
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-stimuli-responsive TiOx/DOX nanodrug system for lung cancer synergistic therapy†
Zideng Dai,
Xue-Zhi Song ,
Junkai Cao,
Yunping He,
Wen Wen,
Xinyu Xu and
Zhenquan Tan*
,
Junkai Cao,
Yunping He,
Wen Wen,
Xinyu Xu and
Zhenquan Tan*
School of Petroleum and Chemical Engineering, Dalian University of Technology, Panjin 124221, P. R. China. E-mail: tanzq@dlut.edu.cn
First published on 14th June 2018
Abstract
Biological applications of nanosheets are rapidly increasing currently, which introduces new possibilities to improve the efficacy of cancer chemotherapy and radiotherapy. Herein, we designed and synthesized a novel nano-drug system, doxorubicin (DOX) loaded titanium peroxide (TiOx) nanosheets, toward the synergistic treatment of lung cancer. The precursor of TiO2 nanosheets with high specific surface area was synthesized by a modified hydrothermal process using the polymer P123 as a soft template to control the shape. TiOx nanosheets were obtained by oxidizing TiO2 nanosheets with H2O2. The anti-cancer drug DOX was effectively loaded on the surface of TiOx nanosheets. Generation of reactive oxygen species, including H2O2, ·OH and ·O2−, was promoted from TiOx nanosheets under X-ray irradiation, which is effective for cancer radiotherapy and drug release in cancer cells. In this way, chemotherapy and radiotherapy were combined effectively for the synergistic therapy of cancers. Our results reinforce the DOX loaded TiOx nanosheets as a pH sensitive and X-ray controlled dual-stimuli-responsive drug release system. The cytotoxicity, cellular uptake, and intracellular location of the formulations were evaluated in the A549 human non-small cell lung cancer cell line. Our results showed that TiOx/DOX complexes exhibited a greater cytotoxicity toward A549 cells than free DOX. This work demonstrates that the therapeutic efficacy of DOX-loaded TiOx nanosheets is strongly dependent on their loading mode and the chemotherapeutic and radiotherapy effect is improved under X-ray illumination, which provides a significant breakthrough for future applications of TiOx as a light activated drug carrier in cancer chemotherapy and radiotherapy.
Introduction
Cancer is one of the most deadly diseases worldwide; for example it is reported that approximately 1.5 million people are killed by lung cancer every year.1 Although modern medical science has achieved notable advancements over the past decade, the long-term survival rates for lung cancer patients are still very poor and even less than 10% for advanced stage lung cancer patients.2 Locally advanced lung cancer is notoriously resistant to many types of cytotoxic chemotherapy and radiotherapy.3 As a result, there are currently no effective therapies for lung cancer. New therapy strategies need to be explored to deal with the problems.Along with the development of nanotechnology, novel targeted drug delivery systems with high efficiency, low toxicity and minimal side effects have been developed to improve the efficacy of chemotherapy and radiotherapy.4 Particularly, nanoparticle sensitized X-ray radiotherapy therapy provides a promising treatment strategy for efficient tumor ablation and minimally invasive treatment.5 Combination of anti-cancer drugs thereby enhancing the sensitivity of chemotherapy, a drug delivery system based on X-ray sensitized nanoparticles show great potential in the synergistic therapy of cancer. Recently, a variety of inorganic nanocarriers, such as iron oxide nanoparticles, mesoporous silica nanoparticles, graphene oxide nanosheets and titanium dioxide (TiO2) nanoparticles have been widely studied for drug delivery and cancer therapy.6
TiO2 is a potential dynamic therapy agent in cancer therapy due to excellent biocompatibility and its unique optical properties.7 The photocatalytic activity and cytotoxicity of anatase TiO2 are higher than those of the rutile forms.8 Recently, TiO2 have been used in the phototherapy of malignant cancer cells and are regarded as potential photosensitizing agents for photodynamic therapy because they play unique phototoxic effects on ultraviolet (UV) irradiation.9 However, this strategy seems to be invalid in treating many cancers and is difficult to apply clinically for two main reasons. Firstly, UV light cannot penetrate the human body to reach internal organs such as the respiratory system, pancreas, and liver, thus limiting the application of this technique to superficial tumors.10 Secondly, the UV-mediated generation of reactive oxygen species (ROS) lasts short enough to provide a continuous and prolonged killing effect.11 Radiation therapy using X-ray is known to be effective in the treatment of various cancers. But, to our knowledge, there is seldom report about use of TiO2 to enhance the therapy effect of X-ray irradiation.12 Wang et al. demonstrated that the potential biological effects of TiO2 depend on their size, crystalline phase, surface coating, and chemical composition.13 For example, TiO2 nanomaterials with a high aspect ratio such as nanorod, nanobelt and nanofilament have much high reactivity, which enables the insoluble TiO2 to agglomerate and affect cellular uptake.14 Anatase TiO2 induces greater ROS production and cell responses, which is more toxic than rutile due to the active sites on its surface.15 So many coating approaches on TiO2 were provided to reduce the pulmonary inflammatory response and cytotoxicity.16 Therefore, some modifications may need to be made in order to make TiO2 suitable as a reagent for enhancing the radiation effect.
Herein, we designed and synthesized TiOx nanosheets derived from TiO2 nanosheets toward the novel drug delivery system by a modified hydrothermal method.17 The designed TiOx nanosheets have an atom-layered thickness, which provides a very large specific surface area comparing to the zero- and one-dimensional TiO2 for small molecular drug loading.18 TiO2 nanosheets have a mixed phase of rutile and anatase, which allows them a high ability to generate ROS than rutile and is less cytotoxic than pure anatase, thus eliminating the need to surface coating with a layer of inorganic or organic substances to reduce its toxicity.15 TiO2 nanosheets were further changed to TiOx nanosheets by the oxidation of H2O2, which enhanced its ability to generate ROS, including ·OH and ·O2−, under X-ray irradiation. Such kind of active oxygen species are known as effective reagents for cancer radiotherapy.19 In addition, the surface of TiOx is more hydrophilic after the oxidation by H2O2, which improves the performance of TiOx as an adsorbent and various carriers, and also provides convenience for surface modification and improve its dispersibility in water.16b In this way, the chemotherapy and radiotherapy were combined effectively for the synergistic therapy of cancers.20
In this study, we investigated the properties of TiOx nanosheets and achieved the effect of efficiently treating cancer by X-ray irradiation in combination with the anticancer drug doxorubicin synergistic effect on lung cancer.21 Scheme 1 demonstrates the schematic approach for the synthesis of TiOx nanosheets by using polyethylene oxide polyoxypropylene–polyethylene oxide (P123) and ethylene glycol (EG) as co-surfactants to control the shape (the details were shown in Experimental section). We achieved the effect of efficiently treating cancer by X-ray irradiation in combination with the anticancer drug DOX synergistic effect on lung cancer.18 Our findings indicate that TiOx can not only act as a drug carrier due to the high specific surface area that can bind to more DOX molecules, but also kill cancer cells by ROS generated under X-ray irradiation. TiOx nanosheets carry DOX into the cells through endocytosis. The high carrier rate of TiOx nanosheets increases the uptake of cells, significantly increases the intracellular DOX concentration, and obviously improves the anti-cancer cell effect. TiOx nanosheets can generate more ROS to activate apoptosis under X-ray irradiation.10 Therefore, TiOx nanosheets loaded DOX improve the efficacy of drug chemotherapy and X-ray radiotherapy.22
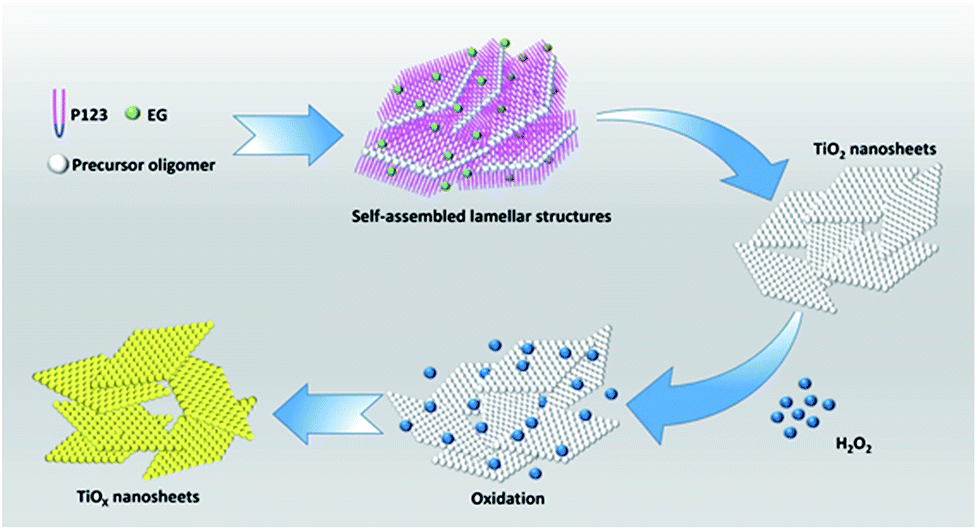 | ||
| Scheme 1 Schematic representation for the synthesis of TiOx nanosheets. Here precursor oligomer is a mixture of titanium tetrachloride and hydrochloric acid. | ||
Results and discussion
Physical characterization and formation of nanosheets
Fig. 1a shows the photo of TiO2 nanosheets before and after oxidation. It can be seen from the figure that the TiO2 nanosheets are pure white before oxidation and yellow after oxidation. TEM investigation was employed to characterize the microstructure of samples. Fig. 1b and c show the morphology of TiO2 nanosheets, which was prepared through the hydrothermal reaction. Fig. 1e and f present the morphology of oxidized TiOx nanosheets. It can be seen that all the products are nanosheets, and their edges roll up due to surface tension that is similar to graphene. TiOx nanosheets seem to be more curly and flexible, probably due to the oxidation treatment by H2O2. This can also be seen in the SEM image of TiO2 and TiOx in Fig. S1 in the ESI.† In order to confirm whether H2O2 changes the crystal structure of TiO2, the prepared TiO2 nanosheets and TiOx nanosheets were characterized by XRD. As shown in Fig. 1d, the XRD pattern of TiO2 nanosheets suggests a mixed phase of anatase and rutile after solvothermal treatment. TiOx also shows the same properties, but with a much lower crystallization after H2O2 oxidation treatment. The deformation and disordering of the lattice under H2O2 oxidation lead to a decrease in the intensity of the XRD peak.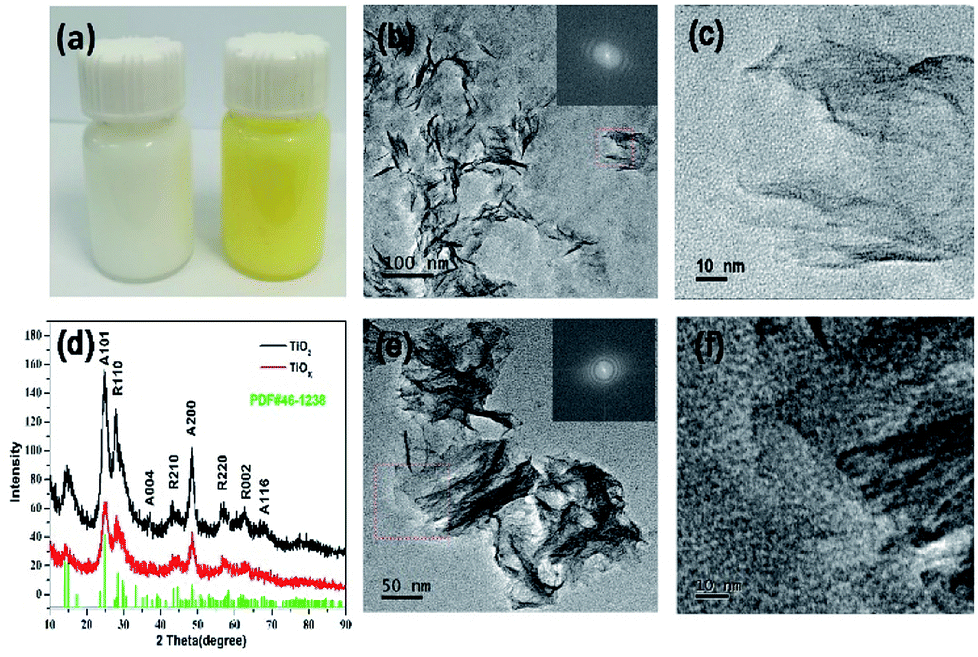 | ||
| Fig. 1 (a) A photo comparing TiO2 and TiOx. (b and c) TEM images of TiO2 nanosheets. (d) XRD patterns of TiO2 and TiOx (A: anatase, R: rutile). (e and f) TEM image of TiOx nanosheets. | ||
N2 adsorption–desorption experiment was conducted to illustrate the effects of H2O2 treatment on the specific surface area and the corresponding pore size distribution. As shown in Fig. S2,† the pore size distribution demonstrated the mesoporous structure. And the H2O2 treatment has little effect on the specific surface area of the sample, and the difference between before and after oxidation is less than 15 m2 g−1. Therefore, TiOx maintains a surface area similar to that of TiO2 and has a similar drug-loading effect, which facilitates the subsequent drug loading process.
Drug loading and pH controlled drug release
The drug loading was confirmed by the UV-vis spectra. The UV-vis absorption spectra of TiO2 nanosheets red shifted after DOX loading showed that the surface of TiO2 was modified by the ligand (DOX) (Fig. S3†). Moreover, the absorbance at 490 nm increased significantly, indicating that DOX has been supported on TiO2. TiOx also showed the same situation.23The successful loading of DOX to the surface of TiO2 was convincingly supported by the change in zeta potential (Fig. S4†). At pH = 7.4, zeta potential of TiO2 was −37.43 mV due to the surface hydroxyl groups. At the same pH condition, the zeta potential of the nanocomposite was −3.57 mV due to the amino group of DOX which was loaded on the surface of TiO2, indicating that the adsorption of DOX on the surface of TiO2 is also driven by the electrostatic interaction. The potential value changes relatively large, indicating that more DOX load to the TiO2 nanosheets. For TiOx nanosheets, the zeta potential was −39.67 mV, which indicates that oxidation treatment changes the surface potential by formation of the more hydroxyl groups and peroxyl groups.24 Typically, both of the prepared TiO2/DOX and TiOx/DOX systems are quite stable in ambient environment free of any special protection for 5 day storage, showing light red color without obvious aggregation in the transparent PBS solution (Fig. S5†).
Fig. S6† shows the FTIR spectra of DOX, TiO2, TiO2/DOX, TiOx and TiOx/DOX. For the DOX sample, the infrared spectrum of DOX contains many absorption peaks, including the O–H stretching vibration mode at 3318 cm−1, the C–H stretching vibration mode at 2910 cm−1, the stretching vibration mode of carbonyl at 1731 cm−1, the stretching vibration mode at 1290 cm−1 of the skeleton vibration, and the C–O stretching vibration at 1000 cm−1. For TiO2 samples, strong absorption was observed at about 3485 cm−1, 1643 cm−1 and 630 cm−1 due to O–H stretching vibration of surface hydroxyl groups, H–O–H bending vibration of physically adsorbed water, and interconnected octahedral [TiO6] Ti–O–Ti stretching vibration.17 In addition, the two weak bands of 2939 and 2865 cm−1 (vCH and vCH2) in the grown TiO2 nanosheets can be attributed to the characteristic frequencies of residual organic species, which are not completely removed by washing with distilled water. Note that the two peaks at 2939 and 2865 cm−1 of TiOx nanosheets almost disappeared after oxidation. This may be due to the decomposition of organic species.9c Similarly, the characteristic infrared band of DOX was also observed for TiO2/DOX, indicating that DOX was immobilized on the TiO2 surface.21 Nevertheless, the absorption peak of TiO2/DOX is mainly the peak of TiO2 or DOX, but no new bands are observed. The results clearly confirm that the DOX loading onto the TiO2 surface is indeed via a non-covalent bonding process. The TiOx/DOX band also shows the same structure.
We measured the isoelectric point of these two kind of nanosheets (TiO2 and TiOx) using the standard zeta potential method. The results are shown in Fig. S7.† The isoelectric point of TiO2 is 3.84, which is a little higher than that of TiOx (2.23). This result indicates that TiOx has more negative charge than TiO2 in the case of larger pH (such as pH = 7.4), which is consistent with the results in Fig. S4.† The more negative charges around the TiOx nanosheets allow the more DOX loading by electrostatic adsorption.
In order to investigate quantitatively the DOX loading of TiOx/DOX and TiO2/DOX nanocomposites, the rates of DOX loading were calculated based on UV-vis absorption spectra results. We measured the UV-vis absorption spectra of free DOX at different concentrations, in which the peaks were located at about 480 nm (Fig. S8a†). By fitting a series of UV-vis characteristic peaks, the standard curves of DOX were obtained, as shown in Fig. S8b.† We quantitatively studied the loading behavior of DOX on TiO2 at pH = 3–7.4. As shown in Fig. 2a, the loading of DOX was the largest at pH = 7.4, indicating a pH effect on the drug loading that pH of the solution may change the surface potential and thus affect the rug loading, which is consistent with the measurement of the isoelectric point (Fig. S7†). Therefore, the condition of pH = 7.4 was chosen for the drug loading. We also studied the change of drug loading rate with time under the condition of pH = 7.4. It can be seen from Fig. 2b that the drug loading rate increases with time. Here, in order to save time, we select 24 h As the medication time. Based on the above results, we investigated the loading of DOX in TiOx and TiO2 at pH = 7.4. Fig. 2c displays the DOX loading efficiency under identical concentration of TiO2 or TiOx but different initial DOX concentrations, revealing that increase of initial DOX concentration leads to enhancement of DOX loading on TiO2 or TiOx. Notably, the loading amount of DOX could reach 100% (w/w). These results validate our hypothesis that a designed system with a higher surface area can effectively conjugate drug molecules.25
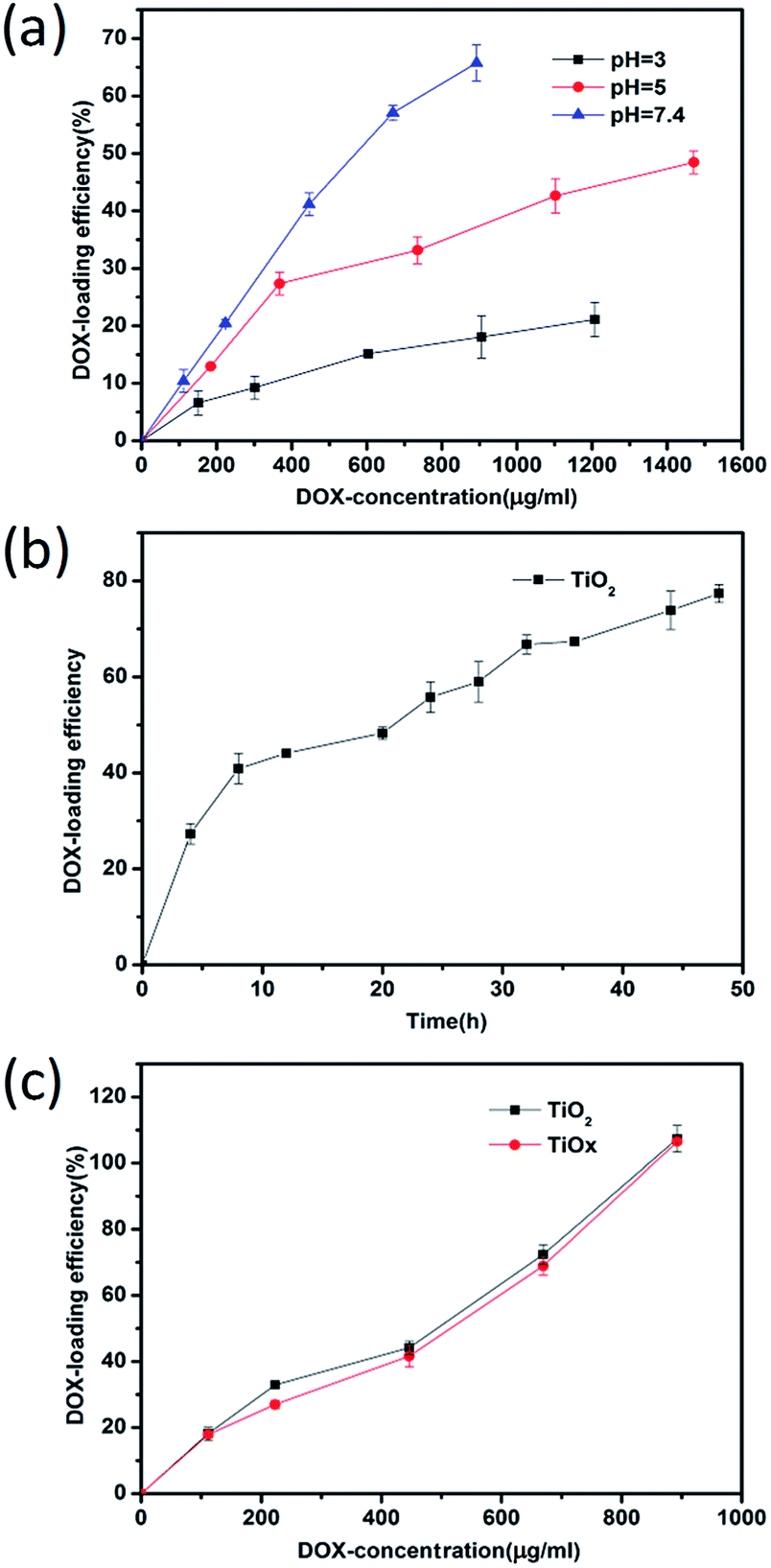 | ||
| Fig. 2 (a) Quantitation of TiO2 on DOX loading with different DOX concentrations at different pH values (loading pHs 3, 5 and 7.4). (b) The drug load changes with time at a pH of 7.4. (c) Comparative study of DOX concentration dependent loading on TiO2 and TiOx at pH = 7.4. | ||
Furthermore, the pH sensitivity of DOX release was also determined. Fig. 3 showed drug release kinetics of TiO2/DOX and TiOx/DOX at different pH values and with X-ray irradiation in buffer solution. Within 48 h, it was observed that the cumulative release rate of TiO2/DOX loaded DOX increased from 14.7% to 50.0% with the pH value decreased from 7.4 to 5.0. In addition, it was observed that the cumulative release rate of DOX loaded on TiOx/DOX increased from 11.4% to 60.0% with the pH value decreased from 7.4 to 5.0. Furthermore, under the X-ray irradiation, DOX release raised up to 75%. The release of DOX from TiO2 or TiOx was accelerated due to both of the pH decrease of PBS buffers and the irradiation of X-ray.26 The results suggested that the as-prepared TiOx/DOX nanocomposites were pH-responsive and X-ray-stimulative to release DOX. Moreover, the DOX release rate of TiOx-based drug system is better than that of TiO2-based system, which is a great benefit to our future cell experiments.
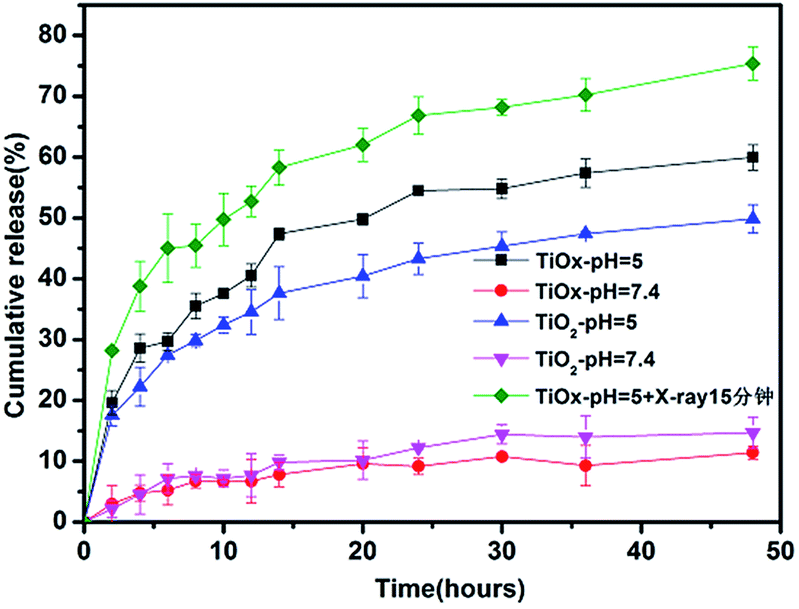 | ||
| Fig. 3 DOX release curves of TiO2/DOX and TiOx/DOX from 0 h to 48 h under the pH values of 7.4, 5.0, and TiOx/DOX under the pH values of 5.0 with X-ray irradiation. | ||
In vitro ROS detection
Quantification of ROS generation from the nanosheets in response to X-ray irradiation in a cell-free system was performed by using ROS fluorescence probe APF (2-[6-(4′-amino)phenoxy-3H-xanthen-3-on-9-yl]benzoic acid).5 10 μM of APF was added to different concentrations of the suspensions of TiOx nanosheets and TiO2 nanosheets prepared in cuvettes. Each cuvettes was then exposed to the same doses of X-ray radiation for 30 min. The APF signals were detected by fluorescence spectrophotometer at excitation/emission wavelengths of 490/510 nm, respectively. As shown in Fig. 4, under the same conditions, the level of ROS generated from TiOx is much higher than that of TiO2. This result suggests that a DOX loaded TiOx drug system, in theory, must be more effective in cancer therapy due to the synergistic effect of radiotherapy and chemotherapy.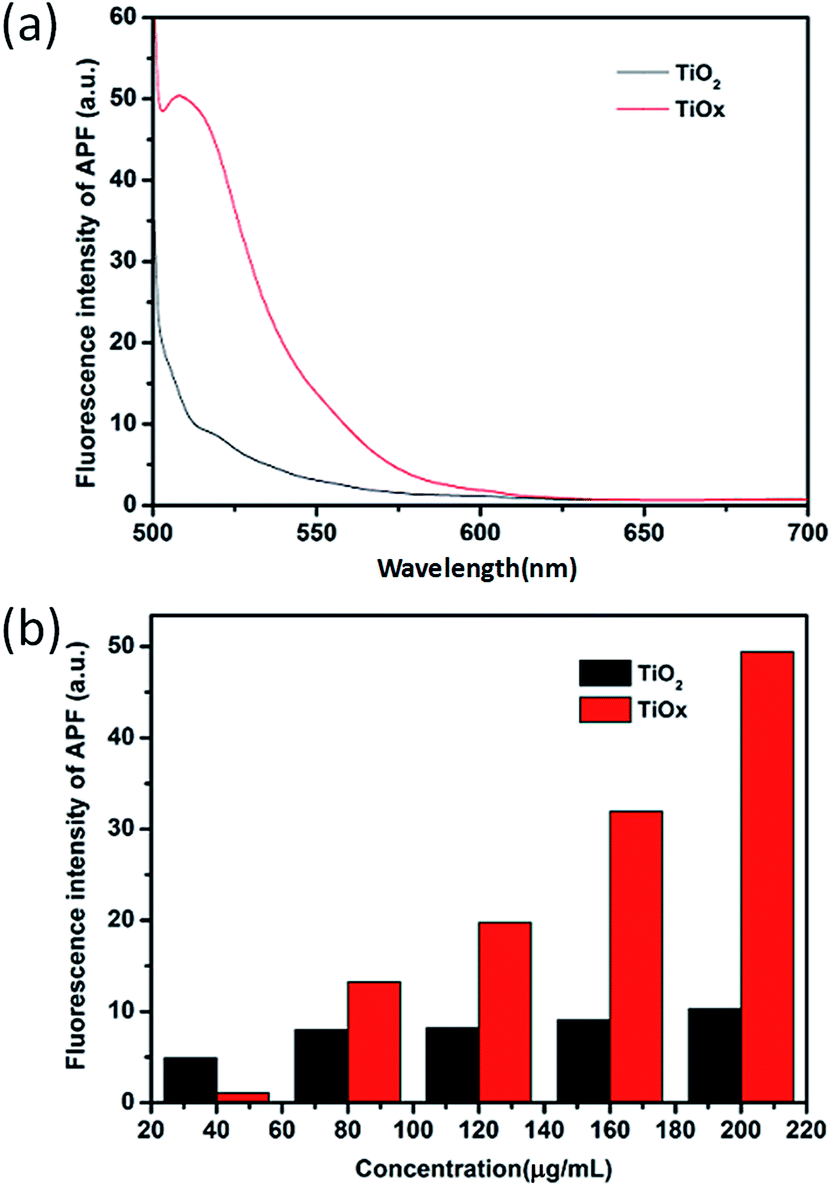 | ||
| Fig. 4 (a) Fluorescence spectrum of ROS with TiO2 and TiOx in vitro under X-ray irradiation. Since ROS production is a fast phenomenon, we measured ROS levels after 2 hours (after treatment). (b) The fluorescence intensity of TiO2 and TiOx under different concentrations. | ||
TiO2 and TiOx served not only as drug carriers for controlled release of X-rays, but also as an agent for enhancing radiation therapy. Intracellular ROS levels are an important indicator of radiation therapy; therefore, we compared the levels of ROS produced by X-ray irradiation in the blank group, TiO2, and TiOx.8a ROS generation with different excitations was observed in the A549 human lung caner cell line using 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescent probe staining. As shown in Fig. 5, an enlarged DCFH signal (green fluorescence) was observed in the group irradiated with X-rays, and the DCFH signal from TiOx was the largest one.
 | ||
| Fig. 5 In vitro studies of X-ray-responsive TiO2 and TiOx detection of intracellular ROS by DCFH-DA staining in A549 cells. (a–c) represent ROS generation in A549 cells by control, TiO2 and TiOx. (d–f) represent ROS generation in A549 cells by control, TiO2 and TiOx after X-ray irradiation. | ||
However, none of the X-ray irradiated groups showed a trace amount of DCFH fluorescence, indicating that ROS generation was affected by both TiOx and X-ray radiation. Note that a small amount of ROS could be produced from background solution after a X-ray irradiation. This is consistent with our cytotoxicity experiments. The DCFH signal (green fluorescence) did not increase much in the existence of TiO2, indicating that TiO2 is not much ROS generated under X-ray irradiation. But after the addition of TiOx, it is clear seen that the DCFH signal remarkably enhanced to a high level, which is consistent with our cytotoxicity experiments.
Cytotoxicity of TiO2 and TiOx on A549 lung cancer cells
It has been demonstrated that TiO2 is environmentally friendly and relatively nontoxic, we evaluated the cytotoxicity of large quantity of TiO2 and TiOx that was determined in vitro by CCK8. Fig. 6 shows the viabilities of A549 cells incubated with TiO2 and TiOx in a variety of concentrations. After 48 hours of incubation with TiO2 and TiOx with a concentration of 500 μg ml−1, the viabilities of A549 cells were 93.64% and 86.74%, respectively. These results verify the low cytotoxicity of TiOx, which is very important for the biomedical applications.27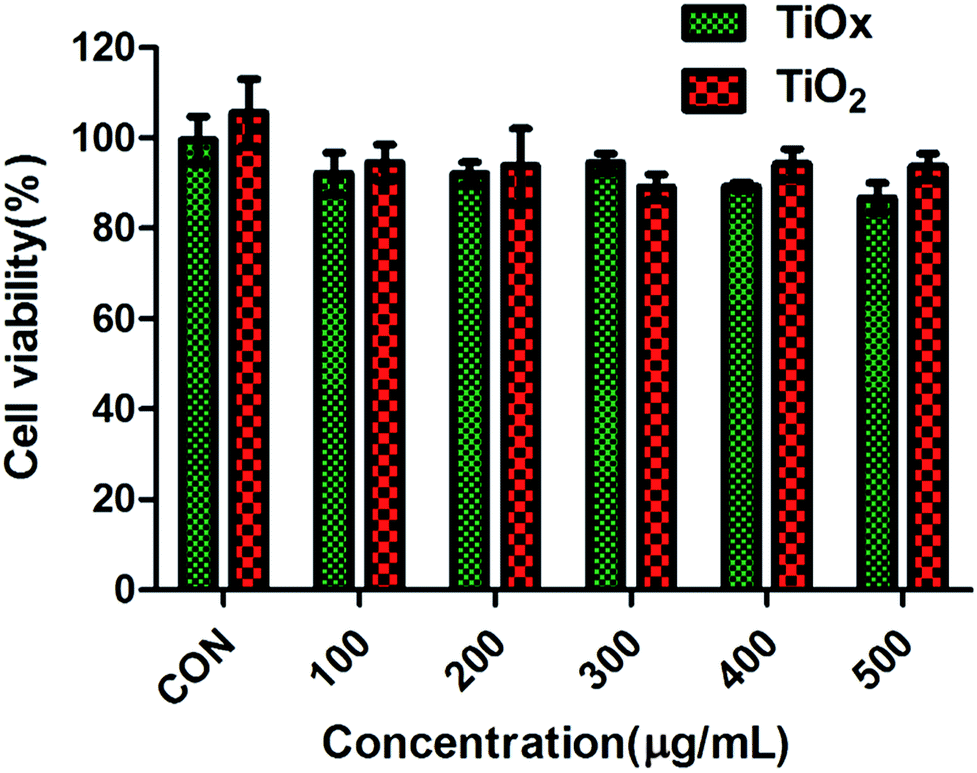 | ||
| Fig. 6 Cytotoxicity of TiO2 and TiOx in lung cancer A549 cells with incubation time of 48 h. Values are expressed as mean ± SD, n = 3. | ||
Drug intracellular localization
To enhance the chemotherapy of DOX, it was critical to release DOX into nucleus. Using laser scanning confocal microscope, the cellular uptake and release of DOX in TiO2/DOX and TiOx/DOX were investigated, respectively. Fig. 7 shows the confocal microscopy images of A549 cells incubated with DOX alone, TiO2/DOX and TiOx/DOX for 8 h, respectively. Amount of DOX was adjusted to the same value (10 μg ml−1) in the treatment of different samples. Most of the DOX were located in the nucleus of A549 cells. This is in good agreement with the widely accepted mechanism of cytotoxicity of DOX in which the cytotoxicity arises mainly from the direct intercalation of DOX into DNA and, subsequently, inhibition of DNA replication in nucleus.28 In the treatment involving in TiO2/DOX and TiOx/DOX nanocomposite, most of the drugs were distributed in the nucleus, while some in granular form were located in the cytoplasm. Since DOX is a water-soluble species and won't aggregate to form granules, the granules may be attributed to the TiO2/DOX and TiOx/DOX nanocomposite that can be invaginated by cells to form endosomes but the drugs were not released completely from the granules. The improvement of accumulation and release of drugs was important to enhance chemotherapeutic efficacy of DOX in cancer therapy.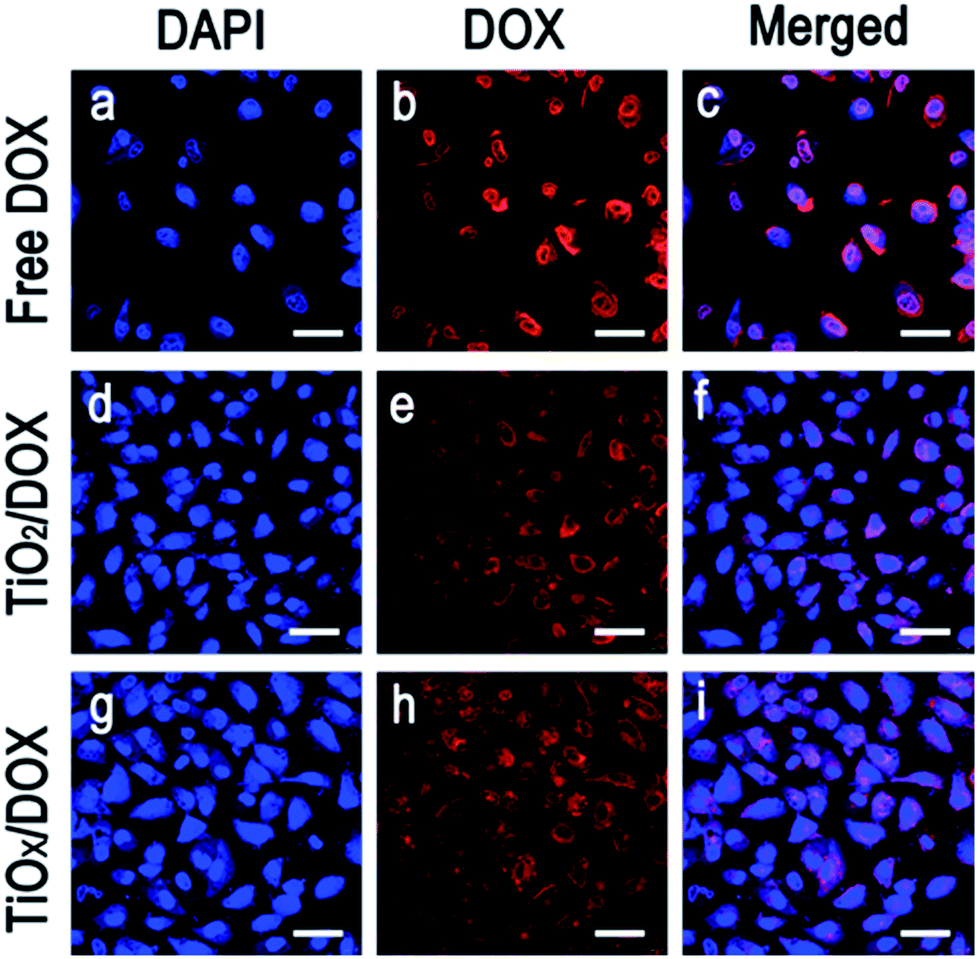 | ||
| Fig. 7 Laser confocal microscopy images of A549 cells incubated for 8 h with (a–c) free DOX (10 μg ml−1 DOX), (d–f) TiO2/DOX system containing 10 μg ml−1 of loaded DOX, and (g–i) TiOx/DOX system containing 10 μg ml−1 of loaded DOX. DAPI were used to label cell membrane nucleus (blue fluorescence) and DOX fluorescence is red. (scale bar is 50 μm). | ||
Fig. 8 shows that DOX is released in a time-dependent manner from TiOx nanodrug system. Typically, TiOx/DOX is first swallowed into the cells and accumulated in the cytoplasm in initial 2 h incubation (Fig. 8a–c). Note that DOX molecules can be efficiently released from TiOx/DOX distributed in lysosome due to acidic environment of the cell (pH 5.0). Meanwhile, the red shiny DOX is initially observed in both the cytoplasm of the cells (2 h incubation, Fig. 8b), and then increasingly accumulated around (4 h incubation, Fig. 8e) and finally entered the nucleus (8 h incubation, Fig. 8h) accompanied with the incubation time. The above results indicate that the drug release depends on the incubation time and the acidic environment of the cell. The drug delivery mechanism involves the internalization of TiOx/DOX, followed by deprotonation in endosomes/lysosomes, and subsequent drug penetration in the nucleus of cells.29
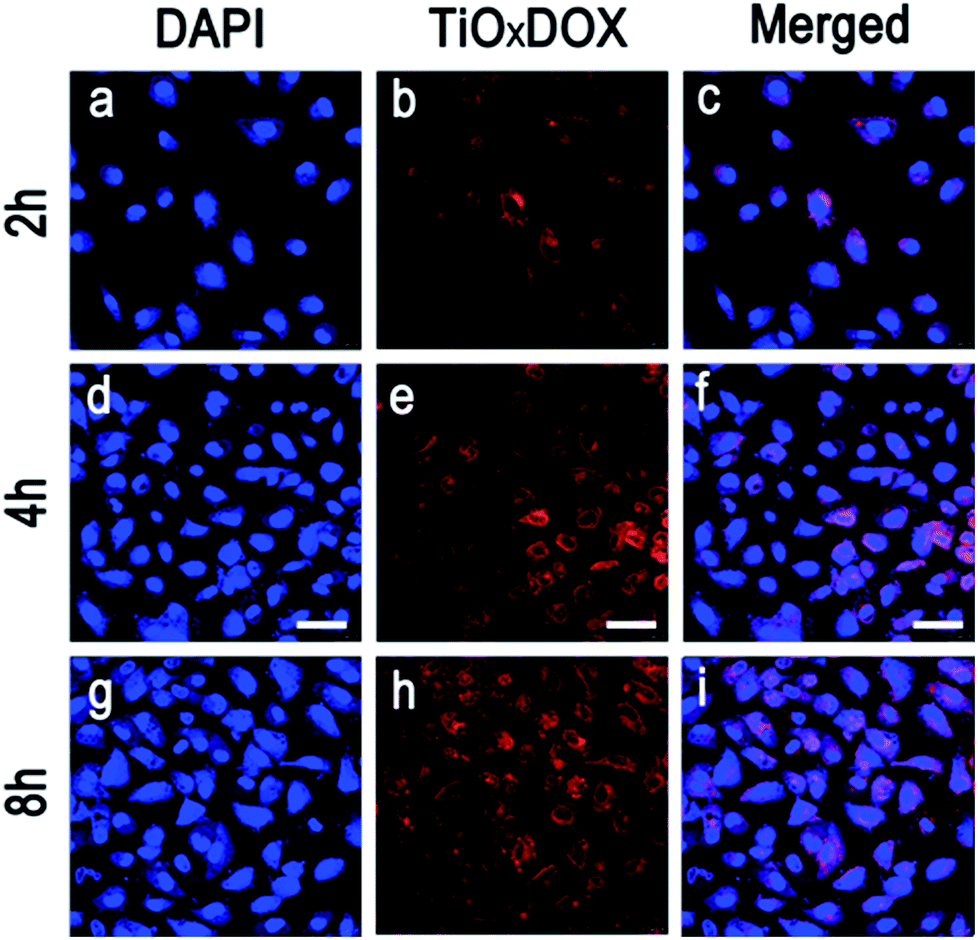 | ||
| Fig. 8 LSCFM images of intracellular internalization of TiOx/DOX (containing 10 μg ml−1 of DOX, loading pH = 7.4) and delivery of DOX to the nucleus in A549 cells. Cells are incubated with TiOx/DOX for specific times at 37 °C. DAPI were used to label cell membrane nucleus (blue fluorescence) and DOX fluorescence is red. Scale bar = 50 μm. | ||
Cytotoxic effect via radiotherapy and chemotherapy of TiOx/DOX
By CCK8 method, the chemotherapy and radiotherapy in vitro were assessed in A549 cells. At first, the chemotherapy performance was investigated by measuring the viabilities of A549 cells incubated with free DOX, TiO2/DOX and TiOx/DOX, respectively. The free DOX and nanocomposites was incubated with cells for 48 h. As shown in Fig. 9a, when the concentration of free DOX reached 10 μg ml−1, 90.64% ± 0.62% of cells were killed. This result indicated that free DOX had strong effect on killing A549 cells. For nanocomposites, 86.24% ± 0.63% and 88.45% ± 0.91% of A549 lung cancer cells were killed with the TiO2/DOX or TiOx/DOX treatment (with similar DOX concentration) at 48 h, respectively. The in vitro cell killing effect of nanocomposites are very near to that of free DOX. These data suggest that TiO2/DOX or TiOx/DOX demonstrate prolonged drug release and higher antitumor cell capabilities. Taken together, the results of our in vitro studies demonstrate that the anticancer activity of DOX is enhanced upon loading onto the TiO2/DOX or TiOx/DOX nanodrug carriers. And TiOx/DOX has a higher killing effect than TiO2/DOX in cancer chemotherapy.30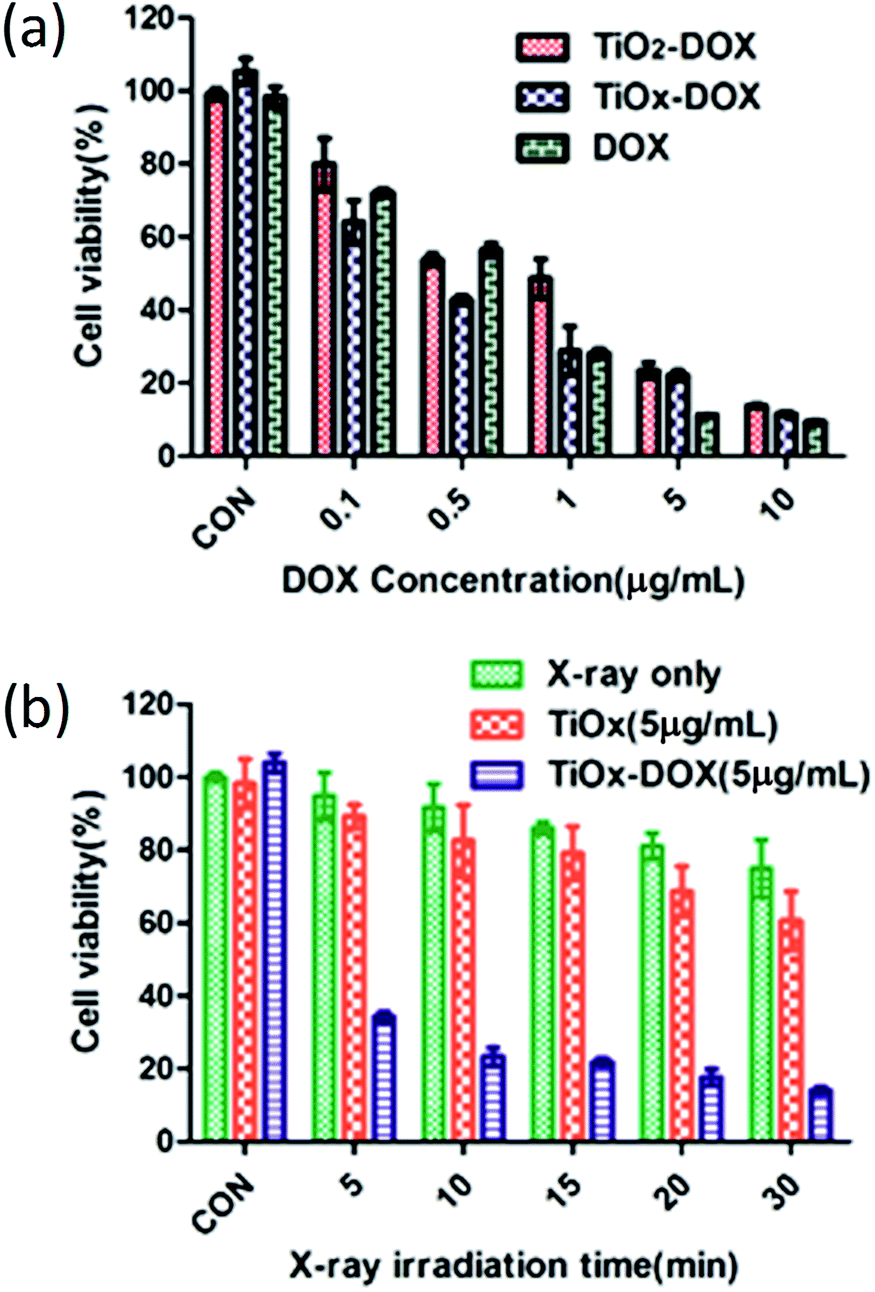 | ||
| Fig. 9 (a) Viability of A549 cells incubated with free DOX, TiO2/DOX and TiOx/DOX for 48 h. (b) Radiotherapy anticancer efficacy of X-ray alone, X-ray with TiOx, and X-ray with TiOx/DOX. Values are expressed as mean ± SD; n = 3. | ||
We studied the effect of TiOx/DOX at a concentration of 5 μg ml−1 (containing 5 μg ml−1 DOX) on X-ray exposure-dependent cancer cell killing. The result we obtained is shown in Fig. 9b. Initially, A549 cells were incubated with TiOx/DOX for 24 hours and then exposed to X-ray irradiation for a period of time. Cancer cell viability was detected after further incubation for 24 hours. The cell viability of the X-ray only group, bare TiOx and TiOx/DOX decreased with the increase of X-ray irradiation time. X-ray irradiation alone showed only small cytotoxic effects on A549 cells with a survival rate of approximately 75% after a 30 min radiation. For the exposed TiOx group, the cell viability was restricted to 60.7% after a 30 min X-ray irradiation, mainly due to the fact that TiOx was promoted to generate ROS internal cancer cells under X-ray irradiation as shown in Fig. 4 and 5. TiOx/DOX showed a much lower cell viability of 14.0% after 30 minutes of X-ray irradiation, which is due to the synergistic effect of ROS caused by X-ray irradiation and the efficient release of DOX from the TiOx/DOX drug system. Therefore, the synergistic effect of X-ray based radiotherapy and drug based chemotherapy plays a dominant role in the anticancer effect of TiOx/DOX.
Conclusions
In summary, we synthesized unique TiOx nanosheets with high specific surface area as drug carriers, and loaded DOX-enhanced chemotherapy and X-ray irradiation-triggered radiotherapy in order to effectively treat lung cancer. In this work, DOX was loaded onto TiOx nanosheets to form DOX loaded TiOx nanocomposites. We determined the nature of the interaction between DOX and TiOx nanosheets by UV-vis, IR and Zeta potential. In addition, our results show that TiOx nanosheets can be loaded with a great deal of DOX due to the high specific surface area. In drug sustained-release studies, TiOx/DOX can release more DOX under acidic conditions and X-ray irradiation. Under normal conditions, TiOx exhibits low anti-cancer effect, which can greatly reduce side effects on normal cells, and TiOx shows effective load and transport on cancer cells. Under the irradiation of X-rays, both TiOx and TiOx-release DOX play important roles in killing cancer cells. Importantly, because exposure in a large dose of X-ray radiation have harmful side effect that does harm to the normal organs during the cancer radiotherapy, our TiOx-based nanodrug system is very effective to reduce dose of X-ray irradiation but achieve much better anti-cancer effect at the same time. We believe that the TiOx nanosheets studied here can be a good treatment for cancer through combination chemotherapy and radiation therapy, which provides a new strategy for the future treatment of cancer.Experimental
Materials and reagents
Unless otherwise noted, all chemicals used are of analytical grade and are used without further purification. Titanium tetrachloride was supplied by Aladdin Chemicals (Shanghai, China) with a purity of 99%. Doxorubicin hydrochloride (DOX) was supplied by Solarbio. polyethylene oxide polyoxypropylene–polyethylene oxide (PEO20-PPO70-PEO20, P123) and ROS Brite™ active oxygen fluorescence probe APF was supplied by AAT Bioquest. Dialysis Bags MD25 and PBS phosphate buffers were purchased from Solarbio (Beijing, China). Ethylene glycol (EG) and ethanol was purchased from Tianjin great Mao Co., Ltd. Hydrogen peroxide (H2O2) was purchased from Tianjin commie chemical reagent co. Cell Counting Kit-8 (CCK-8), 2-[6-(4′-amino)phenoxy-3H-xanthen-3-on-9-yl]benzoic acid (APF), 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) and 4′,6-diamidino-2-phenylindole (DAPI) were supplied by Beyotiome Biotechnology Co. Ltd.Instruments
Samples were characterized by Ultra high resolution field emission scanning electron microscopy (fe-sem, Nova NanoSEM 450) and transmission electron microscopy (TEM, Tecnai G2 F30) at an accelerating voltage of 200 kV. X-ray diffraction (XRD) patterns of the as prepared samples were recorded on a SHIMADZU XRD-7000S diffractometer with Cu K as the radiation source in the a range of 10-90° with a scan speed of 5° min−1. The Brunauer–Emmett–Teller (BET) specific surface areas were measured by JW-BK132F at −196 °C. The pore volumes and pore diameter distributions were calculated with the Barrett–Joyner–Halenda (BJH) model. Particle sizes were acquired on a Malvern Zeta sizer Nano ZS. X-ray irradiation was performed using BJI-1 (Xianwei, Shanghai, China) at a voltage of 60–75 kV and a current of 0.15–0.35 mA. Confocal laser scanning microscopy (CLSM) images were recorded on a Leica TCS SP8 inverted confocal microscope. DOX loading and release studies were carried out in a Shimadzu UV-1800 spectrophotometer. The determination of ROS was measured using Hitachi F-7000 fluorescence spectrophotometer. The cell cytotoxicity was assessed by BioTek SynergyH1 Full-featured microplate detector.Preparation of ultrathin 2D TiOx nanosheets
TiO2 nanosheets were prepared using a solvothermal method. Scheme 1 shows a schematic diagram of our fabrication approach of TiOx nanosheets. We synthesized TiO2 nanosheets by using P123 as a surfactant together with EG as a co-surfactant in ethanol solvent. The surfactant and co-surfactant in solution form an inverted laminar micelle, with the limitation of hydrated inorganic TiO2 precursor oligomers. Afterwards, solvent heat treatment is performed to form 2D nanosheets.31 The resulting TiO2 nanosheets were subsequently oxidized to TiOx nanosheets with hydrogen peroxide. In a typical synthesis, 0.56 g of titanium tetrachloride was added to 0.74 g of concentrated HCl solution in vigorously stirred (A bottle); and 0.2 g of P123 was dissolved in 3.0 g of ethanol (bottle B). After stirring for 10 minutes, the solution in bottle B was added to vial A and stirred for an additional 30 minutes. Then, 2.5 ml of titanium tetrachloride solution having 20 ml of EG was transferred to a 45 ml autoclave and heated at 150 °C for 20 hours. The hydrothermal reaction product was washed three times with water and ethanol, and the white powder was collected and dried at 60 °C for 12 h.32Preparation of TiOx nanosheets
The as-producing TiO2 nanosheets were dissolved in water under stirring. Afterward, 2 ml of hydrogen peroxide solution was added into the TiO2 suspension and reacted for 30 minutes under stirring. The product was washed three times with water to remove hydrogen peroxide for later use.Loading of anticancer drug DOX
The resulting TiOx nanosheets were dissolved in pH = 7.4 buffer solution under stirring condition. DOX was added into the solution and stirred for 24 h. Afterward, the solution was centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm for 10 minutes to remove free DOX. The resulting DOX-loaded TiOx (TiOx/DOX) nanocomposite was washed three times with buffer and then stored at refrigerator.29 The same procedure was used to prepare DOX-loaded TiO2 (TiO2/DOX) as a control.
500 rpm for 10 minutes to remove free DOX. The resulting DOX-loaded TiOx (TiOx/DOX) nanocomposite was washed three times with buffer and then stored at refrigerator.29 The same procedure was used to prepare DOX-loaded TiO2 (TiO2/DOX) as a control.
Drug release
DOX release experiments from TiO2/DOX nanocomposites were investigated at pH = 7.4 and pH = 5.0. TiO2/DOX sample was transferred into two dialysis bags, where each bag contained 4 ml TiO2/DOX. The dialysis bags were then immersed into 20 ml buffer solution at pH 7.4 and 5.0 respectively. The dialysis solution was stirred for 48 h. A 2 ml external dialysis solution was collected at scheduled time intervals, and replaced with the same fresh buffer solution. UV-visible absorptions of DOX with concentration gradients from 0.002 to 0.3 mM were characterized respectively and a standard curve was therefore obtained. Concentration–absorption curve of DOX was calculated according to UV-visible absorptions of DOX concentration gradients. The amount of released DOX in the dialysis solution was then determined by the absorbance measured with UV-visible spectroscopy. The same method was used to investigate drug release for the TiOx/DOX, and the effect of irradiating the X-ray at pH 5 on the TiOx/DOX release was investigated.26Cell culture
The A549 human non-small cell lung carcinoma cell line was obtained from Institute of Basic Medical Science, Chinese Academy of Medical Science (Beijing. China). The cells were cultured in normal DMEM with 10% (v/v) fetal bovine serum (FBS) and 100 U ml−1 of penicillin/streptomycin, and in a humidified incubator at 37 °C in 95% air and 5% CO2.24Cell toxicity study
In vitro cytotoxicity studies of TiO2 and TiOx were performed using A549 cells. A549 cells were plated in 96 well plates and then incubated for 24 h. After to be attached, TiO2 or TiOx with different concentrations (100, 200, 300, 400, and 500 μg ml−1) were added into each well in quadruplicate and further kept for 48 hours. Afterward, the TiO2 or TiOx incubation medium was removed and new medium and 10 μl of CCK8 solution were added into each well, including the control wells without TiO2 or TiOx. The plates were incubated for 1 hour before it was monitored by the absorbance at 450 nm to calculate the cell viability. All experiments were replicated three times, and the data are graphically presented as mean ± standard deviation (SD).33In vitro ROS assay and intracellular ROS detection
The level of ROS production in vitro with and without X-ray treatment bare TiO2 and TiOx was measured using ROS Brite™ active oxygen fluorescence probe APF by means of fluorescence spectrophotometer. We added 2 ml of APF to different concentrations of TiO2 and TiOx suspensions prepared in a cuvette (10 ml). Each cuvette is then exposed to the same dose of X-ray radiation for 30 min. The samples were placed for an hour in the dark. The fluorescent signal was measured at excitation and emission wavelengths of 490 and 510 nm respectively.5Intracellular ROS production following X-ray irradiation was detected using DCFH-DA ROS Assay Kit. A549 cells were seeded at 4 × 105 cells per well in 24-well plates. DCFH-DA was loaded into the cells after 24 hours incubation with TiO2 and TiOx (30 μg ml−1), respectively. After incubation for 30 min, the cells were washed twice with PBS and then exposed to X-ray irradiation for 30 min. After incubation for 1 h, fluorescence images of treated cells were acquired using a fluorescence microscope (LEICA DMI4000 B).29
Cancer cell killing experiments
A549 lung cancer cells were seeded into a 96-well plate at a density of 2 × 104 cells per well. After incubation for 24 h, the cell culture medium was changed to fresh medium containing various doses of TiO2/DOX, TiOx/DOX and free drug DOX (0.1 to 10 μg ml−1). The cells were continuously incubated for 48 h, and a CCK8 assay was performed to detect the killing effect. To study the X-ray irradiation effect on cell killing, the cells were seeded into 96-well plates at a density of 2 × 104 cells per well and incubated for 24 h. Afterward, the same doses of TiOx (5 μg ml−1) or TiOx/DOX (5 μg ml−1) were added into the wells and then incubation for 24 hours. The cells were treated with X-ray irradiation for 5, 10, 15, 20, 30 minutes with control, and continue to incubate for 24 hours. The toxicity assessment was performed according to the above CCK8 protocol. All experiments were replicated three times, and the data are graphically presented as mean ± standard deviation (SD).In vitro cell uptake of drugs
Confocal imaging was used to analyze cellular uptake. A549 cells were plated on 24-well plates at a density of 4 × 105 cells per well and incubated for 24 h. The growth medium was then replaced with free DOX, TiO2/DOX, TiOx/DOX (10 μg ml−1). After incubation for another 8 h, the cells were rinsed three times with PBS, fixed with 4% formaldehyde in PBS for 30 min, and washed three times with PBS. A549 cells were stained with 10 μl of 4′,6-diamidino-2-phenylindole (DAPI) for 10 min in order to visualize the cell nuclei. The cells were washed three times with PBS, and the cells in the micro dishes were observed with a Leica TCS SP8 confocal microscope. DAPI is visible at an excitation wavelength of 405 nm, while DOX is visible at an excitation wavelength of 511 nm.34Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21571028, No. 21601027), the Fundamental Research Funds for the Central Universities (No. DUT15RC(3)055, No. DUT16TD19, No. DUT17LK33), and the Education Department of the Liao Province of China (LT2015007).Notes and references
- N. Soni, N. Soni, H. Pandey, R. Maheshwari, P. Kesharwani and R. K. Tekade, J. Colloid Interface Sci., 2016, 481, 107 CrossRef PubMed.
- (a) T. S. Armstrong, T. Mendoza, I. Gning, I. Gring, C. Coco, M. Z. Cohen, L. Eriksen, M.-A. Hsu, M. R. Gilbert and C. Cleeland, J. Neuro-Oncol., 2006, 80, 27 CrossRef PubMed; (b) E. G. Sarris, M. W. Saif and K. N. Syrigos, Pharmaceuticals, 2012, 5, 1236 CrossRef PubMed; (c) T. G. Whitsett, L. J. Inge, H. D. Dhruv, P. Y. Cheung, G. J. Weiss, R. M. Bremner, J. A. Winkles and N. L. Tran, Transl. Lung Cancer Res., 2013, 2, 273 Search PubMed; (d) R. K. Tekade, S. R. Youngren-Ortiz, H. Yang, R. Haware and M. B. Chougule, Mol. Pharm., 2014, 11, 3671 CrossRef PubMed.
- K. A. Goodman and C. Hajj, J. Surg. Oncol., 2013, 107, 86 CrossRef PubMed.
- H. Chang, L. Tang, Y. Wang, J. Jiang and J. Li, Anal. Chem., 2010, 82, 2341 CrossRef PubMed.
- M. Nakayama, R. Sasaki, C. Ogino, T. Tanaka, K. Morita, M. Umetsu, S. Ohara, Z. Tan, Y. Nishimura, H. Akasaka, K. Sato, C. Numako, S. Takami and A. Kondo, Radiat. Oncol., 2016, 11, 91 CrossRef PubMed.
- (a) E. S. Lee, Z. Gao and Y. H. Bae, J. Controlled Release, 2008, 132, 164 CrossRef PubMed; (b) S. Bhattacharyya, R. A. Kudgus, R. Bhattacharya and P. Mukherjee, Pharm. Res., 2011, 28, 237 CrossRef PubMed; (c) F. M. Kievit, F. Y. Wang, C. Fang, H. Mok, K. Wang, J. R. Silber, R. G. Ellenbogen and M. Zhang, J. Controlled Release, 2011, 152, 76 CrossRef PubMed; (d) R. Gomez-Pastor, E. T. Burchfiel, D. W. Neef, A. M. Jaeger, E. Cabiscol, S. U. McKinstry, A. Doss, A. Aballay, D. C. Lo, S. S. Akimov, C. A. Ross, C. Eroglu and D. J. Thiele, Nat. Commun., 2017, 8, 14405 CrossRef PubMed; (e) C. Leroy, Q. Shen, V. Strande, R. Meyer, M. E. McLaughlin, E. Lezan, M. Bentires-Alj, H. Voshol, D. Bonenfant and L. A. Gaither, Oncogene, 2015, 34, 5593 CrossRef PubMed; (f) A. Zakharchenko, N. Guz, A. M. Laradji, E. Katz and S. Minko, Nat. Catal., 2018, 1, 73 CrossRef; (g) Y. Liu, J. Peng, S. Wang, M. Xu, M. Gao, T. Xia, J. Weng, A. Xu and S. Liu, NPG Asia Mater., 2018, 10, e458 CrossRef.
- (a) C. Chen, L. Kuai, Y. Chen, Q. Wang, E. Kan and B. Geng, RSC Adv., 2015, 5, 98254 RSC; (b) Y. Yin, W.-W. Zhu, L.-P. Guo, R. Yang, X.-S. Li and Y. Jiang, J. Phys. Chem. B, 2013, 117, 125 CrossRef PubMed.
- (a) J. Shi, Z. Chen, B. Wang, L. Wang, T. Lu and Z. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 28554 CrossRef PubMed; (b) Z. Tan, K. Sato and S. Ohara, Adv. Powder Technol., 2015, 26, 296 CrossRef.
- (a) T. Bose, D. Latawiec, P. P. Mondal and S. Mandal, J. Nanopart. Res., 2014, 16, 2527 CrossRef; (b) T. Xu, H. Zheng, P. Zhang, W. Lin and Y. Sekiguchi, J. Mater. Chem. A, 2015, 3, 19115 RSC; (c) J. Yang, Y.-L. Jiang, L.-J. Li, E. Muhire and M.-Z. Gao, Nanoscale, 2016, 8, 8170 RSC.
- B. Liu, C. Li, Z. Cheng, Z. Hou, S. Huang and J. Lin, Biomater. Sci., 2016, 4, 890 RSC.
- C. R. Gordijo, A. Z. Abbasi, M. A. Amini, H. Y. Lip, A. Maeda, P. Cai, P. J. O'Brien, R. S. DaCosta, A. M. Rauth and X. Y. Wu, Adv. Funct. Mater., 2015, 25, 1858 CrossRef.
- P. Retif, S. Pinel, M. Toussaint, C. Frochot, R. Chouikrat, T. Bastogne and M. Barberi-Heyob, Theranostics, 2015, 5, 1030 CrossRef PubMed.
- J. Wang and Y. Fan, Int. J. Mol. Sci., 2014, 15, 22258 CrossRef PubMed.
- (a) D. W. Porter, N. Wu, A. F. Hubbs, R. R. Mercer, K. Funk, F. Meng, J. Li, M. G. Wolfarth, L. Battelli, S. F. M. Andrew, R. Hamilton Jr, K. Sriram, F. Yang, V. Castranova and A. Holian, Toxicol. Sci., 2013, 131, 179 CrossRef PubMed; (b) A. Magrez, L. Horvath, R. Smajda, V. Salicio, N. Pasquier, L. Forro and B. Schwaller, ACS Nano, 2009, 3, 2274 CrossRef PubMed; (c) R. F. Hamilton, N. Wu, D. Porter, M. Buford, M. Wolfarth and A. Holian, Part. Fibre Toxicol., 2009, 6, 35 CrossRef PubMed.
- (a) A. Simon-Deckers, B. Gouget, M. Mayne-L’Hermite, N. Herlin-Boime, C. Reynaud and M. Carrière, Toxicology, 2008, 253, 137 CrossRef PubMed; (b) J. Jiang, G. Oberdorster, A. Elder, R. Gelein, P. Mercer and P. Biswas, Nanotoxicology, 2008, 2, 33 CrossRef PubMed; (c) C. M. Sayes, R. Wahi, P. A. Kurian, Y. P. Liu, J. L. West, K. D. Ausman, D. B. Warheit and V. L. Colvin, Toxicol. Sci., 2006, 92, 174 CrossRef PubMed.
- (a) D. B. Warheit, T. R. Webb, K. L. Reed, S. Frerichs and C. M. Sayes, Toxicology, 2007, 230, 90 CrossRef PubMed; (b) P. Thevenot, J. Cho, D. Wavhal, R. B. Timmons and L. Tang, Nanomedicine: Nanotechnology, Biology and Medicine, 2008, 4, 226 CrossRef PubMed; (c) M. Hamzeh and G. I. Sunahara, Toxicol. In Vitro, 2013, 27, 864 CrossRef PubMed.
- B. Yan, P. Zhou, Q. Xu, X. Zhou, D. Xu and J. Zhu, RSC Adv., 2016, 6, 6133 RSC.
- G. Xiang, D. Wu, J. He and X. Wang, Chem. Commun., 2011, 47, 11456 RSC.
- T. Wang, H. Jiang, L. Wan, Q. Zhao, T. Jiang, B. Wang and S. Wang, Acta Biomater., 2015, 13, 354 CrossRef PubMed.
- (a) P. Xu, R. Wang, J. Ouyang and B. Chen, Nanoscale Res. Lett., 2015, 10, 94 CrossRef PubMed; (b) M. I. Setyawati, V. N. Mochalin and D. T. Leong, ACS Nano, 2016, 10, 1170 CrossRef PubMed; (c) B. Cao, M. Yang, Y. Zhu, X. Qu and C. Mao, Adv. Mater., 2014, 26, 4627 CrossRef PubMed.
- (a) S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211 CrossRef PubMed; (b) R. Subbaiya, M. Saravanan, A. R. Priya, K. R. Shankar, M. Selvam, M. Ovais, R. Balajee and H. Barabadi, IET Nanobiotechnol., 2017, 11, 965 CrossRef PubMed; (c) M. Kima, J. H. Seo, W. I. Jeon, M.-Y. Kim, K. Cho, S. Y. Lee and S.-W. Joo, Talanta, 2012, 88, 631 CrossRef PubMed; (d) U. Ikoba, H. Peng, H. Li, C. Miller, C. Yu and Q. Wang, Nanoscale, 2015, 7, 4291 RSC.
- H. M. Yadav, N. D. Thorat, M. M. Yallapu, S. A. M. Tofail and J.-S. Kim, J. Mater. Chem. B, 2017, 5, 1461 RSC.
- J. Song, X. Yang, O. Jacobson, L. Lin, P. Huang, G. Niu, Q. Ma and X. Chen, ACS Nano, 2015, 9, 9199 CrossRef PubMed.
- W. Ren, L. Zeng, Z. Shen, L. Xiang, A. Gong, J. Zhang, C. Mao, A. Li, T. Paunesku, G. E. Woloschak, N. S. Hosmaned and A. Wu, RSC Adv., 2013, 3, 20855 RSC.
- X. Zhai, M. Yu, Z. Cheng, Z. Hou, P. Ma, D. Yang, X. Kang, Y. Dai, D. Wang and J. Lin, Dalton Trans., 2011, 40, 12818 RSC.
- L. Zeng, Y. Pan, Y. Tian, X. Wang, W. Ren, S. Wang, G. Lu and A. Wu, Biomaterials, 2015, 57, 93 CrossRef PubMed.
- Z. Hou, Y. Zhang, K. Deng, Y. Chen, X. Li, X. Deng, Z. Cheng, H. Lian, C. Li and J. Lin, ACS Nano, 2015, 9, 2584 CrossRef PubMed.
- H. Meng, M. Liong, T. Xia, Z. Li, Z. Ji, J. I. Link and A. E. Nel, ACS Nano, 2010, 4, 4539 CrossRef PubMed.
- X. Ji, F. Peng, Y. Zhong, Y. Su, X. Jiang, C. Song, L. Yang, B. Chu, S.-T. Lee and Y. He, Adv. Mater., 2015, 27, 1029 CrossRef PubMed.
- X. Xue, Y. Zhao, L. Dai, X. Zhang, X. Hao, C. Zhang, S. Huo, J. Liu, C. Liu, A. Kumar, W.-Q. Chen, G. Zou and X.-J. Liang, Adv. Mater., 2014, 26, 712 CrossRef PubMed.
- G. Xiang, T. Li, J. Zhuang and X. Wang, Chem. Commun., 2010, 46, 6801 RSC.
- Z. Sun, T. Liao, Y. Dou, S. M. Hwang, M.-S. Park, L. Jiang, J. H. Kim and S. X. Dou, Nat. Commun., 2014, 5, 1 Search PubMed.
- L. Yu, Y. Chen, M. Wu, X. Cai, H. Yao, L. Zhang, H. Chen and J. Shi, J. Am. Chem. Soc., 2016, 138, 9881 CrossRef PubMed.
- J. Meng, P. Zhang, F. Zhang, H. Liu, J. Fan, X. Liu, G. Yang, L. Jiang and S. Wang, ACS Nano, 2015, 9, 9284 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02899k |
| This journal is © The Royal Society of Chemistry 2018 |