
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA channel-structured Eu-based metal–organic framework with a zwitterionic ligand for selectively sensing Fe3+ ions†
Yong-Qing Huang *a,
Huai-Ying Chena,
Yang Wanga,
Yong-He Rena,
Zong-Ge Lia,
Lu-Chao Lia and
Yan Wangb
*a,
Huai-Ying Chena,
Yang Wanga,
Yong-He Rena,
Zong-Ge Lia,
Lu-Chao Lia and
Yan Wangb
aState Key Laboratory of Mining Disaster Prevention and Control Co-founded by Shandong Province and the Ministry of Science and Technology, College of Chemical and Environmental Engineering, Shandong University of Science and Technology, Qingdao 266590, China. E-mail: yqhuangskd@163.com
bSchool of Chemistry and Chemical Engineering, Anhui Key Laboratory of Functional Coordination Compounds, Anqing Normal University, Anqing 246011, China
First published on 12th June 2018
Abstract
A novel Eu-based MOF [Eu(IMS1)2]Cl·4H2O (1) was successfully constructed based on a semi-rigid zwitterionic 1,3-bis(4-carboxylbenzyl)-imidazolium (IMS1) ligand, featuring a 3-fold interpenetrating dia net structure with a point symbol of 66 and charged permanent micropores. Considering its excellent luminescent property as well as thermal and chemical stability, complex 1 was explored as a potential sensor for detecting Fe3+ ions. The results show that complex 1 has a high sensitivity and selectivity for Fe3+ based on a ‘turn-off’ effect, for which the electrostatic interaction between Fe3+ ions and the inner surface of the micropores may play a critical role. The fluorescence quenching mechanism reveals that dynamic quenching and competitive adsorption between Fe3+ and 1 lead to the quenching effect of 1.
Introduction
It is well known that environmental pollution is a very serious problem that we are currently facing. Recently, with the continuous activity of humans, more and more heavy metal ions and toxic organic molecules have been released into the environment in soil, water and air, threatening the health and life of animals and human beings by way of the food chain.1 Among various pollutants, iron is a special element, being indispensable for all living systems at certain concentrations. However, both its deficiency and excess will cause various biological disorders like hepatic cirrhosis, endotoxemia, and hereditary hemochromatosis.2To accurately detect iron, several analytical techniques have been developed like ICP-MS, FAAS and spectrophotometric detection.3 Among these probing techniques, the method based on luminescent metal–organic frameworks (LMOFs) as fluorescent sensors, combining the advantages of MOFs and luminescence sensors, have attracted considerable attention due to high sensitivity, selectivity and non-destructive nature.4 Compared with traditional luminescent organic conjugate molecules,5 LMOFs as sensing materials have some advantages, such as crystalline nature, diverse and modifiable structures, permanent porosity, tunable band gaps and electronic structures, etc.6 In this respect, Ln-based LMOFs have great potential as fluorescence probes due to their unique optical properties such as large stokes shift, high color purity and relatively long luminescence lifetime.7 Most importantly, for Eu/Tb-based MOF sensors, the pink or green light can be easily observed under a UV lamp by naked eyes, benefitting the low cost quick detection by the ‘turn-off’ effect.8 Up to now, many Eu/Tb-based MOFs have been reported as sensors for detecting cations, anions, small molecules, organic solvents and poisonous gases, etc.7–9
On the other hand, except for metal ions or clusters, selection of organic linkers is another important factor for detection performance improvement of Fe3+ ions. Generally, outstanding organic linkers with functional groups can strengthen host–guest interaction, triggering electron transfer and/or energy transfer, which is responsible for the fluorescence changes.8,10 Therefore, for synthesis of Eu/Tb-based MOF sensors, the design/selection of organic ligands becomes particularly important. Zwitterionic carboxylate ligands, especially functional pyridinium and imidazolium ligands, have been widely employed as bridging ligands to construct MOFs, some of which have exhibited impressive potential applications based on their Lewis acidic property and interaction with guest molecules.11 Moreover, pure zwitterionic ligands have displayed potential application as luminescent sensors for sensing Fe3+ ions.12 Based on the above analysis and taken into account that zwitterionic ligand is capable of forever generating electrostatic field inside the channels, and then reinforcing the interaction between the Eu-based MOF probe and the Fe3+ ions, we designed and synthesized a new semirigid zwitterionic 1,3-bis(4-carboxylbenzyl)-imidazolium (IMS1) ligand to construct Eu-based MOF.13 In this work, we successfully gained a Eu(III)-based channel-structured MOF [Eu(IMS1)2]Cl·4H2O (1) with high stability towards aqueous solution in a wide range of pH values, being a promising fluorescence probe for selectively detecting Fe3+ ions.
Experimental
Materials and methods
All reagents and chemicals are purchased from HEOWNS, which are used without further purification. The 1,3-bis(4-carboxylbenzyl)-imidazolium (IMS1) ligand was synthesized according to the previously reported method.14 Elemental analyses (C, H, N) were carried out using a Perkin-Elmer 240C elemental analyser. Infrared spectra were recorded in the range of 4000–400 cm−1 on a Nicolet 380 FT-IR spectrometer with KBr Pellets. Powder X-ray diffraction (PXRD) were measured at room temperature using a Rigaku Ultima IV diffractometer with graphite monochromatic Cu Kα radiation (λ = 0.15418 nm) at 40 kV and 40 mA. Simulated powder patterns were based on single crystal data and calculated using the Mercury software.15 Thermogravimetric analysis (TGA) was performed using a STA 409 PC thermal analyser under the nitrogen atmosphere with a heating rate of 10 °C min−1. The luminescent spectra for complex 1 were recorded at ambient temperature using a Hitachi F-4600 FL spectrophotometer. UV/Vis spectra were obtained using a Hitachi UH4150 Spectrophotometer within 300–800 nm.Synthesis of [Eu(IMS1)2]Cl·4H2O (1)
A mixture of IMS1 (0.3 mmol, 125.4 mg), Eu(NO3)3·6H2O (0.15 mmol, 66.6 mg), CH3CN (6 mL) and H2O (7 mL) was sealed in a 20 mL Teflon-lined bomb and heated at 150 °C for 3 days. After cooling to room temperature at a rate of 3 °C h−1, light yellow prismatic crystals were obtained. Yield: 78%. Anal. Calcd for C38H38ClEuN4O12: C, 49.07; H, 4.12; N, 6.02. Found: C, 49.10; H, 4.08; N, 6.07. IR (KBr, cm−1): 3468 (m), 3140 (m), 3067 (m), 1617 (s), 1557 (s), 1532 (s), 1416 (vs), 1351 (s), 1183 (w), 1153 (s), 1019 (w), 859 (m), 814 (m), 787 (m), 746 (s), 612 (m), 561 (w), 425 (w).X-ray crystallography
The diffraction data was collected on the Bruker Smart ApexII CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.071073 nm). Empirical absorption correction was applied to the data using the multi-scan program SADABS.16 The structure was solved by direct method (SHELXS-97) and refined by the full-matrix least-square method on F2 using the SHELXL-97 program.17 The coordinates of all non-hydrogen atoms were refined anisotropically. All H atoms of IMS1 ligands were situated at theoretically calculated positions and refined using a riding model isotropically, whereas the H atoms of lattice water molecules have not been ascertained by difference Fourier. All calculations were performed using the SHELTXL crystallographic software package.18 Details of the crystal parameters, data collections and refinements for 1 are summarized in Table 1. Selected bond lengths and angles with their estimated standard deviations are given in Table 2.| a R1 = Σ∥Fo| − |Fc∥/Σ|Fo|; wR2 = {Σ[w(Fo2 − Fc2) 2]/Σ[w(Fo2)2]}1/2. | |
|---|---|
| Complex | 1 |
| Empirical formula | C38H38ClEuN4O12 |
| Formula weight | 930.13 |
| Crystal system | Tetragonal |
| Space group | I41/a |
| a (Å) | 24.0818(8) |
| b (Å) | 24.0818(8) |
| c (Å) | 13.5997(9) |
| α (°) | 90 |
| β (°) | 90 |
| γ (°) | 90 |
| V (Å3) | 7886.9(6) |
| Z | 8 |
| Dcalc (g cm−3) | 1.567 |
| μ (mm−1) | 1.725 |
| T (K) | 293 |
| F (000) | 3760 |
| 2θmax (°) | 55 |
| Independent reflections | 4533 |
| Data/restraints/parameters | 4533/0/258 |
| Rint | 0.0398 |
| Goodness-of-fit on F2 | 1.059 |
| R1, wR2a (I > 2σ(I)) | 0.0306, 0.0815 |
| R1, wR2a (all data) | 0.0412, 0.0870 |
| a Symmetry transformations used to generate equivalent atoms: #1 y + 3/4, −x + 5/4, −z + 5/4; #2 −y + 5/4, x − 3/4, −z + 5/4; #3 −x + 2, −y + 1/2, z + 0; #4 −x + 3/2, −y + 1/2, −z + 1/2; #5 x + 1/2, y, −z + 1/2. | |||
|---|---|---|---|
| Eu(1)-O(2)#1 | 2.290(2) | Eu(1)-O(2)#2 | 2.290(2) |
| Eu(1)-O(1) | 2.435(2) | Eu(1)-O(1)#3 | 2.435(2) |
| Eu(1)-O(4)#4 | 2.443(2) | Eu(1)-O(4)#5 | 2.443(2) |
| Eu(1)-O(3)#5 | 2.503(2) | Eu(1)-O(3)#4 | 2.503(2) |
| O(2)#1-Eu(1)-O(2)#2 | 107.04(13) | O(2)#1-Eu(1)-O(1) | 77.92(9) |
| O(2)#2-Eu(1)-O(1) | 83.97(9) | O(2)#1-Eu(1)-O(1)#3 | 83.97(9) |
| O(2)#2-Eu(1)-O(1)#3 | 77.92(9) | O(1)-Eu(1)-O(1)#3 | 149.34(11) |
| O(2)#1-Eu(1)-O(4)#4 | 152.95(9) | O(2)#2-Eu(1)-O(4)#4 | 90.05(9) |
| O(1)-Eu(1)-O(4)#4 | 125.69(8) | O(1)#3-Eu(1)-O(4)#4 | 79.22(8) |
| O(2)#1-Eu(1)-O(4)#5 | 90.05(9) | O(2)#2-Eu(1)-O(4)#5 | 152.95(9) |
| O(1)-Eu(1)-O(4)#5 | 79.22(8) | O(1)#3-Eu(1)-O(4)#5 | 125.70(8) |
| O(4)#4-Eu(1)-O(4)#5 | 82.87(13) | O(2)#1-Eu(1)-O(3)#5 | 78.76(9) |
| O(2)#2-Eu(1)-O(3)#5 | 149.83(9) | O(1)-Eu(1)-O(3)#5 | 125.88(8) |
| O(1)#3-Eu(1)-O(3)#5 | 73.24(8) | O(4)#4-Eu(1)-O(3)#5 | 76.19(9) |
| O(4)#5-Eu(1)-O(3)#5 | 52.73(8) | O(2)#1-Eu(1)-O(3)#4 | 149.83(9) |
| O(2)#2-Eu(1)-O(3)#4 | 78.76(9) | O(1)-Eu(1)-O(3)#4 | 73.24(8) |
| O(1)#3-Eu(1)-O(3)#4 | 125.88(8) | O(4)#4-Eu(1)-O(3)#4 | 52.73(8) |
| O(4)#5-Eu(1)-O(3)#4 | 76.19(9) | O(3)#5-Eu(1)-O(3)#4 | 111.45(11) |
Results and discussion
Structural description
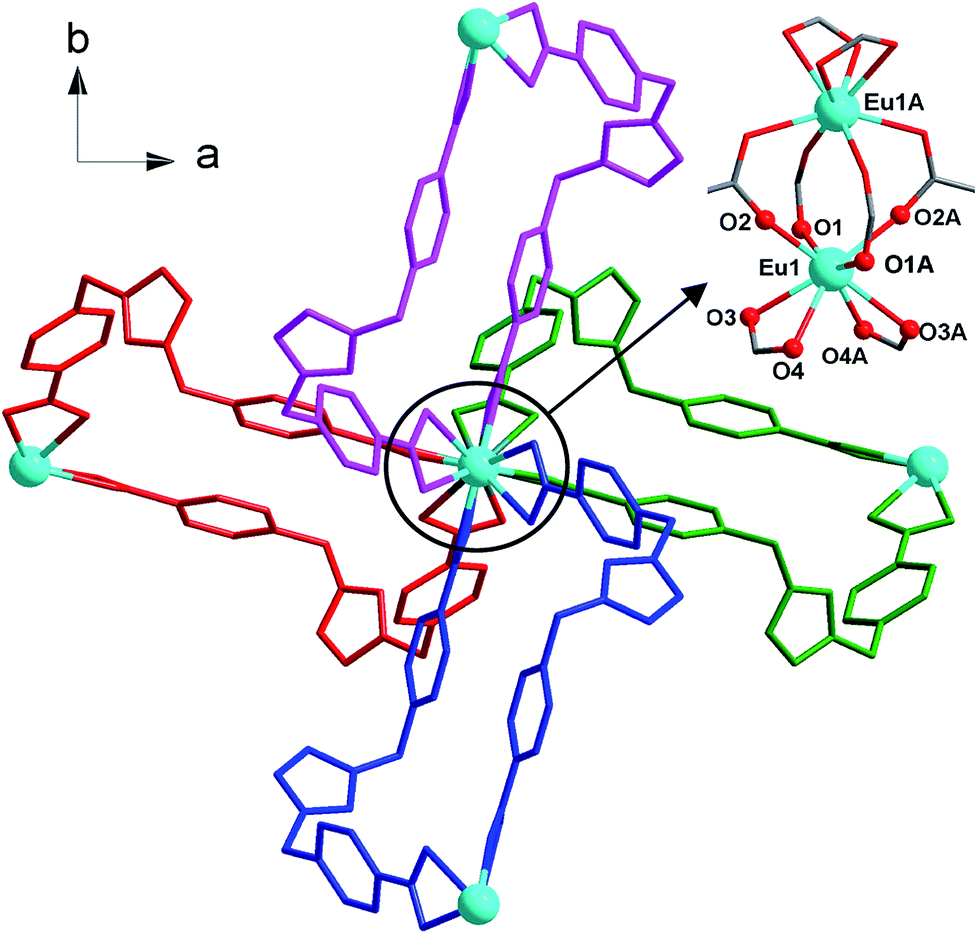
Complex 1 was obtained by the solvothermal reaction of Eu(NO3)3·6H2O and H2(IMS1)Cl in CH3CN–H2O (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, v/v) at 150 °C for 3 days as light yellow crystals, which was confirmed by EA, IR, TGA and single crystal X-ray diffraction. The phase purity of the bulk crystalline samples was independently verified by powder X-ray diffraction (PXRD) (Fig. S1, ESI†). Crystal structure analysis reveals that 1 crystallizes in the tetragonal I41/a space group (Table 1) and features a 3-fold interpenetrating 3D framework with the Eu2(CO2)8 clusters as 4-connected nodes. The crystallographic asymmetric unit of 1 is composed of half europium atom, one IMS1 ligand, half chloride ion and two lattice water molecules. Each Eu3+ ion, sitting on the 41 screw axis, bonds to six IMS1 ligands with O8 donor set, showing a trigondodecahedron geometry as exhibited in Fig. 1. The average Eu–O bond distance is 2.418 Å, and the O–Eu–O bond angles range from 52.73(8)° to 152.95(9)° (Table 2).
:7, v/v) at 150 °C for 3 days as light yellow crystals, which was confirmed by EA, IR, TGA and single crystal X-ray diffraction. The phase purity of the bulk crystalline samples was independently verified by powder X-ray diffraction (PXRD) (Fig. S1, ESI†). Crystal structure analysis reveals that 1 crystallizes in the tetragonal I41/a space group (Table 1) and features a 3-fold interpenetrating 3D framework with the Eu2(CO2)8 clusters as 4-connected nodes. The crystallographic asymmetric unit of 1 is composed of half europium atom, one IMS1 ligand, half chloride ion and two lattice water molecules. Each Eu3+ ion, sitting on the 41 screw axis, bonds to six IMS1 ligands with O8 donor set, showing a trigondodecahedron geometry as exhibited in Fig. 1. The average Eu–O bond distance is 2.418 Å, and the O–Eu–O bond angles range from 52.73(8)° to 152.95(9)° (Table 2).
 | ||
| Fig. 1 Partial structure of 1, showing the coordination environment of Eu(III) and linking fashion of dinuclear Eu(III) clusters. The hydrogen atoms, chloride ions and water molecules were omitted for clarity. | ||
The structure of 1 features dinuclear Eu3+ cores as four-connected tetrahedral topological nodes. Each dinuclear cluster is surrounded by eight bridging IMS1 ligands, accompanying with intermetallic distance of 4.1613(3) Å, similar to that of the reported complex [Eu2(Hsal)8][Zn(phen)3]·(H2sal)(H2O).19 As depicted in Fig. 1, two symmetry-related Eu3+ ions in each cluster are first interconnected via four bidentate bridging carboxyl groups from four different IMS1 ligands, forming a classical paddle-wheel dinuclear unit. Then, another four IMS1 ligands are bound to dinuclear Eu3+ core by four bidentate chelating carboxyl groups. In other words, each IMS1 ligand takes μ3-bridging mode to connect three Eu3+ ions: one carboxyl group take μ2-κ1O:κ1O syn–syn bridging mode to link two Eu3+ ions and the other adopts chelating mode to coordinate to another Eu3+ ion. In fact, in terms of topological viewpoint, each μ3-bridging IMS1 ligand can be regarded as 2-connected linker and, at the same time, each dinuclear Eu3+ core as a whole can be considered as a node.
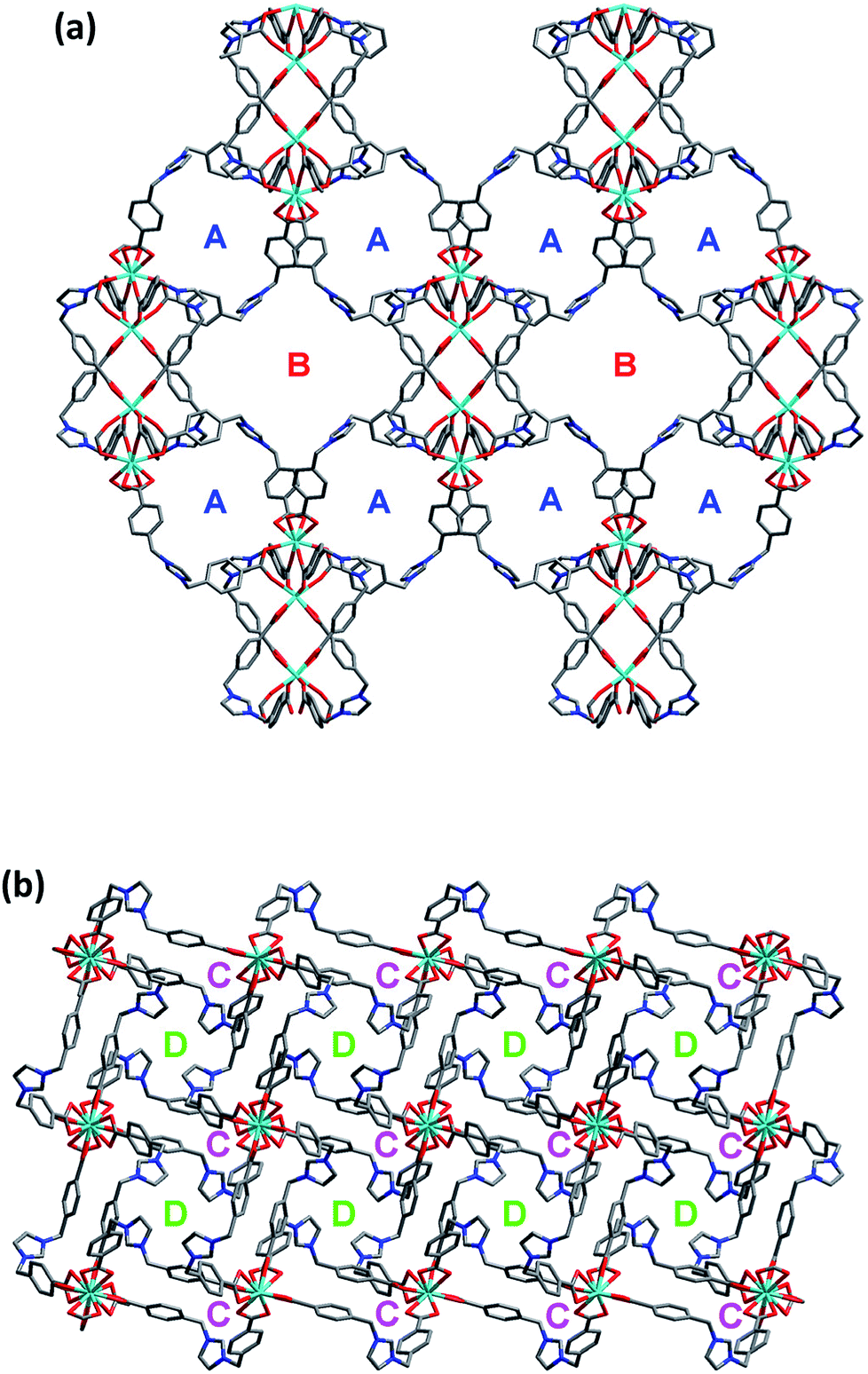
On the other hand, the semirigid IMS1 ligands in 1 adopt cis-configuration, which means that two rigid arms are on the same side of the imidazole ring. As displayed in Fig. 1, two dinuclear cores together with two bent IMS1 ligands make up a M4L2 hexagon metallacycle with chair conformation. That is, although each cluster possesses eight bridging IMS1 ligands, sticking out for inter-cluster connection, it actually link with four same clusters by ‘double bridges’ with intercluster distance of 15.7803(5) Å and can be regarded as 4-connected tetrahedral node (Fig. 1 and 2). Apparently, the 3D framework of 1 can be simplified as usual dia net with a point symbol 66.20 It is noticeable that the single real network of 1 exhibits four types of channel along three axis orientations. As shown in Fig. 2, there are identical two types of porthole (denoted as A and B) along a- and -b axis. Similarly, another two types of channel (denoted as C and D) exist along c-axis, which are parallel to 41 screw axis.
 | ||
| Fig. 2 The wire drawing of single diamondoid network of complex 1 along a- or b-axis (a) and c-axis (b), showing four types of channels. The hydrogen atoms, chloride ions and water molecules were omitted for clarity. | ||
As we all know, the nature hates vacuum for large interface energy. The solid materials, including MOFs, with large vacancy, usually accommodate guest molecules or adopt interpenetration to reduce the free space. Unfortunately, the same thing occurs for compound 1, even if bulky dinuclear Eu3+ clusters together with double bridges fail to prevent interpenetration. At last, 1 presents a 3-fold interpenetrating structure (Fig. 3). Notably, the nodes of three independent nets are aligned and equally spaced along 41 screw axis with the translation of 13.6 Å, equal to the length of c-axis. Additionally, the M4L2 macrocycles remain independent without catenation. Based on this common interpenetration mode, the channels running parallel to a- and b-axis direction disappear. In contrast, the D-type channels along c-axis orientation remain with cross-section of 5.95 × 5.95 Å2 and are filled with guest water molecules. At the same time, the C-type channels are blocked up by counter chloride anions. It is noticeable that after removal of these guest water molecules in the voids, the effective free volume calculated using PLATON is still approximately 11.4% (896.4 Å3 out of the 7886.9 Å3 per unit cell volume).21
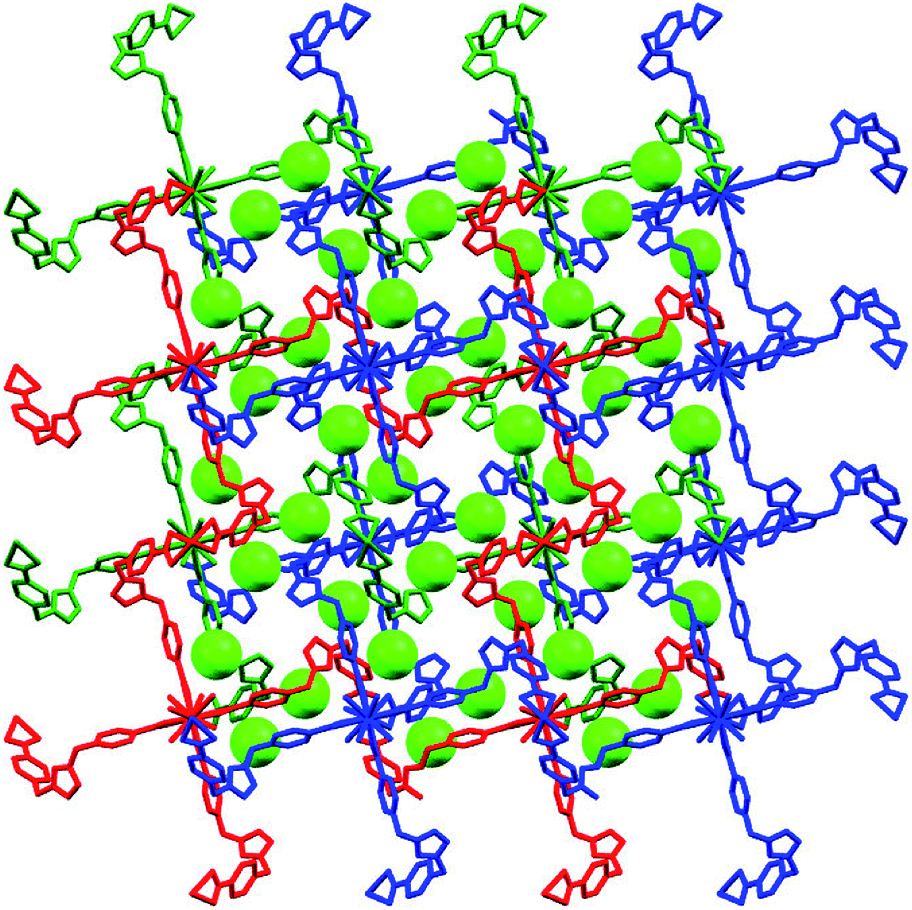 | ||
| Fig. 3 The 3-fold interpenetrating diamondoid framework in 1, showing chloride anions (green spheres) occupy C-type channels along c-axis. The guest water molecules occupying D-type channels and hydrogen atoms were omitted for clarity. | ||
Thermo and chemical stability
To evaluate the thermal stability for further utilization, thermogravimetric analysis (TGA) of 1 was carried out under flowing N2 atmosphere with the temperature range from 30 to 800 °C. The TGA curve displays an incessant weight loss of 7.29% in the low temperature region (<218.7 °C), which corresponds to the removal of lattice water molecules (calcd 7.74%) and the initial decomposition of the framework is observed up to ca. 350.7 °C, exhibiting excellent thermal stability. Needless to say, the high decomposition temperature of complex 1 is mainly attributed to the coordination bonding pattern and threefold interpenetrating structure, which solidified the flexible organic ligand. It is worth noting that, even if the as-synthesized complex 1 was dried under vacuum at 260 °C for 12 h, the crystalline water of 1 remains, which meaning that the guest water molecules are firmly confined in the cavity of 1 (Fig. S1 and S2, ESI†). On the other hand, the chemical stability of 1 in acidic, basic, and neutral aqueous solutions is very important for its practical application as Fe3+ sensor. So, we immersed the crystal samples of 1 into aqueous solutions with different pH values for 24 h to investigate its chemical stability. After filtering, the PXRD patterns of crystal samples are compared with the simulated one (Fig. S3, ESI†), which reveal that this material can maintain its crystallinity in aqueous solutions with pH values between 4 to 13. In one word, complex 1 has presented preeminent thermal and chemical stability, which lays a good foundation for its Fe3+ practical detection.Luminescent sensing of Fe3+ ions
The solid-state fluorescence property of as-synthesized Eu-MOF 1, as well as free H2(IMS1)Cl ligand were both investigated at ambient temperature (Fig. S4, ESI†). The results show that complex 1 has strong characteristic emission at 594 nm and 618 nm upon maximum excitation at 395 nm, which can be attributed to the 5D0 → 7F1 and 5D0 → 7F2 transitions of Eu3+, respectively. Apparently, the electric dipole transition emission at 618 nm is stronger than that of magnetic dipole moment transition at 594 nm, implying the Eu3+ ions of 1 in the tetragonal crystal system deviates from the center of symmetry in agreement with its crystal structure.22 According to the excitation spectrum of 1, the excitation maximum at 395 nm can be definitely attributed to 7F0 → 5L6 transition of Eu3+ (Fig. S5, ESI†). On the other hand, the H2(IMS1)Cl ligand exhibits the emission maximum at 397 nm upon the excitation at 340 nm, whereas there is no detectable luminescence when the excitation wavelength is 395 nm. It is worth noting that the emission intensity of complex 1 become weaker upon excitation at 340 nm than that upon excitation at 395 nm, and the emission peak arising from the free ligand disappears. As shown in Fig. S3 and S4, ESI,† the emission band of H2(IMS1)Cl ligand and the excitation band of 1 is almost completely overlapping, indicating the existence of fluorescence resonance energy transfer (FRET) between the IMS1 ligands and coordinated Eu3+ ions.Based on the microporous and permanent electrostatic field, we investigated the potential ability of complex 1 for sensing metal ions. First, 5 mg of crystalline samples of 1 were ground to powder, and then dispersed into 3 mL of water solution of MClx (M = Mn2+, Ba2+, Ca2+, Co2+, Cu2+, Cd2+, Ni2+, Zn2+, Hg2+, Fe2+, Al3+, Cr3+, Fe3+) (0.1 M) for half an hour of ultrasonic oscillation, forming a series of suspension prior to fluorescence measurements. In contrast to the suspension of complex 1 in water, all suspensions with tested divalent and trivalent metal ions have different extent decrease of luminescence intensity. Among them, Ni2+, Fe2+, and Cr3+ ions display a big intensity reduction, while Fe3+ ion exhibit the drastic quenching effect on the luminescence of 1, accompanied with a remarkable luminescence quenching visible to the naked eye under UV lamp (Fig. 4). Further, competitive experiments demonstrate that the presence of other metal ions have no significant effect on the luminescence response induced by Fe3+ ion (Fig. S6, ESI†). Compared with 1, the ligand H2(IMS1)Cl also showed selective luminescence quenching response to Fe3+ ions in suspensions, but the quenching phenomenon can't be observed to naked eyes under UV lamp (Fig. S7, ESI†). These results preliminarily prove that the electrostatic forces may play a significant role for the detection of Fe3+ ions. To verify the basic framework of the complex 1 treated by the metal aqueous solutions, PXRD patterns were compared with the simulated one (Fig. S8, ESI†). The coincidence of calculated PXRD patterns with the experimental datum can rule out that the fluorescence quenching phenomena resulted from collapse of the framework of 1.
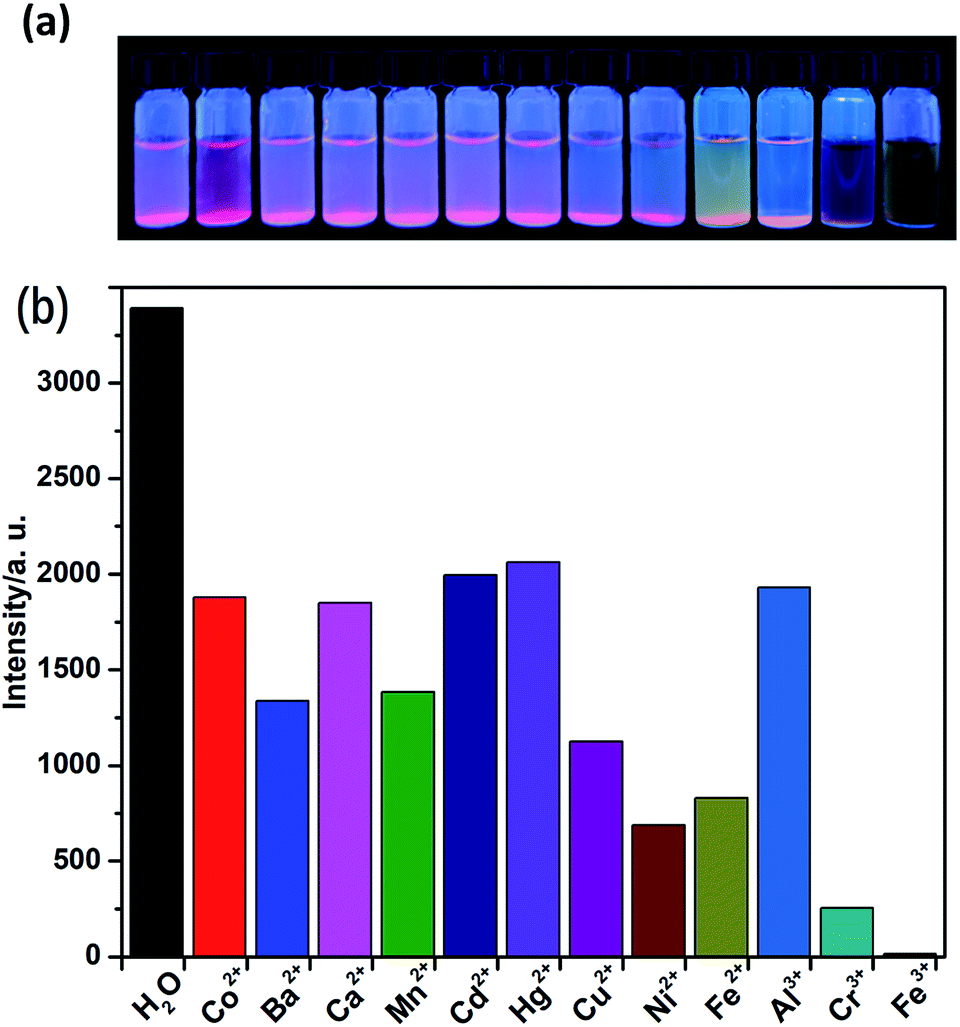 | ||
| Fig. 4 (a) The picture of complex 1 in different metal ion solution at 365 nm UV light; (b) comparison of the luminescence intensity of 1 in different MClx aqueous solutions (0.1 M) upon excitation at 395 nm. | ||
Intrigued by the results, we prepared a series of Fe3+ aqueous solutions with molar concentration ranging from 1 × 10−6 to 1 × 10−1 M to further investigate the relationship between quenching effect, which was quantified by monitoring the emission spectral intensity of 5D0 → 7F2 located in 618 nm, and the concentrations of Fe3+ ion. As shown in Fig. 5, the luminescence intensity of 1 decreased gradually as the concentration of Fe3+ ion increased. When the concentration reaches up to 1 × 10−1 M, the luminescence is completely quenched with 99.61% quenching efficiency. Meanwhile, the dose–response graph was well fitted by the Stern–Volmer equation in the concentration range of 1 × 10−6 to 1 × 10−3 M. As displayed in the inset of Fig. 5, a very good linear correlation (R2 = 0.99504) was observed between the quenching efficiency and the amount of Fe3+ with high KSV value of 5873.4 M−1. Furthermore, the detection limit is calculated to be 2.3 × 10−5 mol L−1 (3.74 ppm) according to 3δ/k, where δ is standard error and k is slope. The high KSV value and low detection limit, which are comparable to the reported values (Table. S1, ESI†), suggest that complex 1 is a powerful candidate for highly detective sensor for Fe3+ ions. On the other hand, the recyclability is one of the important indicator for sensors, which can effectively reduce the cost of practical application. Thus, the related experiments were conducted. Firstly, the crystal samples of 1 were soaked in the 0.1 M Fe3+ solution for minutes to form Fe3+@ 1. Then Fe3+@ 1 was centrifuged and washed with water for three times. The results indicate that the luminescent intensity of recycled 1 was almost unchanged compared to initial value after five recycles (Fig. S9, ESI†). In the meantime, the PXRD patterns of recycled 1 ensure its structure unchangeable.
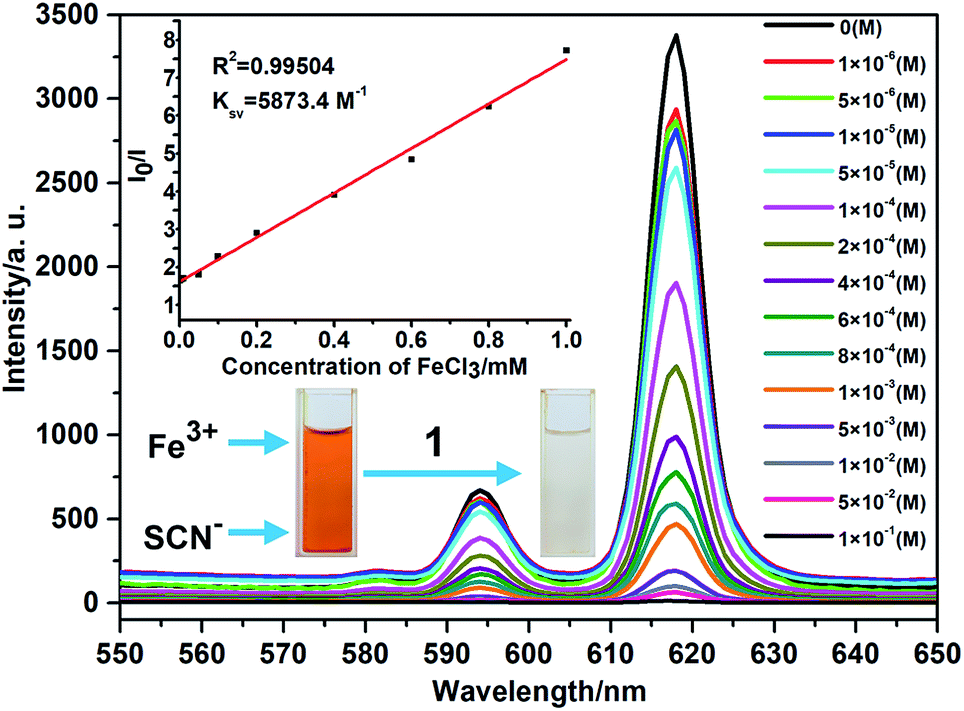 | ||
| Fig. 5 The luminescence spectra of 1 with different concentration of Fe3+ ions. The inset shows that the dose–response graph at 618 nm, revealing the quenching effect and the concentration of Fe3+ ions and photo of Fe(SCN)3 solution treated with complex 1. | ||
To explore the probable quenching reasons caused by Fe3+ ion, the UV-Vis absorption spectra of MClx aqueous solutions (1 × 10−4), suspension of 1 in water, and suspension of 1 with Fe3+ in water were carried out, respectively (Fig. S11, ESI†). The results reveal that Fe3+ aqueous, suspension of 1, and suspension of 1 with Fe3+ ions all have absorption at 395 nm. It is worth noting that the absorption ability of suspension of 1 with Fe3+ ions is stronger than the sum of Fe3+ and suspension of 1, implying the presence of interaction between the charged internal surface of 1 and Fe3+ ions. To prove this point, 1 was added to the brown Fe(SCN)3 solution, and then the solution quickly becomes colorless, further supporting the existence of strong host–guest interaction inside of the micropore. In addition, UV-Vis absorption spectrum of 1 with Fe3+ is similar to that of the Fe3+ aqueous. Based on the fact that, only in the low Fe3+ concentration range of 1 × 10−6 to 1 × 10−3 M, there is a good linear correlation between the I0/I and the concentration of Fe3+, as predicted by the Stern–Volmer law, quenching is considered entirely dynamic23 below the concentration of 1 × 10−3 M, with a large Stern–Volmer constant of 5873.4 M−1. On the contrary, beyond the concentration of 1 × 10−3 M, the luminescence quenching may arise from the competitive adsorption.24 In other words, two types of quenching pathway coexist in a competitive manner with another, which factor playing the key role mainly depends on the Fe3+ concentration.
Conclusions
In summary, a new microporous Eu-MOF 1 with a semirigid zwitterionic IMS1 ligand was designed and synthesized, which displays excellent luminous performance and high chemical stability in aqueous solution with pH value between 4 to 13. Based on these merits together with permanent electrostatic field, the response ability of 1 for different metal ions in aqueous solution was investigated, and the results reveal that 1 had high sensitivity and selectivity for Fe3+. The fluorescence quenching mechanism indicates that dynamic quenching and competitive absorption between Fe3+ and 1 result in the quenching effect of 1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21201111) and the Key Project of Anhui Provincial Fund for Distinguished Young Scholars in Colleges and Universities (No. 2013SQRL059ZD).References
- (a) L. Järup, Br. Med. Bull., 2003, 68, 167–182 CrossRef; (b) S. Cheng, Environ. Sci. Pollut. Res., 2003, 10, 192–198 CrossRef; (c) M. Hakim, Y. Y. Broza, O. Barash, N. Peled, M. Phillips, A. Amann and M. Haick, Chem. Rev., 2012, 112, 5949–5966 CrossRef PubMed; (d) J. L. Martinez, Environ. Pollut., 2009, 157, 2893–2902 CrossRef PubMed; (e) X. Zhang, X. Luo, N. Zhang, J. Wu and Y.-Q. Huang, Inorg. Chem. Front., 2017, 4, 1888–1894 RSC; (f) D. Han and M. J. Currell, Sci. Total Environ., 2017, 580, 602–625 CrossRef PubMed.
- (a) L. L. Dunn, Y. S. Rahmanto and D. R. Richardson, Trends Cell Biol., 2007, 17, 93–100 CrossRef PubMed; (b) N. C. Andrews, N. Engl. J. Med., 1999, 341, 1986–1995 CrossRef PubMed; (c) F. O. Omara and B. R. Blakley, J. Nutr., 1993, 123, 1649–1655 CrossRef PubMed; (d) L. H. Allen, J. Nutr., 2002, 132, 813S–819S CrossRef PubMed.
- (a) L. S. Huang and K. C. Lin, Spectrochim. Acta, Part B, 2001, 56, 123–128 CrossRef; (b) T. B. To, D. K. Nordstrom, K. M. Cunningham, J. W. Ball and R. B. McCleskey, Environ. Sci. Technol., 1999, 33, 807–813 CrossRef; (c) R. Ajlec and J. Stupar, Analyst, 1989, 114, 137–142 RSC; (d) E. Chinoporos, Anal. Chem., 1962, 34, 437–438 CrossRef; (e) P. Wu, Y. Li and X. P. Yan, Anal. Chem., 2009, 81, 6252–5257 CrossRef; (f) Y.-T. Yan, J. Liu, G.-P. Yang, F. Zhang, Y.-K. Fan, W.-Y. Zhang and Y.-Y. Wang, CrystEngComm, 2018, 20, 477–486 RSC.
- (a) K. Müller-Buschbaum, F. Beuerle and C. Feldmann, Microporous Mesoporous Mater., 2015, 216, 171–199 CrossRef; (b) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC; (c) K. Jayaramulu, R. P. Narayanan, S. J. George and T. K. Maji, Inorg. Chem., 2012, 51, 10089–10091 CrossRef PubMed; (d) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef PubMed.
- (a) F. S. K. Sahoo, D. Sharma, R. K. Bera, G. Crisponi and J. F. Callan, Chem. Soc. Rev., 2012, 41, 7195–7227 RSC; (b) C. Pan, K. Wang, S. Ji, H. Wang, Z. Li, H. He and Y. Huo, RSC Adv., 2017, 7, 36007–36014 RSC; (c) B. B. Zhang, H. Y. Liu, F. X. Wu, G. F. Hao, Y. Z. Chen, C. Y. Tan, Y. Tan and Y. Y. Jiang, Sens. Actuators, B, 2017, 243, 765–774 CrossRef; (d) J. Liu and Y. Qian, J. Lumin., 2017, 187, 33–39 CrossRef.
- (a) Z.-F. Wu, L.-K. Gong and X.-Y. Huang, Inorg. Chem., 2017, 56, 7397–7401 CrossRef PubMed; (b) B. Parmar, Y. Rachuri, K. K. Bisht and E. Suresh, Inorg. Chem., 2017, 56, 10939–10949 CrossRef PubMed; (c) Z.-F. Wu and X.-Y. Huang, Dalton Trans., 2017, 46, 12597–12604 RSC; (d) J.-M. Zhou, W. Shi, H.-M. Li, H. Li and P. Cheng, J. Phys. Chem. C, 2014, 118, 416–426 CrossRef; (e) J. Zhao, Y.-N. Wang, W.-W. Dong, Y.-P. Wu, D.-S. Li and Q.-C. Zhang, Inorg. Chem., 2016, 55, 3265–3271 CrossRef PubMed; (f) R. Lv, H. Li, J. Su, X. Fu, B. Yang, W. Gu and X. Liu, Inorg. Chem., 2017, 56, 12348–12356 CrossRef PubMed.
- (a) S. Dang, E. Ma, Z. Sun and H. Zhang, J. Mater. Chem., 2012, 22, 16920–16926 RSC; (b) C. Yang, H. ren and X. Yan, Anal. Chem., 2013, 85, 7441–7446 CrossRef PubMed; (c) J. Hao and B. Yan, J. Mater. Chem. C, 2014, 2, 6758–6764 RSC; (d) X. Song, S. Song, S. Zhao, Z. Hao, M. Zhu, X. Meng, L. Wu and H. Zhang, Adv. Funct. Mater., 2014, 24, 4034–4041 CrossRef; (e) T. W. Duan and B. Yan, J. Mater. Chem. C, 2014, 2, 5098–5104 RSC.
- (a) W. Yan, C. Zhang, S. Chen, L. Han and H. Zheng, ACS Appl. Mater. Interfaces, 2017, 9, 1629–1634 CrossRef PubMed; (b) G.-P. Li, G. Liu, Y.-Z. Li, L. Hou, Y.-Y. Wang and Z. Zhu, Inorg. Chem., 2016, 55, 3952–3959 CrossRef PubMed; (c) M. Zhang, J. Han, H. WU, Q. Wei, G. Xie, S. Chen and S. Gao, RSC Adv., 2016, 6, 94622–94628 RSC; (d) X.-L. Zhao, D. Tian, Q. Gao, H.-W. Sun, J. Xu and X.-H. Bu, Dalton Trans., 2016, 45, 1040–1046 RSC.
- (a) H. Li, Y. Han, Z. Shao, N. Li, C. Huang and H. Hou, Dalton Trans., 2017, 46, 12201–12208 RSC; (b) J. Wang, M. Jiang, L. Yan, R. Peng, M. Huangfu, X. Guo, Y. Li and P. Wu, Inorg. Chem., 2016, 55, 12660–12668 CrossRef PubMed; (c) Z. Hao, X. Song, M. Zhu, X. Meng, S. Zhao, S. Su, W. Yang, S. Song and H. Zhang, J. Mater. Chem. A, 2013, 1, 11043–11050 RSC; (d) S. Xing, Q. Bing, L. Song, G. Li, J. Liu, Z. Shi, S. Feng and R. Xu, Chem. –Eur. J., 2016, 22, 16230–16235 CrossRef PubMed; (e) Z.-Q. Liu, Y.-Q. Huang and W.-Y. Sun, Chin. J. Inorg. Chem., 2017, 33, 1959–1969 Search PubMed; (f) D. Tian, Y. Li, R.-Y. Chen, Z. Chang, G.-Y. Wang and X.-H. Bu, J. Mater. Chem. A, 2014, 2, 1465–1470 RSC; (g) D. Tian, X.-J. Liu, R. Feng, J.-L. Xu, J. Xu, R.-Y. Chen, L. Huang and X.-H. Bu, ACS Appl. Mater. Interfaces, 2018, 10, 5618–5625 CrossRef PubMed.
- (a) S. Pal and P. K. Bharadwaj, Cryst. Growth Des., 2016, 16, 5852–5858 CrossRef; (b) Q. Tang, S. Liu, Y. Liu, J. Miao, S. Li, L. Zhang, Z. Shi and Z. Zheng, Inorg. Chem., 2013, 52, 2799–2801 CrossRef PubMed; (c) M.-L. Han, G.-W. Xu, D.-S. Li, L. M. Azofra, J. Zhao, B. Chen and C. Sun, ChemistrySelect, 2016, 1, 3555–3561 CrossRef; (d) L.-H. Liu, X.-T. Qiu, Y.-J. Wang, Q. Shi, Y.-Q. Sun and Y.-P. Chen, Dalton Trans., 2017, 36, 12106–12113 RSC.
- (a) M. Higuchi, K. Nakamura, S. Horike, Y. Hijikata, N. Yanai, T. Fukushima, J. Kim, K. Kato, M. Takata, D. Watanabe, S. Oshima and S. Kitagawa, Angew. Chem., Int. Ed., 2012, 51, 8369–8372 CrossRef PubMed; (b) Y.-Q. Huang, Z.-G. Li, H.-Y. Chen, H.-D. Cheng, Y. Wang, Y.-H. Ren, Y. Zhao and L. Liu, CrystEngComm, 2017, 19, 6686–6694 RSC; (c) Y.-Q. Huang, H.-D. Cheng, H.-Y. Chen, Y. Wan, C.-L. Liu, Y. Zhao, X.-F. Xiao and L.-H. Chen, CrystEngComm, 2015, 17, 5690–5701 RSC; (d) Y.-Q. Huang, Y. Zhao, P. Wang, Y.-a. Okamura, B. N. Laforteza, Y. Lu, W.-Y. Sun and J.-Q. Yu, Dalton Trans., 2017, 46, 12430–12433 RSC; (e) S. Sen, N. N. Nair, T. Yamada, H. Kitagawa and P. K. Bharadway, J. Am. Chem. Soc., 2012, 134, 19432–19437 CrossRef PubMed.
- (a) K. Vijay, C. Nandi and S. D. Samant, RSC Adv., 2016, 6, 49724–49729 RSC; (b) S. Chaudhary and M. D. Milton, J. Photochem. Photobiol., A, 2018, 356, 595–602 CrossRef; (c) S. Bishnoi and M. D. Milton, J. Photochem. Photobiol., A, 2017, 335, 52–58 CrossRef.
- R.-M. Wen, S.-D. Han, G.-J. Ren, Z. Chang, Y.-W. Li and X.-H. Bu, Dalton Trans., 2015, 44, 10914–10917 RSC.
- S. Sen, N. N. Nair, T. Yamada, H. Kitagawa and P. K. Bharadaj, J. Am. Chem. Soc., 2012, 134, 19432–19437 CrossRef PubMed.
- P. R. Edgington, P. McCabe, C. F. Macrae, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. V. D. Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef.
- G. M. Sheldrick, SADABS, Program for Empirical Absoption Correction of Area Detector Data, University of Göttingen, Germany, 1996 Search PubMed.
- (a) G. M. Sheldrick, SHELX-97, An Integrated System for Solving Crystal Structures from Diffraction Data, University of Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELX-97, An Integrated System for Refining Crystal Structures from Diffraction Data, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXTL, Version 6.10, Bruker AXS Inc., Madison, Wisconsin, USA, 2000 Search PubMed.
- M.-C. Yin, L.-J. Yuan, C.-C. Ai, C.-W. Wang, E.-T. Yuan and J.-T. Sun, Polyhedron, 2004, 23, 529–536 CrossRef.
- M. Du, X.-H. Bu, Y.-M. Guo, H. Liu, S. R. Batten, J. Ribas and T. C. W. Mak, Inorg. Chem., 2002, 41, 4904–4908 CrossRef PubMed.
- A. L. Spec, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef.
- (a) H. Wang, L. Lyu, S. Zhang, J. Feng, R. Pang, L. Jiang, D. Li and C. Li, Chin. J. Lumin., 2017, 38, 1429–1435 CrossRef; (b) G. Blasse and A. Bril, J. Chem. Phys., 1966, 45, 3327–3332 CrossRef.
- (a) M. A. Kesseler, Anal. Chim. Acta, 1998, 364, 125–129 CrossRef; (b) J. R. Lakowicz, H. Szmacinski and M. Karakelle, Anal. Chim. Acta, 1993, 272, 179–186 CrossRef; (c) H. Xu, H. C. Hu, C. S. Cao and B. Zhao, Inorg. Chem., 2015, 54, 4585–4587 CrossRef PubMed.
- (a) B. Liu, W. Wu, A. Dou and Y. Wang, Dalton Trans., 2015, 44, 4423–4427 RSC; (b) J. Hao and B. Yan, Chem. Commun., 2015, 51, 7737–7740 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns, TGA curves, Fluorescent spectra, UV/Vis spectra and X-ray crystallographic files for complex 1 in CIF format. CCDC 1821918. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra02809e |
| This journal is © The Royal Society of Chemistry 2018 |