS state to 119° for the HS cobalt(II) center, respectively. For compound 3ClO4, the angular distortion parameters, Σ (deg), about the cobalt atom shrank from 113.2(1)° at room temperature corresponding to HS Co(II) to the value of 102.8(4)° at 150 K in a LS structure, indicating the heavier deviations from the ideal octahedral for the LS cobalt(II) center in this system. The low-spin Σ value was unusually large for this complex and, to the best of our knowledge, is at the high end among all cobalt(II) spin-crossover active complexes.20–24
| Comp. | 1 | 3ClO4 | 3BF4 | 4 | |
|---|---|---|---|---|---|
| a The value corresponds to the bond distance of Co–O.b av means the average bond distance.c Σ (deg) is defined as the sum of deviation from 90° of twelve cis-N–Fe–N angles about the cobalt atom. | |||||
| Temp. | 150 K | 150 K | 298 K | 298 K | 296 K |
| Co–Nimine | 1.866(3) | 1.884(9) | 2.020(7) | 2.007(6) | 2.047(5) |
| 1.871(3) | 1.962(9) | 2.053(6) | 2.038(6) | ||
| Co–Npyrazine | 1.901(3) | 1.934(1) | 2.028(7) | 2.012(7) | 2.163(5) |
| 1.902(3) | 2.086(8) | 2.114(6) | 2.127(5) | ||
| Co–Npyridine | 1.932(3) | 1.988(1) | 2.037(7) | 2.034(6) | 2.089(6)a |
| 1.934(3) | 2.119(7) | 2.122(5) | 2.143(5) | ||
| Co–N/Oavb | 1.901(3) | 1.996(1) | 2.061(3) | 2.067(5) | 2.100(2) |
| Σ (deg)c | 66.4(5) | 102.8(4) | 113.2(1) | 111.6(5) | 131.4(8) |
Cobalt(II) complex 4 crystallized in the orthorhombic system Aba2 with structural data collected at room temperature. The central CoII, which lies on a center of inversion, is surrounded by two deprotonated L4 ligands binding via the chelating N2O pocket (Fig. 2(D)). Compound 4 featured a distorted octahedral sphere due to the chelating character of the L4 ligand with cis-N–Co–N and cis-N–Co–O angles ranging between 75.70(4)–110.15(2)° and 75.12(4)–100.10(2)°. The Co–N (pyrazine) bond distance was 2.160(2) Å, while the Co–N (imine) and Co–O (carboxylate) bond distances were 2.050(2) Å and 2.091(2) Å. The bond valence sum (BVS) value of 2.66 for the cobalt center in 4 supported the 2+ valence assignment on the basis of bond lengths.42,43 There were no solvent molecules in the outer coordination sphere or the anions in the lattice, indicating that deprotonation occurs on the N4 atom upon metal coordination.
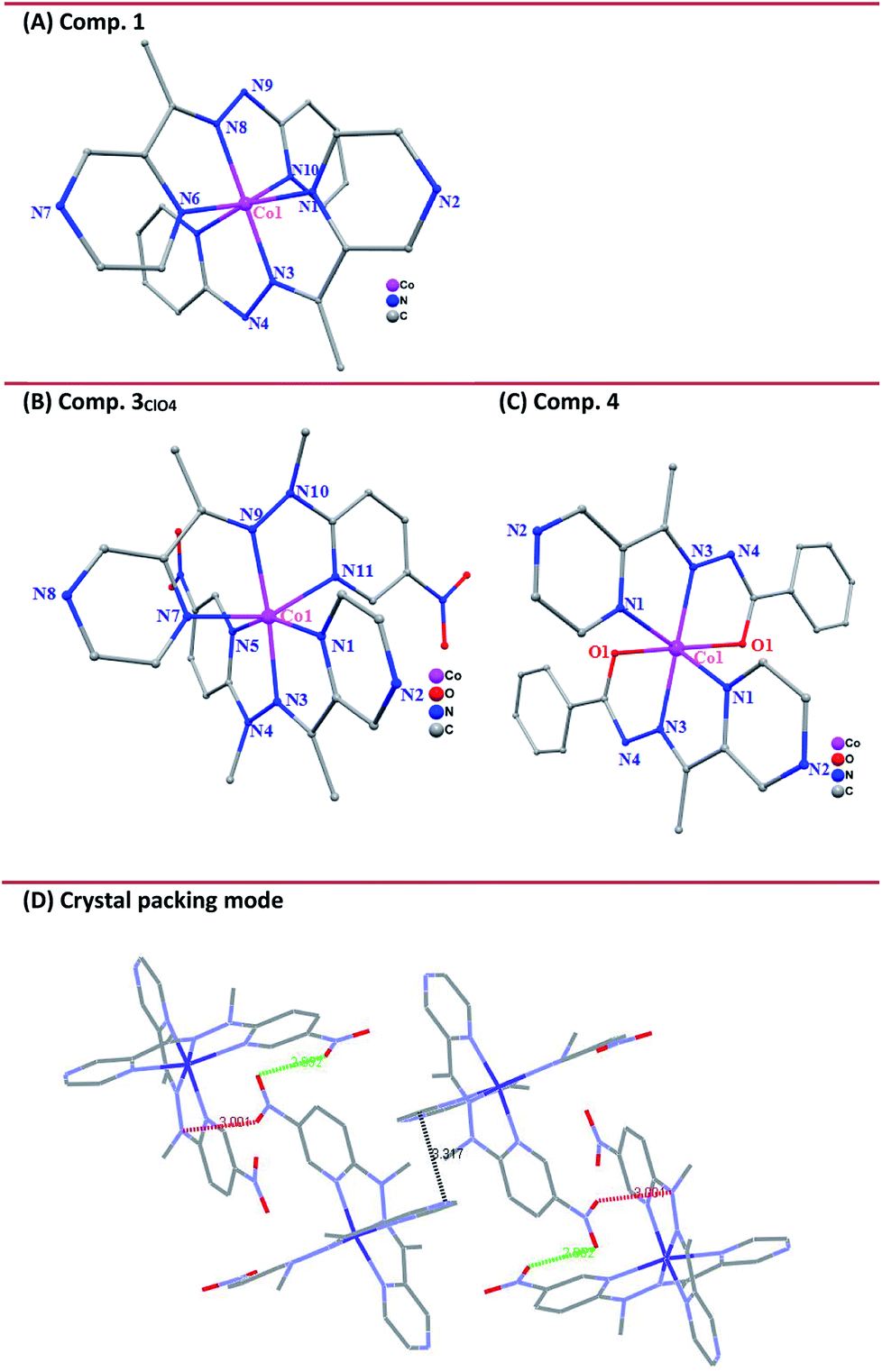
On the basis of X-ray diffraction data and electronic absorption spectra, magnetic susceptibility data of all the complexes were further collected in the studied temperature ranges in the solid state (Fig. 3(a)). As expected, complex 1 was diamagnetic over the measured temperature region, corresponding to the LS CoIII ions. The room temperature χMT value of compound 4 was equal to 2.34 cm3 K mol−1, which was obviously higher than the expected values of 1.875 cm3 K mol−1 corresponding to HS d6 (5T2g) ground state, reflecting the substantial orbital contribution to the magnetic moment. As the temperature decreased, χMT did not significantly decrease until 40 K, then decreased more rapidly to reach a value of 1.8 cm3 K mol−1 at 5 K, due to zero field splitting effects on the resultant 5A2g state. However, the magnetic moment of complexes 2 and 3 with the different anions showed different results. The sample was first measured from room temperature to 5 K, followed by the sequential heating/cooling cycles. The temperature-dependent magnetic data showed the gradual spin-crossover phenomenon for all three compounds. Compared to compound 2, compound 3 evidenced the more complete transition from the spin state (s = 1/2) at low temperature to high spin (s = 3/2) at high temperature. As a representative, complex 3ClO4 showed an initial χMT value of 2.08 cm3 mol−1 K at 400 K, corresponding to the main population of the HS CoII ground state, then a decrease in χMT occurred as the sample was cooled to 150 K where a χMT value of 0.88 cm3 mol−1 K was observed. Further cooling beyond this point gave rise to a sloping plateau in χMT, reaching 0.58 cm3 mol−1 K at 5 K. The plot is typical of a gradual S = 1/2 ↔ S = 3/2 spin crossover. No obvious hysteresis occured upon the sequential heating–cooling cycles in the temperature range of 5–400 K. The investigation of magnetic data for compound 3BF4 gave a similar result. However, at low temperature, the compound showed a more complete spin transition. For comparison, the magnetic data for the grid compound were aligned with the data of 3BF4.(Fig. S10, ESI†) In the reported tetranuclear cobalt grid complexes,15 the typical χMT value at low temperature is ca. 2.2 cm3 mol−1 K, corresponding to four low-spin states (s = 1/2). With increasing temperature, but the χMT value could not reach the saturation value even at the highest temperature of 400 K, indicating the rather incomplete spin transition in the Co4 grid complex. However, for 3BF4, the high temperature χMT value above 350 K almost plateaued at ca. 2.1 cm3 mol−1 K, indicating the complete spin transition for the mononuclear complex.
 | ||
| Fig. 3 (a) Temperature dependence of the product χMT for solids 1–4 under an applied magnetic field of 2500 Oe. (b) Solid UV-visible absorption spectra of 3ClO4 powder at temperatures between 77 K and 348 K. | ||
Given the potential of temperature-dependent metal–ligand interactions occurring on the SCO complex 3ClO4, there should be a partial population of the ligand-center ground state and concomitant π → π* transition within a wide temperature range.44 To further check this point, we collected UV-vis absorption spectra in the solid state as a function of temperature over the range 77−348 K. As expected, the ultraviolet absorption feature increased in intensity as the sample was heated. On subsequent cooling to room temperature, the spectra could reversibly recover to the original one (Fig. 3(b)). This temperature-dependence behavior reflected an equilibrium between the high- and low-spin populations, where the low-spin state is favored enthalpically and the high-spin state dominate at increasing temperature due to the contributions of entropy to the total Gibbs free energy.
This electrochemical switching provides an alternative stimuli for spin crossover from ca. 4 μB for CoII-HS by oxidation to 0 μB for CoIII-LS.24,45–47 Cyclic voltammetric studies of a 0.1 mM solution of all the cobalt complexes under transient conditions were performed by using cyclic voltammetry (CV) and differential pulse voltammetry (DPV) at 25 ± 2 °C in acetonitrile containing 0.05 M of [Bu4N][PF6] as the supporting electrolyte, and showed one to three well-separated processes labeled as I, II, and III, respectively, in Fig. 4 (Table 3). The formal reversible potentials (E(II) = (Eoxp + Eredp)/2) were −946 mV and −1010 mV versus [Fc]0/+ for complexes 1 and 2, which could be attributed to the one-electron reduction of Co(III) to Co(II) for process I. The peak current is proportional to the square root of the scan rates in the range of 0.05 V s−1 to 0.5 V s −1, indicating this process is diffusion controlled at the peak potential (Fig. S11–14, ESI†). A second potentially chemically reversible reduction process (II) with an approximate E(I) = −1706 and −1554 mV for 1 and 2, respectively, was observed when the potential was scanned to more negative values, which was associated with the reduction of the Co(II) to Co(I). The Ioxp/Iredp ratio at all scan rates studied was almost unity and ΔEp (ΔEp = Eoxp − Eredp, where the superscripts ox and red denote the oxidation and reduction peaks respectively) was close to the theoretical value of 57 mV at 25 °C for a simple one-electron process, indicating all the processes were one-electron chemically and electrochemically reversible. However, complex 3 evidenced only one redox process, II, with the formal potential of −1698 mV. Interestingly, an additional process, III, was observed for complex 4 with the formal potentials (E(n), n = III, II, I) versus [Fc]0/+ of −2140, −1510, and −510 mV. The current magnitude for processes I, II, and III were similar. Process III, (E1/2(III) = −2140 mV), exhibited a negative or reduction current, and hence was ascribed to a ligand-centered redox process on the basis of previous assignments of pyrazine-substituted cobalt complexes.48,49 From the electrochemical data, there existed a slight difference in the electronic behavior between complex 1 and 2 and the highest oxidation state corresponding to CoIII ion required a quite negative oxidation potential. In this situation, complex 2 was still isolated as CoII species and did not induce any partial oxidation, as evidenced by magnetic characterization and X-ray diffraction analysis. However, the oxidation of the Co(II) complex required the most negative potential in complex 2 compared to those in the cases of 1 and 4, suggesting that the ligand in 2 provided the most weak ligand field in the order of L2Me < L1 < L4. This is normal because deprotonation should render the delocalized aromatic system of the ligand as a weaker π acceptor, thus weakening the ligand strength in 1 and 4. This result is consistent with the observation of cobalt metallogrid complexes in the previous report.15
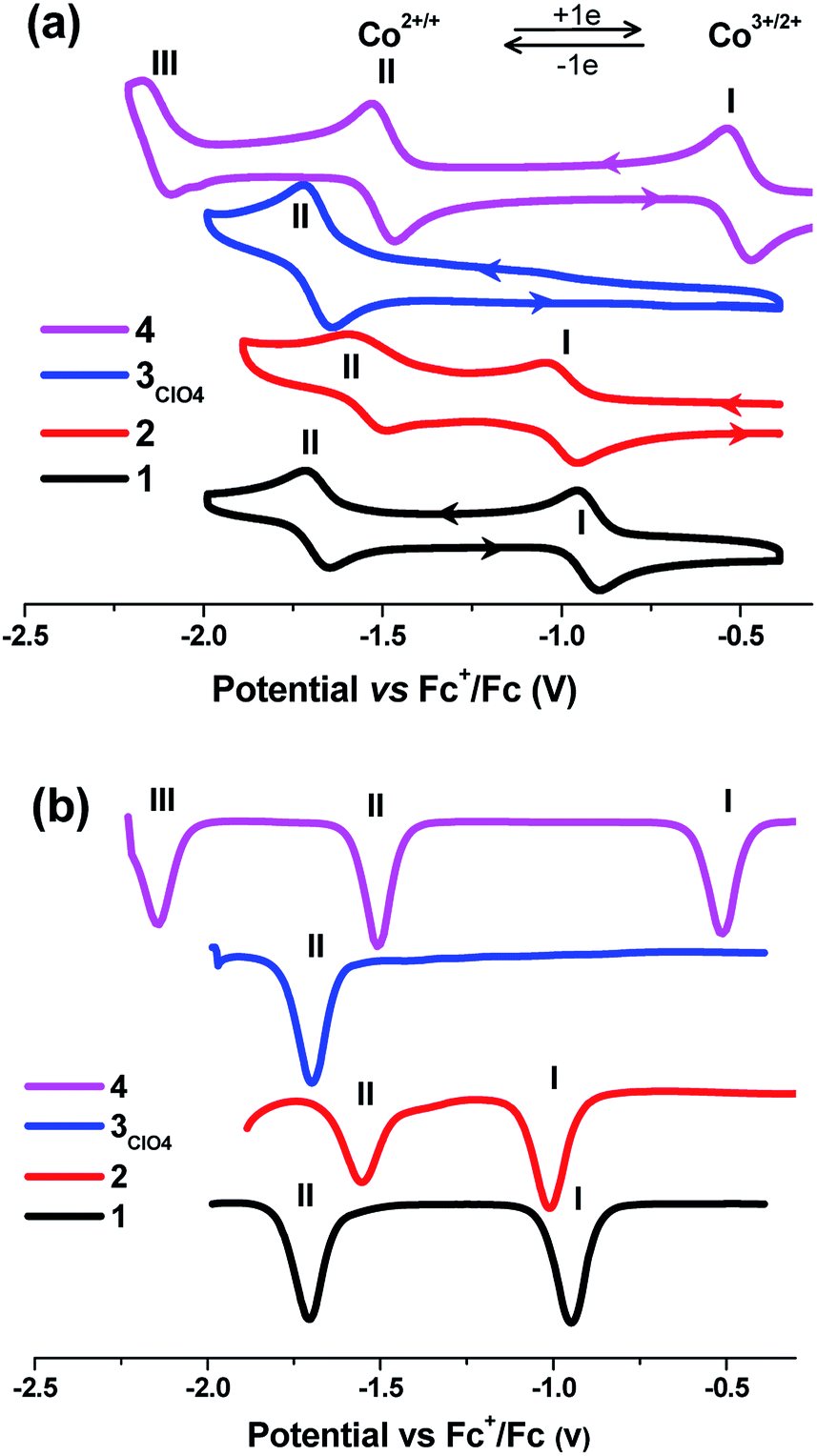 | ||
| Fig. 4 Cyclic voltammogram (a) and DPV (b) of complexes 1–4 (0.1 mM) in MeCN/0.05 mM NBu4PF6 referenced against Fc+/Fc at a scan rate of 100 mV s−1. | ||
| Solvent | Comp. | E1/2 (V)a | ||||
|---|---|---|---|---|---|---|
| CoIII/II | CoII/I | Ligand-base | ||||
| a Scan rate = 100 mV s−1.b Concentration in all cases = 0.1 mM.c – Not observed.d M = Na+, K+, Ca2+, Sr2+, and Ba2+.e The potential values were referenced against Ag/AgNO3. | ||||||
| CH3CN | [CoIII(L1)2]ClO4 (1) | −0.93 | −1.66 | –c | This workb | |
| CoII(L2Me)2 (2) | −0.98 | −1.53 | — | |||
| [CoII(L3NO2)2]·CH3OH (3ClO4) | — | −1.67 | — | |||
| CoII(L4)2 (4) | −0.49 | −1.52 | −2.14 | |||
| CH3CN | C1–C8 | −0.21 to +0.84 | −0.82 to −1.92 | −1.61 to −2.49 | Ref. 10 | |
| [Co(BTSC) (L)2]+ | −0.69 to −1.10 | −1.00 to −1.19 | — | Ref. 12 | ||
| DMF | Co(salen–OMe) | — | −1.71 | — | Ref. 14 | |
| 2Md | — | −1.58 to −1.41 | — | |||
| CH3CN | [CoII(dpzca)2]e | −0.22 | − | −1.46, −1.84 | Ref. 24 | |
| CoIIICo3II/Co4II | Co2IIICo2II/CoIIICo3II | Co3IIICoII/Co2IIICo2II | CoIII4/CoIII3CoII | Ref. 15 | ||
| CH3CN | [CoIII4(LH)4](ClO4)4 | 0.63 | 0.52 | −0.77 | −0.83 | |
| [CoII4(LMe)4](ClO4) | 0.30 | 0.02 | −0.60 | −0.74 | ||
| [CoII4(LBr)4](ClO4)8 | −0.40 | −0.49 | −0.62 | −0.87 | ||
Conclusion
In conclusion, we prepared a series of new tridentate Schiff-base ligands bearing a variety of substituting moieties through a condensation reaction between 2-acetylpyrazine and different hydrazone derivatives. The corresponding cobalt complexes with bis-substitute were isolated and structurally characterized. The analysis of the crystal structures provided insights into understanding the differences in the Co–N bond lengths observed in the crystal structures, assigning them to differences in terms of the spin state and valence state. In particular, the Co–N bond length can be diagnostic for differentiating the low-spin doublet from the high-spin quartet state. In addition, we showed that the redox potential of the Co(III)/Co(II), Co(II)/Co(I), and pyrazine-base couples could be easily modulated over a wide range by tuning the substituents on the ligands, allowing a broad range of potential applications. Study of the catalytic proton reduction by applying this class of complexes as a catalyst is currently ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support by PAPD of Jiangsu Higher Education Institutions and by NSFC programs (Grants 21671027 and 21471023) and sponsored by the Jiangsu Provincial QingLan Project.Notes and references
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed.
- Y. Kuramochi and O. Ishitani, Inorg. Chem., 2016, 55, 5702–5709 CrossRef CAS PubMed.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, J. Am. Chem. Soc., 2016, 138, 5765–5768 CrossRef CAS PubMed.
- Z. Guo, S. Cheng, C. Cometto, E. Anxolabéhère-Mallart, S. M. Ng, C. C. Ko, G. J. Liu, L. J. Chen, M. Robert and T. C. Lau, J. Am. Chem. Soc., 2016, 138, 9413–9416 CrossRef CAS PubMed.
- S. M. Fatur, S. G. Shepard, R. F. Higgins, M. P. Shores and N. H. Damrauer, J. Am. Chem. Soc., 2017, 139, 4493–4505 CrossRef CAS PubMed.
- R. G. Miller, S. Narayanaswamy, J. L. Tallon and S. Brooker, New J. Chem., 2014, 38, 1932–1941 RSC.
- M. W. Bezpalko, A. M. Poitras, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2017, 56, 503–510 CrossRef CAS PubMed.
- K. Y. Chen, C. Du, B. O. Patrick and C. P. Berlinguette, Inorg. Chem., 2017, 56, 2383–2386 CrossRef CAS PubMed.
- S. Aroua, T. K. Todorova, P. Hommes, L. M. Chamoreau, H. U. Reissig, V. Mougel and M. Fontecave, Inorg. Chem., 2017, 56, 5930–5940 CrossRef CAS PubMed.
- J. Palion-Gazda, B. Machura, R. Kruszynski, T. Grancha, N. Moliner, F. Lloret and M. Julve, Inorg. Chem., 2017, 56, 6281–6296 CrossRef CAS PubMed.
- A. P. King, H. A. Gellineau, J. E. Ahn, S. N. MacMillan and J. J. Wilson, Inorg. Chem., 2017, 56, 6609–6623 CrossRef CAS PubMed.
- H. S. Scott, C. J. Gartshore, S. X. Guo, B. Moubaraki, A. M. Bond, S. R. Batten and K. S. Murray, Dalton Trans., 2014, 43, 15212–15220 RSC.
- A. H. Reath, J. W. Ziller, C. Tsay, A. J. Ryan and J. Y. Yang, Inorg. Chem., 2017, 56, 3713–3718 CrossRef CAS PubMed.
- F. X. Shen, W. Huang, D. Y. Wu, Z. Zheng, X. C. Huang and O. Sato, Inorg. Chem., 2016, 55, 902–908 CrossRef CAS PubMed.
- H. Kon and T. Nagata, Inorg. Chem., 2009, 48, 8593–8602 CrossRef CAS PubMed.
- D. Ruiz-Molina, J. Veciana, K. Wurst, D. N. Hendrickson and C. Rovira, Inorg. Chem., 2000, 39, 617–619 CrossRef CAS PubMed.
- P. Ghosh, S. Samanta, S. K Roy, S. Joy, T. Krämer, J. E. McGrady and S. Goswami, Inorg. Chem., 2013, 52, 14040–14049 CrossRef CAS PubMed.
- C. C. Lu, T. Weyhermüller, E. Bill and K. Wieghardt, Inorg. Chem., 2009, 48, 6055–6064 CrossRef CAS PubMed.
- Y. Guo, X. L. Yang, R. J. Wei, L. S. Zheng and J. Tao, Inorg. Chem., 2015, 54, 7670–7672 CrossRef CAS PubMed.
- S. Hayami, Y. Komatsu, T. Shimizu, H. Kamihata and Y. H. Lee, Coord. Chem. Rev., 2011, 255, 1981–1990 CrossRef CAS.
- S. Hayami, K. Kato, Y. Komatsu, A. Fuyuhiro and M. Ohba, Dalton Trans., 2011, 40, 2167–2169 RSC.
- Y. Komatsu, K. Kato, Y. Y. Amamoto, H. Kamihata, Y. H. Lee, A. Fuyuhiro, S. Kawata and S. Hayami, Eur. J. Inorg. Chem., 2012, 16, 2769–2775 CrossRef.
- M. G. Cowan, J. Olguín, S. Narayanaswamy, J. L. Tallon and S. Brooker, J. Am. Chem. Soc., 2012, 134, 2892–2894 CrossRef CAS PubMed.
- S. Hayami, M. R. Karim and Y. H. Lee, Eur. J. Inorg. Chem., 2013, 5–6, 683–696 CrossRef.
- J. Zarembowitch and O. Kahn, Inorg. Chem., 1984, 23, 589–593 CrossRef CAS.
- A. V. Vologzhanina, A. S. Belov, V. V. Novikov, A. V. Dolganov, G. V. Romanenko, V. I. Ovcharenko, A. A. Korlyukov, M. I. Buzin and Y. Z. Voloshin, Inorg. Chem., 2015, 54, 5827–5838 CrossRef CAS PubMed.
- (a) M. G. Cowan and S. Brooker, Dalton Trans., 2012, 41, 1465–1474 RSC; (b) R. G. Miller, S. Narayanaswamy, S. M. Clark, P. Dera, G. B. Jameson, J. L. Tallon and S. Brooker, Dalton Trans., 2015, 44, 20843–20849 RSC; (c) M. G. Cowan, R. G. Miller and S. Brooker, Dalton Trans., 2015, 44, 2880–2892 RSC.
- M. Graf, G. Wolmershäuser, H. Kelm, S. Demeschko, F. Meyer and H. J. Krüger, Angew. Chem., 2010, 122, 962–965 CrossRef.
- D. Leung and E. V. Anslyn, Org. Lett., 2011, 13, 2298–2301 CrossRef CAS PubMed.
- C. J. O'Connor, Magnetochemistry–Advances in Theory and Experimentation, in Progress in Inorganic Chemistry, ed. S. J. Lippard, John Wiley & Sons, Inc., New York, 1982, Vol. 29, pp. 203–283 Search PubMed.
- SAINT, version 8.34A, Bruker AXS Inc., Madison, WI, 2013 Search PubMed.
- APEX2, version 2014.5–0; Bruker AXS Inc., Madison, WI, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- SADABS, Bruker AXS Inc., Madison, WI, 2014 Search PubMed.
- C. K. Johnson, in Crystallographic Computing, ed. F. R Ahmed, Munksgaard, Copenhagen, Denmark, 1970 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands. 1998 CrossRef CAS PubMed; A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- O. Sato, J. Tao and Y. Z. Zhang, Angew. Chem., 2007, 46, 2200–2236 CrossRef.
- D.-Y. Wu, G.-H. Wu, W. Huang and C.-Y. Duan, Polyhedron, 2008, 27, 947–954 CrossRef CAS.
- S. Q. Wu, Y. T. Wang, A. L. Cui and H. Z. Kou, Inorg. Chem., 2014, 53, 2613–2618 CrossRef CAS PubMed.
- M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493–2514 CrossRef CAS and references therein.
- (a) I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef; (b) N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–199 CrossRef.
- W. Huang, F. Pan, Z. Wang, Y. Bai, X. Feng, J. Gu, Z.-W. Ouyang and D. Wu, Dalton Trans., 2017, 46, 5069–5075 RSC.
- S. M. Fatur, S. G. Shepard, R. F. Higgins, M. P. Shores and N. H. Damrauer, J. Am. Chem. Soc., 2017, 139, 4493–4505 CrossRef CAS PubMed.
- R. Chant, A. R. Hendrickson, R. L. Martin and N. M. Rohde, Inorg. Chem., 1975, 14, 1894–1902 CrossRef CAS.
- I. A. Gass, C. J. Gartshore, D. W. Lupton, B. Moubaraki, A. Nafady, A. M. Bond, J. F. Boas, J. D. Cashion, C. Milsmann, K. Wieghardt and K. S. Murray, Inorg. Chem., 2011, 50, 3052–3064 CrossRef CAS PubMed.
- K. G. Alley, G. Poneti, P. S. D. Robinson, A. Nafady, B. Moubaraki, J. B. Aitken, S. C. Drew, C. Ritchie, B. F. Abrahams, R. K. Hocking, K. S. Murray, A. M. Bond, H. H. Harris, L. Sorace and C. Boskovic, J. Am. Chem. Soc., 2013, 135, 8304–8323 CrossRef CAS PubMed.
- J. W. Jurss, R. S. Khnayzer, J. A. Panetier, K. A. El Roz, E. M. Nichols, M. Head-Gordon, J. R. Long, F. N. Castellano and C. J. Chang, Chem. Sci., 2015, 6, 4954–4972 RSC.
- L. Chen, A. Khadivi, M. Singh and J. W. Jurss, Inorg. Chem. Front., 2017, 4, 1649–1653 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectra characterization, X-ray crystallography data and additional electrochemical data. CCDC 1571714–1571718. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra02963f |
| This journal is © The Royal Society of Chemistry 2018 |