
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe3O4 nanoclusters highly dispersed on a porous graphene support as an additive for improving the hydrogen storage properties of LiBH4†
Guang Xu‡
 a,
Wei Zhang‡b,
Ying Zhang*a,
Xiaoxia Zhaoa,
Ping Wena and
Di Maa
a,
Wei Zhang‡b,
Ying Zhang*a,
Xiaoxia Zhaoa,
Ping Wena and
Di Maa
aDepartment of Materials Science and Engineering, College of Science, China University of Petroleum (Beijing), Changping District, Beijing 102249, PR China. E-mail: Y.Zhang@cup.edu.cn; Fax: +86-010-89732273; Tel: +86-010-89732273
bHebei Key Laboratory of Applied Chemistry, School of Environmental and Chemical Engineering, Yanshan University, Qinhuangdao 066004, PR China
First published on 25th May 2018
Abstract
Fe3O4 nanoclusters anchored on porous reduced graphene oxide (Fe3O4@rGO) have been synthesized by a one-step hydrothermal route, and then ball milled with LiBH4 to prepare a hydrogen storage composite with a low onset dehydrogenation temperature, and improved dehydrogenation kinetics and rehydrogenation reversibility. The LiBH4–20 wt% Fe3O4@rGO composite begins to release hydrogen at 74 °C, which is 250 °C lower than for ball-milled pure LiBH4. Moreover, the composite can release 3.36 wt% hydrogen at 400 °C within 1000 s, which is 2.52 times as high as that of pure LiBH4. Importantly, it can uptake 5.74 wt% hydrogen at 400 °C under 5 MPa H2, and its hydrogen absorption capacity still reaches 3.73 wt% after 5 de/rehydrogenation cycles. The activation energy (Ea) of the hydrogen desorption of the composite is decreased by 79.78 kJ mol−1 when 20 wt% Fe3O4@rGO is introduced into LiBH4 as a destabilizer and catalyst precursor, showing enhanced thermodynamic properties. It could be claimed that not only the destabilization of Fe3O4, but also the active Li3BO3 species formed in situ, as well as the wrapping effect of the graphene, synergistically improve the hydrogen storage properties of LiBH4. This work provides insight into developing non-noble metals supported on functional graphene as additives to improve the hydrogen storage properties of LiBH4.
1. Introduction
Hydrogen is considered to be a promising energy carrier capable of solving global energy and environmental crises on account of its clean combustion and high energy density.1 However, the major barrier for realizing its large-scale utilization is the shortage of safe hydrogen storage and transportation techniques.2 Extensive efforts have been centered on developing safe and efficient solid hydrogen storage materials with high hydrogen density, low dehydrogenation temperature, and favorable reversibility.3 Lithium borohydride (LiBH4) has been regarded as one of the materials with the most potential in solid state hydrogen storage systems due to its high gravimetric hydrogen capacity (18.4 wt%).4 However, it is too thermodynamically stable and kinetically slow at releasing hydrogen under moderate conditions, and high temperature and pressure are required for its rehydrogenation.5 To overcome the poor thermodynamic and slow kinetic properties of LiBH4, several strategies have been adopted to reduce its dehydrogenation temperature and improve its sorption rate, as well as its reversibility under benign conditions.6One conventional strategy is doping with appropriate additives or catalysts, such as transition metals,7,8 transition metal oxides9, or halides,10,11 which can effectively improve the hydrogen storage properties of LiBH4. Among them, iron-based compounds have been found to be a class of additives with high catalytic activity. Yu et al.12 investigated the effects of various oxides on the dehydrogenation behavior of LiBH4, wherein Fe2O3 showed the best destabilization effect among the studied oxides (TiO2, Nb2O5, Fe2O3, V2O5, and SiO2). The LiBH4–2Fe2O3 mixture started to release hydrogen below 100 °C, and 6 wt% hydrogen could be released after heating to 200 °C. Zhang et al.13 found that the dehydrogenation temperature of LiBH4 was significantly reduced by doping with small amounts of FeCl2, and the complete hydrogen desorption of LiBH4 could be achieved due to the formation of metal boride. Recently, a superior destabilization effect of LiBH4 was realised through the addition of nano-sized nickel ferrite NiFe2O4, and the in situ-formed Fe3O4, NiB, and Fe3B from the reaction between LiBH4 and NiFe2O4 acted as the actual destabilization agents in accelerating the dehydrogenation properties of LiBH4.14 More recently, boron-based compounds like H3BO3,15 HBO2, and B2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 have been reported to effectively promote the dehydrogenation of LiBH4 due to the interactions between protonic and hydridic hydrogen. Cai et al.17 found that nano-sized metal borides MBx (M = Mg, Ti, Fe, and Si) played an important catalytic role in enhancing the hydrogen storage performance of LiBH4, and the catalytic effects of MBx were influenced largely by the Pauling electronegativity of M in MBx and the interfacial contact character between LiBH4 and MBx.
16 have been reported to effectively promote the dehydrogenation of LiBH4 due to the interactions between protonic and hydridic hydrogen. Cai et al.17 found that nano-sized metal borides MBx (M = Mg, Ti, Fe, and Si) played an important catalytic role in enhancing the hydrogen storage performance of LiBH4, and the catalytic effects of MBx were influenced largely by the Pauling electronegativity of M in MBx and the interfacial contact character between LiBH4 and MBx.
Of particular interest is the fact that porous and nanostructured carbon materials as efficient catalysts or nanoscale frameworks can effectively lower the hydrogen evolution temperature and ameliorate the rehydrogenation conditions of LiBH4.18 In the case where LiBH4 was ball milled with various carbons (graphite, single-walled carbon nanotubes, and activated carbon), all the carbon additives could improve the H-exchange kinetics and H-capacity of LiBH4 due to heterogeneous nucleation and micro-confinement effects.19 Besides this, other carbon hosts like activated carbon nanofibers,20 fluorographite,21 and nitrogen-doped hierarchically porous carbon22 have also been used as substrates to destabilize or catalyze LiBH4. Nevertheless, relying solely on this method still makes it difficult to achieve rapid reversible hydrogen absorption and desorption processes under mild conditions. Furthermore, nearly (or even more than) a 50 wt% capacity loss in the first dehydrogenation and rehydrogenation cycle was repeatedly observed in the carbon-added systems due to the addition of plenty of carbon materials to disperse or confine LiBH4.23,24
Graphene has attracted great interest and has been applied in various areas, such as energy storage, sensors, and catalysis, on account of its unique characteristics.25 The addition of functional graphene to LiBH4 could result in a high hydrogen storage capacity, rapid kinetics, and excellent reversibility. Xu et al.26,27 reported that precious metal (Pt and Pd) nanoparticles anchored onto graphene sheets could greatly improve the hydrogen desorption behavior and uptake reversibility of LiBH4. The artful combination of catalysis and nanoconfinement has become a promising way to improve the dehydrogenation and rehydrogenation performance of LiBH4. More recently, the hydrogen storage properties of LiBH4 have been shown to be improved tremendously by ball milling with graphene-supported highly-dispersed nickel nanoparticles at a low addition amount.28 Zhang et al.29 reported that a remarkable improvement in the hydrogen sorption, thermodynamic, and kinetic performances of LiBH4 could be realized by modification with three-dimensional porous fluorinated graphene.
Based on the previous studies, it is anticipated that Fe-based compounds can weaken the B–H bond and thus improve the hydrogen storage properties of LiBH4. Fe3O4 has attracted considerable attention due to its low cost, eco-friendliness, and natural abundance, which are widely applicable in energy storage, environmental governance, and photocatalytic fields.30,31 In this work, Fe3O4 nanoclusters assembled on porous reduced graphene oxide sheets (Fe3O4@rGO) have been prepared successfully by a facile one-step hydrothermal route, and then were characterized by scanning electron microscopy, transmission electron microscopy, X-ray diffraction, X-ray photoelectron spectroscopy, etc. Subsequently, the composite was employed as a destabilizer and catalytic additive for improving the dehydrogenation and rehydrogenation behavior of LiBH4. The LiBH4–20 wt% Fe3O4@rGO composite prepared by high-energy ball milling showed an enhanced hydrogen desorption performance and uptake reversibility compared with ball-milled pure LiBH4. The catalytic effects of Fe3O4@rGO on the hydrogen storage properties of LiBH4 were investigated and discussed in detail.
2. Experimental section
2.1 Materials
Commercially available LiBH4 (95%, Aladdin), FeCl3·6H2O (AR, Aladdin), ascorbic acid (AR, Sinopharm Chemical Reagent Beijing Co., Ltd), N2H4·H2O (99%, Aladdin), and Fe3O4 (AR, Aladdin) were used as received, without further purification.2.2 Synthesis of Fe3O4 nanoclusters supported on porous reduced graphene oxide sheets (Fe3O4@rGO)
The graphene oxide (GO) used in this work was prepared according to Hummer’s method.32 Fe3O4@rGO was synthesized on the basis of the reported procedure by Hu et al.33 with a slight modification. Typically, 0.12 g GO was dispersed in 60 mL distilled water under ultrasound at room temperature, followed by slowly adding 0.90 g of FeCl3·6H2O and 1.76 g of ascorbic acid. Then, 10 mL N2H4·H2O was quickly added into the solution under vigorous stirring. Subsequently, the mixture was transferred to a 100 mL Teflon-lined autoclave and heated at 180 °C for 8 h. After filtration, washing, and freeze drying, the final product was designated as Fe3O4@rGO. For comparison, the reduced graphene oxide (rGO) was obtained by a simple hydrogen thermal reduction method. In brief, the GO was heated from room temperature to 500 °C under 1 MPa hydrogen with a heating rate of 5 °C min−1, and held for 5 h in a home-made temperature programmed apparatus.2.3 Preparation of the hydrogen storage composites
LiBH4 was ball milled with the as-prepared Fe3O4@rGO nanohybrid with a mass ratio of 4:1 at 500 rpm for 10 h under an Ar atmosphere using a planetary Fritsch-P6 mill. The weight ratio of the ball to powder mixture was about 40:1. The ball-milling procedure was carried out by alternating 15 min of milling and 15 min of resting to avoid the temperature of the sample container rising. For comparison, pure LiBH4 or the LiBH4 composites with the addition of Fe3O4 and rGO were prepared following the same procedure. It should be pointed out that before the ball milling of Fe3O4@rGO, rGO, and Fe3O4 with LiBH4, they were all dried at 120 °C for 12 h in a vacuum oven to remove any possible absorbed water on their surfaces, and were then stored in an Ar-filled glove box. The samples are hereafter labeled as LiBH4, LiBH4–Fe3O4, LiBH4–rGO, and LiBH4–Fe3O4@rGO. All the sample handling was performed in an Ar-filled glove box. The amounts of oxygen and water vapour inside the glove-box were kept below 1 ppm.
2.4 Characterization
The crystal structure and morphology of the samples were characterized by scanning electron microscopy (SEM, Hitachi SU8010), transmission electron microscopy (TEM, Hitachi HT7700), X-ray diffraction (XRD, D/Max-2500/PC, Cu Kα radiation), and X-ray photoelectron spectrometry (XPS, Thermo Scientific K-Alpha+, Al Kα radiation). For the XRD studies, all the samples were smeared on a glass slide in the Ar-filled glove box, and then were covered by a layer of plastic film to avoid moisture and oxygen contact during measurement. This resulted in a scattering peak at 2θ = 22° in the XRD pattern attributed to the plastic film coverage. The specific surface area and porosity of the Fe3O4@rGO nanohybrid were measured by nitrogen adsorption–desorption at 77 K using a Micromeritics TriStar II 3020 Analyzer. Fourier transform infrared (FTIR) spectra were obtained by a Nicolet 6700 FTIR spectrometer equipped with a MCT detector to record the features of the chemical bonding states of the samples. For the FTIR studies, the samples were milled with dry KBr with a mass ratio of 1:100 and pressed into one thin slice in the Ar-filled glove box, and then they were placed into a lab-built Ar-filled container to protect the samples from air and moisture during the transfer process. The thermodynamic analysis of the samples was carried out using a simultaneous thermal analyzer (DTG-60A) in air from room temperature to 900 °C, at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) measurements for the dehydrogenation of the samples were carried out using a Mettler Toledo TGA/DSC1 system at heating rates of 5, 10, 15, and 20 °C min−1 from 50 to 500 °C under an Ar flow (50 mL min−1).
The hydrogen desorption and absorption properties were investigated using a Sievert-type apparatus manufactured by the General Research Institute for Nonferrous Metals. Temperature-programmed-desorption (TPD) experiments were carried out in a temperature range from room temperature to 620 °C, at a heating rate of 5 °C min−1. The isothermal dehydrogenation and rehydrogenation measurements were performed at the desired temperature under initial hydrogen pressures of 0.01 MPa and 5 MPa, respectively. For the rehydrogenation measurement of the samples, the samples were first evacuated continuously until they did not release any more hydrogen, in order to make sure that they had completely released their hydrogen. Then, the isothermal rehydrogenation was performed at the desired temperature with an initial hydrogen pressure of 5 MPa for 1 h. In this work, the hydrogen contents of all the samples are presented based on the total weight of the composites, in order to make a comparison with the as-milled LiBH4.
3. Results and discussion
3.1 Synthesis and characterization of as-prepared Fe3O4@rGO
The schematic synthesis procedure of the Fe3O4@rGO nanohybrid is illustrated in Scheme 1. Firstly, GO was sonicated in water to obtain nanosheets of a few layers, and the liquid was brown-yellow. Then, iron(III) chloride hexahydrate and ascorbic acid were added to the liquid under continuous stirring, so that the Fe3+ ions were thoroughly dispersed onto the GO surface by electrostatic interaction. Later, hydrazine hydrate was added dropwise into the liquid under vigorous stirring. After the liquid became dark, it was transferred into a Teflon-lined container and reacted at 180 °C for 8 h. Lastly, the black product was isolated, washed, and freeze-dried.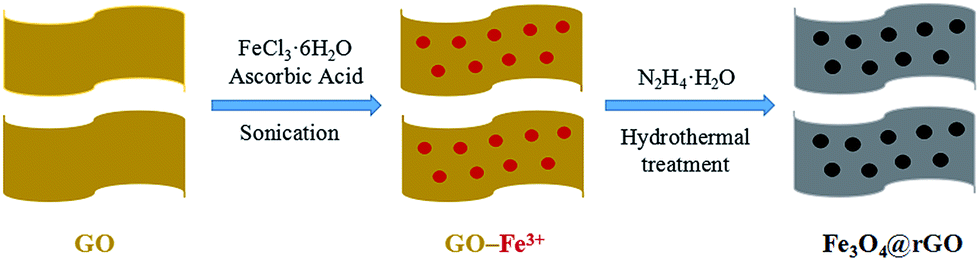 | ||
| Scheme 1 Schematic illustration of the synthesis of the Fe3O4@rGO nanohybrid. | ||
The microstructure of the as-prepared Fe3O4@rGO nanohybrid was studied using SEM and TEM. From the SEM image shown in Fig. 1a, it is clearly observed that tiny Fe3O4 nanoparticles are evenly distributed onto the surface of the large, disordered, and over-lapped graphene sheets. Fig. S1a† shows the typical morphology of GO, revealing the wrinkled sheet-like structure, and large, smooth surface. After the hydrogen thermal treatment, rGO with abundant wrinkles and defects is obtained (Fig. S1b†). Fig. 1b shows the TEM image of the Fe3O4@rGO nanohybrid, in which it can been found that the snowflake-like Fe3O4 nanoclusters consist of plentiful small particles of 5–6 nm in size, which are anchored uniformly onto the reduced graphene surface. The resultant Fe3O4@rGO nanohybrid is mainly composed of Fe, O, and C elements, according to the EDS analysis (Fig. S2†). N2 adsorption–desorption isotherms and the corresponding pore size distribution curve of the nanohybrid are exhibited in Fig. 1c and d. Its specific surface area is calculated to be 119.3 m2 g−1 based on the BET method. The pore volume is 0.374 cm3 g−1, with major pores located at about 3.7 nm and 14.7 nm in diameter, indicating the mesoporous characteristics of the as-prepared Fe3O4@rGO nanohybrid.
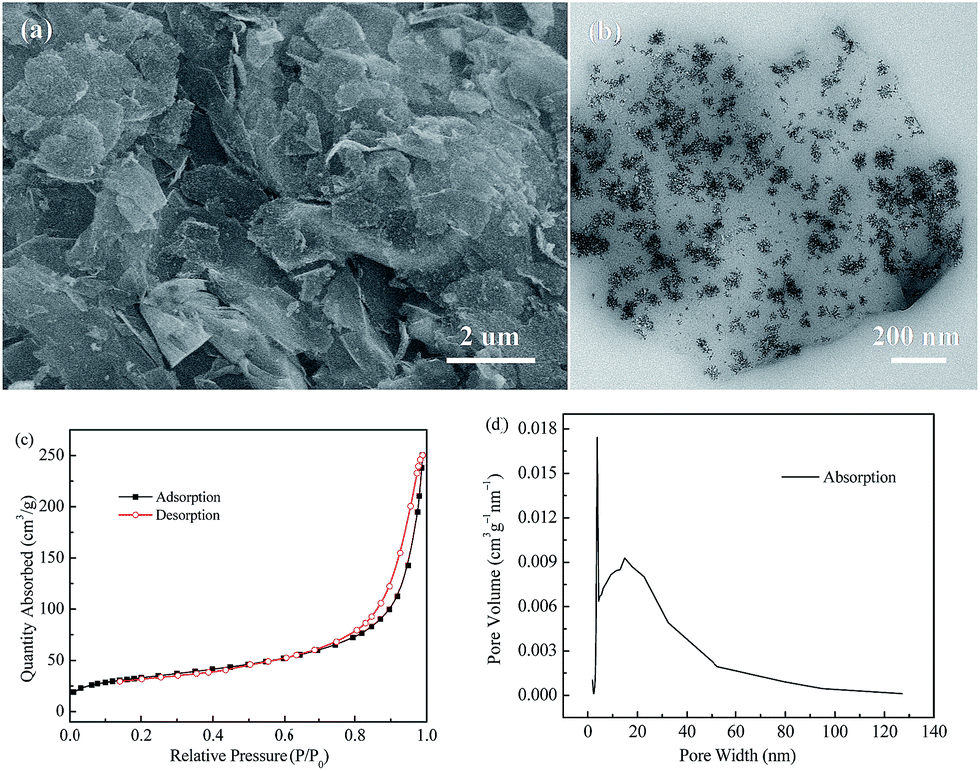 | ||
| Fig. 1 (a) SEM image, (b) TEM image, (c) nitrogen adsorption–desorption isotherms, and (d) BJH pore-size distribution pattern of the as-prepared Fe3O4@rGO nanohybrid. | ||
Fig. S3† shows the XRD patterns of the GO and rGO. For the GO, a well-defined peak is located at 2θ = 10.8° with 0.87 nm d-spacing, indicating the presence of abundant oxygen-containing functional groups on the sides of the GO nanosheets.34 However, the rGO has only a very low intensity peak around 25.8°, which indicates a distorted graphite structure and hence suggests the formation of rGO sheets.35 The XRD pattern of the as-prepared Fe3O4@rGO nanohybrid is displayed in Fig. 2a. Some diffraction peaks occur at 30.1°, 35.6°, 43.1°, 57.0°, and 62.7°, which are in good agreement with the characteristic peaks of pure cubic Fe3O4 (JCPDF 65-3107). To further verify the surface chemical composition of Fe3O4@rGO, XPS analysis was carried out. Two typical characteristic peaks situated at the binding energies of 709.81 and 723.46 eV are attributed to the Fe 2p3/2 and Fe 2p1/2 of Fe3O4 (Fig. 2b).36 The peak at 530.27 e V in the O 1s spectrum (Fig. 2c) can be assigned to the Fe–O bonds of Fe3O4@rGO.37 TGA was used to quantify the loading amount of Fe3O4 in the Fe3O4@rGO nanohybrid, and was conducted from room temperature to 900 °C in air, at a heating rate of 10 °C min−1. The TGA curves of Fe3O4@rGO and pure Fe3O4 are shown in Fig. 2d. There are two stages of weight loss for Fe3O4@rGO. The weight loss below 150 °C is ascribed to the evaporation of absorbed moisture. The abrupt weight loss from 192 °C to 425 °C is due to the burning of graphene in air. Based on the weight loss, 51.4 wt% graphene and 48.6 wt% Fe3O4 are estimated for the Fe3O4@rGO nanohybrid.
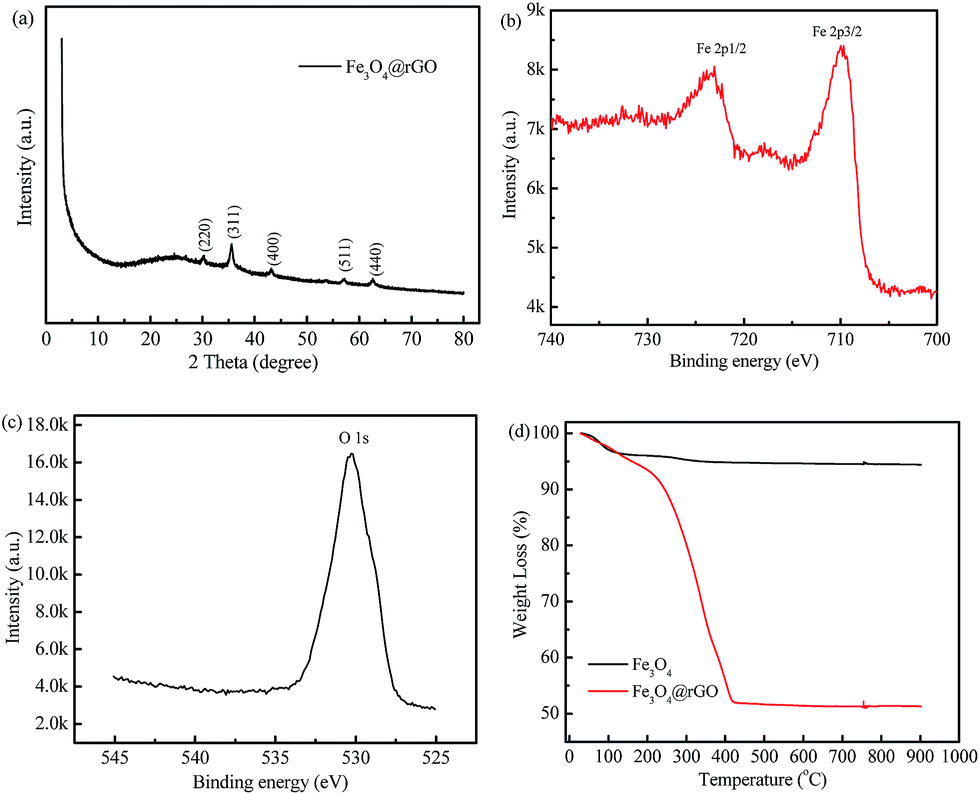 | ||
| Fig. 2 (a) XRD pattern, (b) Fe 2p and (c) O 1s XPS spectra of the as-prepared Fe3O4@rGO nanohybrid, (d) TGA curves of Fe3O4@rGO and Fe3O4 in air ranging from room temperature to 900 °C. | ||
3.2 Hydrogen sorption, thermodynamic, and kinetic properties of the composites
It is well-known that the hydrogen storage properties of LiBH4 can be altered by varying the amount of catalyst added. The preliminary experiments of LiBH4 with different rGO addition amounts were performed and the results are shown in Fig. S4 and S5.† It can be seen that all the LiBH4–x wt% rGO composites (x = 11, 20, and 33) exhibit improved dehydrogenation performances with respect to the ball-milled pure LiBH4. The three composites have almost the same initial dehydrogenation temperature, being able to release hydrogen at 220.5 °C, but their final dehydrogenation capacity is different. With an increase in the addition amount, the final dehydrogenation capacity decreases gradually. Furthermore, the isothermal hydrogen desorption profiles at 400 °C of the as-milled LiBH4 with and without the rGO composites are presented in Fig. S5.† The LiBH4–20 wt% rGO composite exhibits the quickest hydrogen desorption rate among the LiBH4-x wt% rGO composites, and it can release 3.46 wt% hydrogen after 3600 s, which is twice as large as the dehydrogenated capacity of ball-milled pure LiBH4 under identical conditions. Taking the dehydrogenation temperature and kinetics into account, the addition of 20 wt% carbon materials is regarded as the best choice and LiBH4–20 wt% y composites (y = Fe3O4, rGO, and Fe3O4@rGO) are prepared and further investigated in the later work.Fig. 3 illustrates the TPD performances of ball-milled pure LiBH4, LiBH4–20 wt% Fe3O4, LiBH4–20 wt% rGO, and LiBH4–20 wt% Fe3O4@rGO composites from room temperature to 620 °C. The pure LiBH4 starts to slowly release hydrogen at 324 °C. However, the LiBH4–Fe3O4 and LiBH4–rGO samples can release hydrogen at 78 °C and 220.5 °C, which are 246 °C and 103.5 °C lower than that of pure LiBH4, respectively. The initial dehydrogenation temperature of the LiBH4–20 wt% Fe3O4@rGO sample is as low as 74 °C, and a total dehydrogenation capacity of 8.88 wt% is obtained. It is also found that the LiBH4–20 wt% Fe3O4 and LiBH4–20 wt% Fe3O4@rGO samples showed quicker dehydrogenation rates than the pure LiBH4 and LiBH4–20 wt% rGO samples below 70 min, indicating that the destabilization effect of Fe3O4 is predominant. However, the LiBH4–20 wt% Fe3O4@rGO and LiBH4–20 wt% rGO samples showed quicker dehydrogenation rates than LiBH4–20 wt% Fe3O4 and pure LiBH4 after 70 min. The improvement of the dehydrogenation rate is attributed to the wrapping effect of the rGO and the catalytic effect of the in situ-formed Li3BO3. However, the formation of the stable species of Li5FeO4 in the LiBH4–20 wt% Fe3O4@rGO composite may affect the dehydrogenation of LiBH4, leading to the slightly reduced dehydrogenation performance when comparing it with the LiBH4–20 wt% rGO composite. The significantly decreased initial dehydrogenation temperature and improved dehydrogenation rate of LiBH4 indicate that the addition of Fe3O4@rGO by ball milling can clearly improve the desorption properties of LiBH4.
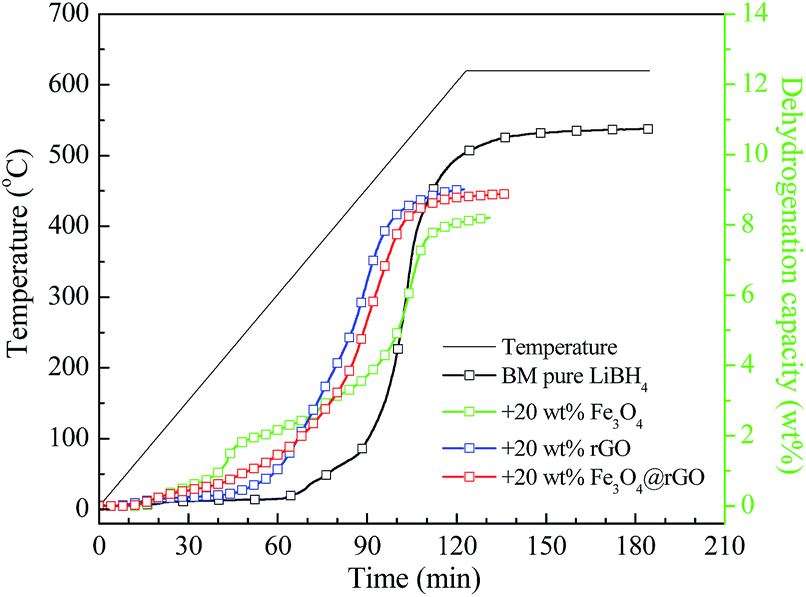 | ||
| Fig. 3 TPD curves of ball-milled pure LiBH4, LiBH4–20 wt% Fe3O4, LiBH4–20 wt% rGO, and LiBH4–20 wt% Fe3O4@rGO composites. | ||
To further study the desorption kinetics of the LiBH4–20 wt% y composites (y = Fe3O4, rGO, and Fe3O4@rGO), DSC tests at different heating rates were performed to evaluate the activation energy (Ea) of these composites using Kissinger’s method:38
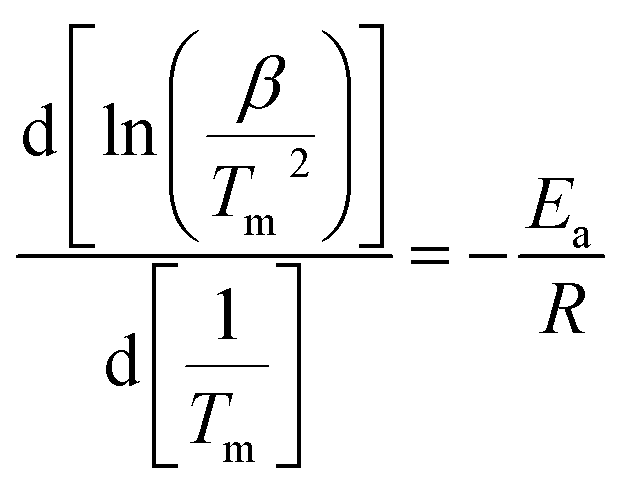
in which β is the heating rate, Tm is the peak temperature, and R is the ideal gas constant. In this work, Tm is calculated using the DSC results obtained under the diverse heating rates of 5, 10, 15, and 20 °C min−1. The DSC curves of the LiBH4–20 wt% Fe3O4@rGO composite are shown in Fig. 4a. Fig. S6† presents the DSC curves of the LiBH4–20 wt% Fe3O4 and LiBH4–20 wt% rGO composites. The dependence of ln(β/Tm) vs. 1/Tm of the composites is shown in Fig. 4b. The value of Ea is calculated to be 102.02 kJ mol−1 for the LiBH4–Fe3O4@rGO composite, which is much smaller than that of ball-milled pure LiBH4 (181.80 kJ mol−1).39 This suggests that the Ea is significantly affected when using Fe3O4@rGO as a destabilizer and catalytic additive, which effectively decreases the energy barrier of LiBH4 in the dehydrogenation process and thus causes the rapid dehydrogenation behavior of the composite.
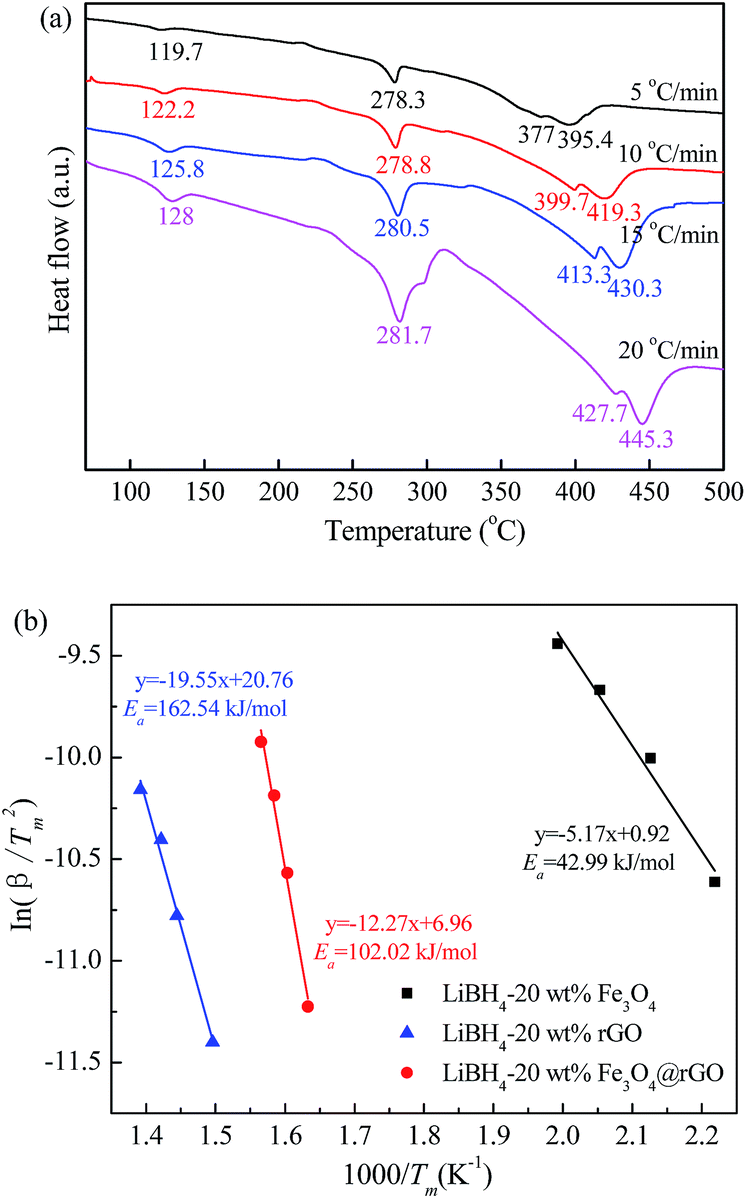 | ||
| Fig. 4 (a) DSC curves for the LiBH4–20 wt% Fe3O4@rGO composite at various heating rates and (b) Kissinger’s plots of Ea for the LiBH4–20 wt% y composites (y = Fe3O4, rGO, and Fe3O4@rGO). | ||
Isothermal dehydrogenation measurements were carried out to explore the effect of Fe3O4@rGO on the dehydrogenation kinetics of LiBH4. Fig. 5a shows the isothermal dehydrogenation curves of the LiBH4–20 wt% y composites (y = Fe3O4, rGO, and Fe3O4@rGO) at 400 °C, under an initial hydrogen pressure of 0.01 MPa. The LiBH4–20 wt% Fe3O4@rGO sample shows a faster hydrogen desorption rate than the other three hydrogen storage materials, and it can release 3.84 wt% hydrogen within 3600 s, whereas the pure LiBH4, LiBH4–20 wt% Fe3O4, and LiBH4–20 wt% rGO samples can release 1.71, 2.04, and 3.47 wt% hydrogen under the same conditions, respectively. In addition, the dehydrogenation curves of the LiBH4–20 wt% Fe3O4@rGO composite at different temperatures are shown in Fig. 5b. The composite is capable of releasing 2.28, 3.84, and 5.93 wt% hydrogen at 350, 400, and 450 °C, respectively. Remarkably, Fe3O4@rGO has a superior catalytic effect on the dehydrogenation kinetics and capacity of LiBH4 compared with Fe3O4 or rGO alone. The improvement in the hydrogen storage performance not only comes from the destabilization of the Fe3O4 NPs, but also from the confinement of the rGO.18,40 It is speculated that their synergistic effects will assist in improving the dehydrogenation performance of LiBH4. The newly formed structural defects and the close contact between the Fe3O4@rGO and LiBH4 during the ball milling process will also help achieve an enhanced dehydrogenation performance of LiBH4.
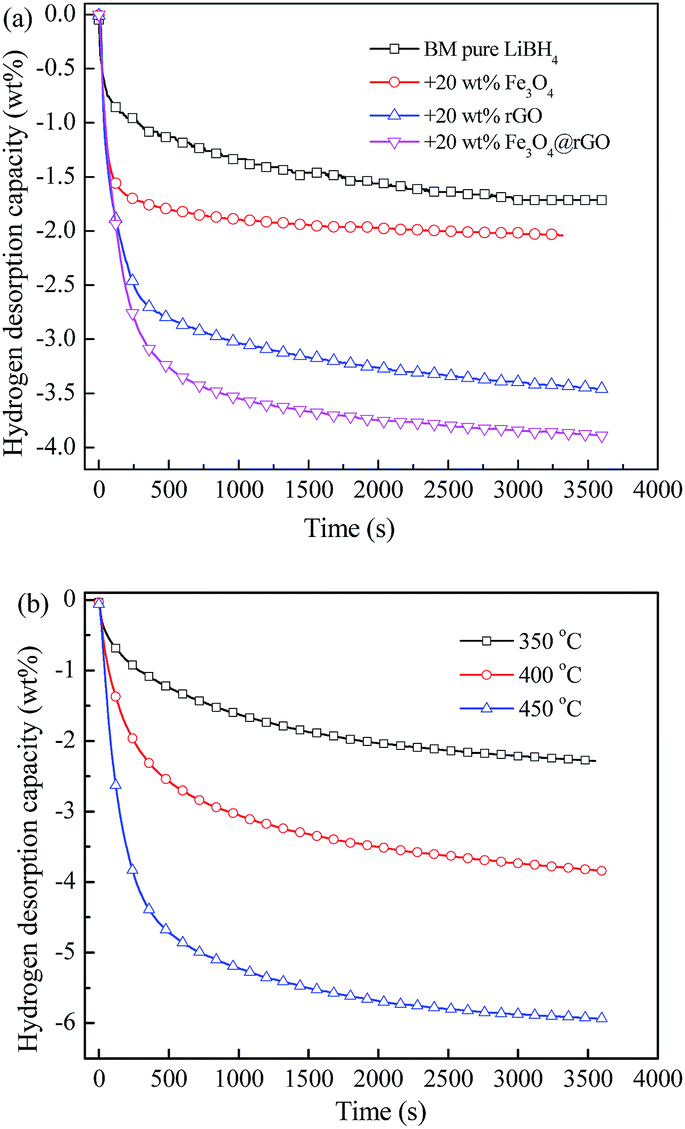 | ||
| Fig. 5 Isothermal dehydrogenation curves of ball-milled (a) pure LiBH4 and LiBH4–20 wt% y composites (y = Fe3O4, rGO, and Fe3O4@rGO) at 400 °C, and (b) the LiBH4–20 wt% Fe3O4@rGO composite at different temperatures. | ||
Fig. 6a demonstrates the isothermal rehydrogenation curves at 400 °C for ball-milled pure LiBH4 and the LiBH4 composites with 20 wt% Fe3O4, rGO, and Fe3O4@rGO, under an initial hydrogen pressure of 5 MPa. The LiBH4–20 wt% Fe3O4@rGO composite can absorb 5.45 wt% hydrogen within 3600 s. However, the pure LiBH4, LiBH4–20 wt% Fe3O4, and LiBH4–20 wt% rGO composites can only absorb 2.36, 4.18, and 4.86 wt% hydrogen under the same conditions, respectively. Therefore, it is concluded that adding Fe3O4@rGO into LiBH4 can result in a higher hydrogen storage capacity and quicker ab/desorption kinetics. In order to study the reversible hydrogen storage performance, the LiBH4–20 wt% Fe3O4@rGO composite is rehydrogenated at 400 °C under 5 MPa H2 for 1 h after its complete dehydrogenation, and the cyclic rehydrogenation curves are shown in Fig. 6b. The composite exhibits superior rehydrogenation behavior and an absorption capacity of 3.73 wt% after 5 cycles of hydrogen de/absorption is achieved, which is up to 69.1% of the initial hydrogen absorption capacity for the composite. However, pure LiBH4 requires extremely harsh conditions of 600 °C and 35 MPa hydrogen pressure to rehydrogenate.41 Obviously, the LiBH4–Fe3O4@rGO composite shows a higher hydrogen absorption capacity than the composites with Fe3O4 or rGO. Thus, Fe3O4@rGO can simultaneously ameliorate the hydrogen release and uptake reversibility for the LiBH4 system under moderate conditions, which is largely attributed to the combined effects of the reduced graphene oxide and Fe3O4 NPs.
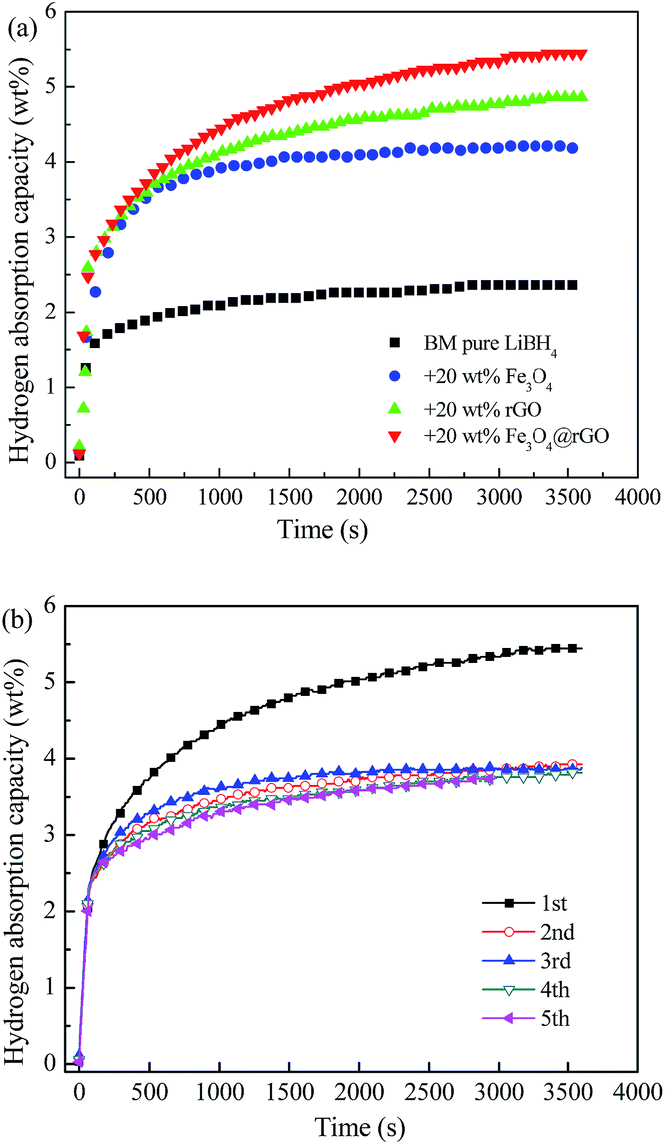 | ||
| Fig. 6 (a) Isothermal rehydrogenation curves of the pure LiBH4 and LiBH4–20 wt% y composites (y = Fe3O4, rGO, and Fe3O4@rGO), and (b) the rehydrogenation cycling stability of the LiBH4–20 wt% Fe3O4@rGO composite at 400 °C under 5 MPa H2. | ||
3.3 Discussion and deduction of the reaction mechanism
To investigate the effect of morphology on the improvement of the hydrogen storage properties of the LiBH4–20 wt% Fe3O4@rGO composite, microstructural analysis of the composite was performed using SEM techniques. Fig. 7a shows that the composite has many defects and pores on its surface, with some particles, in the range of several tens of nanometers to several microns, encapsulated by graphene sheets. A high resolution SEM image is shown in Fig. S7† to further demonstrate the pores and particles of the as-milled LiBH4–20 wt% Fe3O4@rGO sample. It can be observed that many small particles are wrapped tightly by the rGO sheets. After dehydrogenation at 400 °C (Fig. 7b), numerous wrinkles and channels randomly scattered on the sample surface appear. Furthermore, the dehydrogenated sample is still wrapped tightly by the rGO sheets. It is speculated that Fe3O4@rGO might play the role of a heterogeneous nucleation site for the LiBH4 decomposition by providing more hydrogen diffusion pathways and it may also prevent agglomeration during the dehydrogenation process. The uniform dispersion and smaller particle size will benefit the hydrogen desorption and absorption behavior of LiBH4.42 | ||
| Fig. 7 SEM images of the LiBH4–20 wt% Fe3O4@rGO composite: (a) as-milled and (b) after desorption at 400 °C. | ||
In order to further analyze the effect of Fe3O4@rGO on the hydrogen storage properties of LiBH4, the LiBH4–20 wt% Fe3O4@rGO composite in different states was assessed using XRD and FTIR techniques. In order to investigate the effects of rGO and Fe3O4 on LiBH4 individually, XRD patterns of the LiBH4–20 wt% rGO and LiBH4–20 wt% Fe3O4 composites were also obtained (Fig. S7 and S8†). As shown in Fig. S8a,† the intense characteristic peaks of LiBH4 can be clearly seen after ball milling, and the LiBO2 phase is formed due to the reaction between LiBH4 and the O in the rGO. After it dehydrogenates at 400 °C (Fig. S8b†), LiBH4 can completely decompose, the LiBO2 converts to Li3BO3, and the Li3BO3 exists in the later hydrogenation process (Fig. S8c†). As for the ball-milled LiBH4–20 wt% Fe3O4 composite (Fig. S9a†), apart from the characteristic peaks of LiBH4 and Fe3O4, LiBO2 and Li5FeO4 peaks are also detected. For its dehydrogenated sample (Fig. S9b†), Li5FeO4 still exists. LiBH4, Fe3O4, and LiBO2 disappear, while LiH and Li3BO3 phases appear, which indicates there are some reactions between LiBH4 and Fe3O4 during the dehydrogenation reaction. Similar results are observed for the LiBH4–20 wt% Fe3O4@rGO composite (Fig. 8a and b). The characteristic peaks of the LiBH4, Fe3O4, LiBO2, and Li5FeO4 species are verified for the ball-milled LiBH4–Fe3O4@rGO sample, confirming that LiBH4 can react with Fe3O4 during the ball milling process. It is believed that Fe3O4 can react with LiBH4 to form LiBO2 and Li5FeO4. After dehydrogenation at 400 °C, the formed LiBO2 can react further with the remaining LiBH4 to generate Li3BO3.43 The Li3BO3 and Li5FeO4 still exist in the hydrogenation reaction (Fig. 8c), wherein Li3BO3 acts as the actual active species for improving the reversible hydrogen storage properties of LiBH4.44 However, the formation of thermodynamically stable compounds like Li3BO3 and Li5FeO4 is an important reason for the reduced rehydrogenation capacity. The possible hydrogen desorption reactions of the LiBH4–20 wt% Fe3O4@rGO composite may be expressed as follows:
| 7LiBH4 + 2Fe3O4 → 2LiBO2 + Li5FeO4 + 5FeB + 14H2 |
| 2LiBH4 → 2LiH + 2B + 3H2 |
| 3LiBH4 + 3LiBO2 → 2Li3BO3 + 4B + 6H2 |
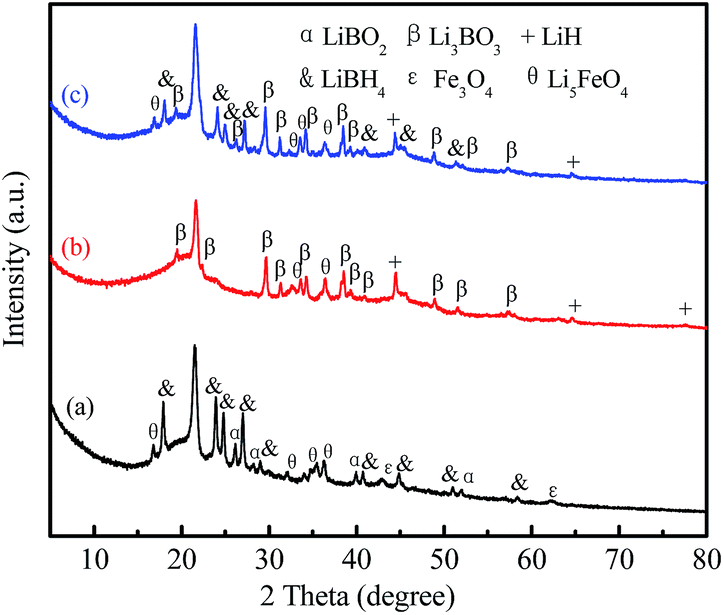 | ||
| Fig. 8 XRD patterns of the LiBH4–20 wt% Fe3O4@rGO composite: (a) as-milled, (b) after desorption at 400 °C, and (c) after absorption at 400 °C. | ||
Although LiBH4 can reform in the rehydrogenation reaction, the existence of LiH in this state indicates that the rehydrogenation reaction is incomplete, which may be another reason for the degradation of the cyclic hydrogen absorption capacity of the LiBH4–Fe3O4@rGO composite.
To further prove that LiBH4 is generated again after rehydrogenation, FTIR analysis of the as-milled, dehydrogenated, and rehydrogenated LiBH4–20 wt% Fe3O4@rGO samples was carried out. From Fig. 9a, characteristic peaks of B–H stretching bands at 2227, 2292, and 2387 cm−1 and B–H bending bands at 1127 cm−1 are revealed. For the dehydrogenated sample (Fig. 9b), these characteristic bands of B–H are absent, indicating the complete decomposition of LiBH4 in the hydrogen desorption process. In contrast, the characteristic peaks of the B–H bands are visible in the spectrum of the rehydrogenated sample (Fig. 9c). The disappearance and re-emergence of the B–H bands are strong evidence for the reversibility of the hydrogen desorption/absorption reactions. However, we also observe a new absorption peak at 2478 cm−1, which can be assigned to Li2B12H12.45 Although Li2B12H12 can react with LiH to generate LiBH4, this reaction requires harsh conditions of 500 °C and 100 MPa H2 for 72 h.46,47 In our work, the rehydrogenation was carried out under the moderate conditions of 400 °C and 5 MPa H2 for 1 h. Therefore, some Li2B12H12 and LiH still remained in the rehydrogenated state for the LiBH4–20 wt% Fe3O4@rGO sample, suggesting that the reaction between Li2B12H12 and LiH was not complete, thus affecting its cyclic rehydrogenation behavior. Furthermore, FTIR spectra of the LiBH4–20 wt% rGO composite in different states were also obtained and are shown in Fig. S10.† The LiBH4 can decompose and regenerate likewise after the dehydrogenation and rehydrogenation of the LiBH4–20 wt% rGO composite (Fig. S10b and c†). The peak of Li2B12H12 at 2477 cm−1 is also detected in the rehydrogenated LiBH4–rGO sample. The surface morphology of the LiBH4–20 wt% Fe3O4@rGO composite after the first hydrogenation changed greatly, as shown in Fig. S11.† Although the rehydrogenated sample is still wrapped tightly by the rGO nanosheets, the amount of wrinkles and channels on its surface apparently decreases. The morphology change can be another reason for the degradation of the cyclic hydrogen absorption capacity of the LiBH4–20 wt% Fe3O4@rGO composite, but its rehydrogenation performance is still superior to that of pure LiBH4.
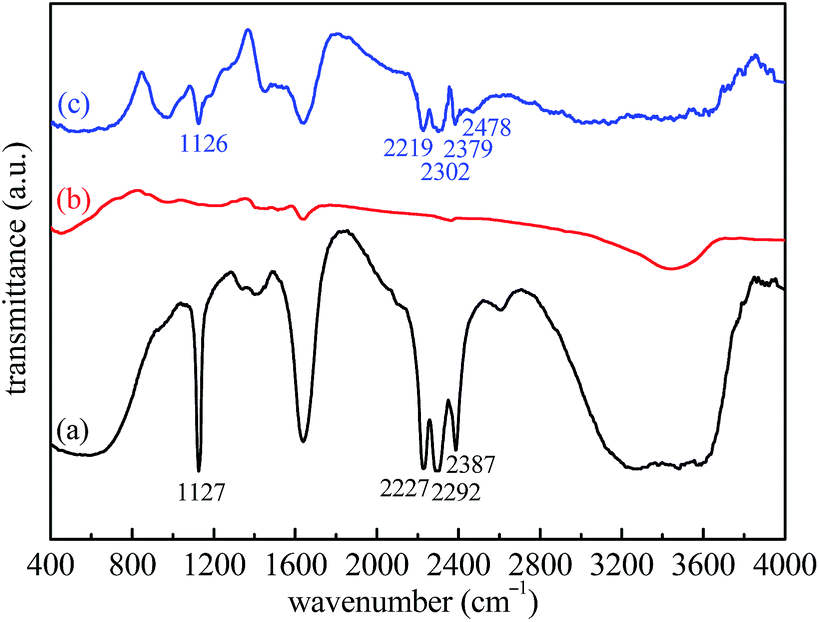 | ||
| Fig. 9 FTIR spectra of the LiBH4–20 wt% Fe3O4@rGO composite: (a) as-milled, (b) after desorption at 400 °C, and (c) after absorption at 400 °C. | ||
Based on the above analyses, we conclude that some LiBH4 can be confined in the pores of the reduced graphene oxide sheets, while the remaining LiBH4 spreads evenly over the surface after ball milling due to the loose porous structure of the sheets. Fe3O4@rGO acts as a destabilization agent by reacting with LiBH4 to decrease its onset dehydrogenation temperature. The in situ-formed active Li3BO3 species during the dehydrogenation process has an actual catalytic effect on improving the dehydrogenation kinetics and rehydrogenation reversibility of LiBH4. Simultaneously, abundant structural defects and channels are created during this process. The wrapping effect of the rGO nanosheets is proven to be able to effectively prevent the nucleated particles from agglomerating during the dehydrogenation and rehydrogenation cycles.48 In this regard, the destabilization, catalysis, and wrapping effect of the Fe3O4@rGO nanohybrid contribute jointly to the improved hydrogen storage performance of LiBH4.
4. Conclusions
In this study, porous reduced graphene oxide sheets decorated with Fe3O4 nanoclusters were synthesized by a facile hydrothermal method. The Fe3O4@rGO nanohybrid, serving as a destabilizer and catalyst precursor, is added to LiBH4, resulting in the improvement of its hydrogen storage properties. The onset dehydrogenation temperature is reduced to 74 °C for the LiBH4–20 wt% Fe3O4@rGO composite, which is 250 °C lower than that of ball-milled pure LiBH4. Rehydrogenation with a promoted cycling stability is achieved at 400 °C under 5 MPa H2. The addition of Fe3O4@rGO also decreases the Ea of the dehydrogenation reaction of LiBH4 to 102.02 kJ mol−1, suggesting excellent desorption kinetics, and it can release 3.89 wt% hydrogen at 400 °C after 3600 s, which is 2.27 times as high as pure LiBH4. The significantly enhanced hydrogen storage properties of the composite are not only attributed to the destabilization derived from Fe3O4, but also the in situ-formed active Li3BO3 species in the decomposition process, as well as the wrapping of the rGO sheets. This work may provide insight into new applications of Fe3O4@rGO and the addition of other functional graphenes decorated with non-noble metals into LiBH4, in order to fabricate hydrogen storage materials of desirable performance.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51571173, 216762896 and U1162118), the Specialized Research Fund for the Doctoral Program of Higher Education of China (20130007110008), and the Beijing Young Talents Plan (YEPT0687).References
- X. B. Yu, Z. W. Tang, D. L. Sun, L. Z. Ouyang and M. Zhu, Prog. Mater. Sci., 2017, 88, 1–48 CrossRef.
- R. Chamoun, U. B. Demirci and M. Philippe, Energy Technol., 2015, 3, 100–117 CrossRef.
- H. Wang, H. J. Lin, W. T. Cai, L. Z. Ouyang and M. Zhu, J. Alloys Compd., 2016, 658, 280–300 CrossRef.
- Y. Wu, X. J. Jiang, J. Chen, Y. Qi, Y. X. Zhang, H. Fu, J. Zheng and X. G. Li, Dalton Trans., 2017, 46, 4499–4503 RSC.
- H. Q. Liu, L. F. Jiao, Y. P. Zhao, K. Z. Cao, Y. C. Liu, Y. J. Wang and H. T. Yuan, J. Mater. Chem. A, 2014, 2, 9244–9250 Search PubMed.
- J. W. Huang, Y. R. Yan, L. Z. Ouyang, H. Wang, J. W. Liu and M. Zhu, Dalton Trans., 2014, 43, 410–413 RSC.
- P. Ngene, M. R. V. Zwienen and P. E. D. Jongh, Chem. Commun., 2010, 46, 8201–8820 RSC.
- M. Meggouh, D. M. Grant and G. S. Walker, J. Phys. Chem. C, 2011, 115, 22054–22061 Search PubMed.
- X. L. Si, F. Li, L. X. Sun, F. Xu, S. S. Liu, J. Zhang, M. Zhu, L. Z. Ouyang and D. L. Sun, J. Phys. Chem. C, 2011, 19, 9780–9786 Search PubMed.
- F. Fang, Y. T. Li, Y. Song, D. L. Sun, Q. G. Zhang, L. Z. Ouyang and M. Zhu, J. Phys. Chem. C, 2011, 115, 13528–13533 Search PubMed.
- W. T. Cai, H. Wang, L. F. Jiao, Y. J. Wang and M. Zhu, Int. J. Hydrogen Energy, 2013, 38, 3304–3312 CrossRef.
- X. B. Yu, D. M. Grant and G. S. Walker, J. Phys. Chem. C, 2009, 113, 17945–17949 Search PubMed.
- B. J. Zhang and B. H. Liu, Int. J. Hydrogen Energy, 2010, 35, 7288–7294 CrossRef.
- J. Zhang, P. Li, Q. Wan, F. Q. Zhai, A. A. Volinsky and X. H. Qu, RSC Adv., 2015, 5, 81212–81219 RSC.
- Y. Wu, X. J. Jiang, J. Chen, Y. Qi, Y. X. Zhang, H. Fu, J. Zheng and X. G. Li, Dalton Trans., 2017, 46, 4499–4503 RSC.
- W. T. Cai, J. Chen, L. Y. Liu, Y. Z. Yang and H. Wang, J. Mater. Chem. A, 2017, 6, 1171–1180 Search PubMed.
- W. T. Cai, Y. Z. Yang, P. J. Tao, L. Z. Ouyang and H. Wang, Dalton Trans., 2018, 47, 4987–4993 RSC.
- J. Xu, R. R. Meng, J. Y. Cao, X. F. Gu, Z. Q. Qi, W. C. Wang and Z. D. Chen, Int. J. Hydrogen Energy, 2013, 38, 2796–2803 CrossRef.
- Z. Z. Fang, X. D. Kang and P. Wang, Int. J. Hydrogen Energy, 2010, 35, 8247–8252 CrossRef.
- S. Thiangviriya and R. Utke, Int. J. Hydrogen Energy, 2015, 40, 4167–4174 CrossRef.
- L. T. Zhang, L. X. Chen, X. Z. Xiao, Z. W. Chen, S. K. Wang, X. L. Fan, S. Q. Li, H. W. Ge and Q. D. Wang, Int. J. Hydrogen Energy, 2014, 39, 896–904 CrossRef.
- Y. Zhao, Y. C. Liu, H. Y. Kang, K. Z. Cao, Y. J. Wang and L. F. Jiao, Int. J. Hydrogen Energy, 2016, 41, 17175–17182 CrossRef.
- P. A. Ward, J. A. Teprovich Jr, B. Peters, J. Wheeler, R. N. Compton and R. R. Zidan, J. Phys. Chem. C, 2013, 117, 22569–22575 Search PubMed.
- K. K. Wang, X. D. Kang, J. W. Ren and P. Wang, J. Alloys Compd., 2016, 685, 242–247 CrossRef.
- Y. W. Zhu, S. Murali, W. W. Cai, X. S. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef PubMed.
- J. Xu, Z. Q. Qi, J. Y. Cao, R. R. Meng, X. F. Gu, W. C. Wang and Z. D. Chen, Dalton Trans., 2013, 42, 12926–12933 RSC.
- J. Xu, R. R. Meng, J. Y. Cao, X. F. Gu, W. L. Song, Z. Q. Qi, W. C. Wang and Z. D. Chen, J. Alloys Compd., 2013, 564, 84–90 CrossRef.
- J. Xu, Y. Li, J. Y. Cao, R. R. Meng, W. C. Wang and Z. D. Chen, Catal. Sci. Technol., 2015, 5, 1821–1828 Search PubMed.
- W. Zhang, G. Xu, L. J. Chen, S. Y. Pan, X. Jing, J. S. Wang and S. M. Han, Int. J. Hydrogen Energy, 2017, 42, 15262–15270 CrossRef.
- G. M. Zhou, D. W. Wang, F. Li, L. L. Zhang, N. Li, Z. S. Wu, L. Wen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef.
- B. P. Lu, Z. Zhang, J. H. Hao and J. L. Tang, RSC Adv., 2014, 4, 21909–21917 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- J. Su, M. H. Cao, L. Ren and C. W. Hu, J. Phys. Chem. C, 2011, 115, 14469–14477 Search PubMed.
- J. Z. Wang, G. H. Zhao, L. Y. Jing, X. M. Peng and Y. F. Li, Biochem. Eng. J., 2015, 98, 75–83 CrossRef.
- A. K. Mishra and S. Ramaprabhu, J. Appl. Phys., 2014, 116, 064306 CrossRef.
- L. H. Zhuo, Y. Q. Wu, L. Y. Wang, J. Ming, Y. C. Yu, X. B. Zhang and F. Y. Zhao, J. Mater. Chem. A, 2013, 1, 3954–3960 Search PubMed.
- R. Kumar, R. K. Singh, A. R. Vaz, R. Savu and S. A. Moshkalev, ACS Appl. Mater. Interfaces, 2017, 9, 8880–8890 Search PubMed.
- G. P. Tu, X. Z. Xiao, T. Qin, Y. Q. Jiang, S. Q. Li, H. W. Ge and L. X. Chen, RSC Adv., 2015, 5, 51110–51115 RSC.
- J. S. Wang, Z. B. Wang, Y. Li, D. D. Ke, X. Z. Lin, S. M. Han and M. Z. Ma, Int. J. Hydrogen Energy, 2016, 41, 13156–13162 CrossRef.
- A. Gasnier and F. C. Gennari, RSC Adv., 2017, 7, 27905–27912 RSC.
- W. T. Cai, H. Wang, J. W. Liu, L. F. Jiao, Y. J. Wang, L. Z. Ouyang, T. Sun, D. L. Sun, H. H. Wang, X. D. Yao and M. Zhu, Nano Energy, 2014, 10, 235–244 CrossRef.
- J. Shao, X. Z. Xiao, X. L. Fan, L. T. Zhang, S. Q. Li, H. W. Ge, Q. D. Wang and L. X. Chen, J. Phys. Chem. C, 2014, 118, 11252–11260 Search PubMed.
- L. L. Guo, Y. Li, Y. F. Ma, Y. Liu, D. D. Peng, L. Zhang and S. M. Han, Int. J. Hydrogen Energy, 2016, 42, 2215–2222 CrossRef.
- Y. F. Ma, Y. Li, T. Liu, X. Zhao, L. Zhang, S. M. Han and Y. J. Wang, J. Alloys Compd., 2016, 689, 187–191 CrossRef.
- M. P. Pitt, M. Paskevicius, D. H. Brown, D. A. Sheppard and C. E. Buckley, J. Am. Chem. Soc., 2013, 135, 6930–6941 CrossRef PubMed.
- Y. G. Yan, H. Wang, M. Zhu, W. T. Cai, D. Rentsch and A. Remhof, Crystals, 2018, 8, 131–138 CrossRef.
- J. L. White, R. J. Newhouse, J. Z. Zhang, T. J. Udovic and V. Stavila, J. Phys. Chem. C, 2016, 120, 25725–25731 Search PubMed.
- Y. Y. Zhu, J. X. Zou and X. Q. Zeng, RSC Adv., 2015, 5, 82916–82923 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02762e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |