
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe fundamental roles of monovalent and divalent cations with sulfates on molybdenite flotation in the absence of flotation reagents
Yubiao Li *ab,
Clement Lartey*a,
Shaoxian Songa,
Yingjie Lic and
Andrea R. Gersond
*ab,
Clement Lartey*a,
Shaoxian Songa,
Yingjie Lic and
Andrea R. Gersond
aSchool of Resources and Environmental Engineering, Wuhan University of Technology, Wuhan, 430070, Hubei, China. E-mail: Yubiao.Li@whut.edu.cn; cle.lartey@yahoo.com
bSchool of Natural and Built Environments, University of South Australia, Mawson Lakes, SA 5095, Australia
cShaanxi Provincial Academy of Environmental Science, Xi'an, 710061, Shaanxi, China
dBlue Minerals Consultancy, Wattle Grove, Tasmania, Australia 7109
First published on 27th June 2018
Abstract
Due to regional shortage of freshwater, the use of saline/seawater for Cu–Mo sulfide ore flotation has received considerable attention. However, the effects of various salts, especially the cations present in seawater, on molybdenite flotation and the mechanisms involved remain unclear due to the complexity of the solutions applied. In this work, the influence of some common cations (i.e., Na+, K+, Ca2+ and Mg2+) with sulfate (SO42−) anions on molybdenite flotability was investigated in the absence of flotation reagents (i.e., frothers and collectors) at pH 10. Flotation results indicated a greater depression of molybdenite recovery with increased sulfate salt concentration. The underlying mechanisms responsible for the deleterious effects in the presence of Na+ and K+ can be attributed to the increased repulsive forces between molybdenite particles and bubbles owing to increased molybdenite oxidation to produce e.g., MoO42− and HMoO4−. However, the increased depression observed in the presence of Mg2+ and Ca2+ is likely due to the adsorption of precipitated Mg(OH)2 and CaMoO4, respectively, onto molybdenite surfaces. These clearly show the different depressing mechanisms due to monovalent and divalent sulfates on molybdenite flotation in the absence of flotation reagents, to reveal the influence of these sulfate salts on its natural flotability.
1. Introduction
Mineral flotation is a water-intensive process consuming vast amounts of freshwater every year.1 The scarcity of freshwater in some arid areas (e.g., Mt Keith and Leinster Mines in Western Australia, Grasberg Mine in Indonesia, Xstrata Nickel Raglan Mine in Canada, Las Luces Mine in Chile) coupled with economic and environmental concerns has prompted alternatives to freshwater.2,3 Ideally, saline, seawater or recycled water would serve as sustainable water sources for future flotation processes, especially for those located near the sea and/or lacking freshwater.4,5 However, seawater having a salinity of 3.5 wt% and containing various ions including Na+, K+, Mg2+, Ca2+, Cl− and SO42− may affect mineral flotation processes.6Many studies have shown improved mineral recovery in the presence of inorganic electrolytes.7–9 For instance, Zhang, et al.10 found that seawater modified the network structure of bentonite, enhancing copper and gold recovery. Ozdemir11 showed that coal recovery in a salt water system depended on the type and concentration of electrolytes. In addition, the presence of inorganic salts inhibited bubble coalescence, producing a smaller bubble and enhancing sulfide mineral recovery.12
The chief source of molybdenum (Mo), molybdenite (MoS2), is normally associated with porphyry copper minerals. Enhanced molybdenite recovery has been reported in salt water.4,7,13 For instance, Lucay, et al.14 reported that Na+ decreased electrostatic repulsion between bubbles and anionic edges of molybdenite, thus improving molybdenite recovery. However, several other studies have reported deleterious effects of saline or seawater on molybdenite flotability,15–18 e.g., Raghavan and Hsu19 reported that molybdenite depression in saline or seawater may be caused by the adsorption of hydrolyzed Ca2+ species on molybdenite surface. Recently, Wan, et al.20 studied the interactions between Ca2+ and molybdenite edges and found that preferential oxidation of molybdenite edges produced MoO42− which made the molybdenite edges negatively charged and attracted Ca2+, leading to the formation of CaMoO4 which was responsible for the depression of molybdenite flotation. Other studies have shown that the precipitates formed in saline or seawater at pH > 9.5 dominated molybdenite depression.8,21–23
Nevertheless, no generally accepted understanding of the influence of various salts has been available to explain why saline water improves or reduces molybdenite recovery. Most studies have indicated that the anisotropic features of molybdenite with van de Waal forces occurring within its basal planes (face) and Mo–S covalent bonds at edges11,13,24,25 play an important role in influencing molybdenite flotation. Lu, et al.26 investigated the anisotropic surface properties of molybdenite by direct surface force measurements using atomic force measurement (AFM) in 10 mM NaCl solution at various pH and concluded that the faces and edges of molybdenite displayed hydrophobic and hydrophilic features, respectively. They further postulated that small particles with a small face-edge ratio were less hydrophobic. Although many researchers have studied the surface properties of the face and edge of natural molybdenite,26–28 no convincing premise has been achieved to clearly explain salt effects on the faces and edges during flotation process.
To date, although several studies have attempted to investigate the influence of chlorides on molybdenite flotation, the roles and contributing effects of sulfate salts have not attracted sufficient attention.5–7,29 As molybdenite is normally associated with other sulfide minerals, the oxidation of these sulfides in air or water would produce sulfate. In addition, the recycling of flotation water results in various cations in recycled solution. The accumulated cations and sulfates influence molybdenite flotation significantly. Furthermore, the flotation reagents that are normally applied to flotation process at least partially hide the effects of these cations on the natural flotability of molybdenite. Therefore, this work aimed to better understand the underlying flotation mechanisms of naturally hydrophobic molybdenite in the presence of sulfate salts (i.e. Na+, K+, Ca2+, and Mg2+) in the absence of flotation reagents.
2. Materials and methods
2.1 Materials
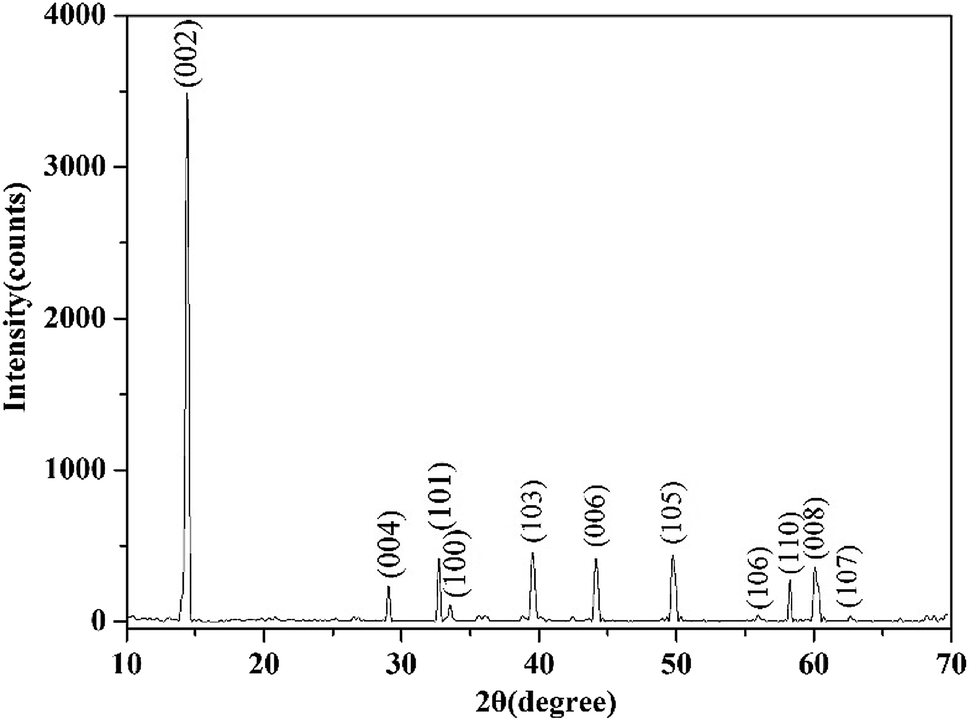 | ||
| Fig. 1 XRD pattern of molybdenite sample. | ||
2.2 Flotation experiments
Mineral flotation tests were conducted using a hanging trough flotation machine (XFG, Wuhan Exploration Machinery Factory, China) with a 25 mL micro flotation cell, without (control) and with various concentrations of Na2SO4, K2SO4, CaSO4, and MgSO4. 0.2 g molybdenite powder was conditioned in the flotation cell for 6 min and the pulp pH was adjusted using 0.1 M NaOH during this period. The froth products were collected every 10 s at 1, 3, 5, 8 and 10 min for 1 min, at an airflow rate of 0.1 L min−1 at 1200 rpm. Froth concentrate and residue were collected and dried in a vacuum oven at 35 °C for 24 h and subsequently weighed to determine cumulative molybdenite recovery.2.3 Contact angle measurements
Fresh molybdenite surfaces were obtained by peeling off the top layer of molybdenite sample. These surfaces were then conditioned in a salt solution for 6 min. The sessile drop method (JC2000C1, Shanghai Zhongchen Digital Technology Company, China) was employed for contact angle measurements between molybdenite surface and a 0.25 μL drop.29,30 Measurements were conducted in the pH 10 solution with salt concentrations ranging from 10−4 to 10−2 M. The average of three different measurements was recorded as the final contact angle.2.4 Zeta potential measurements
Zeta potential measurements (Zetasizer Nano-ZS90, Malvern Co., Ltd.) were conducted in simulated solution using Na2SO4, K2SO4, CaSO4, and MgSO4 at 10−4 M, 10−3 M, and 10−2 M. A fresh molybdenite suspension (−5 μm) was prepared and the pH was adjusted to 10 using 0.1 M NaOH.16,29 The average of three measurements was reported as the final zeta potential.2.5 Solution concentration analyses
The solution concentrations were analysed by Inductively Coupled Plasma-Optical Emission Spectroscopy (Prodigy 7, Teledyne Leeman Labs, USA).3. Results
3.1 Effects of pH
Fig. 2 shows the pH influence on molybdenite flotation recovery in 0 M and 10−2 M Na2SO4 solution for 3 min with pH ranging from 4 to 12. The application of 10−2 M Na2SO4 solution improved molybdenite recovery in acidic medium but performed poorly under high alkaline conditions, compared to the absence of Na2SO4 (0 M). Molybdenite recovery was reached 91% in 10−2 M Na2SO4 at pH 4, and gradually reduced to 89%, 86%, 73% and 60% at pH 6, 8, 10 and 12, respectively. In the absence of Na2SO4, 78% recovery was observed at pH 4 which was increased to 87% at pH 8. However, a further increase in pH resulted in reduced recovery, indicating an adverse effect under highly alkaline conditions. Considering that pH 10 was normally used to depress pyrite flotation in molybdenite flotation plants,8,23 pH 10 was selected for further flotation.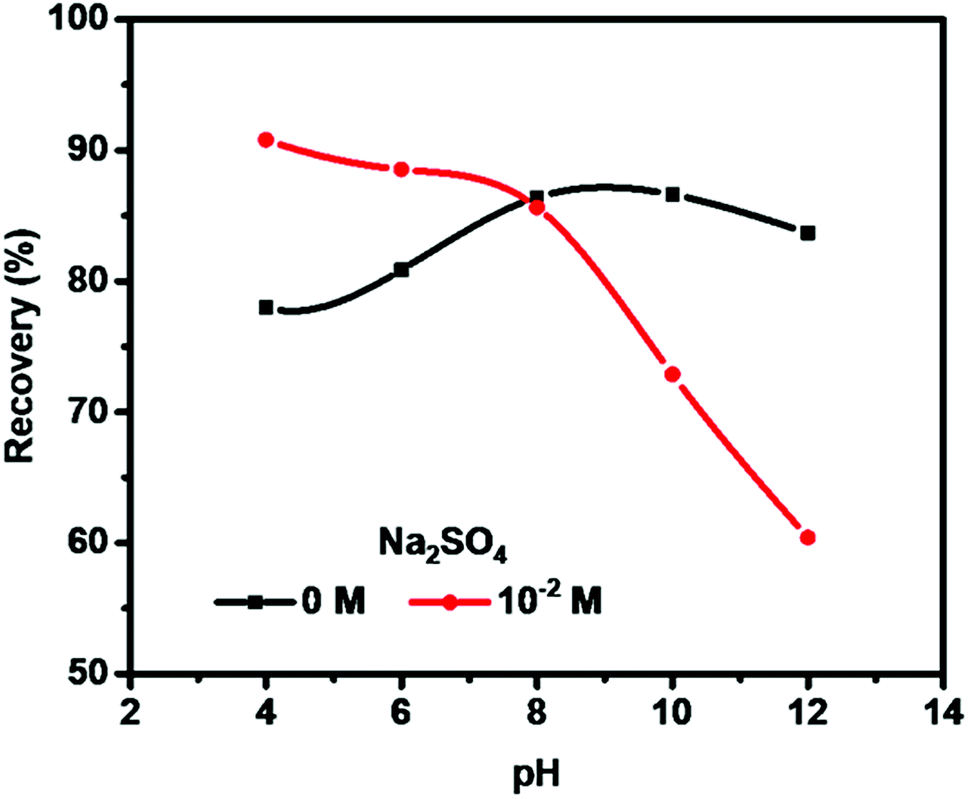 | ||
| Fig. 2 Effects of pH on molybdenite recovery. | ||
3.2 Effects of sulfate salts
Fig. 3 shows the cumulative molybdenite recovery as a function of flotation time in four sulfate salts within 10 min. The molybdenite recovery was increased rapidly within the first 3 min but thereafter only a slight increase in recovery was observed up to 10 minutes. Specifically, a 90% recovery was achieved in the absence of sulfate salts, which agrees well with other studies.20,28 Molybdenite flotability was depressed to various extents in the presence of sulfate salts within the concentrations investigated in an order of Mg2+ > Ca2+ > K+ > Na+.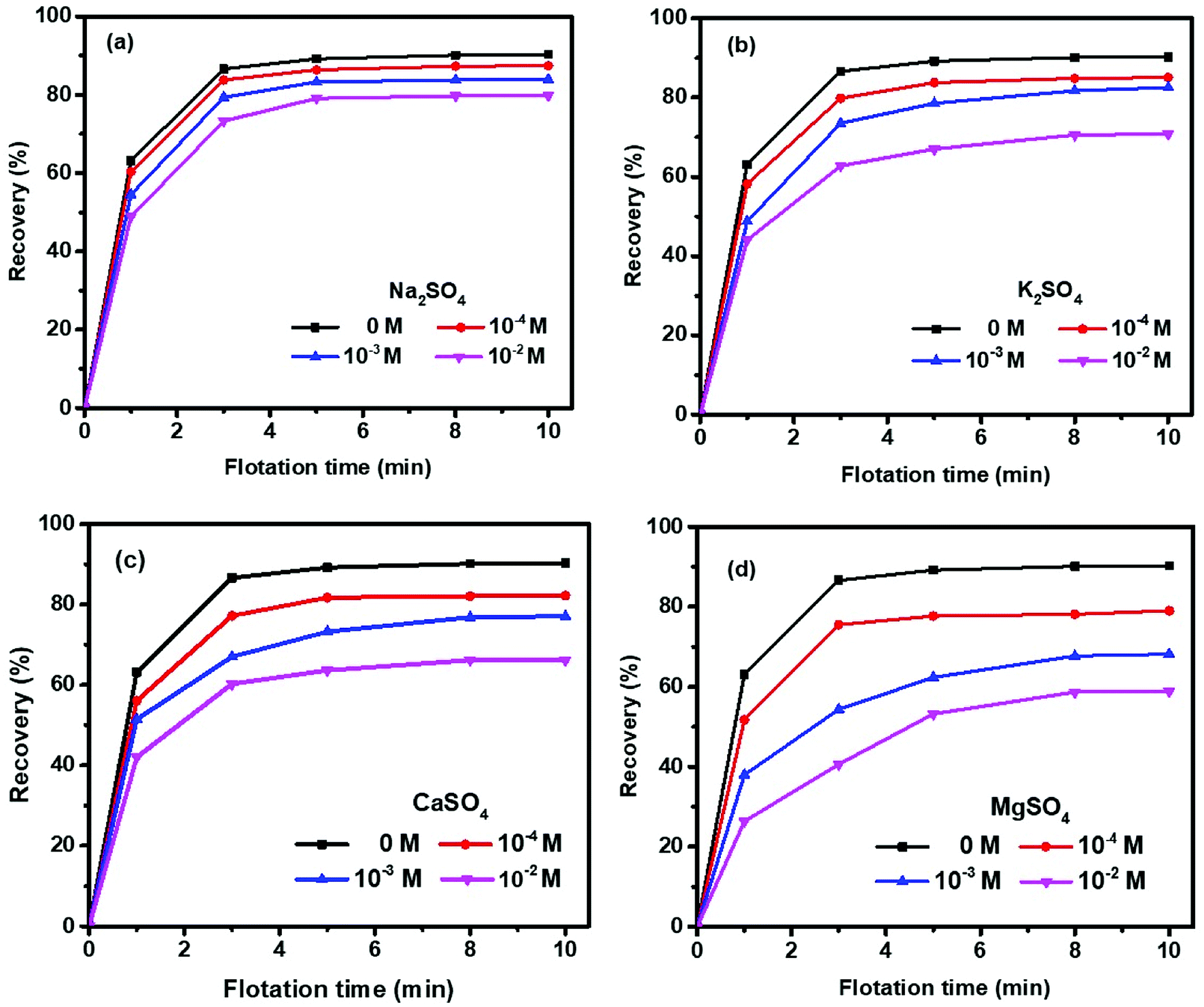 | ||
| Fig. 3 Molybdenite recovery at pH 10: (a) Na2SO4, (b) K2SO4, (c) CaSO4, and (d) MgSO4. | ||
A recovery of 87%, 84%, and 80% in Na2SO4 solution while 85%, 83%, 71% in K2SO4 solution was observed at 10−4, 10−3 and 10−2 M, respectively, indicating that the presence of K+ resulted in greater decrease in molybdenite flotability compared to Na+ over the entire concentrations investigated (Fig. 3(a) and (b)). Moreover, as shown in Fig. 3(c) and (d), both Ca2+ and Mg2+ sulfates resulted in significantly greater negative effects on molybdenite recovery than either Na+ or K+. Although a rapid flotation recovery was observed within 3 min, the initial molybdenite recovered within this period was smaller in Mg2+ solution than that in Ca2+ solution. Clearly, the presence of Mg2+ resulted in greater depressions than Ca2+, dropping from 90% (without salts) to 79%, 68%, 59% and 82%, 77%, 66% when Ca2+ and Mg2+ were controlled at 10−4, 10−3 and 10−2 M, respectively.
The classical first-order rate model,31–33 a generally accepted model for analyzing and interpreting flotation kinetics, was applied to determine the flotation rate constant of molybdenite, as shown in eqn (1):
| R = Rmax (1 − e(−kt)) | (1) |
Fig. 4 shows the flotation kinetics of molybdenite in 10−4 to 10−2 M sulfate solution. The rate constants (k) of molybdenite flotation was decreased with increased sulfate concentration. In addition, with increased flotation time, the k values were significantly decreased, indicating slower flotation rates during the latter flotation stage.
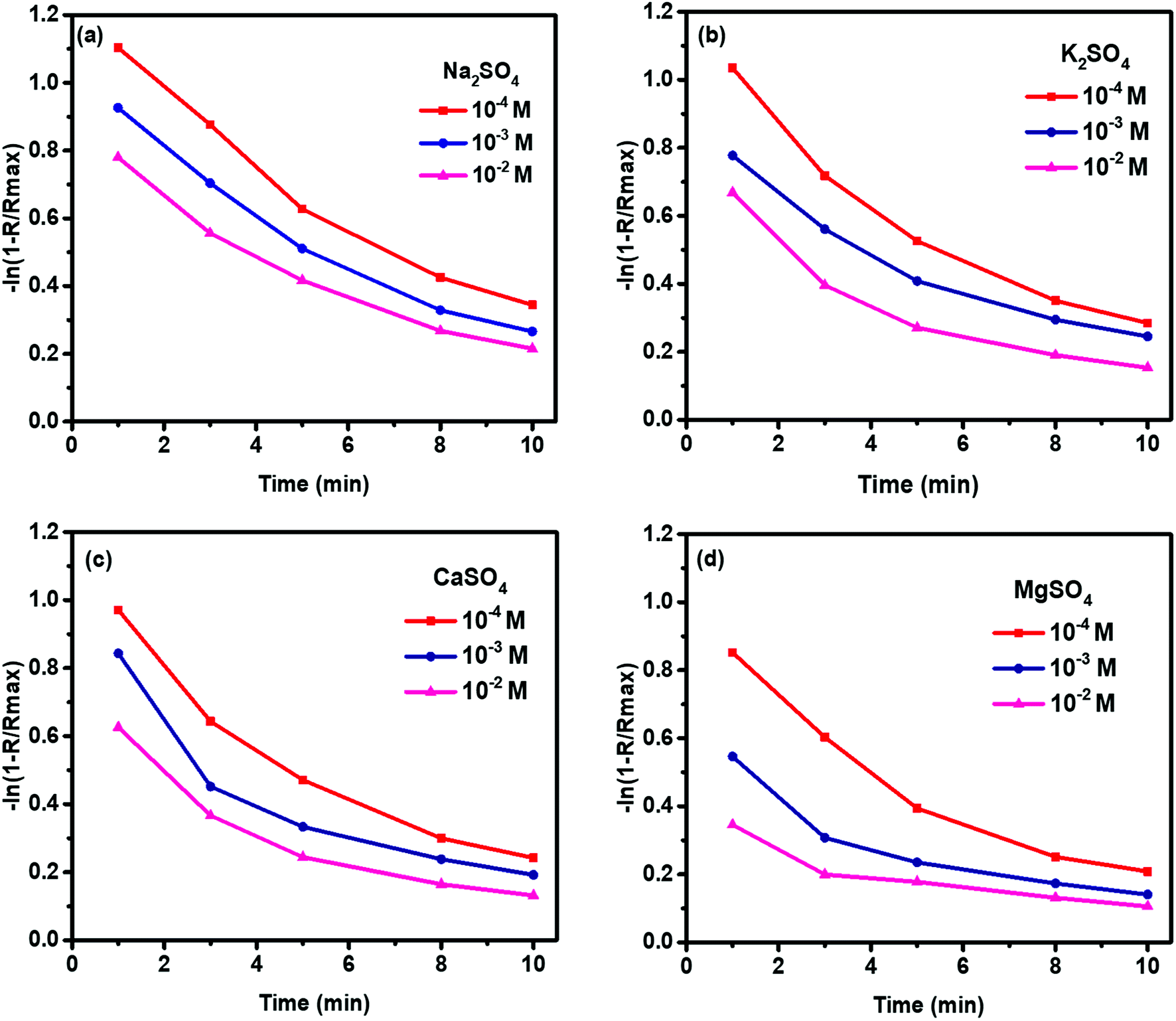 | ||
| Fig. 4 Molybdenite flotation kinetics in 10−4 to 10−2 M (a) Na2SO4, (b) K2SO4, (c) CaSO4, and (d) MgSO4 at pH 10. | ||
3.3 Contact angle measurements
Fig. 5 shows the contact angle measurements of molybdenite conditioned in Na2SO4, K2SO4, CaSO4, and MgSO4 at pH 10. Prior to the addition of these salts, the fresh molybdenite surface exhibited was inherently hydrophobic with a contact angle of 89°, which agrees well with that reported previously.34 A gradual decrease in contact angle with increased sulfate salt concentrations indicated that sulfate salts disrupted the natural hydrophobicity of molybdenite surface, with smaller contact angles being observed at greater sulfate concentrations. Final contact angles of 82.0°, 80.8°, 78.0° and 77.5° were observed for 10−2 M Na2SO4, K2SO4, CaSO4 and MgSO4 solutions, respectively, showing an order of MgSO4 < CaSO4 < K2SO4 < Na2SO4.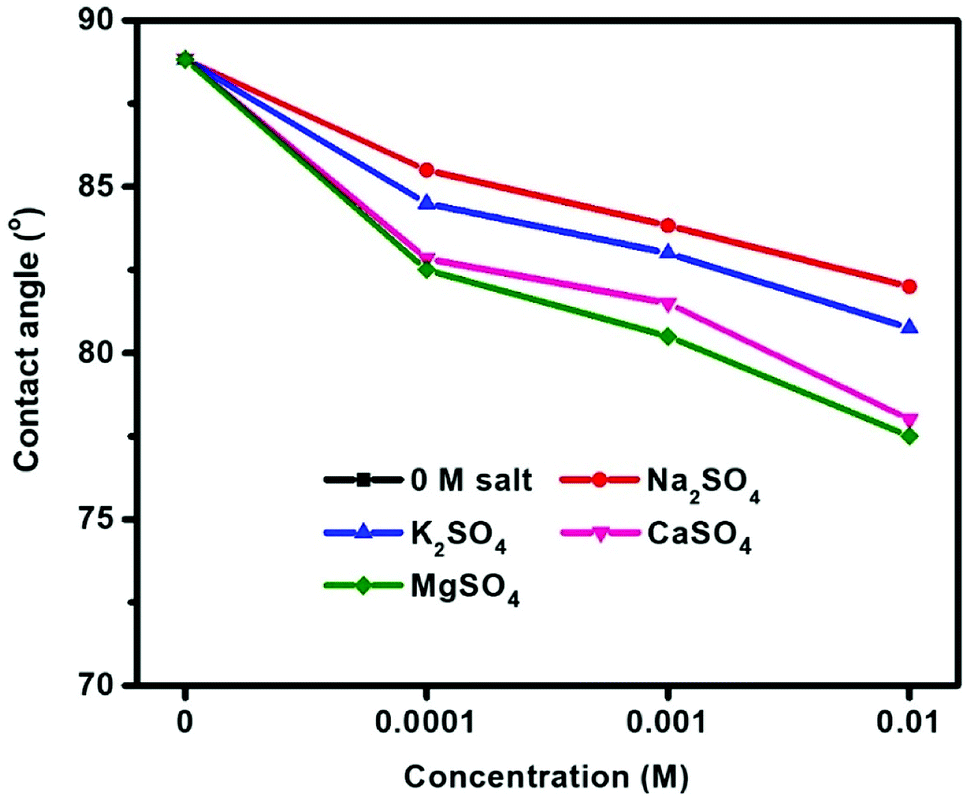 | ||
| Fig. 5 Contact angle of molybdenite in sulfate solution at pH 10. | ||
3.4 Zeta potential measurements
Fig. 6 shows the zeta potentials of molybdenite as a function of sulfate salt concentration at pH 10. The zeta potentials were negative for all salt concentrations investigated, similar to that reported in Hirajima, et al.8 In the absence of sulfate salts, the zeta potential was measured as −32.6 mV, consistent with other studies.19,24 Clearly, the increased concentration of Na2SO4 and K2SO4 from 10−4 to 10−2 M resulted in more negative molybdenite zeta potential values, e.g. −41.2 mV, −62.6 mV, −66.6 mV and −41.0 mV, −60.1 mV, −65.65 mV in 10−4, 10−3 and 10−2 M Na2SO4 and K2SO4 solutions, respectively.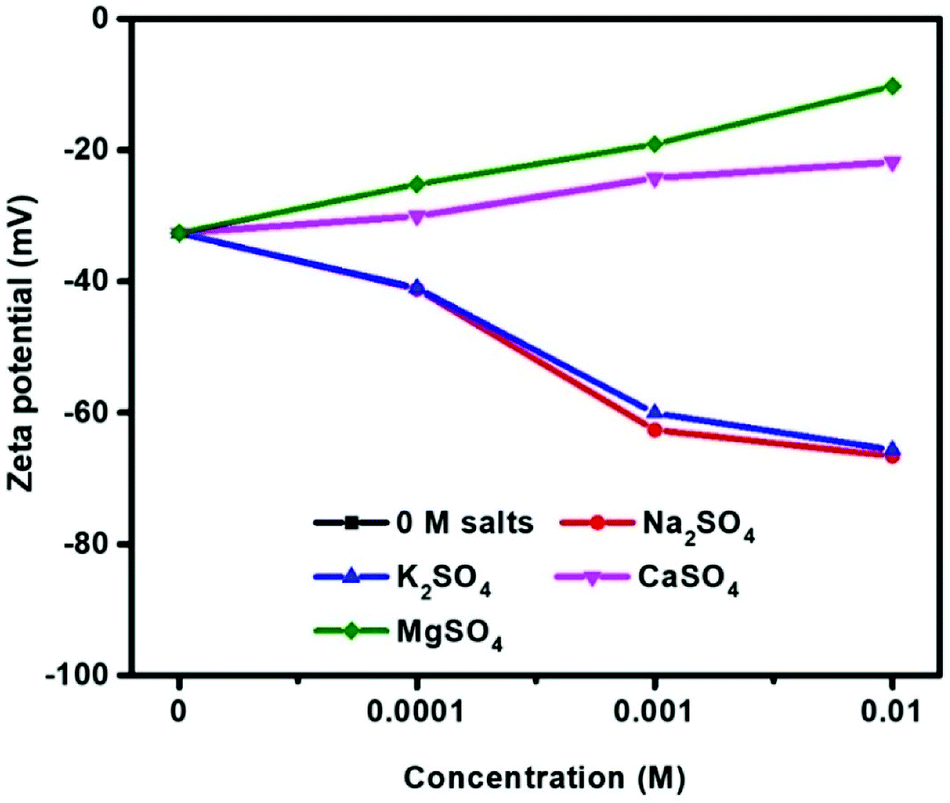 | ||
| Fig. 6 Zeta potential of molybdenite in sulfate salts at pH 10. | ||
In contrast, the zeta potentials in divalent sulfate solution became less negative with increasing sulfate salt concentrations. This effect was most pronounced for MgSO4 solutions with zeta potentials of −25.2 mV, −19.1 mV and −10.3 mV for 10−4, 10−3 and 10−2 M as compared to −30.1 mV, −24.3 mV and −21.8 mV for CaSO4 solutions.
4. Discussion
Most previous studies have only demonstrated negative effects due to chlorides, or Ca2+ and Mg2+ on molybdenite flotation, but have not examined sulfate, or Na+ or K+.6,20,22 This study, however, has examined the roles of four common sulfate salts existing in seawater or recycled water systems in mineral processing plants.The flotation results as a function of pH (Fig. 2, 0 and 10−2 M Na2SO4) agree well with results reported by Qiu, et al.6 who observed that molybdenite depression was started from pH 9.5 on increasing pH with strong depression being observed at pH 11 in seawater flotation. Lucay, et al.14 reported a considerable repulsive force between molybdenite particles and bubbles in alkaline solution. The decrease in molybdenite flotability with increased pH, in the presence of chloride salts, has been attributed to strong electric charge repulsive forces between molybdenite and air bubbles with increasing repulsion with increasing pH.35,36
In this study, the addition of all sulfates reduced flotation recovery in the absence of flotation reagents (pH 10, 0 to 10−2 M, Fig. 3) with increasing depression in an order of Mg2+ > Ca2+ > K+ > Na+. The degree of depression was increased with increased sulfate concentration, probably due to the increased electrostatic repulsion between solid surfaces and air bubbles, similar to that in the chloride solution. Contact angle measurements (Fig. 5) were consistent with flotation results, e.g. increased surface wettability corresponded to reduced recovery. In contrast, less negative zeta potentials (Fig. 6) in the presence of CaSO4 and MgSO4 as compared to no salt addition indicated declining electrostatic repulsion, which should improve molybdenite flotability.
Some published works have indicated that molybdenite faces are not perfectly smooth, with hydrophilic micro-edges present on hydrophobic face.24,37,38 These micro-edges exhibit similar characteristics as molybdenite edges.14,20 López-Valdivieso, et al.36 proposed that the faces of molybdenite particles were heterogeneous in nature with clusters of micro-crystals, giving rise to nano-edges and nano-faces. Therefore, not only are the edges of molybdenite hydrophilic but also the hydrophobic surfaces contain hydrophilic micro-edges capable of adsorbing inorganic electrolytes,7,39 both resulting in detrimental effects on molybdenite flotation.
Lu, et al.26 reported that both surfaces and edges become more negatively charged under alkaline conditions, with the charge on the latter being relatively greater than the former. Wan, et al.20 recently reported that zeta potential of molybdenite was predominantly determined by the edges rather than faces, especially for fine particles. Moreover, compared to hydrophobic faces, molybdenite edges were more easily oxidized in solutions containing O2 and OH− to form, e.g., MoO42− and HMoO4−,13,19 according to eqn (2) and (3).
| 2MoS2 + 9O2 + 10OH− → 2HMoO4− + 4SO42− + 4H2O | (2) |
| HMoO4− + OH− → MoO42− + H2O | (3) |
The oxidation of molybdenite to produce HMoO4− normally occurs across the pH range of 2 to 6, with increased pH, MoO42− predominates.26,40 Therefore, MoO42− will be the primary oxidation products on molybdenite edges in the flotation process controlled at pH 10, as shown in Fig. 7(a).
 | ||
| Fig. 7 Schematic of molybdenite oxidation and flotation in the presence of various cations. (a) Oxidised molybdenite edge, in the presence of (b) Na+, (c) K+, (d) Ca2+, and (e) Mg2+. | ||
Some studies have shown that the presence of NaCl and KCl improves molybdenite flotation. It has also been reported that both Na2SO4 and K2SO4 are beneficial to chalcopyrite oxidation/leaching due to easier breakage of S–S bonds when these two sulfates are available.41 Solubilised Mo in 0 M and 10−2 M Na2SO4, and K2SO4 was examined to understand the oxidation of molybdenite under flotation conditions. After 10 min flotation, the Mo concentrations were 797, 804, and 812 μg L−1, respectively, suggesting that Na2SO4 and K2SO4 increased molybdenite dissolution during flotation, with greater leaching being observed in K2SO4. This is consistent with flotation results shown in Fig. 3(a) and (b), i.e. the depressant effect due to K2SO4 was more significant than that of Na2SO4. Moreover, the oxidation/leaching occurring at micro-edges present on molybdenite faces or edges increased molybdenite wettability, consistent with contact angles shown in Fig. 4 and other studies.26 Therefore, the presence of Na2SO4 and K2SO4 catalysed surface oxidation, giving rise to more negative charges, i.e. more negative zeta potential (Fig. 6), consistent with those observed in Ozdemir, et al.42 In addition, more negative zeta potential of molybdenite leads to greater electrostatic repulsion between molybdenite surfaces and air bubbles which overrides the van der Waals and hydrophobic forces of attraction.20,24
The mechanisms of flotation depression due to the presence of CaSO4 and MgSO4 are different from those due to Na2SO4 and K2SO4. As indicated in many other studies,8,29,39,43 the presence of Ca2+ and Mg2+ may result in precipitation, thereby depressing sulfide mineral flotation. Fig. 8(a) to (c) show that typical Ca2+ species present in 10−4 M, 10−3 M and 10−2 M CaSO4 solutions are Ca2+, CaOH+ and Ca(OH)2(aq) when the solution pH is less than 12. In addition, no Ca-containing precipitation is expected at pH 10. Notably, the molybdenite surface was less negatively charged (zeta potential measurements, Fig. 6) at pH 10 in the presence of CaSO4 suggesting both Ca2+ and CaOH+ could have been adsorbed unto molybdenite edges or micro-edges, forming for instance CaMoO4. The adsorption of these species is likely to cover molybdenite edges and reduce its hydrophobicity, leading to decreased contact angles on increasing sulfate concentration (Fig. 5), in agreement with findings reported in López-Valdivieso, et al.36 The stabilisation of liquid layer on less hydrophobic surface results in increased induction time for bubble-particle attachment, thereby decreasing molybdenite recovery44 consistent with that observed in Wan, et al.20
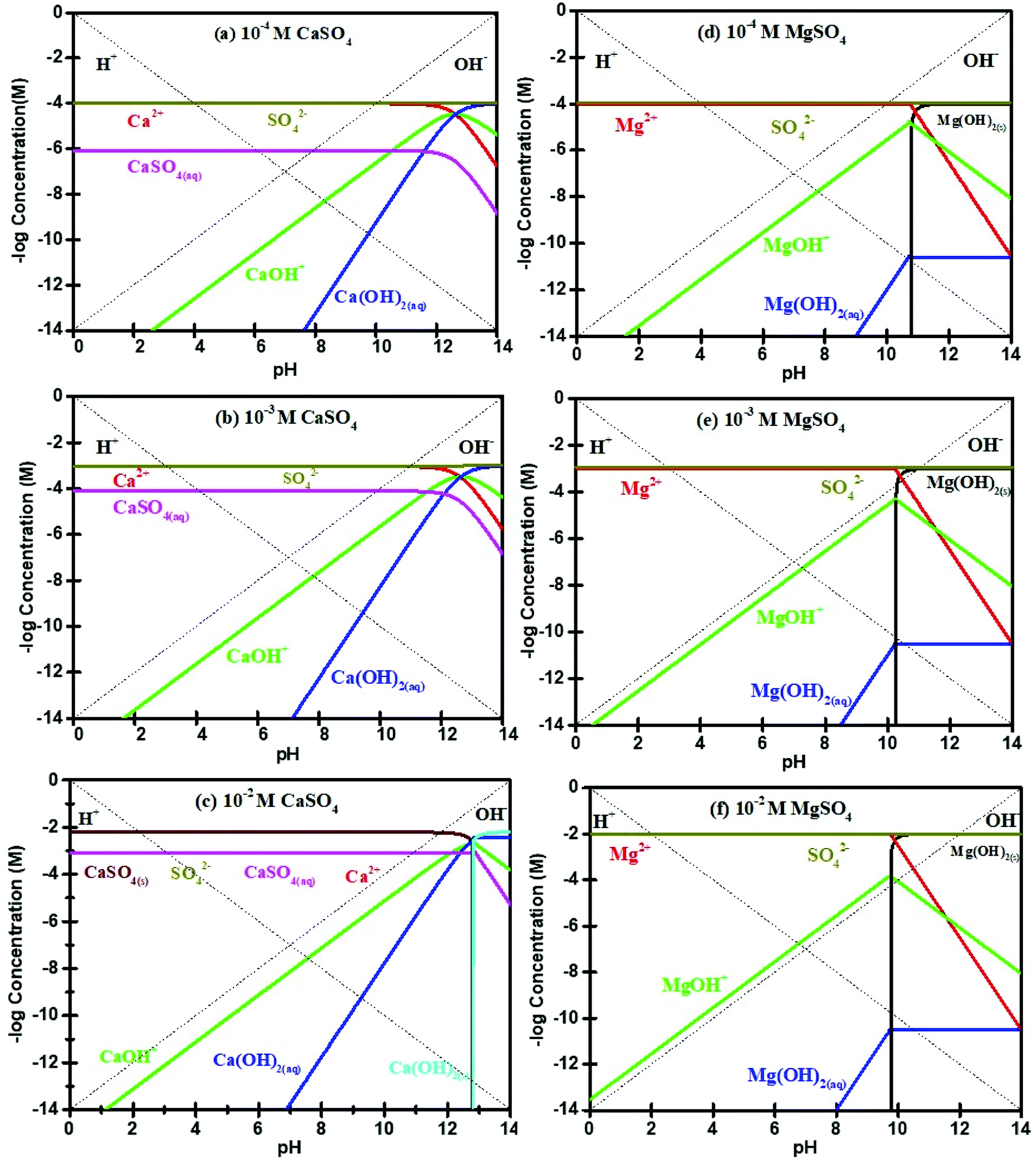 | ||
| Fig. 8 Solution speciation diagrams for Ca2+ at concentrations of (a) 10−4 M, (b) 10−3 M and (c) 10−2 M and Mg2+ concentrations at (d) 10−4 M, (e) 10−3 M and (f) 10−2 M. | ||
Fig. 8(d)–(f) show that the pH at which Mg(OH)2(s) precipitates decreases from pH 10.4 to pH 9.4 as MgSO4 concentration is increased from 10−4 M to 10−2 M, in agreement with Hirajima, et al.8 Li and Somasundaran39 observed that Mg(OH)2(s) precipitated with pH ranging from 9.2 to 11 where Mg2+ concentrations were decreased from 10−2 to 10−5 M. The adsorption of Mg(OH)2(s) onto molybdenite faces can make the hydrophobic surface to be hydrophilic, thereby depressing molybdenite flotation. Mg(OH)2 adsorbed onto molybdenite faces results in increased surface wettability and reduced molybdenite recovery under alkaline conditions.8 Therefore, the observed flotation depression in the presence divalent cation sulfate salts was associated with adsorption of hydrophilic and positive complexes and/or precipitation onto molybdenite surfaces.2,22,23
5. Conclusions
The effects of four sulfate salts on molybdenite recovery were investigated. Both monovalent and divalent sulfate salts were detrimental to molybdenite flotability. The presence of Na+ and K+ salts resulted increased molybdenite oxidation/leaching, most likely at the edges and micro-edges, resulting in more negative zeta potentials. The increased electrostatic repulsion between negatively charged bubble and molybdenite surface therefore decreaseed molybdenite recovery. However, in the presence of Ca2+ and Mg2+, the depressed molybdenite was attributed to the adsorption of positively charged complexes and/or precipitation of their hydroxides, e.g. the adsorption of Ca2+, Ca(OH)+, Mg(OH)+, Mg(OH)2. As the zeta potential of molybdenite was increased in the presence of Ca2+ and Mg2+, the edge species of CaMoO4 and the adsorption of Mg(OH)2 might predominate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from National Natural Science Foundation of China under projects of 51604205 and 51774223, Natural Science Foundation of Hubei Province (2016CFB268). The supports from Fundamental Research Funds for Central Universities (WUT: 2016IVA046 and 2017IVB018) and undergraduate research foundation for independent innovation from Wuhan University of Technology (2017-ZH-A1-03 and 2017-ZH-C1-18) are gratefully acknowledged. In addition, Yingjie Li thanks the supports from Social Development Science and Technology Breakthrough Project of Shaanxi province (2015SF293).References
- G. Levay, R. S. C. Smart and W. Skinner, J. South. Afr. Inst. Min. Metall., 2001, 101, 69–75 Search PubMed.
- R. I. Jeldres, L. Forbes and L. A. Cisternas, Miner. Process. Extr. Metall. Rev., 2016, 37, 369–384 CrossRef.
- J. Drelich and J. D. Miller, Water in Mineral Processing, 2012, pp. 73–85 Search PubMed.
- S. Castro and J. S. Laskowski, KONA Powder Part. J., 2011, 29, 4–15 CrossRef.
- P. A. Moreno, H. Aral, J. Cuevas, A. Monardes, M. Adaro, T. Norgate and W. Bruckard, Miner. Eng., 2011, 24, 852–858 CrossRef.
- Z. Qiu, G. Liu, Q. Liu and H. Zhong, Colloids Surf., A, 2016, 509, 123–129 CrossRef.
- J. Laskowski, S. Castro and O. Ramos, Physicochem. Probl. Miner. Process., 2014, 50, 17–29 Search PubMed.
- T. Hirajima, G. P. W. Suyantara, O. Ichikawa, A. M. Elmahdy, H. Miki and K. Sasaki, Miner. Eng., 2016, 96, 83–93 CrossRef.
- G. Bournival, R. Pugh and S. Ata, Miner. Eng., 2012, 25, 47–53 CrossRef.
- M. Zhang, Y. Peng and N. Xu, Miner. Eng., 2015, 77, 93–98 CrossRef.
- O. Ozdemir, Physicochem. Probl. Miner. Process., 2013, 49, 511–524 Search PubMed.
- J. Quinn, W. Kracht, C. Gomez, C. Gagnon and J. Finch, Miner. Eng., 2007, 20, 1296–1302 CrossRef.
- S. Chander and D. Fuerstenau, Trans. Am. Inst. Min., Metall. Pet. Eng., 1972, 252, 62–69 Search PubMed.
- F. Lucay, L. Cisternas, E. Gálvez and A. López-Valdivieso, Miner. Metall. Process., 2015, 32, 203–208 Search PubMed.
- S. Castro, P. Rioseco and J. Laskowski, Proceedings of the 26th International Mineral Processing Congress, New Delhi, 2012 Search PubMed.
- A. Kasha, H. Al-Hashim, W. Abdallah, R. Taherian and B. Sauerer, Colloids Surf., A, 2015, 482, 290–299 CrossRef.
- O. Ramos, S. Castro and J. Laskowski, Miner. Eng., 2013, 53, 108–112 CrossRef.
- M. C. Fuerstenau, J. D. Miller and M. C. Kuhn, Chemistry of flotation, Society of Mining Engineers of American Institute of Mining, Metallurgical and Petroleum Engineers, 1985 Search PubMed.
- S. Raghavan and L. L. Hsu, Int. J. Miner. Process., 1984, 12, 145–162 CrossRef.
- H. Wan, W. Yang, W. Cao, T. He, Y. Liu, J. Yang, L. Guo and Y. Peng, Minerals, 2017, 7, 141 CrossRef.
- S. Castro, Water in Mineral Processing–Proceedings. of the First International Symposium, ed. J. Drelich, Society of Mining, Metallurgy, and Exploration, Seattle, WA, USA, 2012 Search PubMed.
- R. I. Jeldres, M. P. Arancibia-Bravo, A. Reyes, C. E. Aguirre, L. Cortes and L. A. Cisternas, Miner. Eng., 2017, 109, 10–13 CrossRef.
- E. Rebolledo, J. S. Laskowski, L. Gutierrez and S. Castro, Miner. Eng., 2017, 100, 71–74 CrossRef.
- J. O. Tabares, I. M. Ortega, J. R. Bahena, A. S. López, D. V. Pérez and A. L. Valdivieso, Proceedings of 2006 China-Mexico Workshop on Minerals Particle Technology, San Luis Potosi, Mexico, 2006 Search PubMed.
- T. Hirajima, M. Mori, O. Ichikawa, K. Sasaki, H. Miki, M. Farahat and M. Sawada, Miner. Eng., 2014, 66, 102–111 CrossRef.
- Z. Lu, Q. Liu, Z. Xu and H. Zeng, Langmuir, 2015, 31, 11409–11418 CrossRef PubMed.
- H. El-Shall, G. Zucker and K. Lofftus, Proceedings of the International Symposium on Electrochemistry in Mineral and Metal Processing, ed. P. E. Richardson, S. Srinivasan and R. Woods, 1984, pp. 96–111 Search PubMed.
- M. Zanin, I. Ametov, S. Grano, L. Zhou and W. Skinner, Int. J. Miner. Process., 2009, 93, 256–266 CrossRef.
- Y. Li, W. Li, Q. Xiao, N. He, Z. Ren, C. Lartey and A. R. Gerson, Minerals, 2017, 7, 111 CrossRef.
- E. Nowak, P. Robbins, G. Combes, E. H. Stitt and A. W. Pacek, Powder Technol., 2013, 250, 21–32 CrossRef.
- S. Castro and E. Mayta, IV Meeting of the Southern Hemisphere on Mineral Technology, and III Latin American Congress on Froth Flotation, ed. S. Castro and J. Alvarez, Chile, 1994 Search PubMed.
- W. Chimonyo, K. Corin, J. Wiese and C. O'Connor, Miner. Eng., 2017, 110, 57–64 CrossRef.
- B. Yang, S. Song and A. Lopez-Valdivieso, Physicochem. Probl. Miner. Process., 2015, 51, 181–189 Search PubMed.
- S. Kelebek, J. Colloid Interface Sci., 1988, 124, 504–514 CrossRef.
- S. Castro, A. Lopez-Valdivieso and J. Laskowski, Int. J. Miner. Process., 2016, 148, 48–58 CrossRef.
- A. López-Valdivieso, I. Madrid-Ortega, D. Valdez-Pérez, B. Yang and S. Song, Proceedings of the 9th International Mineral Processing Conference, 2012 Search PubMed.
- B. Triffett, C. Veloo, B. Adair and D. Bradshaw, Miner. Eng., 2008, 21, 832–840 CrossRef.
- M. Komiyama, K. Kiyohara, Y. Li, T. Fujikawa, T. Ebihara, T. Kubota and Y. Okamoto, J. Mol. Catal. A: Chem., 2004, 215, 143–147 CrossRef.
- C. Li and P. Somasundaran, J. Colloid Interface Sci., 1991, 146, 215–218 CrossRef.
- P. Cannon and F. Norton, Nature, 1964, 203, 750–751 CrossRef.
- Z. Wei, Y. Li, Q. Xiao and S. Song, Minerals, 2016, 6, 89 CrossRef.
- O. Ozdemir, E. Taran, M. Hampton, S. Karakashev and A. Nguyen, Int. J. Miner. Process., 2009, 92, 177–183 CrossRef.
- D. Nagaraj and R. Farinato, Proceedings of the Processing Congress Presented at the XXVII International Mineral, Santiago, Chile, 2014 Search PubMed.
- G. P. W. Suyantara, T. Hirajima, A. M. Elmahdy, H. Miki and K. Sasaki, Colloids Surf., A, 2016, 501, 98–113 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |