DOI:
10.1039/C8RA02961J
(Paper)
RSC Adv., 2018,
8, 24760-24772
Evaluation of superficially and fully porous particles for HILIC separation of lanthanide–polyaminocarboxylic species and simultaneous coupling to ESIMS and ICPMS
Received
6th April 2018
, Accepted 3rd July 2018
First published on 9th July 2018
Abstract
In this work, amide-bonded columns packed with fully porous particles (FPP) and superficially porous particles (SPP) were evaluated to separate lanthanide–polyaminocarboxylic species by hydrophilic interaction liquid chromatography (HILIC), using two model samples of interest in nuclear and other industrial applications. We assessed the gains achieved by reducing the dimensions of the columns along with the size of the FPPs to sub-2 μm and by using sub-3 μm SPP-packed columns. The FPP-packed Acquity column (100 × 2.1 mm; 1.7 μm) performed better than the SPP-packed Accucore column (150 × 2.1 mm; 2.6 μm), with a separation that was two times more efficient and three times shorter, while generating around 30% less in effluent volumes. This column was also coupled simultaneously to electrospray ionisation mass spectrometry (ESIMS) and inductively coupled plasma mass spectrometry (ICPMS). The instrumental set-up was performed in a conventional laboratory, by taking into account the geometrical constraints existing in the laboratory dedicated to radioelement analysis. Furthermore, separation of the series of lanthanide (Ln) species was demonstrated for the first time thanks to the separation mode of hydrophilic interaction liquid chromatography.
Introduction
Green analytical chemistry has gained increasing attention over the last few years, incited by the ‘three Rs’ concept (reduce, replace, recycle).1 This approach encourages users to replace hazardous solvents with greener alternatives, and to reduce the consumption of solvent and reagents, as well as the waste generated during analytical procedures. In nuclear applications, this approach is a major step forward in that it helps reduce radioactive waste volumes and sample consumption, not to mention associated facility and cost constraints. With the aim of studying the speciation of lanthanides (Ln) in the framework of spent fuel treatment processes, this approach was initiated in a previous work2 and focused on the separation conditions by hydrophilic interaction liquid chromatography (HILIC) of Ln species containing strong chelating agents such as ethylenediaminetetraacetic acid (EDTA) and diethylenetriaminepentaacetic acid (DTPA). In this preliminary study, the selected Ln species were Nd, Eu, Gd, and Er, to be representative of the Ln series, from the light to the heavy. We showed that among several stationary phases functionalized with cross-linked diol, cyano, zwitterionic and amide functions, the first three led to the co-elution of Ln species, known to be difficult to separate due to their very close physico-chemical properties. Only amide-bonded stationary phases allowed the separation of Ln species.
In order to reduce the effluent volumes produced by these HILIC separations while maintaining or improving their speed and efficiency, two sustainable chromatographic strategies3 were applied in the current work. We took advantage of the gains achieved by reducing the diameter of fully porous particles (FPP) to sub-2 μm along with the dimensions of the columns, and by using columns packed with superficially porous particles (SPP). Sub-2 μm FPP and SPP technologies4–6 represent a major advance in chromatographic techniques these past few years, driven by the growing demand of high sample throughput, as well as fast and high efficient separation techniques, prompted by several industrial sectors. However, most applications of sub-2 μm FPP- and SPP-packed columns lie in the analysis of organic compounds by reversed phase liquid chromatography (RPLC).7–10 Although hydrophilic interaction liquid chromatography is devoted to separating polar, hydrophilic and/or charged compounds and is well suited for separating metallic complexes, including non-covalent species,11 application of this separation mode for speciation analysis is still relatively limited. Studies on the speciation of lanthanides have focused so far on gadolinium (Gd) species containing linear or cyclic polyaminocarboxylic acids, used as magnetic resonance imaging contrast agents.12 Dedicated analytical methods have been developed to monitor and quantify Gd species in the environment using HILIC stationary phases in narrow-bore or classical format, most widely functionalised by zwitterionic groups, and packed with fully porous particles of diameters exceeding 3 μm.13–15 To the best of our knowledge and excluding our previous work, sub-2 μm FPP-packed columns have only been used in one recent study dealing with speciation,16 while various polar stationary phases have been developed with this technology since its commercial introduction in 2004.17 Furthermore, the majority of polar SPP-packed stationary phases dedicated to hydrophilic interaction liquid chromatography are unmodified silica,18 despite numerous columns packed with SPP of different sizes, chemistries and porous shell thickness, have become available. The only example of separation by hydrophilic interaction liquid chromatography, involving a SPP-packed column for speciation analysis is with a bare silica SPP-packed column.19 Therefore, sub-2 μm FPPs and shell technologies are currently almost never used for speciation investigations using hydrophilic interaction liquid chromatography.
In this work, amide-bonded columns of different lengths and diameters, packed with 3.5 μm and sub-2 μm FPPs, as well as sub-3 μm SPP-packed columns, were evaluated to separate Ln species by hydrophilic interaction liquid chromatography using two model samples. The first sample concerns the development of nuclear spent fuel treatment processes based on liquid–liquid extraction, being a model of actinide species that can be found in aqueous back extraction phases. It was composed of natural neodymium (Nd) and samarium (Sm), exhibiting natural isobaric interferences also found in irradiated fuel solutions, in the presence of EDTA and DTPA. The second sample contained the entire lanthanide series with EDTA, leading to the formation of the corresponding 14 Ln–EDTA complexes. The separation of these kind of chemical species is of interest for nuclear applications, as well as for environmental and rare-earth recycling purposes.20,21 We determined the best compromise between the efficiency, the resolution, the analysis time, and the effluent volumes generated by separation of the species contained in the two samples, in order to select the most suited chromatographic technology. These experiments were run in a conventional laboratory by coupling the separations by hydrophilic interaction liquid chromatography to electrospray ionisation mass spectrometry (ESIMS). In a further step, the selected column was coupled simultaneously to ESIMS and inductively coupled plasma mass spectrometry (ICPMS), also in a conventional laboratory. This integrated mass spectrometric approach will allow to provide, in a single step, the structural characterization of the separated Ln species, along with the elemental and isotopic characterization of the Ln within the species. In addition, the overall effluent volumes, the consumption of the samples and the analysis times are even more reduced compared with the savings already achieved through a single coupling, which is of great interest for the analysis of nuclear or precious samples.
Experimental part
1. Chemicals and samples
Ethylenediaminetetraacetic acid (EDTA) tetrasodium salt dihydrate (C10H12O8N2Na4, 2H2O, purity ≥ 99.5%), diethylenetriaminepentaacetic acid (DTPA) (C14H23O10N3, purity ≥ 98%) and toluene were supplied by Sigma Aldrich (Saint Quentin Fallavier, France). Ultrapure water (resistivity 18.2 MΩ cm) was obtained from a Milli-Q water purification system (Millipore, Guyancourt, France). Acetonitrile (ACN, quality LC-MS grade), formic acid (Normapur grade), ammonium acetate (CH3CO2NH4, Normapur grade) and ammonia solution 25% were purchased from VWR Prolabo (Briare-le-Canal, France). Elemental standard solutions at 1000 μg mL−1 (lanthanum (La), cerium (Ce), praseodymium (Pr), neodymium (Nd), samarium (Sm), europium (Eu), gadolinium (Gd), terbium (Tb), dysprosium (Dy), holmium (Ho), erbium (Er), thulium (Tm), ytterbium (Yb) and lutetium (Lu)) were provided by the Spex Certiprep Group (Longjumeau, France), diluted in nitric acid media (HNO3, 2% w/w).
The stock solutions of EDTA and DTPA were prepared by dissolving the appropriate amount of ligand in ultrapure water to obtain a concentration of 5 × 10−3 mol L−1. Each Ln stock solution was prepared by diluting the corresponding elemental standard solution in ultrapure water to obtain a concentration of 5 × 10−3 mol L−1. The samples were prepared by mixing the appropriate volume of the stock solutions to obtain Ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EDTA:DTPA ratios of 2:1.75:0.75 (Ln = Nd, Sm), with each Ln concentration at 10−3 mol L−1 for sample 1, and a Ln:EDTA ratio of 1:1.6 (Ln = the series of 14 Ln) with each Ln concentration at 1.5 × 10−4 mol L−1 for sample 2. Then the pH of each sample was adjusted to 3.2 by adding an ammonia solution (25%) to reach a common pH in the aqueous back-extraction phases of treatment processes. The samples were further diluted in the mobile phase to reach a final concentration of 10−4 mol L−1 for each Ln in sample 1 and a concentration of 5 × 10−5 mol L−1 for each Ln in sample 2.
:EDTA:DTPA ratios of 2:1.75:0.75 (Ln = Nd, Sm), with each Ln concentration at 10−3 mol L−1 for sample 1, and a Ln:EDTA ratio of 1:1.6 (Ln = the series of 14 Ln) with each Ln concentration at 1.5 × 10−4 mol L−1 for sample 2. Then the pH of each sample was adjusted to 3.2 by adding an ammonia solution (25%) to reach a common pH in the aqueous back-extraction phases of treatment processes. The samples were further diluted in the mobile phase to reach a final concentration of 10−4 mol L−1 for each Ln in sample 1 and a concentration of 5 × 10−5 mol L−1 for each Ln in sample 2.
2. Hydrophilic interaction liquid chromatography conditions
Separations were carried out in a conventional laboratory using an Ultimate 3000 UHPLC+ focused system (Dionex/ThermoFisher, Courtaboeuf, France), which consisted of a degasser, a dual RS pump, an RS auto sampler, a column compartment and a model RS diode array detector. All stationary phases were functionalized with amide groups (Table 1). The FPP-packed columns were obtained from Waters (Milford, MA, USA): XBridge amide (150 × 2.1 mm; 3.5 μm), Acquity BEH amide (100 × 2.1 mm; 1.7 μm and 100 × 1.0 mm; 1.7 μm). Two Accucore-amide columns (150 × 2.1 mm; 2.6 μm and 100 × 2.1 mm; 2.6 μm) packed with SPPs were provided by Thermo Scientific (Waltham, MA, USA). A Sunshell HILIC amide column (100 × 2.1 mm; 2.6 μm) packed with SPPs was obtained from ChromaNik Technologies (Osaka, Japan). The column dead times were determined using toluene as marker and UV detection at 254 nm. The separations were run in isocratic mode, with a mobile phase composed of 70/30 (v/v) acetonitrile/water, 0.5% formic acid and 15 mM ammonium acetate. The optimal range of the mobile phase flow rates for the Accucore column (150 × 2.1 mm; 2.6 μm) was determined by plotting the Knox curve at flow rates varying between 35 and 1500 μL min−1 and injecting 3 μL of the Gd–DTPA complex at 10−4 mol L−1. The Gd–DTPA complex was chosen because of its location in the middle of the Ln series and its retention factor of around ten. The discrimination factor d0 was defined as 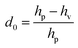 where hp: the smallest peak height between two consecutive peaks and hv: the valley height between two peaks.22 As chromatographic peaks are asymmetrical, efficiency values N were calculated for each species from the extracted ion chromatogram (XIC), with the Foley–Dorsey equation:23
where hp: the smallest peak height between two consecutive peaks and hv: the valley height between two peaks.22 As chromatographic peaks are asymmetrical, efficiency values N were calculated for each species from the extracted ion chromatogram (XIC), with the Foley–Dorsey equation:23 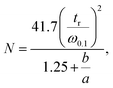 where tr: the retention time of analyte, ω0.1: peak width at 10% of the peak height, a and b: peak width before and after the apex, measured at 10% of the peak height. Analyses were performed two times with the XBridge (150 × 2.1 mm; 3.5 μm), Acquity (100 × 2.1 mm; 1.7 μm) and Accucore (150 and 100 × 2.1 mm; 2.6 μm) columns. Similar efficiencies were obtained in all cases, and the average of N is given in the tables. For the Acquity column of internal diameter of 1 mm, as well as for the tests of the Sunshell column, analyses were performed one time.
where tr: the retention time of analyte, ω0.1: peak width at 10% of the peak height, a and b: peak width before and after the apex, measured at 10% of the peak height. Analyses were performed two times with the XBridge (150 × 2.1 mm; 3.5 μm), Acquity (100 × 2.1 mm; 1.7 μm) and Accucore (150 and 100 × 2.1 mm; 2.6 μm) columns. Similar efficiencies were obtained in all cases, and the average of N is given in the tables. For the Acquity column of internal diameter of 1 mm, as well as for the tests of the Sunshell column, analyses were performed one time.
Table 1 Physico-chemical properties of the different columns used in this study and analysis conditions (NA: non-available)
| Name (manufacturer) |
Dimensions, particle size, flow rate (F); injection volume (Vinj) |
Functionalisation and support |
Specific surface (m2 g−1) |
Pore size (Å) |
Amide grafting, carbon load (%) |
| XBridge amide (Waters) |
150 × 2.1 mm; 3.5 μm, F 150 μL min−1; Vinj 5 μL |
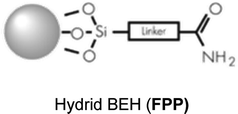 |
182 |
138 |
7.34 μmol m−2, 12% C |
| Acquity BEH amide (Waters) |
100 × 2.1 mm; 1.7 μm, F 300 μL min−1; Vinj 3 μL |
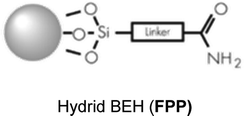 |
186 |
134 |
7.37 μmol m−2, 12% C |
| 100 × 1.0 mm; 1.7 μm, F 70 μL min−1; Vinj 1 μL |
| Sunshell HILIC amide (ChromaNik) |
100 × 2.1 mm; 2.6 μm, F 300 μL min−1; Vinj 3 μL |
 |
150 |
89 |
NA, 3.3% C |
| Accucore-amide (Thermo-Scientific) |
150 × 2.1 mm; 2.6 μm, F 150 μL min−1; Vinj 5 μL |
 |
80 |
150 |
NA, NA |
| 100 × 2.1 mm; 2.6 μm, F 150 μL min−1; Vinj 3 μL |
3. ESIMS parameters
The ESIMS instrument was a TSQ Quantum Ultra™ Stage Quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham MA, USA) equipped with an H-ESI II ionization source. ESIMS data was processed with XCalibur software from Thermo Fisher. All ESIMS spectra were recorded in negative ionization mode with the spray voltage at −3.7 kV. The vaporization and capillary transfer temperatures, the sheath gas, the auxiliary gas and the skimmer offset were optimized for each sample and flow rate, so as to obtain optimal signal stability and intensity. Full-scan mass spectra were acquired in the mass range m/z 200–700 and single ion monitoring (SIM) mass spectra were acquired by adjusting the scan width to the isotopic pattern of each species. In negative ionization mode, the Ln species were observed as [Ln–EDTA]− and [(Ln–DTPA) + H]−, and are noted Ln–EDTA and Ln–DTPA throughout the manuscript.
4. ICPMS parameters
The ICPMS instrument used was an XSeriesII (Thermo Electron, UK) equipped with platinum skimmer and sampler cones, a perfluoroalkoxy PFA-ST nebulizer at 200 μL min−1, a quartz cyclonic spray chamber thermostated at 3 °C and a 1 mm inner diameter injector. In order to prevent any carbon deposition linked to the use of organic solvents, a 8.2 mL min−1 oxygen flow rate was injected in the plasma tanks to an oxygen port located just after the spray chamber.24,25 ICP-MS data was processed with Plasmalab software from Thermo Fisher. Experimental parameters, such as the torch position, flow rates and optical lens system, were optimized daily with a Sm–EDTA solution obtained by diluting the Sm–EDTA sample at 10−4 mol L−1 prepared according to part 1, by a factor 150 in the mobile phase. This solution was further re-diluted in the appropriate volume of HNO3 2% to reach a concentration of 3.4 ppb for the Sm. For each species, the most abundant Ln isotope was monitored with a dwell time of 30 ms.
5. Simultaneous coupling of HILIC to ESIMS and ICPMS
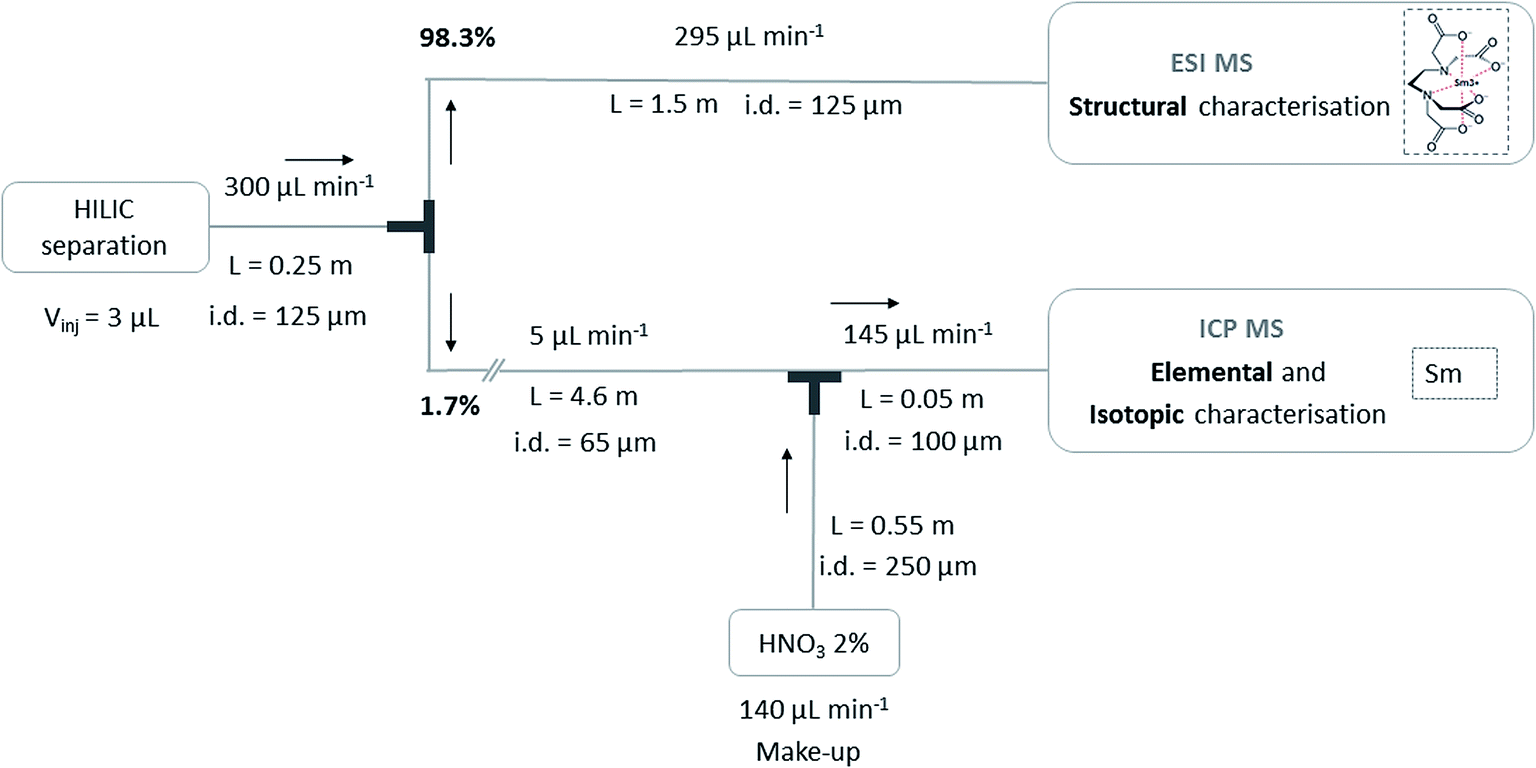
The instrumental set-up for the simultaneous coupling of HILIC to ESIMS and ICPMS was performed in a conventional laboratory, taking into account the geometrical constraints of the current configuration of the instruments equipped with glove boxes in the laboratory dedicated to the analysis of radioactive samples. The column output stream was split using a T-connector without a dead volume (MicroTee Assy Peek – 1/16in, IDEX Health & Science, USA) (Fig. 1), connected to the ESIMS through a 1.5 m × i.d. 125 μm tubing on the one hand, and to the ICPMS through a 4.6 m × i.d. 65 μm tubing on the other hand. Experimental flow rates were 295 μL min−1 for the flow entering the ESIMS and 5 μL min−1 for the flow entering the ICPMS, corresponding to 98.3% for ESIMS and 1.7% for ICPMS. Before entering the ICPMS source, a make-up made of HNO3 2% was mixed at 140 μL min−1 with the stream at 5 μL min−1 using a mixing tee (Micro Static Mixing Tee Assy, IDEX Health & Science, USA).
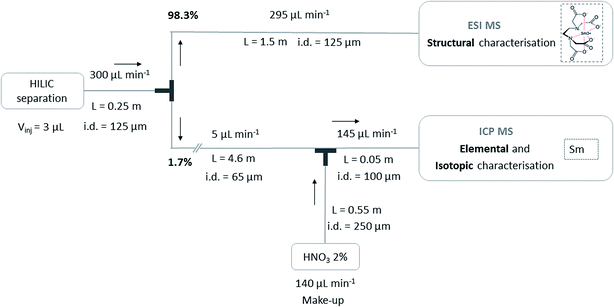 |
| | Fig. 1 Schematic representation of the simultaneous coupling of HILIC separation to ESIMS and ICPMS. | |
Results and discussion
The mobile phase composition was defined in our previous study2 on the separation of Ln species by hydrophilic interaction liquid chromatography in isocratic mode, with amide-bonded stationary phase. This mobile phase (70/30 v/v acetonitrile/water, 0.5% v/v formic acid, 15 mM ammonium acetate) was therefore used in the present study.
1. Separation of lanthanide–polyaminocarboxylic species using amide columns packed with fully porous particles (FPP)
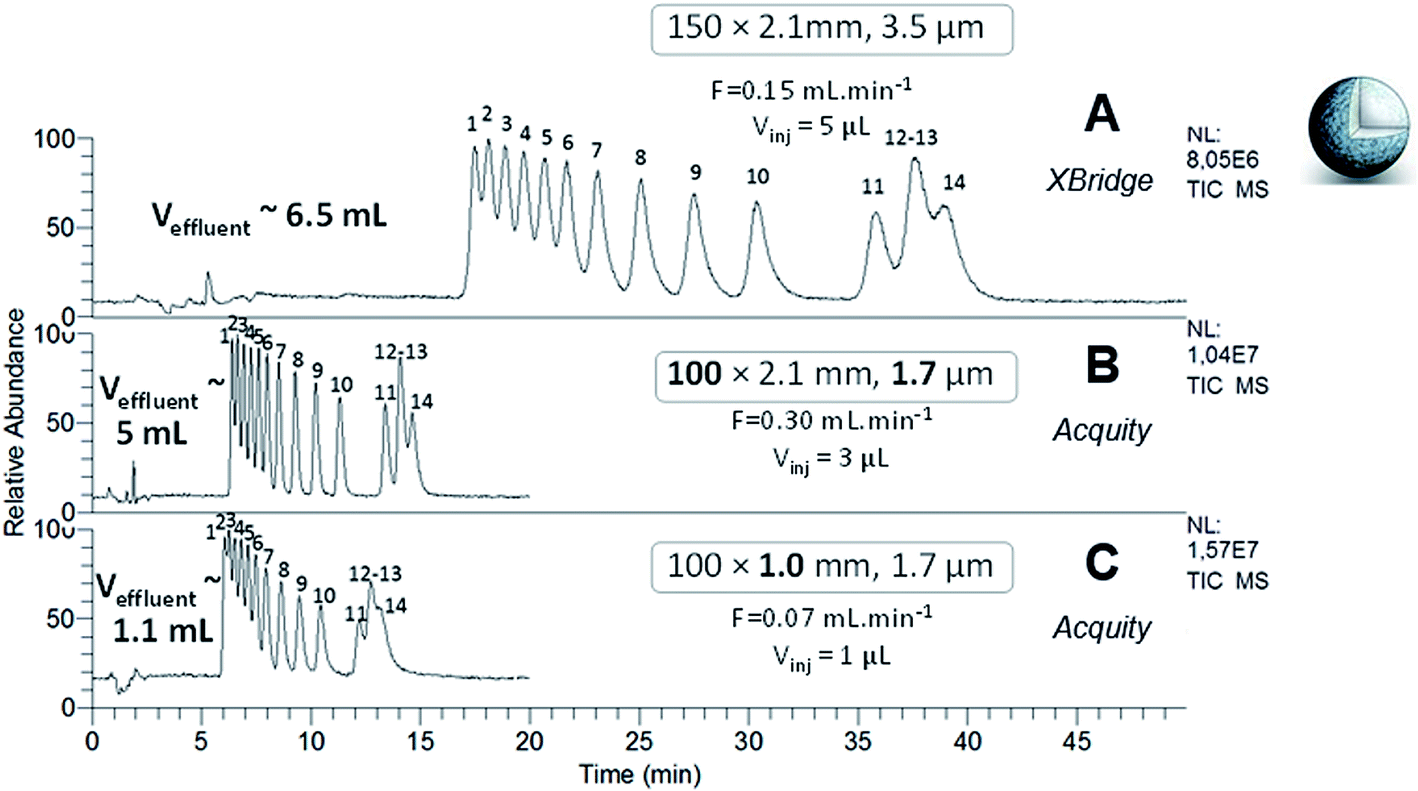
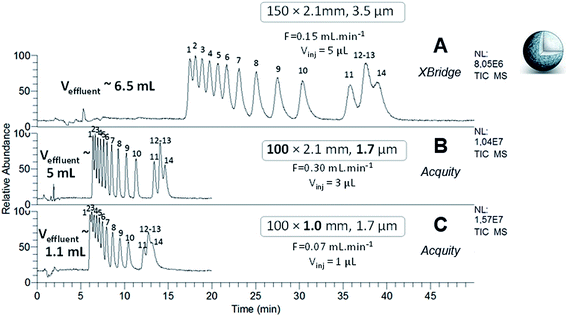
The separation of Ln–polyaminocarboxylic species contained in samples 1 and 2 was performed with the XBridge column in narrow-bore format (150 × 2.1 mm; 3.5 μm) at the optimal flow rate which was determined in our previous study, i.e. 150 μL min−1 (Fig. 2A and 3A). The separation performance was evaluated with the efficiency (N) and the discrimination factor (d0) values shown in Tables 2 and 3. The discrimination factor was considered instead of the classical resolution Rs because of peak asymmetry. The calculation to obtain N and d0 are given in the experimental part. As specified in Fig. 2A and 3A, the effluent volumes generated by these separations were higher than 5 mL. A relevant way to reduce the volume of chromatographic effluents while improving the separation efficiency, is to use sub-2 μm fully porous particles combined with a reduced column length. Therefore, the same experiments were performed with the Acquity column, packed with 1.7 μm particles made of the same material and functionalized with the same amide group as that of the XBridge column. The Acquity column was shorter, but the internal diameter was kept constant (100 × 2.1 mm). According to the rules of geometrical down-scaling26 for isocratic elution mode, the calculated mobile phase flow rate for the Acquity column was 300 μL min−1 and the injection volume was 3 μL, instead of 5 μL for the XBridge column. We expected the separation process to be 1.5 more efficient and 3 times faster, as well as around 30% less waste volume with the Acquity column. The chromatograms obtained for the two samples are given in Fig. 2B and 3B. From these experiments, we can see that decreasing the particle diameter and shortening the column length by switching from the XBridge to the Acquity column, resulted in a separation that was 2.6 times faster, while reducing waste by around 20%, with a maximum volume of 5 mL. For sample 2, the discrimination factor was improved about 1.5 times with the Acquity column (Table 3). For the two samples, the separation efficiency provided by the Acquity column was around 2 times higher than for the XBridge (Tables 2 and 3), which is higher than expected. By calculating N values per meter, the Acquity column was even around 3 times more efficient than the XBridge column. The difference between theoretical and experimental values regarding efficiency, duration and effluent volumes provided by the separations, may be explained by a slight difference in packing quality and functionalization degree between both columns. Overall, our results are in agreement with the trends observed in the literature based on the comparison of the XBridge (150 × 2.1 mm; 3.5 μm) and the Acquity (100 × 2.1 mm; 1.7 μm) columns packed with silica hybrid FPPs for the HILIC separation of polar organic compounds.27,28 The authors found that the analysis time was divided by a factor 3 with the Acquity column, achieving the same plate count N as with the XBridge column.
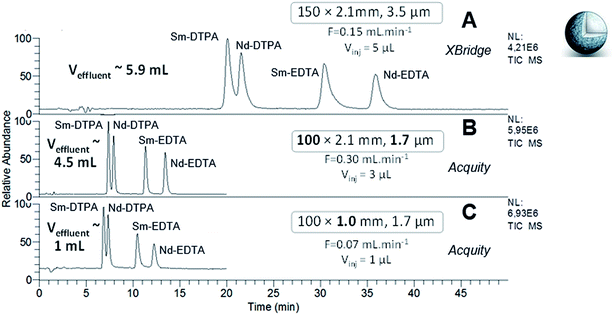 |
| | Fig. 2 HILIC separation of Ln species contained in sample 1, in isocratic mode with the three FPP-packed columns. ESI-MS detection with an acquisition in SIM mode, centred on m/z 436 and m/z 537 with a scan width of 20 u. | |
 |
| | Fig. 3 HILIC separation of Ln species contained in sample 2, in isocratic mode with the three FPP-packed columns. ESI-MS detection with an acquisition in SIM mode, centred on m/z 444 with a scan width of 46 u. Elution order of Ln–EDTA complexes with: (1) Lu, (2) Yb, (3) Tm, (4) Er, (5) Ho, (6) Dy, (7) Tb, (8) Gd, (9) Eu, (10) Sm, (11) Nd, (12) La, (13) Pr, (14) Ce. | |
Table 2 Chromatographic data (discrimination factor d0 and efficiency N) associated with sample 1, obtained with FPP-packed columns: XBridge amide column (150 × 2.1 mm; 3.5 μm); Acquity BEH amide column (100 × 2.1 mm; 1.7 μm) and Acquity BEH amide column (100 × 1.0 mm; 1.7 μm). d0 and N were calculated from the XIC for each species. Flow rate of the mobile phase and injection volume are specified in Table 1 for each column
| |
d0 |
N |
| XBridge i.d. 2.1 mm |
Acquity i.d. 2.1 mm |
Acquity i.d. 1.0 mm |
XBridge i.d. 2.1 mm |
Acquity i.d. 2.1 mm |
Acquity i.d. 1.0 mm |
| Sm-DTPA |
|
|
|
1937 |
3392 |
2245 |
| |
0.76 |
0.87 |
0.78 |
|
|
|
| Nd-DTPA |
|
|
|
2382 |
3780 |
2364 |
| |
1.00 |
1.00 |
1.00 |
|
|
|
| Sm-EDTA |
|
|
|
1825 |
4952 |
1763 |
| |
1.00 |
1.00 |
1.00 |
|
|
|
| Nd-EDTA |
|
|
|
4810 |
7447 |
3242 |
Table 3 Chromatographic data (discrimination factor d0 and efficiency N) associated with sample 2, obtained with FPP-packed columns: XBridge amide column (150 × 2.1 mm; 3.5 μm); Acquity BEH amide column (100 × 2.1 mm; 1.7 μm) and Acquity BEH amide column (100 × 1.0 mm; 1.7 μm). d0 and N were calculated from the XIC for each species. Flow rate of the mobile phase and injection volume are specified in Table 1 for each column
| |
d0 |
N |
| XBridge i.d. 2.1 mm |
Acquity i.d. 2.1 mm |
Acquity i.d. 1.0 mm |
XBridge i.d. 2.1 mm |
Acquity i.d. 2.1 mm |
Acquity i.d. 1.0 mm |
| Lu–EDTA |
|
|
|
2934 |
5659 |
2864 |
| |
0.34 |
0.48 |
0.14 |
|
|
|
| Yb–EDTA |
|
|
|
3938 |
5836 |
3742 |
| |
0.47 |
0.63 |
0.31 |
|
|
|
| Tm–EDTA |
|
|
|
2839 |
6248 |
2649 |
| |
0.55 |
0.72 |
0.41 |
|
|
|
| Er–EDTA |
|
|
|
2311 |
3675 |
3703 |
| |
0.57 |
0.77 |
0.47 |
|
|
|
| Ho–EDTA |
|
|
|
3160 |
6546 |
2882 |
| |
0.62 |
0.82 |
0.55 |
|
|
|
| Dy–EDTA |
|
|
|
3390 |
6300 |
3313 |
| |
0.81 |
0.94 |
0.75 |
|
|
|
| Tb–EDTA |
|
|
|
3140 |
6720 |
3142 |
| |
0.94 |
0.98 |
0.92 |
|
|
|
| Gd–EDTA |
|
|
|
3402 |
6632 |
2810 |
| |
0.96 |
0.98 |
0.91 |
|
|
|
| Eu–EDTA |
|
|
|
2976 |
6798 |
2253 |
| |
0.97 |
0.99 |
0.90 |
|
|
|
| Sm–EDTA |
|
|
|
2881 |
7386 |
1817 |
| |
1.00 |
1.00 |
0.84 |
|
|
|
| Nd–EDTA |
|
|
|
3192 |
7238 |
1776 |
| |
0.65 |
0.87 |
0.35 |
|
|
|
| La–EDTA |
|
|
|
2337 |
2178 |
973 |
| |
0.00 |
0.00 |
0.00 |
|
|
|
| Pr–EDTA |
|
|
|
1771 |
4308 |
956 |
| |
0.15 |
0.37 |
0.03 |
|
|
|
| Ce–EDTA |
|
|
|
2904 |
5400 |
918 |
In order to further reduce the effluent volumes and sample consumption, we used the Acquity column in micro-bore format with an internal diameter of 1 mm, keeping the length and the particle size constant, i.e. 100 mm and 1.7 μm respectively. Using the equations of geometrical down-scaling,26 the adjusted flow rate was found to be 70 μL min−1. We expected a similar analysis time and separation efficiency as those obtained with the 100 × 2.1 mm Acquity column, but a waste volume around 80% lower. A sample volume of 1 μL was injected instead of the 0.7 μL theoretical one, since it is the smallest volume that can be injected with the autosampler in question. Since extra-column volumes could have a significant impact on band broadening when reducing the internal diameter of the column, we minimized the length and the internal diameter of the connectors, and bypassed the UV detector and the diverting valve. The resulting chromatograms are shown in Fig. 2C and 3C. For the two samples and in agreement with the theoretical values, the Acquity column in micro-bore format generated a waste volume of around 1 mL, which is around 80% less than that produced with the Acquity column with an inner diameter of 2.1 mm, and the separation durations were similar. Unfortunately, the separation efficiencies were degraded by a factor of at least 2 for the two samples and poor discrimination factors were obtained for the Ln–EDTA complexes contained in sample 2 (Tables 2 and 3). The advantages expected by reducing the column's inner diameter to 1 mm could not fully be exploited despite minimizing the extra-column volumes and using UHPLC instrumentation which is designed to provide the smallest dead volumes. Some papers have stressed the detrimental impact of extra-column effects on the efficiency of sub-2 μm FPP-packed columns with an internal diameter of 1.0 mm, and operating with an UHPLC system.29,30 The major source of dispersion has been attributed to the connectors interfacing with the chromatographic instrument and the mass spectrometer.31,32
To summarize, among the FPP-packed columns, the Acquity BEH amide (100 × 2.1 mm; 1.7 μm) provided the best trade-off between separation efficiency, discrimination factor, analysis time and waste volume produced. With this stationary phase, it must be pointed out that some Ln complexes were not separated with a return to the baseline, including d0 values less than 1. However, we were able to extract the mass spectra from the co-eluted or partially co-eluted peaks, allowing the unambiguous identification of the species. Another remarkable point is that the highest pressure reached only 240 bar, in agreement with the limited impact of decreasing the particle diameter on the pressure in hydrophilic interaction liquid chromatography. Since mobile phases contain a significant fraction of organic solvent, less viscous than water, lower pressures are reached compared with RPLC. This advantage makes it possible to use longer columns or to couple two columns in hydrophilic interaction liquid chromatography, for applications requiring high resolution,27 but at the cost of producing more effluent.
2. Lanthanide polyaminocarboxylic species separation using amide columns with superficially porous particles (SPP)
SPPs are composed of a solid core surrounded by a porous shell, showing restricted axial diffusion associated with the lower internal porosity of the particles.33 SPPs also show reduced dispersion and band broadening consistent with a narrow particle size distribution, as well as improved mass transfer from shorter diffusion path lengths through the porous layer.33 Because of these features, SPP-packed columns are expected to provide similar or better performance levels than those of sub-2 μm FPP-packed columns, while generating lower back-pressures.34,35 Several generations of shell particles have been marketed in various diameters but fit especially with RPLC applications,5,6 while the variety and the size of polar shell particles are more restricted for the hydrophilic interaction liquid chromatography.18 In our study, we intended to follow the same approach with the SPP-packed columns as with the FPP-packed columns. However, the smallest diameter of amide-bonded SPPs on the market was 2.6 μm and the smallest internal diameter of columns packed with these particles for a use with an UHPLC system, was 2.1 mm. Therefore, we were able to use only 2.1 mm i.d. amide columns of different lengths, and packed with SPP of 2.6 μm. Several tests were conducted with a Sunshell column (100 × 2.1 mm; 2.6 μm) (Table 1), at 300 μL min−1 for a first fast screening. DTPA-complexes were not observable and the heavier EDTA-complexes, containing elements ranging from Tb to Lu, eluted at a shorter time of 27 minutes, while the lighter complexes were too retained. Neither increasing the elution strength by increasing the water percentage, nor increasing the salt concentration, allowed us to improve the quality of the separations. These results highlighted the extremely retentive property of this stationary phase, which could be attributed to the silica-based packing material, together with the structure of the grafted chemical group. This latter is a primary amide that contains a hydrophilic group on the carbonyl function, rendering the stationary phase more polar than the common amide columns. This Sunshell stationary phase was therefore unsuitable for our applications. The Accucore column (150 × 2.1 mm; 2.6 μm) (Table 1), was then tested and revealed retention times comparable to those obtained with the FPP-packed columns. The optimal flow rate was found to be 150 μL min−1 as can be seen in the associated Knox plot (Fig. 4). A non-flat plot at higher flow rates was obtained, reflecting a loss of efficiency for flow rates above the optimal value. This is unusual with shell particles, since one of the well-known properties of the SPP-packed columns is to easily operate at high flow rates without losing separation efficiency.33 The lower efficiencies observed at higher flow rates have been explained by the complex mechanism of the HILIC separation mode and the associated mobile phase compositions.36 The water layer adsorbed at the surface of the stationary phase, combined with the adsorption and/or electrostatic interaction of analytes with the stationary phase, result in decreased diffusivity and greater resistance to the mass transfer, which in turn adversely affect the separation efficiencies.27,28,37 It must be pointed out that we obtained the same results with the XBridge column packed with FPPs (data not shown), indicating that the decrease in efficiency with the increasing mobile phase flow rate can be assigned to the features of the HILIC mechanism rather than to the particle technology and the support material. Furthermore, we have shown that with amide-bonded stationary phases,2 an adsorption mechanism played an important role in the HILIC retention of the Ln species, which likely promoted the non-flat trend at high flow rates. Overall, our results confirm that the gains offered by the SPP technology, in connection with an improved mass transfer and shorter path length at higher flow rates, are not clear-cut in hydrophilic interaction liquid chromatography.
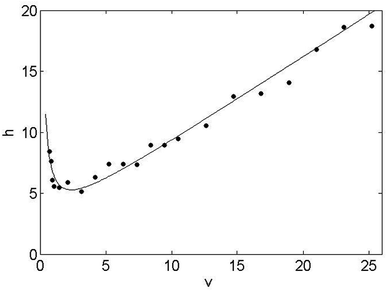 |
| | Fig. 4 Experimental Knox curve obtained with the SPP-packed column Accucore-amide (150 × 2.1 mm; 2.6 μm). h represents the height equivalent to a reduced theoretical plate and v the reduced linear velocity. | |
The chromatograms of the two samples were recorded with Accucore columns of lengths of 150 and 100 mm at 150 μL min−1. The length and the diameter of the tubing connections had to be minimized to avoid dispersion of the chromatographic peak. As could be expected, the 100 mm column generated around 30% less effluent than the 150 mm column (data not shown), but with a loss of separation efficiency by at least a factor of 1.5. Since this parameter is essential and must be retained or even improved, the sub-3 μm SPP-packed column Accucore (150 × 2.1 mm; 2.6 μm) was used for the comparative study with the Acquity column.
3. Comparison of sub-2 μm FPP and sub-3 μm SPP technologies for lanthanide polyaminocarboxylic species separation by HILIC
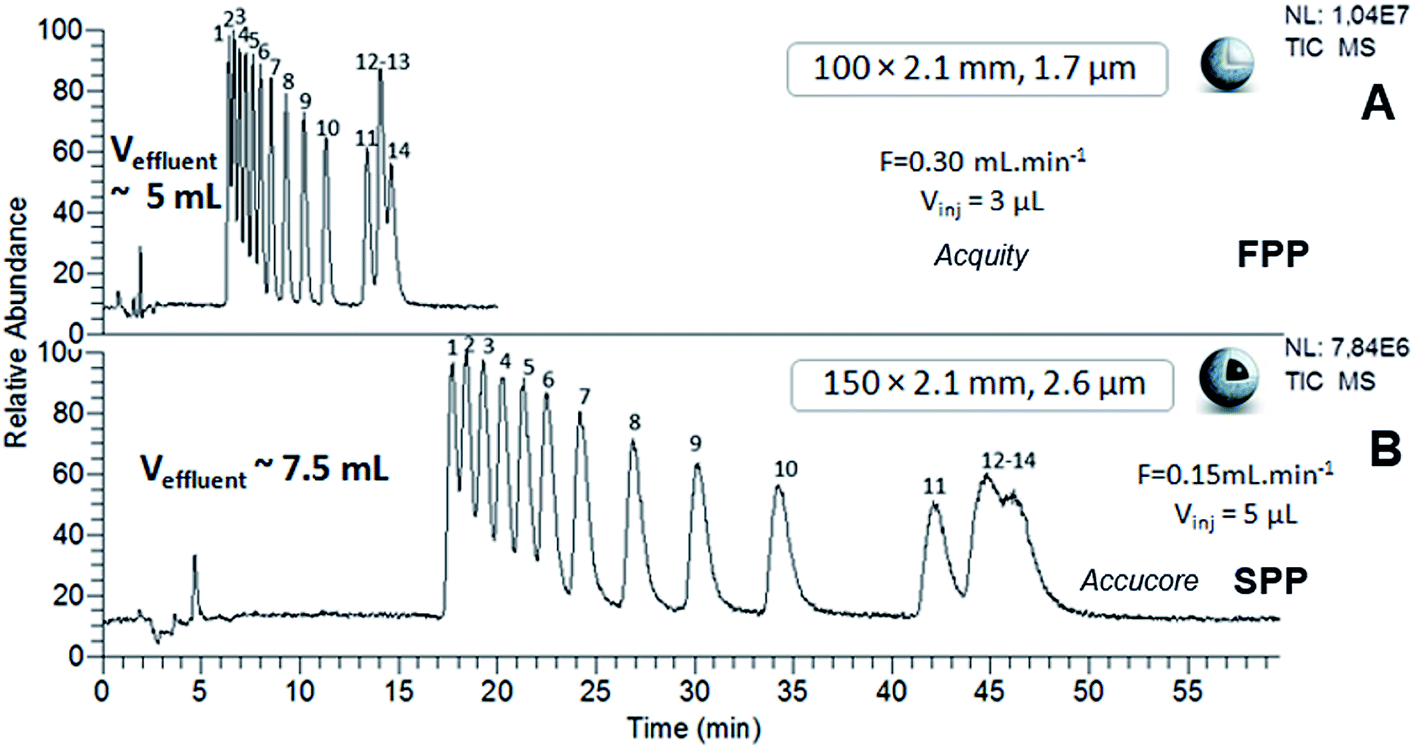
The chromatograms of the two samples obtained using sub-2 μm FPP and sub-3 μm SPP-packed columns, the Acquity (100 × 2.1 mm; 1.7 μm) and the Accucore (150 × 2.1 mm; 2.6 μm), at their respective optimal flow rates, are shown in Fig. 5 and 6. Considering the differences in column lengths and flow rates, the separations carried out with the SPP-packed Accucore column were three times more time-consuming and generated around 30% more waste than the FPP-packed Acquity column. Moreover, the Accucore column was about two times less efficient than the Acquity column (Tables 4 and 5). By working with columns of the same dimensions (100 × 2.1 mm) and at the same flow rate of 300 μL min−1, we obtained slightly longer separations and slightly higher effluent volumes for the Accucore column compared with the Acquity column (data not shown). The two stationary phases were made of different packing material, which was untreated bare silica for the Accucore column and hybrid material for the Acquity column. This resulted in a lower retention capability and therefore less waste production for the latter. In these conditions, the sub-3 μm SPP-packed Accucore column also provided less efficient separations than the sub-2 μm FPP-packed Acquity column, despite the reduction in the extra-column volumes for the former column. These results are not consistent with what could be expected at least in RPLC, since it is well-recognized that sub-3 μm shell particles generally generate at least an equivalent efficiency level to that of the sub-2 μm porous particles, while inducing lower pressure and solvent consumption.34 Studies comparing polar SPP- and FPP-packed columns in hydrophilic interaction liquid chromatography are not as widespread, and almost always limited to silica-based stationary phases and exclusively for organic compound separation.36–40 With columns of the same dimensions, using shell technology led to higher efficiency levels than with porous particles, with shorter analysis times.36 This has particularly been observed for underivatized silica columns packed with SPPs and FPPs with comparable sub-2 μm diameters.37 By contrast, in the study evaluating hybrid and nude silica-based columns of the same dimensions and packed with sub-2 μm FPPs and sub-3 μm SPPs, the shell technology was shown to be 30% less efficient regarding the HILIC separation of organic compounds exhibiting different properties.39 The authors attributed this result to the different quality of the column packing, the different support material and the complex mechanism of hydrophilic interaction liquid chromatography. Our results are in agreement with the latter study and similar explanations can be provided to interpret them. In spite of the benefits claimed for the shell technology, our work shows that sub-2 μm FPPs are most suited for achieving the best trade-off between efficiency, discrimination factor, analysis time and effluent volumes regarding the HILIC separation of Ln–polyaminocarboxylic species. Separation levels reached by the FPP-packed Acquity column (100 × 2.1 mm; 1.7 μm) were two times more efficient than those obtained with the SPP-packed Accucore column (150 × 2.1 mm; 2.6 μm), and three times more efficient when using N values per meter. The Acquity column also generated around 30% less waste, lower than 5 mL, in three times shorter analysis time, i.e. a maximum of 15 minutes. In addition, thanks to the HILIC mobile phase composition, the back-pressure did not exceed 240 bar even using sub-2 μm FPP particles, which is particularly interesting to implement chromatographic separations in glove box for the study of radioactive samples.
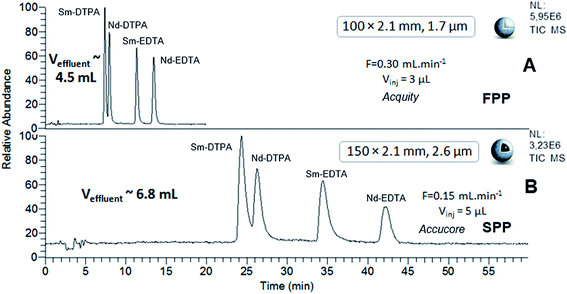 |
| | Fig. 5 HILIC separation of Ln species contained in sample 1, in isocratic mode with sub-2 μm FPP- and sub-3 μm SPP-packed columns. ESI-MS detection with an acquisition in SIM mode, centred on m/z 436 and m/z 537 with a scan width of 20 u. | |
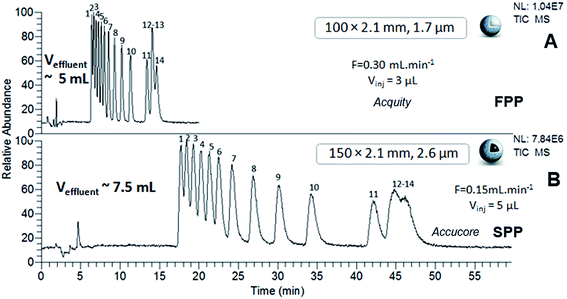 |
| | Fig. 6 HILIC separation of Ln species contained in sample 2, in isocratic mode with sub-2 μm FPP- and sub-3 μm SPP-packed columns. ESI-MS detection with an acquisition in SIM mode, centred on m/z 444 with a scan width of 46 u. Elution order of Ln–EDTA complexes with: (1) Lu, (2) Yb, (3) Tm, (4) Er, (5) Ho, (6) Dy, (7) Tb, (8) Gd, (9) Eu, (10) Sm, (11) Nd, (12) La, (13) Pr, (14) Ce. | |
Table 4 Chromatographic data (discrimination factor d0 and efficiency N) associated with sample 1, obtained with the SPP-packed Accucore-amide column (150 × 2.1 mm; 2.6 μm) and FPP-packed Acquity BEH amide column (100 × 2.1 mm; 1.7 μm). d0 and N were calculated from the XIC for each species. Flow rate of the mobile phase and injection volume are specified in Table 1 for each column
| |
d0 |
N |
| Accucore 150 × 2.1 mm; 2.6 μm |
Acquity 100 × 2.1 mm; 1.7 μm |
Accucore 150 × 2.1 mm; 2.6 μm |
Acquity 100 × 2.1 mm; 1.7 μm |
| Sm–DTPA |
|
|
1348 |
3392 |
| |
0.74 |
0.87 |
|
|
| Nd–DTPA |
|
|
1771 |
3780 |
| |
1.00 |
1.00 |
|
|
| Sm–EDTA |
|
|
2034 |
4952 |
| |
1.00 |
1.00 |
|
|
| Nd–EDTA |
|
|
5377 |
7447 |
Table 5 Chromatographic data (discrimination factor d0 and efficiency N) associated with sample 2, obtained with SPP-packed Accucore-amide column (150 × 2.1 mm; 2.6 μm) and FPP-packed Acquity BEH amide column (100 × 2.1 mm; 1.7 μm). d0 and N were calculated from the XIC for each species. Flow rate of the mobile phase and injection volume are specified in Table 1 for each column
| |
d0 |
N |
| Accucore 150 × 2.1 mm; 2.6 μm |
Acquity 100 × 2.1 mm; 1.7 μm |
Accucore 150 × 2.1 mm; 2.6 μm |
Acquity 100 × 2.1 mm; 1.7 μm |
| Lu–EDTA |
|
|
2498 |
5659 |
| |
0.47 |
0.48 |
|
|
| Yb–EDTA |
|
|
3658 |
5836 |
| |
0.57 |
0.63 |
|
|
| Tm–EDTA |
|
|
2575 |
6248 |
| |
0.66 |
0.72 |
|
|
| Er–EDTA |
|
|
2606 |
3675 |
| |
0.70 |
0.77 |
|
|
| Ho–EDTA |
|
|
2773 |
6546 |
| |
0.73 |
0.82 |
|
|
| Dy–EDTA |
|
|
2912 |
6300 |
| |
0.88 |
0.94 |
|
|
| Tb–EDTA |
|
|
2982 |
6720 |
| |
0.93 |
0.98 |
|
|
| Gd–EDTA |
|
|
2839 |
6632 |
| |
0.96 |
0.98 |
|
|
| Eu–EDTA |
|
|
2648 |
6798 |
| |
0.96 |
0.99 |
|
|
| Sm–EDTA |
|
|
2666 |
7386 |
| |
0.98 |
1.00 |
|
|
| Nd–EDTA |
|
|
2574 |
7238 |
| |
0.82 |
0.87 |
|
|
| La–EDTA |
|
|
2237 |
2178 |
| |
0.00 |
0.00 |
|
|
| Pr–EDTA |
|
|
2691 |
4308 |
| |
0.04 |
0.37 |
|
|
| Ce–EDTA |
|
|
3043 |
5400 |
4. Simultaneous coupling of HILIC separation to ESIMS and ICPMS
The selected Acquity column was used to perform the simultaneous coupling to ESIMS and ICPMS (Fig. 1). This single-step approach exhibits outstanding advantages. Firstly, comprehensive speciation information can be collected through the simultaneous acquisition of structural, elemental and isotopic data. Secondly, an additional decrease in the overall effluent volumes, sample consumption and analysis times is afforded with respect to independent coupling experiments. All these advantages are of prime importance, in particular for nuclear applications.
Fig. 7A and B shows the chromatograms of the sample containing the entire series of Ln–EDTA complexes, recorded simultaneously by ESIMS and ICPMS. Each Ln–EDTA species was identified by ESIMS through the extraction of the mass spectra from the chromatographic peaks and by comparison with the theoretical isotopic patterns of the species (Fig. 7A). ICPMS allowed us to monitor the Ln isotopes contained in the species associated with each chromatographic peak (Fig. 7B). The ICPMS chromatographic profiles were comparable to those obtained by ESIMS, but with a 2.4 minutes delay due to the difference in the connector lengths from the outlet of the column to the ICPMS on the one hand, and to the ESIMS in the other hand. The separation efficiencies calculated with ESIMS and ICPMS chromatograms were comparable. These preliminary results demonstrate that the 4.6 m tubing with a small inner diameter of 65 μm made it possible to compensate the performance loss expected with such connector lengths, which is paramount to implement this coupling with instruments equipped with glove boxes. As indicated above, in addition to species identification, comprehensive speciation includes both quantitative and isotopic measurements of separated species. The associated analytical methods are currently under development. It must be noted that because of the numerous Ln industrial applications, the separation of the series of lanthanide ions has been reported with other separation techniques such as ionic chromatography,41 ion pairing reversed-phase chromatography,42 capillary electrophoresis43 and isotachophoresis.44 All these techniques require the addition of ion pairing and/or complexing agents in the mobile phases for Ln elution and the use of complexing agents as constituents of background electrolyte to insure differential Ln migration. To the best of our knowledge, this is the first time that the separation of the entire series of Ln chemical species is performed in hydrophilic interaction liquid chromatography and isocratic mode. This is a great advantage since this separation technique can be directly coupled with ICPMS and ESIMS, without requiring the addition of any extra reagent in the mobile phase or after separation in order to be compatible with the mass spectrometers.
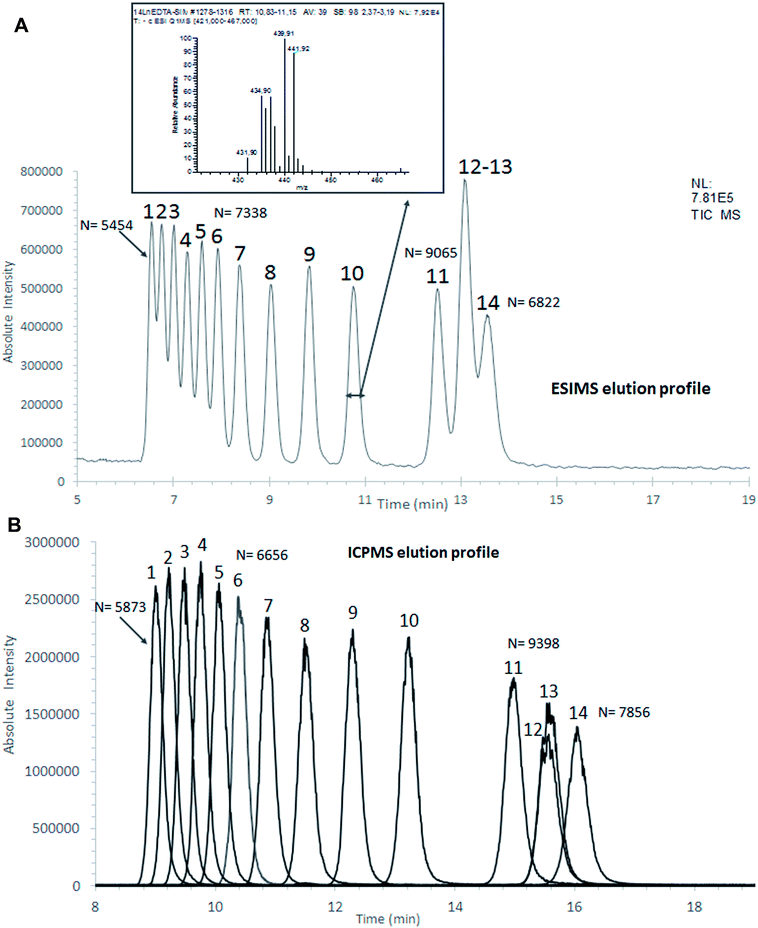 |
| | Fig. 7 HILIC separation of Ln species contained in sample 2, in isocratic mode with the FPP-packed column Acquity BEH amide (100 × 2.1 mm; 1.7 μm). (A) ESI MS detection with an acquisition in SIM mode centred on m/z 444 with a scan width of 46 u. Elution order of Ln–EDTA complexes with: (1) Lu, (2) Yb, (3) Tm, (4) Er, (5) Ho, (6) Dy, (7) Tb, (8) Gd, (9) Eu, (10) Sm, (11) Nd, (12) La, (13) Pr, (14) Ce. (B) ICP MS detection, by monitoring the most abundant lanthanide in each species. The corresponding intensities were normalized to 100%. Elution order (relative abundance indicated in brackets): (1) 175Lu [97.4%], (2) 172Yb [21.8%], (3) 169Tm [100%], (4) 166Er [33.5%], (5) 165Ho [100%], (6) 163Dy [24.9%], (7) 159Tb [100%], (8) 156Gd [20.5%], (9) 153Eu [52.2], (10) 152Sm [26.8%], (11) 146Nd [17.2%], (12) 139La [99.9%], (13) 141Pr [100%], (14) 140Ce [88.5%]. N was calculated for each species based on the extracted ion chromatogram. | |
Conclusion
Amide-bonded columns packed with FPPs and SPPs were evaluated for the separation of Ln–polyaminocarboxylic species by hydrophilic interaction liquid chromatography. The efficiency, discrimination factor, analysis time and effluent volumes yielded by 3.5 μm FPP-packed columns were compared with those obtained with sub-2 μm FPP- and sub-3 μm SPP-packed columns. Regarding the FPP technology, the gains afforded by decreasing the particle size, column length and internal diameter were assessed, leading to the selection of the Acquity (100 × 2.1 mm; 1.7 μm) column. For the SPP-packed columns, the most satisfactory conditions were obtained with the Accucore column (150 × 2.1 mm; 2.6 μm). By comparing the two technologies, an efficiency level multiplied by a factor two, an analysis time three times shorter and around 30% less effluent waste were obtained with the sub-2 μm FPP Acquity column compared with the sub-3 μm SPP Accucore column, demonstrating that the totally porous technology was the most suitable for our purpose. Such an approach, i.e. evaluating sustainable chromatographic techniques, had never been undertaken for speciation analysis, by investigating the separation of metal species by hydrophilic interaction liquid chromatography. Finally, we selected the Acquity column for simultaneous coupling to ICPMS and ESIMS in order to carry out speciation studies of lanthanides in a single step. The instrumental set-up was implemented in a conventional laboratory using the sample containing the entire series of Ln–EDTA complexes, taking into account the geometrical constraints existing in the laboratory dedicated to radioactive sample analysis. The separation of the entire Ln series under their complexed forms, in hydrophilic interaction liquid chromatography and isocratic mode, was demonstrated for the first time. The next stage will involve developing isotopic measurements and quantification methods. Simultaneous coupling will then be implemented in glove box for the speciation analysis of radioelements.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors acknowledge CEA for financial support.
References
- C. J. Welch, N. Wu, M. Biba, R. Hartman, T. Brkovic, X. Gong, R. Helmy, W. Schafer, J. Cuff and Z. Pirzada, TrAC, Trends Anal. Chem., 2010, 29, 667 CrossRef.
- L. Beuvier, C. Bresson, A. Nonell, L. Vio, N. Henry, V. Pichon and F. Chartier, RSC Adv., 2015, 5, 92858 RSC.
- A. I. Olives, V. González-Ruiz and M. A. Martín, ACS Sustainable Chem. Eng., 2017, 5, 5618 CrossRef.
- F. T. Mattrey, A. A. Makarov, E. L. Regalado, F. Bernardoni, M. Figus, M. B. Hicks, J. Zheng, L. Wang, W. Schafer, V. Antonucci, S. E. Hamilton, K. Zawatzky and C. J. Welch, TrAC, Trends Anal. Chem., 2017, 95, 36 CrossRef.
- N. Tanaka and D. V. McCalley, Anal. Chem., 2016, 88, 279 CrossRef PubMed.
- S. Fekete, E. Olah and J. Fekete, J. Chromatogr. A, 2012, 1228, 57 CrossRef PubMed.
- E. M. Borges, M. A. Rostagno and M. A. A. Meireles, RSC Adv., 2014, 4, 22875 RSC.
- K. M. Kalili, D. Cabooter, G. Desmet and A. de Villiers, J. Chromatogr. A, 2012, 1236, 63 CrossRef PubMed.
- V. R. J. Acquaro, F. M. Lancas and M. E. Queiroz, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1048, 1 CrossRef PubMed.
- J. J. DeStefano, B. E. Boyes, S. A. Schuster, W. L. Miles and J. J. Kirkland, J. Chromatogr. A, 2014, 1368, 163 CrossRef PubMed.
- J. Koster, R. Shi, N. von Wiren and G. Weber, J. Chromatogr. A, 2011, 1218, 4934 CrossRef PubMed.
- L. Telgmann, M. Sperling and U. Karst, Anal. Chim. Acta, 2013, 764, 1 CrossRef PubMed.
- J. Künnemeyer, L. Terborg, S. Nowak, A. Scheffer, L. Telgmann, F. Tokmak, A. Günsel, G. Wiesmüller, S. Reichelt and U. Karst, Anal. Chem., 2008, 80, 8163 CrossRef PubMed.
- C. S. Kesava Raju, A. Cossmer, H. Scharf, U. Panne and D. Lück, J. Anal. At. Spectrom., 2010, 25, 55 RSC.
- M. Birka, C. A. Wehe, O. Hachmoller, M. Sperling and U. Karst, J. Chromatogr. A, 2016, 1440, 105 CrossRef PubMed.
- F. Xie, P. Colin and J. Van Bocxlaer, Talanta, 2017, 174, 171 CrossRef PubMed.
- A. Periat, B. Debrus, S. Rudaz and D. Guillarme, J. Chromatogr. A, 2013, 1282, 72 CrossRef PubMed.
- L. Nováková, L. Havlíková and H. Vlčková, TrAC, Trends Anal. Chem., 2014, 63, 55 CrossRef.
- M. Birka, C. A. Wehe, L. Telgmann, M. Sperling and U. Karst, J. Chromatogr. A, 2013, 1308, 125 CrossRef PubMed.
- A. Fisher and D. Kara, Anal. Chim. Acta, 2016, 935, 1 CrossRef PubMed.
- M. K. Jha, A. Kumari, R. Panda, J. Rajesh Kumar, K. Yoo and J. Y. Lee, Hydrometallurgy, 2016, 161, 77 CrossRef.
- M. El Fallah and M. Martin, Chromatographia, 1987, 24, 115 CrossRef.
- J. P. Foley and J. G. Dorsey, Anal. Chem., 1983, 55, 730 CrossRef.
- A. Leclercq, A. Nonell, J. L. Todoli Torro, C. Bresson, L. Vio, T. Vercouter and F. Chartier, Anal. Chim. Acta, 2015, 885, 33 CrossRef PubMed.
- A. Leclercq, A. Nonell, J. L. T. Torró, C. Bresson, L. Vio, T. Vercouter and F. Chartier, Anal. Chim. Acta, 2015, 885, 57 CrossRef PubMed.
- D. Guillarme, D. T.-T. Nguyen, S. Rudaz and J.-L. Veuthey, Eur. J. Pharm. Biopharm., 2007, 66, 475 CrossRef PubMed.
- H. Song, E. Adams, G. Desmet and D. Cabooter, J. Chromatogr. A, 2014, 1369, 83 CrossRef PubMed.
- J. C. Heaton, X. Wang, W. E. Barber, S. M. Buckenmaier and D. V. McCalley, J. Chromatogr. A, 2014, 1328, 7 CrossRef PubMed.
- N. Wu and A. C. Bradley, J. Chromatogr. A, 2012, 1261, 113 CrossRef PubMed.
- F. Lestremau, D. Wu and R. Szücs, J. Chromatogr. A, 2010, 1217, 4925 CrossRef PubMed.
- D. Spaggiari, S. Fekete, P. J. Eugster, J.-L. Veuthey, L. Geiser, S. Rudaz and D. Guillarme, J. Chromatogr. A, 2013, 1310, 45 CrossRef PubMed.
- M. Rodriguez-Aller, R. Gurny, J. L. Veuthey and D. Guillarme, J. Chromatogr. A, 2013, 1292, 2 CrossRef PubMed.
- R. Hayes, A. Ahmed, T. Edge and H. Zhang, J. Chromatogr. A, 2014, 1357, 36 CrossRef PubMed.
- V. González-Ruiz, A. I. Olives and M. A. Martín, TrAC, Trends Anal. Chem., 2015, 64, 17 CrossRef.
- J. J. Kirkland, S. A. Schuster, W. L. Johnson and B. E. Boyes, J. Pharm. Anal., 2013, 3, 303 CrossRef PubMed.
- M. D. Dolzan, D. A. Spudeit, Z. S. Breitbach, W. E. Barber, G. A. Micke and D. W. Armstrong, J. Chromatogr. A, 2014, 1365, 124 CrossRef PubMed.
- J. C. Heaton and D. V. McCalley, J. Chromatogr. A, 2014, 1371, 106 CrossRef PubMed.
- D. V. McCalley, J. Chromatogr. A, 2008, 1193, 85 CrossRef PubMed.
- B. Chauve, D. Guillarme, P. Cleon and J. L. Veuthey, J. Sep. Sci., 2010, 33, 752 CrossRef PubMed.
- D. V. McCalley, in UHPLC in Life Sciences, ed. D. Guillarme and J. L. Veuthey, Royal Society of Chemistry, Cambridge, 1st edn, 2012, 6, p. 164 Search PubMed.
- T. Ishikawa, K. Sugimoto and K. Nagaishi, Geochem. J., 2003, 37, 671 CrossRef.
- A. Datta, N. Sivaraman, T. G. Srinivasan and P. R. Vasudeva Rao, Radiochim. Acta, 2010, 98, 277 Search PubMed.
- P. Janos, Electrophoresis, 2003, 24, 1982 CrossRef PubMed.
- L. Vio, G. Crétier, F. Chartier, V. Geertsen, A. Gourgiotis, H. Isnard, P. Morin and J.-L. Rocca, J. Anal. At. Spectrom., 2012, 27, 850 RSC.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a
 where hp: the smallest peak height between two consecutive peaks and hv: the valley height between two peaks.22 As chromatographic peaks are asymmetrical, efficiency values N were calculated for each species from the extracted ion chromatogram (XIC), with the Foley–Dorsey equation:23
where hp: the smallest peak height between two consecutive peaks and hv: the valley height between two peaks.22 As chromatographic peaks are asymmetrical, efficiency values N were calculated for each species from the extracted ion chromatogram (XIC), with the Foley–Dorsey equation:23  where tr: the retention time of analyte, ω0.1: peak width at 10% of the peak height, a and b: peak width before and after the apex, measured at 10% of the peak height. Analyses were performed two times with the XBridge (150 × 2.1 mm; 3.5 μm), Acquity (100 × 2.1 mm; 1.7 μm) and Accucore (150 and 100 × 2.1 mm; 2.6 μm) columns. Similar efficiencies were obtained in all cases, and the average of N is given in the tables. For the Acquity column of internal diameter of 1 mm, as well as for the tests of the Sunshell column, analyses were performed one time.
where tr: the retention time of analyte, ω0.1: peak width at 10% of the peak height, a and b: peak width before and after the apex, measured at 10% of the peak height. Analyses were performed two times with the XBridge (150 × 2.1 mm; 3.5 μm), Acquity (100 × 2.1 mm; 1.7 μm) and Accucore (150 and 100 × 2.1 mm; 2.6 μm) columns. Similar efficiencies were obtained in all cases, and the average of N is given in the tables. For the Acquity column of internal diameter of 1 mm, as well as for the tests of the Sunshell column, analyses were performed one time.