
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApatinib inhibits macrophage-mediated epithelial–mesenchymal transition in lung cancer
Shuliang Liua,
Lingfei Sub,
Xuri Mua,
Yubo Shia,
Aifeng Zhangc and
Xingping Ge *b
*b
aDepartment of Thoracic Surgery, Yantaishan Hospital, Yantai, 264001, China
bDepartment of Radiotherapy, Yantaishan Hospital, No. 167 Jichang Road, Zhifu District, 264025, Yantai, Shandong, P. R. China. E-mail: Gexingping1968@qq.com; Tel: +86 13589807433
cDepartment of Outpatient, Yantaishan Hospital, Yantai, 264001, China
First published on 12th June 2018
Abstract
Chemotherapy is one of the main treatment approaches for lung cancer. However, few drugs can be used in the post-first-line treatment of lung cancer. Apatinib is a small molecule inhibitor of vascular endothelial growth factor receptor-2 (VEGFR-2) and is widely used in advanced gastric cancer. This study aimed to investigate the therapeutic effect of apatinib in the second-line treatment of lung cancer and explore its underlying mechanism from the aspect of macrophage-mediated epithelial–mesenchymal transition (EMT). The results showed that apatinib attenuated macrophage-induced EMT and migration of lung cancer cells but not normal lung cells, as demonstrated by the loss of epithelial properties and gain of mesenchymal characteristics. Moreover, apatinib selectively decreased hepatocyte growth factor (HGF) secretion in polarized macrophages. Furthermore, apatinib down-regulated the expression of the HGF-Met signaling pathway in polarized macrophage-stimulated lung cancer cells. Taken together, our study has identified a novel pathway through which apatinib exerts its anti-cancer functions, and provided a molecular basis for apatinib potential applications in the post-first line treatment of lung cancer.
Introduction
Lung cancer is the most frequently diagnosed malignant tumor and the leading cause of cancer death worldwide.1,2 Histologically, lung cancer can be divided into small-cell lung cancer and non-small-cell lung cancer (NSCLC), and the latter accounts for more than 70% of lung cancer.3,4 In NSCLC, tumor-associated macrophages located in lung carcinomas increase tumor recurrence after surgery and reduce patient survival.5,6 Thus, interference with macrophage polarization may shift macrophage function from a tumor facilitator to a tumor suppressor, suggesting a potential mechanism for lung cancer therapy.To date, EGFR tyrosine kinase inhibitors (EGFR-TKIs) and ALK inhibitors such as gefitinib and afatinib have been suggested as the first-line treatment for NSCLC patients with epidermal growth factor receptor (EGFR) mutation and anaplastic lymphoma kinase (ALK) rearrangements.7–9 In the NSCLC patients without genetic driver mutation, platinum-based chemotherapy is the best option for first-line treatment.
Tumor angiogenesis is an essential step for tumor proliferation and metastasis. Vascular endothelial growth factor/vascular endothelial growth factor receptor (VEGF/VEGFR) signaling pathway has been proved to play crucial role in tumor angiogenesis.10,11 Thus, VEGF/VEGFR was considered as a target in cancer therapy. In fact, multiple drugs which target VEGF/VEGFR have received the Food and Drug Administration approval and have shown encouraging efficacy in several solid tumors. For example, it was reported that bevacizumab or ramucirumab combined with chemotherapy significantly improved the progression-free survival (PFS) and overall survival (OS) in NSCLC.12,13 However, these anti-angiogenesis drugs were suggested adding to platinum-based doublet chemotherapy as first-line treatment for advanced NSCLC without driver mutation, and there was little evidence for their application in post-first-line monotherapy of advanced lung cancer.
Apatinib is an oral small molecule inhibitor of VEGFR-2, and displays efficiency and safety in breast and gastric cancer therapy.14–16 However, its application on other malignant tumors, such as lung carcinoma is just getting started. Recent studies have reported that high dose (500–825 mg d−1) and low dose (250–500 mg d−1) of apatinib showed an efficiency and safety in lung cancer patients.17–19 In the present study, we used apatinib as a post-first-line therapy to investigate the underlying mechanism in the aspect of macrophage-mediated EMT. We found that apatinib inhibits macrophage infiltration in vivo and suppresses macrophage-induced EMT in vivo and in vitro. Further studies suggested that apatinib inhibits macrophage-mediated EMT through HGF-Met signaling pathway.
Materials and methods
Reagents
Following reagents (suppliers) were used in this study: apatinib (Selleck, Texas, USA); hepatocyte growth factor (HGF) (PeproTech, Suzhou, China); antibodies to CD68, ZO-1, N-cadherin, vimentin, Slug and β-actin (Santa Cruz Biotechnology, USA); antibodies to fibronectin, E-cadherin, Snail, p-Met, Met (Abcam, Cambridge, MA). HRP-conjugated secondary antibodies (Vazyme, Nanjing, China). ELISA kit for HGF, EGF, TGF-β1 and IL-6 (R&D Systems, Minneapolis, MN).Cell culture
A549, H1975 and normal human lung cell line BEAS-2B (ATCC, Manassas, USA) were grown in DMEM complemented with 10% FBS (Life Technologies, Grand Island, USA), 100 IU ml−1 penicillin (Sigma, USA), and 100 μg ml−1 streptomycin (Sigma, USA). All cells were cultured at 37 °C in a 5% CO2 incubator. Human monocyte cell line THP1 was stimulated with phorbol myristate acetate (PMA, 0.1 μM, Thermo Scientific, USA) for 3 hours to generate polarized macrophages. Apatinib or DMSO was added to polarized macrophages for 24 hours, followed by a medium exchange and stimulate with LPS for 24 hours. The supernatant was collected and centrifugated for further studies.Immunohistochemistry
Paraffin-embedded tissue sections were cut to a thickness of 4 μm. After dewaxing, rehydration and blocking, the sections were incubated with CD68 and fibronectin antibody overnight at 4 °C, followed by incubation with fluorophore-conjugated secondary antibody for 1 h. Sections were visualized with a fluorescent microscope and quantified by ImageJ software as before.42Quantitative real-time polymerase chain reaction (qPCR)
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, Carlsbad, USA) according to the manufacturer's instructions. Equal amounts of RNA were reversely-transcribed to cDNA with M-MLV Transcriptase (Invitrogen). Then PCR was performed by Premix Ex Taq (Takara, Japan) in a Fast Real-time PCR 7300 System (Applied Biosystems). The PCR specific primers were: 5′-CGAATGGATGAAAGACCCATCC-3′ (forward) and 5′-GGAGCCACTGCCTTCATAGTC-3′ (reverse) for N-cadherin; 5′-TTAAACTCCTGGCCTCAAGCAATC-3′ (forward) and 5′-TATCTTGGGCAAAGCAACTG-3′ (reverse) for E-cadherin; 5′-TGAGTACCGGAGACAGGTGCAGTAGCAG-3′ (forward) and 5′-CTTCAACGGCAAAGTTC-3′ for vimentin; 5′-GCACTGTGATGCCCAGTCTA-3′ (forward) and 5′-CAGTGAGGGCAAGAGAAAGG-3′ (reverse) for slug; 5′-GTT CAATGTGGGACAAGAACATGG-3′ (forward) and 5′-GGATTTCGGCAGTAATTCTCATTCA-3′ (reverse) for HGF; 5′-CCTCACCATAGCTAATCTTGGGACA-3′ (forward) and 5′-CACAATCACTTCTGGAGACACTGGA-3′ (reverse) for MET; 5′-TCGACATGGAGCTGGTGAAA-3′ (forward) and 5′-GGGACTGGCGAGCCTTAGTT-3′ (reverse) for TGF-β1; 5′-GCTTCACTCTGGAAGATGCC-3′ (forward) and 5′-AAGGAGTGTGGTCACTGTGC-3 (reverse) for TGF-βR2; 5′-TGTGCAATGGCAATTCTGAT-3′ (forward) and 5′-GGTACTCCAGAAGACCAGAGGA-3′ (reverse) for IL-6; 5′- AAGGACCTCCAGCATCACTGTGTCA-3′ (forward) and 5′-CCTTCAGAGCCCGCAGCTTCCACGT-3′ (reverse) for IL-6R; 5′-CCTGGCACCCAGCACAAT-3′ (forward) and 5′-GCTGATCCACATCTGCTGGAA-3′ (reverse) for β-actin. The original Ct values were adjusted to GAPDH.Western blot
Total protein samples from cells were prepared by using RIPA buffer. Equal amounts of protein sample were separated by 10% SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, USA). Membranes were blocked with 5% non-fat milk, and then incubated with primary antibodies and corresponding secondary antibodies. The targeted bands in the membranes were visualized using enhanced chemical luminescence (ECL) and quantificated by ImageJ software.Wound healing assay
Confluent cell monolayer were cultured in a 24-well chamber and stroked with a plastic pipette tip. Cells were washed to remove detached and damaged cells. After treated with the supernatant of polarized macrophages (which have treated with apatinib or DMSO) for 24 h, the cell migrations were monitored microscopically and the migration distance was measured from 5 points per 1 wound area by the ImageJ software.Transwell invasion assay
Cell invasive capacity was detected using a 24-well plate with transwell chamber (0.4 μm pore size, Corning) inserts precoated with Matrigel. Briefly, cells (1 × 105) were seeded into the upper chamber in serum-free medium. Then, complete medium (containing 10% FBS) was added into the lower chamber. After 24 h of indicated treatment, cells in the upper surface of the membranes were removed with cotton swab. The cells that had migrated to the outer side of the membranes were stained with hematoxylin. The stained cells were photographed and counted in 10 random fields.Enzyme linked immunosorbent assay (ELISA)
The expression of HGF, epidermal growth factor (EGF), Transforming growth factor (TGF)-β and interleukin (IL)-6 were quantified by ELISA according to the manufacturer's instructions (Sigma, St. Louis, USA). Polarized macrophages induced by different concentrations of apatinib were precoated onto ELISA plates and served as the antigen. o-Dianisidine was used as substrate and the absorbance of the colored horseradish peroxidase (HRP) product was measured spectrophotometrically at 490 nm by an automated microplate reader (Thermo, Waltham, USA).Statistical analysis
Data were analyzed by SPSS 19.0 software and the results were expressed as mean ± standard deviation (SD). The statistical significance of the studies was analyzed using one way ANOVA. The difference was considered significant at P < 0.05.Results
Apatinib suppresses polarized macrophage-induced EMT in lung cancer cells
To investigate the effect of apatinib on macrophages, human monocyte cell line THP1 was treated with PMA to generate polarized macrophage. Then, human lung cancer cell line A549 and H1975 and normal human lung cell line BEAS-2B were cultured with the supernatant from polarized macrophages for 24 h. The expressions of EMT-related genes were detected by qPCR and western blot. As shown in Fig. 1A–D, the supernatant from polarized macrophages significantly increased the mRNA expression of N-cadherin, vimentin and Slug and decreased the level of E-cadherin in A549 and H1975 cells. However, apatinib treated macrophage supernatant effectively corrected these abnormalities. Consistent with the mRNA changes, the supernatant from polarized macrophages significantly decreased the protein expression of epithelial markers (ZO-1 and E-cadherin) and increased the protein levels of N-cadherin, vimentin, Slug and Snail in A549 and H1975 cells. These changes were reversed when polarized macrophages were pretreated with apatinib (Fig. 1E–K). Similarly, no obvious changes were observed in BEAS-2B cells. These results indicate that apatinib suppresses polarized macrophage-induced EMT only in lung cancer cells.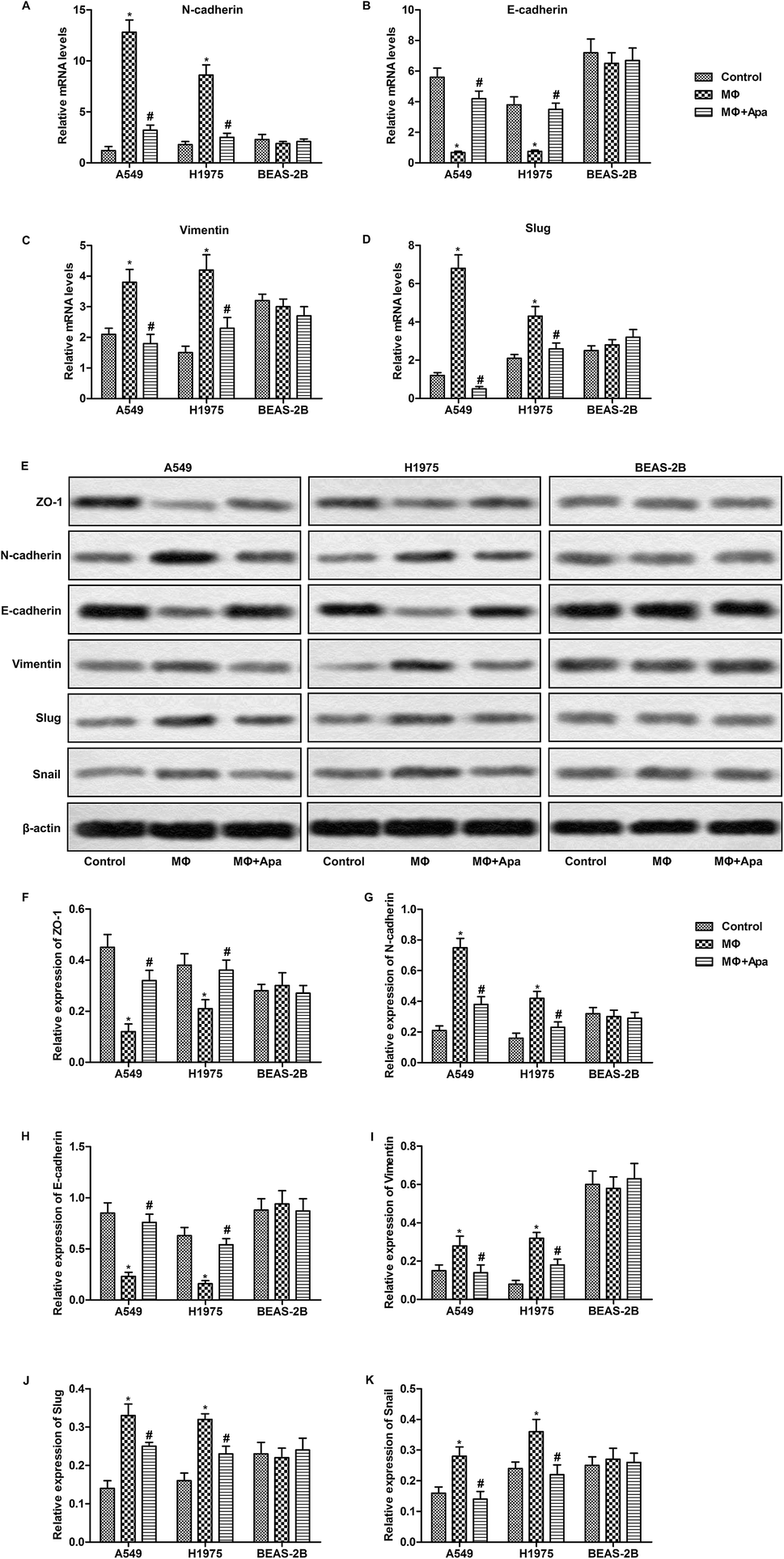 | ||
| Fig. 1 Apatinib suppresses polarized macrophage-induced EMT in lung cancer cells. A549, H1975 and BEAS-2B cells were treated with the supernatant of polarized macrophages (MΦ) which had been stimulated with LPS (1 ng ml−1) and/or apatinib (10 μM) for 24 h. (A–D) The mRNA levels of N-cadherin (A), E-cadherin (B), vimentin (C) and Slug (D) were assayed by qPCR. (E) The expressions of ZO-1, N-cadherin, E-cadherin, vimentin, Slug and Snail were detected by western blot. (F–K) Statistical analysis of Fig. 2E.*P < 0.05 versus control. #P < 0.05 versus MΦ. | ||
Apatinib inhibits polarized macrophage-induced migration and invasion of lung cancer cells
To determine whether the effect of apatinib on polarized macrophages leads to an inhibition of cellular migration, wound healing assay and transwell invasion assay were carried out. As shown in Fig. 2A and B, polarized macrophages promoted the migration of A549 and H1975 cells. However, apatinib pretreatment significantly decreased the migration distance in polarized macrophage-stimulated A549 and H1975 cells. In addition, transwell assay showed that polarized macrophages stimulation promoted the invasion of A549 and H1975 cells, which was blocked by apatinib treatment (Fig. 2C and D). In addition, the increased percentage of fibroblast-like cells in MΦ were strongly decreased under apatinib treatment (Fig. 2E and F). These data suggest that apatinib inhibits macrophage-induced migration in lung cancer cells.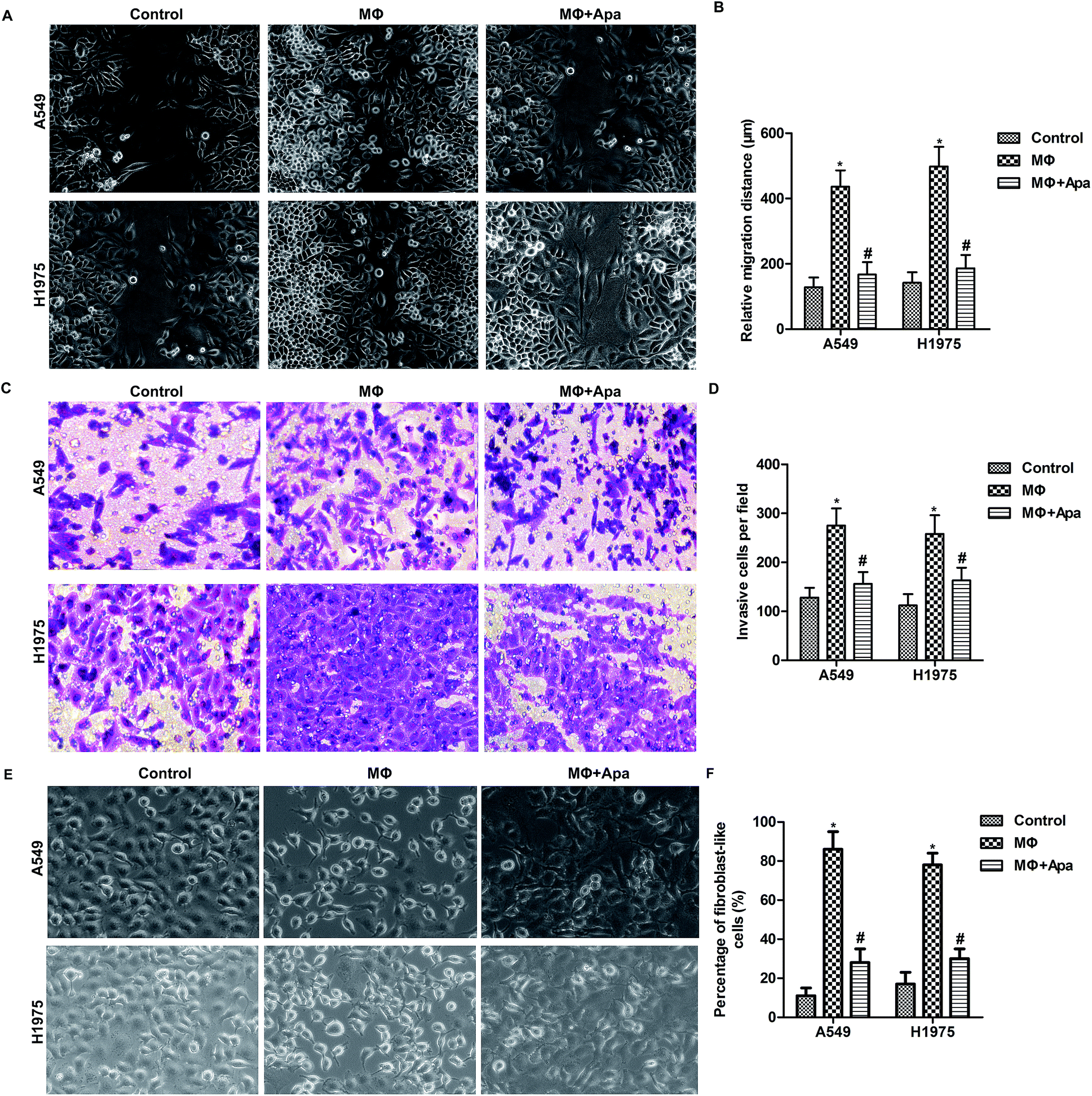 | ||
| Fig. 2 Apatinib inhibits polarized macrophage-induced migration and invasion of lung cancer cells. A549 and H1975 cells in 12-well plate or transwell chamber were treated with the supernatant of polarized macrophages (MΦ) which had been stimulated with LPS (1 ng ml−1) and/or apatinib (10 μM) for 24 h. (A) Wound healing assay. (B) Quantification of migration distance. (C) Transwell invasion assay. (D) Quantification of invaded cells. (E) The morphologic characteristics of EMT were detected under microscope. (F) Quantification of EMT cells. *P < 0.05 versus control. #P < 0.05 versus MΦ. | ||
Apatinib inhibits the expression of HGF in polarized macrophages
To investigate whether cytokine secretion of polarized macrophages contributes to the EMT progression, the expression of EMT-related cytokines in polarized macrophages was analyzed by qPCR and ELISA. The results showed that apatinib notably decreased the level of HGF in polarized macrophages. However, other EMT-related cytokines, such as EGF, TGF-β1 and IL-6 had no obvious changes in polarized macrophages with or without apatinib stimulation (Fig. 3). Thus, apatinib inhibits macrophage-mediated EMT probably through regulating HGF secretion.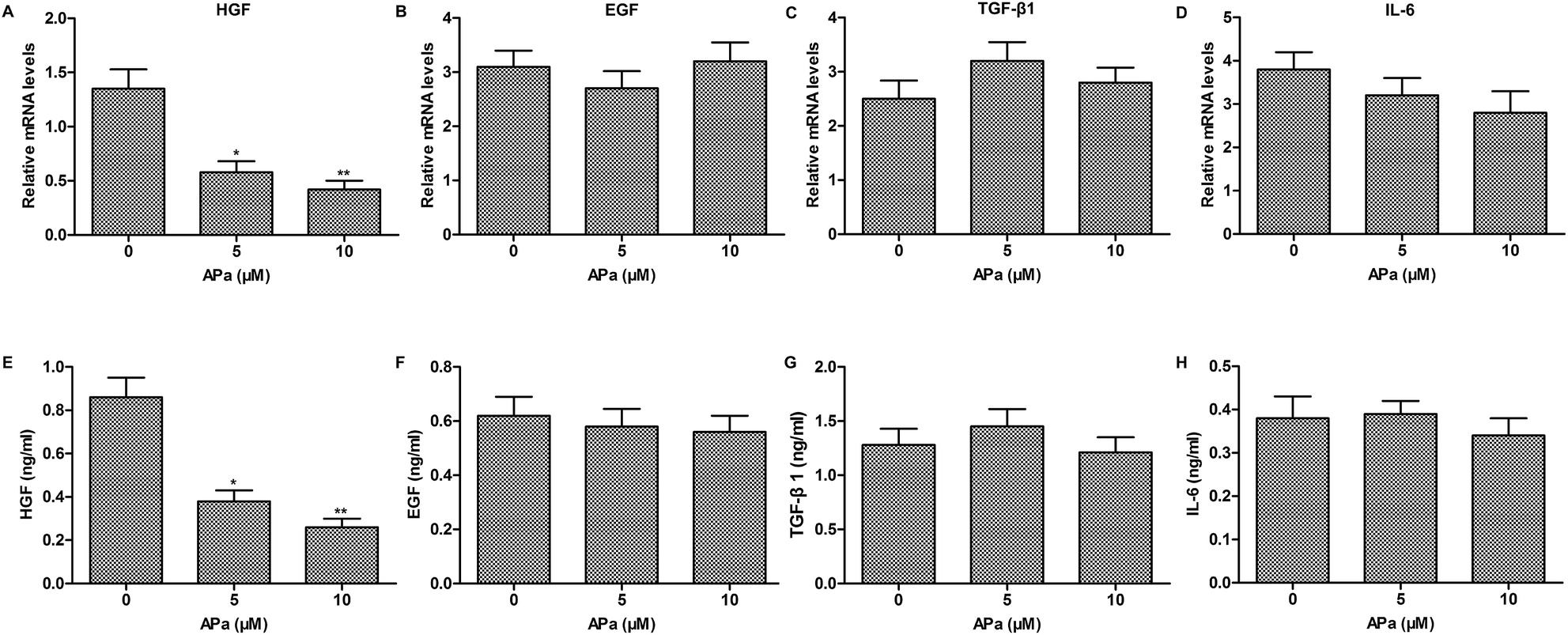 | ||
| Fig. 3 Cytokine profiles in apatinib-treated macrophages. Polarized macrophages were stimulated with LPS (1 ng ml−1) for 24 h and treated with a series dose of apatinib for 3 h. (A–D) The mRNA levels of HGF, EGF, TGF-β1 and IL-6 were assayed by qPCR. (E–H) The concentrations of HGF, EGF, TGF-β1 and IL-6 were detected by ELISA. *P < 0.05, **P < 0.01 versus control. | ||
Apatinib inhibits HGF-induced migration and invasion of lung cancer cells
Based on the above observations, the migration and invasion capacities of lung cancer cells were examined after HGF stimulation. As shown in Fig. 4, HGF significantly increased the migration distance and invasion capacity and the percentage of fibroblast-like cells of A549 and H1975 cells. As expected, apatinib treatment effectively corrected these abnormalities. These results further indicate that HGF is involved in the macrophage-mediated EMT.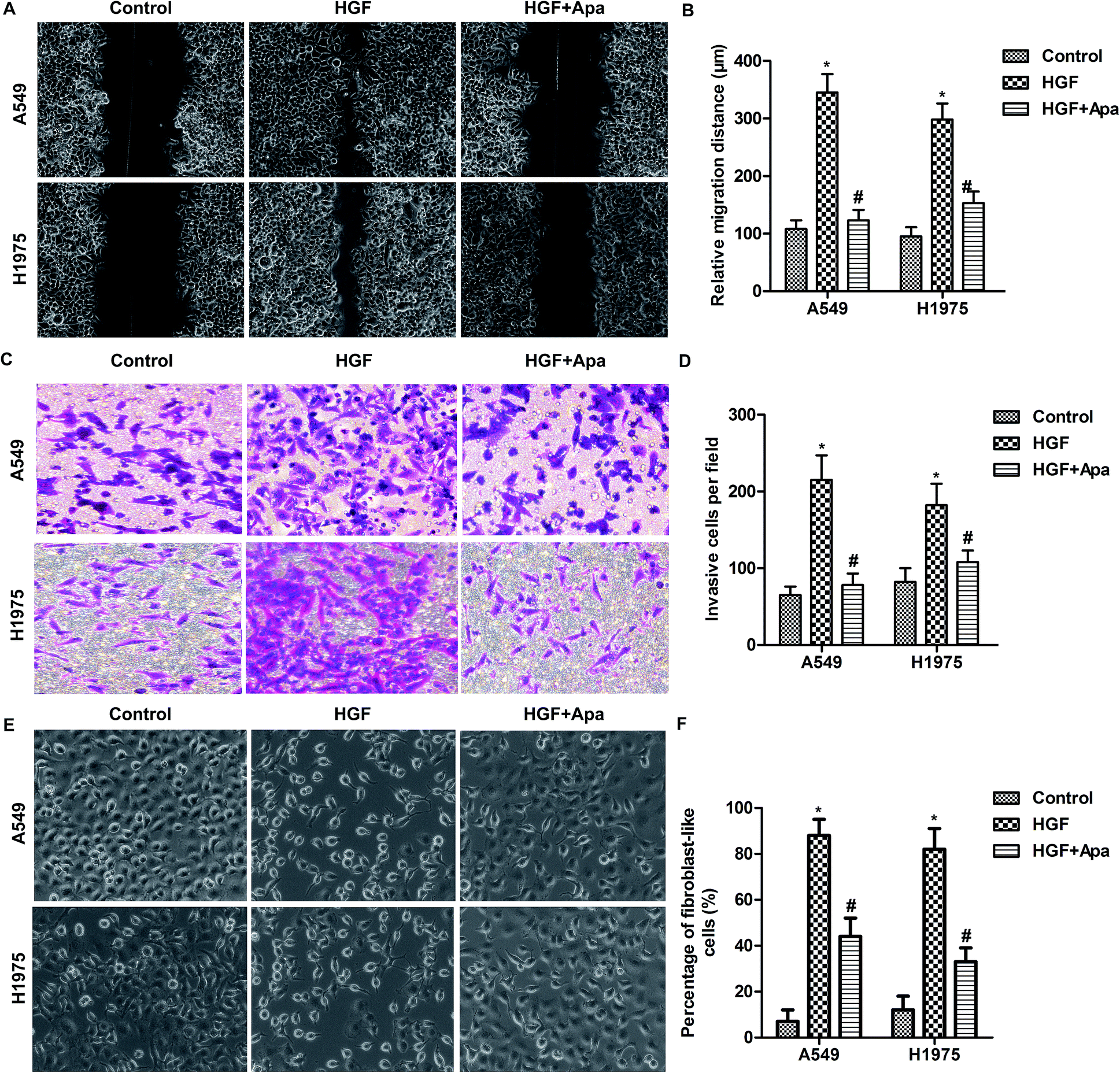 | ||
| Fig. 4 Apatinib inhibits HGF-induced migration and invasion of lung cancer cells. A549 and H1975 cells in 12-well plate or transwell chamber were treated with HGF (5 ng ml−1) and/or apatinib (10 μM) for 24 h. Cells without any treatment were considered as control group. (A) Wound healing assay. (B) Quantification of migration distance. (C) Transwell invasion assay. (D) Quantification of invaded cells. (E) The morphologic characteristics of EMT were detected under microscope. (F) Quantification of EMT cells. *P < 0.05 versus control. #P < 0.05 versus HGF treated alone. | ||
Apatinib down-regulates HGF-Met signaling in polarized macrophage supernatant-treated lung cancer cells
We next investigated the expression of the downstream receptors of HGF, EGF, TGF-β1 and IL-6 in polarized macrophage supernatant-treated lung cancer cells. As expected, apatinib dose-dependently inhibited the expression of Met in polarized macrophage-stimulated A549 and H1975 cells but not in BEAS-2B cells (Fig. 5A). However, the expression of EGFR, TGFβR and IL6R was not changed in lung cancer cells or normal lung cells (Fig. 5B–D). Since HGF-Met signaling mainly depends on Met phosphorylation, we next examined the expression of phosphorylated Met. The results showed that apatinib effectively decreased polarized macrophage-induced phosphorylation of Met in A549 and H1975 cells (Fig. 5E and F). Taken together, these results suggest that apatinib blocks polarized macrophage-activated HGF-Met signaling in lung cancer cells.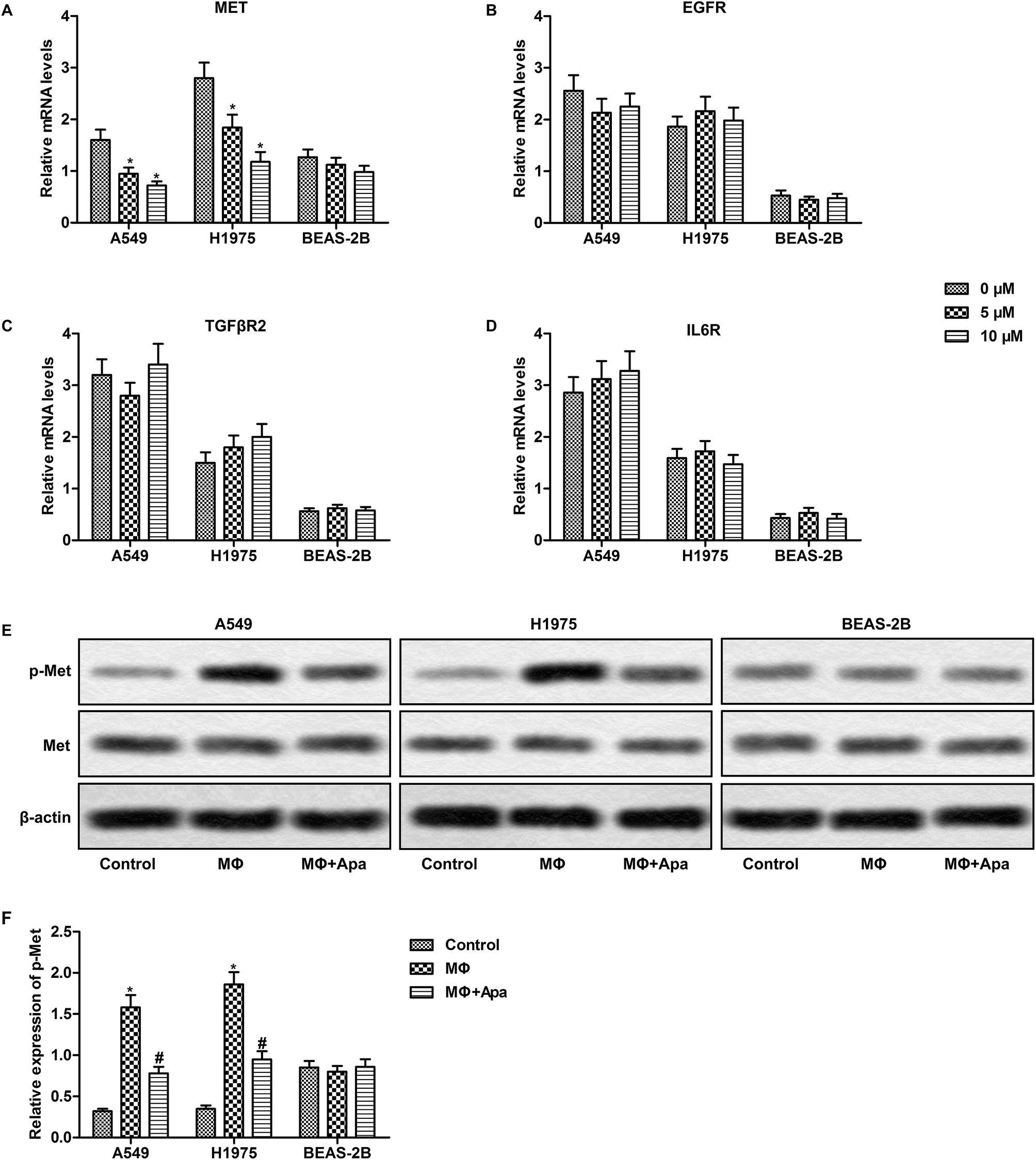 | ||
| Fig. 5 Apatinib down-regulates HGF-Met signaling in polarized macrophage-treated lung cancer cells. A549, H1975 and BEAS-2B cells were treated with the supernatant of polarized macrophages (MΦ) which had been stimulated with LPS (1 ng ml−1) and/or apatinib (0–10 μM) for 24 h. (A–D) The mRNA level of MET, EGFR, TGFβR2 and IL6R was detected by qPCR. (E) The phosphorylation of Met was detected by western blot. (F) Statistical analysis of Fig. 2E. *P < 0.05 versus control. #P < 0.05 versus MΦ. | ||
Discussion
To date, platinum-based chemotherapy is the preferred treatment of advanced lung cancer without genetic driver. However, the efficiency of first-line chemotherapy has reached a threshold and there are fewer options for post-first-line therapy. Recent studies have reported that combination of anti-angiogenic drugs and chemotherapy could significantly improve PFS and overall OS in advanced NSCLC.12,13 It was reported that apatinib inhibits the invasion and migration of lung cancer cells in vitro via RET/Src signaling pathway.20 Some clinical cases reports demonstrated that apatinib prolonged the PFS in NSCLC patients.16,17 In this study, we found that apatinib treatment significantly attenuated macrophage infiltration and EMT of lung tissue. EMT is a developmental cellular process by which polarized epithelial cells lose epithelial properties and cell–cell adhesions, and gain mesenchymal characteristics,21,22 which allows the cancer cell to become more invasive, metastatic and form a secondary metastasis.23 E-cadherin and ZO-1 are required for the formation of stable adherens junction. During EMT, epithelial cells lose E-cadherin and convert into spindle shaped mesenchymal cells by acquiring N-cadherin.24,25 It was reported that E/N-cadherin switch play a crucial role in cancer progression. Slug and Snail are key regulators of E-cadherin and it was demonstrated that all these factors promotes tumor growth, recurrence and metastasis.26,27 Vimentin and fibronectin are mesenchymal markers and are overexpressed in cancer cells.28–30 In the present study, we found that apatinib significantly reversed macrophage-induced increase of N-cadherin, vimentin, Slug and Snail and decrease of ZO-1 and E-cadherin. Furthermore, apatinib also attenuated EMT-mediated migration and invasion of lung cancer cells.Previous studies have reported that macrophage derived cytokines stimulate the progression of EMT.31 Thus, the expression of EMT-related cytokines was examined in polarized macrophages. Among these cytokines, TGF-β1 is considered to be the most common activator of EMT.32 It promotes EMT through various pathways, such as smad2/3 and Wnt/β-catenin.33,34 However, the expression of TGF-β1 is not changed in polarized macrophages after apatinib stimulation. TGF-β1 interacts with other cytokines to regulate the progression of EMT, such as IL-6 and EGF, and EGF also directly induces EMT.35,36 Similarly, the expression of EGF and IL-6 was not significantly altered by apatinib stimulation. Only the production of HGF was consistent with EMT progression. These data indicated that apatinib attenuated macrophage-induced EMT through inhibiting HGF expression.
HGF is a multifunctional growth factor originally isolated from rat platelets.37 HGF-Met activation is considered to promote tumor cell proliferation and survival and EMT.38 HGF and Met are also localized at sites of pathological angiogenesis and are a potent endothelial mitogens.39 Many studies have reported that inhibition of HGF-Met pathway could attenuate the progression of EGFR-TKI-resistant lung cancer.40,41 Overexpression of Met activates ERBB3/PI3K/AKT signaling in EGFR mutant lung cancers, which lead to EGFR-TKI-resistance. In the present study, the results showed that apatinib not only inhibited HGF secretion in polarized macrophages, but also suppressed Met activation in polarized macrophage-stimulated lung cancer cells, suggesting that apatinib attenuated macrophage-induced EMT through HGF-Met signaling pathway. This study reported that apatinib may act as a potential VEGFR inhibitor in suppressing cancer progression. We are now applying a commonly used inhibitor of MET and VEGFRs, Foretinib (GSK1363089), as a positive control of apatinib in lung cancer cells A549 and H1975, in order to convince the effect of apatinib on HGF expressions and EMT in lung cancer. In this study we first reported that apatinib inhibits macrophage-mediated EMT in lung cancers. Our study not only provided a new insight into apatinib's application in post-first-line therapy of lung cancer, but also elaborated a novel mechanism of apatinib's tumor-suppressive effect in treatment of lung cancer.
Conclusions
In this study we first reported that apatinib inhibits macrophage-mediated EMT in lung cancers. Our study not only provided a new insight into apatinib's application in post-first-line therapy of lung cancer, but also elaborated a novel mechanism of apatinib's tumor-suppressive effect in treatment of lung cancer.Conflicts of interest
There are no conflicts to declare.Abbreviations
| VEGFR | Vascular endothelial growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| HGF | Hepatocyte growth factor |
| NSCLC | Non-small-cell lung cancer |
| EGFR | Epidermal growth factor receptor |
| ALK | Anaplastic lymphoma kinase |
| PFS | Progression-free survival |
| OS | Overall survival |
References
- A. Jemal, R. Siegel, J. Xu and E. Ward, Cancer statistics, Ca-Cancer J. Clin., 2010, 60, 277–300 CrossRef PubMed.
- Y. Y. Soon, M. R. Stockler, L. M. Askie and M. J. Boyer, Duration of chemotherapy for advanced non-small-cell lung cancer: a systematic review and meta-analysis of randomized trials, J. Clin. Oncol., 2009, 27, 3277–3283 CrossRef PubMed.
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Global cancer statistics, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed.
- C. M. Tsai, J. T. Chen, C. H. Chiu, C. L. Lai, S. Y. Hsiao and K. T. Chang, Combined epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor and chemotherapy in non-small-cell lung cancer: chemo-refractoriness of cells harboring sensitizing-EGFR mutations in the presence of gefitinib, Lung Cancer, 2013, 82, 305–312 CrossRef PubMed.
- D. Che, S. Zhang, Z. Jing, L. Shang, S. Jin, F. Liu, J. Shen, Y. Li, J. Hu, Q. Meng and Y. Yu, Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE2/beta-catenin signalling pathway, Mol. Immunol., 2017, 90, 197–210 CrossRef PubMed.
- L. Pang, S. Han, Y. Jiao, S. Jiang, X. He and P. Li, Bu Fei Decoction attenuates the tumor associated macrophage stimulated proliferation, migration, invasion and immunosuppression of non-small cell lung cancer, partially via IL-10 and PD-L1 regulation, Internet J. Oncol., 2017, 51, 25–38 CrossRef PubMed.
- C. Gridelli, F. de Marinis, F. Cappuzzo, M. Di Maio, F. R. Hirsch, T. Mok, F. Morgillo, R. Rosell, D. R. Spigel, J. C. Yang and F. Ciardiello, Treatment of Advanced Non-Small-Cell Lung Cancer with Epidermal Growth Factor Receptor (EGFR) Mutation or ALK Gene Rearrangement: Results of an International Expert Panel Meeting of the Italian Association of Thoracic Oncology, Clin. Lung Cancer, 2014, 15, 173–181 CrossRef PubMed.
- L. Shi, J. Tang, L. Tong and Z. Liu, Risk of interstitial lung disease with gefitinib and erlotinib in advanced non-small cell lung cancer: a systematic review and meta-analysis of clinical trials, Lung Cancer, 2014, 83, 231–239 CrossRef PubMed.
- Y. Shi, Y. Sun, J. Yu, C. Ding, Z. Wang, C. Wang, D. Wang, C. Wang, Z. Wang, M. Wang, X. Zhi, Y. Lu, J. Feng, Y. Liu, X. Liu, W. Liu, G. Wu, X. Li, K. Li, E. Li, W. Li, G. Chen, Z. Chen, P. Yu, N. Wu, M. Wu, W. Xiao, L. Zhang, Y. Zhang, S. Zhang, S. Yang, X. Song, D. Lin, R. Luo, L. Shan, C. Zhou, Z. Zhou, Q. Zhao, C. Hu, Y. Hu, Q. Guo, J. Chang, C. Huang, X. Zeng, B. Han, X. Han, B. Jia, Y. Han and Y. Huang, China experts consensus on the diagnosis and treatment of advanced stage primary lung cancer (2016 version), Asia Pac. J. Clin. Oncol., 2017, 13, 87–103 CrossRef PubMed.
- P. K. Kopparapu, S. A. Boorjian, B. D. Robinson, M. Downes, L. J. Gudas, N. P. Mongan and J. L. Persson, Expression of VEGF and its receptors VEGFR1/VEGFR2 is associated with invasiveness of bladder cancer, Anticancer Res., 2013, 33, 2381–2390 Search PubMed.
- Q. Zhang, C. Yu, S. Peng, H. Xu, E. Wright, X. Zhang, X. Huo, E. Cheng, T. H. Pham, K. Asanuma, K. J. Hatanpaa, D. Rezai, D. H. Wang, V. Sarode, S. Melton, R. M. Genta, S. J. Spechler and R. F. Souza, Autocrine VEGF signaling promotes proliferation of neoplastic Barrett's epithelial cells through a PLC-dependent pathway, Gastroenterology, 2014, 146, 461–472.e466 CrossRef PubMed.
- J. Bennouna, L. Falchero, R. Schott, F. Bonnetain, M. Coudert, B. Ben Hadj Yahia and C. Chouaid, Bevacizumab in Combination with Platinum-Based Chemotherapy in Patients with Advanced Non-Squamous Non-Small Cell Lung Cancer with or without Brain Metastases: A French Cohort Study (EOLE), Oncology, 2018, 94(1), 55–64 CrossRef PubMed.
- E. B. Garon, T. E. Ciuleanu, O. Arrieta, K. Prabhash, K. N. Syrigos, T. Goksel, K. Park, V. Gorbunova, R. D. Kowalyszyn, J. Pikiel, G. Czyzewicz, S. V. Orlov, C. R. Lewanski, M. Thomas, P. Bidoli, S. Dakhil, S. Gans, J. H. Kim, A. Grigorescu, N. Karaseva, M. Reck, F. Cappuzzo, E. Alexandris, A. Sashegyi, S. Yurasov and M. Perol, Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial, Lancet, 2014, 384, 665–673 CrossRef.
- J. Li, S. Qin, J. Xu, W. Guo, J. Xiong, Y. Bai, G. Sun, Y. Yang, L. Wang, N. Xu, Y. Cheng, Z. Wang, L. Zheng, M. Tao, X. Zhu, D. Ji, X. Liu and H. Yu, Apatinib for chemotherapy-refractory advanced metastatic gastric cancer: results from a randomized, placebo-controlled, parallel-arm, phase II trial, J. Clin. Oncol., 2013, 31, 3219–3225 CrossRef PubMed.
- J. Li, S. Qin, J. Xu, J. Xiong, C. Wu, Y. Bai, W. Liu, J. Tong, Y. Liu, R. Xu, Z. Wang, Q. Wang, X. Ouyang, Y. Yang, Y. Ba, J. Liang, X. Lin, D. Luo, R. Zheng, X. Wang, G. Sun, L. Wang, L. Zheng, H. Guo, J. Wu, N. Xu, J. Yang, H. Zhang, Y. Cheng, N. Wang, L. Chen, Z. Fan, P. Sun and H. Yu, Randomized, Double-Blind, Placebo-Controlled Phase III Trial of Apatinib in Patients With Chemotherapy-Refractory Advanced or Metastatic Adenocarcinoma of the Stomach or Gastroesophageal Junction, J. Clin. Oncol., 2016, 34, 1448–1454 CrossRef PubMed.
- X. Hu, J. Zhang, B. Xu, Z. Jiang, J. Ragaz, Z. Tong, Q. Zhang, X. Wang, J. Feng, D. Pang, M. Fan, J. Li, B. Wang, Z. Wang, Q. Zhang, S. Sun and C. Liao, Multicenter phase II study of apatinib, a novel VEGFR inhibitor in heavily pretreated patients with metastatic triple-negative breast cancer, Int. J. Cancer, 2014, 135, 1961–1969 CrossRef PubMed.
- L. Ding, Q. J. Li, K. Y. You, Z. M. Jiang and H. R. Yao, The Use of Apatinib in Treating Nonsmall-Cell Lung Cancer: Case Report and Review of Literature, Medicine, 2016, 95, e3598 CrossRef PubMed.
- S. C. Fang, H. T. Zhang, Y. M. Zhang and W. P. Xie, Apatinib as post second-line therapy in EGFR wild-type and ALK-negative advanced lung adenocarcinoma, OncoTargets Ther., 2017, 10, 447–452 CrossRef PubMed.
- D. X. Zeng, C. G. Wang, W. Lei, J. A. Huang and J. H. Jiang, Efficiency of low dosage apatinib in post-first-line treatment of advanced lung adenocarcinoma, Oncotarget, 2017, 8, 66248–66253 Search PubMed.
- C. Lin, S. Wang, W. Xie, R. Zheng, Y. Gan and J. Chang, Apatinib inhibits cellular invasion and migration by fusion kinase KIF5B-RET via suppressing RET/Src signaling pathway, Oncotarget, 2016, 7, 59236–59244 Search PubMed.
- J. P. Thiery and J. P. Sleeman, Complex networks orchestrate epithelial–mesenchymal transitions, Nat. Rev. Mol. Cell Biol., 2006, 7, 131–142 CrossRef PubMed.
- J. P. Thiery, Epithelial–mesenchymal transitions in development and pathologies, Curr. Opin. Cell Biol., 2003, 15, 740–746 CrossRef PubMed.
- T. Brabletz, F. Hlubek, S. Spaderna, O. Schmalhofer, E. Hiendlmeyer, A. Jung and T. Kirchner, Invasion and metastasis in colorectal cancer: epithelial–mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin, Cells Tissues Organs, 2005, 179, 56–65 CrossRef PubMed.
- S. S. Nagaraja, V. Krishnamoorthy, R. Raviraj, A. Paramasivam and D. Nagarajan, Effect of Trichostatin A on radiation induced epithelial–mesenchymal transition in A549 cells, Biochem. Biophys. Res. Commun., 2017, 493, 1534–1541 CrossRef PubMed.
- X. Lin, B. W. Lin, X. L. Chen, B. L. Zhang, X. J. Xiao, J. S. Shi, J. D. Lin and X. Chen, PAI-1/PIAS3/Stat3/miR-34a forms a positive feedback loop to promote EMT-mediated metastasis through Stat3 signaling in non-small cell lung cancer, Biochem. Biophys. Res. Commun., 2017, 493, 1464–1470 CrossRef PubMed.
- A. Cano, M. A. Perez-Moreno, I. Rodrigo, A. Locascio, M. J. Blanco, M. G. del Barrio, F. Portillo and M. A. Nieto, The transcription factor Snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression, Nat. Cell Biol., 2000, 2, 76–83 CrossRef PubMed.
- V. Bolos, H. Peinado, M. A. Perez-Moreno, M. F. Fraga, M. Esteller and A. Cano, The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors,, J. Cell Sci., 2003, 116, 499–511 CrossRef PubMed.
- R. C. Dourado, L. P. A. Porto, A. Leitao, P. S. G. Cerqueira, J. N. Dos Santos, L. M. P. Ramalho and F. C. A. Xavier, Immunohistochemical Characterization of Cancer-associated Fibroblasts in Oral Squamous Cell Carcinoma, Appl. Immunohistochem. Mol. Morphol., 2017 DOI:10.1097/PAI.0000000000000486.
- Z. Niknami, A. Eslamifar, A. Emamirazavi, A. Ebrahimi and R. Shirkoohi, The association of vimentin and fibronectin gene expression with epithelial–mesenchymal transition and tumor malignancy in colorectal carcinoma, EXCLI J., 2017, 16, 1009–1017 Search PubMed.
- C. P. Tang, H. J. Zhou, J. Qin, Y. Luo and T. Zhang, MicroRNA-520c-3p negatively regulates EMT by targeting IL-8 to suppress the invasion and migration of breast cancer, Oncol. Rep., 2017, 38, 3144–3152 CrossRef PubMed.
- Y. R. Deng, W. B. Liu, Z. X. Lian, X. Li and X. Hou, Sorafenib inhibits macrophage-mediated epithelial–mesenchymal transition in hepatocellular carcinoma, Oncotarget, 2016, 7, 38292–38305 Search PubMed.
- H. A. Chapman, Epithelial–mesenchymal interactions in pulmonary fibrosis, Annu. Rev. Physiol., 2011, 73, 413–435 CrossRef PubMed.
- A. Nawshad, D. Lagamba, A. Polad and E. D. Hay, Transforming growth factor-beta signaling during epithelial–mesenchymal transformation: implications for embryogenesis and tumor metastasis, Cells Tissues Organs, 2005, 179, 11–23 CrossRef PubMed.
- A. Gal, T. Sjoblom, L. Fedorova, S. Imreh, H. Beug and A. Moustakas, Sustained TGF beta exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis, Oncogene, 2008, 27, 1218–1230 CrossRef PubMed.
- A. Abulaiti, Y. Shintani, S. Funaki, T. Nakagiri, M. Inoue, N. Sawabata, M. Minami and M. Okumura, Interaction between non-small-cell lung cancer cells and fibroblasts via enhancement of TGF-beta signaling by IL-6, Lung Cancer, 2013, 82, 204–213 CrossRef PubMed.
- S. Uttamsingh, X. Bao, K. T. Nguyen, M. Bhanot, J. Gong, J. L. Chan, F. Liu, T. T. Chu and L. H. Wang, Synergistic effect between EGF and TGF-beta1 in inducing oncogenic properties of intestinal epithelial cells, Oncogene, 2008, 27, 2626–2634 CrossRef PubMed.
- T. A. Martin and W. G. Jiang, Hepatocyte growth factor and its receptor signalling complex as targets in cancer therapy, Anti-Cancer Agents Med. Chem., 2010, 10, 2–6 CrossRef PubMed.
- H. J. Hu, X. L. Lin, M. H. Liu, X. J. Fan and W. W. Zou, Curcumin mediates reversion of HGF-induced epithelial–mesenchymal transition via inhibition of c-Met expression in DU145 cells, Oncol. Lett., 2016, 11, 1499–1505 CrossRef PubMed.
- S. Ding, T. Merkulova-Rainon, Z. C. Han and G. Tobelem, HGF receptor up-regulation contributes to the angiogenic phenotype of human endothelial cells and promotes angiogenesis in vitro, Blood, 2003, 101, 4816–4822 CrossRef PubMed.
- S. Takeuchi, W. Wang, Q. Li, T. Yamada, K. Kita, I. S. Donev, T. Nakamura, K. Matsumoto, E. Shimizu, Y. Nishioka, S. Sone, T. Nakagawa, T. Uenaka and S. Yano, Dual inhibition of Met kinase and angiogenesis to overcome HGF-induced EGFR-TKI resistance in EGFR mutant lung cancer, Am. J. Pathol., 2012, 181, 1034–1043 CrossRef PubMed.
- T. Nakagawa, S. Takeuchi, T. Yamada, S. Nanjo, D. Ishikawa, T. Sano, K. Kita, T. Nakamura, K. Matsumoto, K. Suda, T. Mitsudomi, Y. Sekido, T. Uenaka and S. Yano, Combined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancer, Mol. Cancer Ther., 2012, 11, 2149–2157 CrossRef PubMed.
- D. R. Mane, A. D. Kale and C. Belaldavar, Validation of immunoexpression of tenascin-C in oral precancerous and cancerous tissues using ImageJ analysis with novel immunohistochemistry profiler plugin: An immunohistochemical quantitative analysis, J. Oral Maxillofac. Pathol., 2017, 21, 211–217 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |