
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly conductive and stable graphene/PEDOT:PSS composite as a metal free cathode for organic dye-sensitized solar cells†
Jae Cheon Kima,
Md. Mahbubur Rahmanb,
Myung Jong Ju*c and
Jae-Joon Lee *a
*a
aDepartment of Energy Materials and Engineering, Research Center for Photoenergy Harvesting & Conversion Technology (phct), Dongguk University, 26 Pil-dong, 3-ga, Jung-gu, Seoul, 04620, Republic of Korea. E-mail: jjlee@dongguk.edu
bNanotechnology Research Center & Department of Energy and Materials, Konkuk University, Chungju 27478, Republic of Korea
cUlsan National Institute of Science and Technology (UNIST), School of Energy and Chemical Engineering/Centre for Dimension-Controllable Organic Frameworks, 50, UNIST, Ulsan 44919, Republic of Korea. E-mail: jmj12@unist.ac.kr
First published on 23rd May 2018
Abstract
Platinum (Pt) as the counter electrode (CE) in dye-sensitized solar cells (DSSCs) is expensive and non-optimal for cobalt(II/III) redox couples, which can tune and improve the performance of DSSCs, thus motivating the search for replacements of the Pt CE. Graphene nanoplatelets (GnPs) are possible alternatives to Pt CEs but they are mechanically unstable as CEs due to their poor substrate adhesion. Here we report a new type of PEDOT:PSS (PP)/GnP (PPG) composite that maintains the catalytic performance of GnPs with enhanced adhesion to the substrate via a conductive PEDOT matrix. The resultant PPG exhibited extremely low charge-transfer resistance (Rct) compared to Pt in its role as an electrocatalyst toward a Co(bpy)32+/3+ (bpy = 2,2′-bipyridine) redox couple, and displayed extremely high electrochemical stability for Co(bpy)33+ reduction even after 1000 cycles. The inter-stacking of GnP layers between PEDOT and PSS was confirmed by XPS and Raman spectra. It helps to delocalize charges in the PEDOT backbone and rapidly transfers electrons from the external circuit to Co(bpy)33+. This reduces the Rct and ultimately improves the photovoltaic performance. The DSSC based on Y123 sensitizer and PPG-CE showed a higher photovoltaic performance of 8.33% than its Pt counterpart does (7.99%) under the optimized conditions.
1. Introduction
Economical photovoltaic (PV) technologies are urgently needed to meet the global energy crisis of the 21st century. Although several PV technologies including dye-sensitized solar cells (DSSCs),1,2 quantum-dots,3,4 and perovskite based cells5,6 have been developed to date, their practical applications will hardly be realized unless we have large-scale capability for their low-cost fabrication with long-term stability. Among them, DSSCs are indeed a ubiquitous choice due to their low cost, easy fabrication, and relatively high power conversion efficiency (PCE).1,2 The counter electrode (CE) is a key component of a DSSC, which catalyzes the reduction of oxidized ions generated after electron injection from the redox mediator into the oxidized sensitizer. Platinum (Pt) has been widely employed as the standard choice for the CE in DSSCs due to its high catalytic activity toward iodide/triiodide (I−/I3−) and high conductivity. However, the risk of Pt corrosion by the redox species, high cost, and the rarity of Pt have highlighted the need for low-cost, easily scalable, and high photo-corrosion stable materials for use as CEs. In addition, Pt exhibits relatively poor electrocatalytic activity for cobalt (II/III)-based redox couples compared to I−/I3−.7,8 Further, the charge-transfer resistance (Rct) at the Pt/cobalt(II/III)-based electrolyte interface is generally high with poor electrochemical stability, which leads to loss of fill factor (FF), long-term stability, and PCE. Consequently, many efforts have been made to find suitable alternatives to the Pt CE and these include many carbon-based materials such as nanocarbons,9 carbon nanotubes,10,11 fullerenes,11 and graphene nanoplatelets (GnPs),11–19 metal–oxides,20,21 and conducting polymers (CPs).22,23 Carbon-based materials and conducting polymers have attracted more attention because of their abundance, and greater affordability than precious metal catalysts such as Pt and gold (Au). Moreover, they greatly reduce the Rct at the CE/electrolyte interfaces in the case of cobalt(II/III)-based electrolyte.Among them, several functionalized graphene-based nanomaterials have been reported recently as highly efficient CEs for DSSCs with cobalt(II/III)-based electrolytes, which can replace Pt.11–19 GnPs have abundant edge sites24 that are catalytically active, which allow them to display a remarkably low Rct when used with cobalt(II/III)-based electrolytes.13–16 Meanwhile, previous reports have shown that many functionalized GnPs have good physical and electrochemical stability but the adhesion capability to form a robust film formation on various substrates was not thoroughly investigated. Indeed, most GnPs only weakly adhered onto conducting substrates such as fluorine-doped tin oxide (FTO)/glass after high-temperature annealing process, and they were easily removed by slight mechanical perturbations such as wiping, blowing, and collision. This loss of catalyst would clearly reduce the electrochemical performance and stability. In this regard, PEDOT:PSS/GnP (PPG) composites have been developed to enhance the conductivity, stability, and catalytic activity of the CEs in DSSCs.25,26 PP has attracted significant interest as a prospective material for the development of CEs in DSSCs because of its high electrical and thermal conductivity, high specific surface area, good chemical stability, optical transparency, and excellent carrier mobility of graphene.25,26 Hong et al. developed a GnP/PP composite CE for N719 sensitizer- and I−/I3− mediator-based DSSCs with PCE of 4.5%, which was not compatible with that of Pt CE-based system (6.3%).27
In the present study, we developed PP/GnP (0.02, 0.1, 0.5, and 1 wt% of GnP) composite-based CEs for DSSCs by an electrospray (e-spray) method and used them in conjunction with Y123 sensitizer and Co(bpy)32+/3+ (bpy = 2,2′-bipyridine) redox couple. PP/GnPs with higher amount of GnPs were found to be more active toward Co(bpy)32+/3+ redox reaction (CRR) than Pt and possessed extremely high electrochemical stability of CRR even after 1000 cyclic voltammetric (CV) potentials. Accelerated mechanical aging causes complete detachment of the GnP electrodes without PP whereas the optimized PP/GnP CE exhibited significantly high conductivity and catalytic activity, lower Rct, and improved PCE.
2. Experimental section
2.1 Materials
Conductive grade PEDOT:PSS (PP) aqueous solution (1.3 wt% dispersion in water) was purchased from Sigma-Aldrich. Graphene nanoplatelets (GnPs) were purchased from XG Science, Inc. (USA). According to the manufacturer's specifications, they consisted of several sheets of graphene of few nanometers with particle diameters less than 2 μm and surface area of approximately 500 m2 g−1. The oxygen and nitrogen functional groups present on the edges of GnPs were ∼5.5% and ∼1.5%, respectively. Organic sensitizer of 3-{6-{4-[bis(2′,4′-dihexyloxybiphenyl-4-yl)amino-]phenyl}-4,4-dihexyl-cyclopenta-[2,1-b:3,4-b']dithiophene-2-yl}-2-cyanoacrylic acid (Y123) was purchased from Dyenamo and used without purification. Co-complexes were prepared by the procedure reported in a previous study.16 All other solvents were supplied by Aldrich Chemical Inc. and used without further purification, unless otherwise specified.2.2 Device fabrication
The steps performed in the device fabrication are summarized in the supplementary section.2.3 Characterization
The transmittance spectra of the CEs were measured by a UV-Visible spectrophotometer (Scinco, S-3100, Korea). Field emission scanning electron microscopy (FE-SEM, JSM-6700F, JEOL) and atomic force microscopy (AFM, PSIA, XE100, Korea) were used to characterize the surface morphology and topography of the CEs, respectively. The binding energy of the PP/GnP composite materials was measured by X-ray photoelectron spectroscopy (XPS, K-alpha, Thermo U.K.) equipped with a monochromatic Al Kα X-ray source (1486.6 eV). Raman shift spectra of the PP/GnP composite films were obtained by Raman spectrometer (LabRam Aramis, Horriba Jovin Yvon) using a ND:YAG laser source. Electrochemical experiments were performed using a potentiostat (CHI430A, CH Instruments Inc., USA). Cyclic voltammetry (CV) was performed with a conventional three-electrode system, where Pt-, GnP-, and PP-thin films, or PP/GnP composite films on FTO, Pilkington TEC Glass-TEC 8, Solar, 2.3 mm thickness)/glass electrodes were used as the working electrode. A Pt wire and Ag/AgCl were used as counter and reference electrodes, respectively. Electrochemical impedance spectroscopy (EIS) was measured in the frequency range of 106–0.1 Hz with the ac amplitude of 5 mV (IM6ex, Zahner-Elektrik GmbH & CoKG), and their EIS spectra were analyzed by using a Z-view software (Scribner Associates Inc., version 3.1).3. Results and discussions
The well-dispersed solution of graphene in the polymer solution plays the pivotal role of obtaining a highly conductive graphene/polymer composite film with improved electrical and physical properties by avoiding the aggregation of graphene particles in the composite film.27 In this study, different wt% of GnPs were dispersed by sonication into the aqueous-ethanolic (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) solution of PP. The dispersed GnP, PP, and PP/GnP (PPGx, x = 1, 2, 3, 4 for 0.02, 0.1, 0.5, and 1 wt% of GnP, respectively) solutions exhibited excellent stability for over one month (ESI, Fig. S1†). During the mixing and sonication of GnPs with PP, the GnP bundle was broken up and exfoliated into smaller pieces,27,28 and thus it could be deposited on FTO/glass substrates by e-spray technique (Fig. 1a). Thus, the thin films could be easily deposited on various substrates by a simple process.12,29 The thin film fabrication details are described in the (ESI†). For comparison, Pt on an FTO electrode was also fabricated, as described in the ESI. The optical transmittances of the thin films of GnPs, PP, and PPGx on the FTO are shown in Fig. 1b. The optical transmittance of the PP film was nearly 84.2% at 550 nm, whereas it was nearly 34% for the GnP film. The transmittances of the PPGx (x = 1–4) electrodes at 550 nm were decreased with the increase in the wt% of GnP, and they were approximately 65, 30, 2.4, and 0.04%, respectively as shown in Fig. 1b. Fig. 1c shows the representative photographic images of GnP, PP, and PPG4 films deposited on the FTO, and their corresponding top and cross-sectional SEM images are shown in Fig. 1d. The photographic and SEM images of other samples are shown in ESI (Fig. S2a and S2b†). The homogenous intermixing of GnPs with PP, and the incorporation of PP into the GnP interlayer increased the compactness of the films. PPG4 exhibited lower film thickness than the GnP film under the same deposition conditions (such as the amount of dispersed GnPs (1 wt%) and the deposition time (2 min) for both GnPs and PPG4). The thicknesses of the GnP and PP films were approximately 0.72 and 0.25 μm, respectively, whereas they were 0.27, 0.40, 0.48, and 0.57 μm for the PPGx (x = 1–4) films, respectively. The entire surface of the FTO was compactly covered with a homogeneous multilayer-type GnP/PP composite in the case of PPG4, which is expected to increase the charge carrier mobility in the CE of the DSSCs.
:3 v/v) solution of PP. The dispersed GnP, PP, and PP/GnP (PPGx, x = 1, 2, 3, 4 for 0.02, 0.1, 0.5, and 1 wt% of GnP, respectively) solutions exhibited excellent stability for over one month (ESI, Fig. S1†). During the mixing and sonication of GnPs with PP, the GnP bundle was broken up and exfoliated into smaller pieces,27,28 and thus it could be deposited on FTO/glass substrates by e-spray technique (Fig. 1a). Thus, the thin films could be easily deposited on various substrates by a simple process.12,29 The thin film fabrication details are described in the (ESI†). For comparison, Pt on an FTO electrode was also fabricated, as described in the ESI. The optical transmittances of the thin films of GnPs, PP, and PPGx on the FTO are shown in Fig. 1b. The optical transmittance of the PP film was nearly 84.2% at 550 nm, whereas it was nearly 34% for the GnP film. The transmittances of the PPGx (x = 1–4) electrodes at 550 nm were decreased with the increase in the wt% of GnP, and they were approximately 65, 30, 2.4, and 0.04%, respectively as shown in Fig. 1b. Fig. 1c shows the representative photographic images of GnP, PP, and PPG4 films deposited on the FTO, and their corresponding top and cross-sectional SEM images are shown in Fig. 1d. The photographic and SEM images of other samples are shown in ESI (Fig. S2a and S2b†). The homogenous intermixing of GnPs with PP, and the incorporation of PP into the GnP interlayer increased the compactness of the films. PPG4 exhibited lower film thickness than the GnP film under the same deposition conditions (such as the amount of dispersed GnPs (1 wt%) and the deposition time (2 min) for both GnPs and PPG4). The thicknesses of the GnP and PP films were approximately 0.72 and 0.25 μm, respectively, whereas they were 0.27, 0.40, 0.48, and 0.57 μm for the PPGx (x = 1–4) films, respectively. The entire surface of the FTO was compactly covered with a homogeneous multilayer-type GnP/PP composite in the case of PPG4, which is expected to increase the charge carrier mobility in the CE of the DSSCs.
 | ||
| Fig. 1 (a) Schematic of GnP, PP and GnP-PP composite thin films fabrication using e-spray technique, (b) transmittance of PP and PP/GnPs (PPGx, x = 1–4) thin films on FTO/glass substrate, (c) photographs of e-sprayed GnP, PP and PPG4 thin films deposited on FTO/glass substrates, and (d) SEM images of e-sprayed GnP, PP and PPG4 thin films deposited on FTO/glass substrates (Insets shows corresponding cross section images). | ||
XPS and Raman spectroscopy analysis of the GnP, PP, and PPG4 thin films were performed to study the chemical species and energetic vibrations of the elements (Fig. 2a and b, S3†). The XPS spectra clearly show difference in binding nature between the GnP and PP films. The C 1s spectra of PP revealed the existence of several C groups including C–C, C–O–C, and C–S at 284.5, 285.8, and 287.7 eV, respectively. The GnPs exhibited similar C 1s spectra with different C groups including C–C, C–O–C, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 284.5, 285.8, and 287.3 eV, respectively.27,28,30 The PP, GnP, and PPGx films showed similar C 1s spectra of different C groups including C–C, C–O–C, CO, and C–S at 284.5, 285.8, 287.3, and 286.7 eV, respectively. However, an additional unique peak in the PPG films appeared between the binding energy range of 289 to 294 eV (Fig. 2a and S3†). This was due to the strong π–π interaction on charge-delocalized PEDOT backbone and the existence of a non-charge doping phase in the sample.31–33 The binding energy peak due to the formation of π–π interaction did not appear in the GnP and PP films, whereas it was observed at approximately 290.8 eV for PPG1, PPG2, and PPG3 films. On the other hand, a broader and stronger peak further appeared between 289 eV to 293 eV in the PPG4 film, which could be ascribed to the delocalized charge of PEDOT due to the separation of PEDOT and PSS by the intercalation of the GnP particles.
O at 284.5, 285.8, and 287.3 eV, respectively.27,28,30 The PP, GnP, and PPGx films showed similar C 1s spectra of different C groups including C–C, C–O–C, CO, and C–S at 284.5, 285.8, 287.3, and 286.7 eV, respectively. However, an additional unique peak in the PPG films appeared between the binding energy range of 289 to 294 eV (Fig. 2a and S3†). This was due to the strong π–π interaction on charge-delocalized PEDOT backbone and the existence of a non-charge doping phase in the sample.31–33 The binding energy peak due to the formation of π–π interaction did not appear in the GnP and PP films, whereas it was observed at approximately 290.8 eV for PPG1, PPG2, and PPG3 films. On the other hand, a broader and stronger peak further appeared between 289 eV to 293 eV in the PPG4 film, which could be ascribed to the delocalized charge of PEDOT due to the separation of PEDOT and PSS by the intercalation of the GnP particles.
 | ||
| Fig. 2 (a) C 1s XPS peaks of the GnPs, PP, and PPG4 thin films. (b) Raman spectra of the GnPs, PP, and PPG4 thin films. (c) Sheet resistance, conductivity and surface area of PP/GnPs composite thin films on FTO/glass substrates. (d) AFM images of GnPs, PP and representative PP/GnPs composite thin films on FTO/glass substrates. | ||
To confirm the geometry and aggregation form of GnPs in the PPG4 film, we investigated the Raman spectra, which can provide further evidence of the molecular level interaction between the GnPs and PP (Fig. 2b). As expected, the GnPs exhibited the dominant G band at approximately 1583 cm−1. In the case of PPG4, the G and D bands were blue shifted up to 7.87 and 10.3 cm−1, respectively. This Raman shift might have been induced from the enhanced electron–phonon activity.34 The electron distribution on the interconnecting GnP geometry improves,34 which implies that intermolecular electron transfer is faster between GnPs and PP.27,35 This suggests that the use of the PPG4 film as the CE of DSSCs would be advantageous. In addition, the conductivities of the GnP and PP films were 1.28 and 0.55 S cm−1, whereas they were 0.16, 1.29, 2.58, and 6.24 S cm−1 for PPGx (x = 1–4) films, respectively. The PPG1 film showed lower conductivity than PP and GnP films because of the delocalized PP due to the intercalating GnPs, as mentioned above. However, conductivity of PPG2 to PPG4 significantly enhanced, and thus PPG4 showed approximately five times higher conductivity than GnP CE because of the inter-stacking of GnPs particles. AFM analysis was performed to confirm the surface topography of the thin films (Fig. 2d and S4†). The surface area of the GnP and PP films were 122.65 and 103.75 cm2, respectively, whereas the PPGx (x = 1–4) films exhibited the surface area of 106.92, 110.63, 118.4, and 123.5 cm2, respectively. Additionally, mechanical stress test was performed for all films by the ‘scotch tape test’. The photographic images and surface morphological SEM images before and after the ‘scotch tape test’ are shown in Fig. S5 and S6,† respectively. After the tape test, the GnPs were completely peeled off from the FTO surface, whereas PP and PPGx (x = 1–4) showed strong adhesion to FTO, which confirmed the suitability of PPGx (x = 1–4) CEs for DSSCs.
Prior to their application as CEs in DSSCs, the electrocatalytic activity of the as-deposited Pt, GnPs, PP, and PPGx (x = 1–4) on FTO/glass was systematically examined for the Co(bpy)32+/3+ (bpy = 2,2′-bipyridine) redox mediator. Fig. 3a shows the CVs of the Pt, GnP, PP, and PPG4 films, and the CVs of the PPGx (x = 1–3) films are shown in Fig. S7a.† The concentration of the Co(bpy)32+/3+ redox mediator was 100 times lower than the concentration of the electrolyte used to evaluate the DSSC performance. LiClO4 of 0.1 M concentration was used as the supporting electrolyte for the CV measurement. The PPG4 film exhibited the highest anodic (Ipa, Table 1) and cathodic (Ipc) peak currents compared to all other electrodes. The lower peak potential separation (ΔEpp) revealed that the PPG4 film had better electrocatalytic activity for the reduction of Co(bpy)33+ to Co(bpy)32+, and therefore it was considered as the optimized CE among the PPGx (x = 1–4) electrodes.
 | ||
| Fig. 3 (a) Cyclic voltammograms (CV) of the Co(bpy)32+/3+ redox couple at the Pt, GnP, PP, and PPG4 CEs; scan rate 10 mV s−1. (b) Nyquist plots of the Pt, GnP, PP and PPG4 symmetrical dummy cells; the inset showed the magnified Nyquist plot of the Pt dummy cell in the high-frequency region, and the equivalent circuit model for fitting the EIS data. (c) Linear sweep voltammograms (LSV) of the Co(bpy)32+/3+ redox couples at the Pt, GnP, PP, and PPG4 CEs-based dummy cells; scan rate 10 mV s−1. (d) Chrono-amperometric (CA) plots of the Pt, GnP, PP, and PPG4 CEs-based dummy cells. | ||
| CEs | Ipa (mA cm−2) | Rct (Ω cm2) | J0, EIS (mA cm−2) | RLSV (Ω cm2) | J0, LSV (mA cm−2) | A (cm2) |
|---|---|---|---|---|---|---|
| Pt | 0.087 | 1.19 ± 0.06 | 21.7 ± 1.08 | 4.77 ± 0.24 | 5.42 ± 0.27 | 0.58 |
| GnPs | 0.085 | 1.29 ± 0.06 | 20.02 ± 1.00 | 4.47 ± 0.22 | 7.70 ± 0.38 | 0.56 |
| PP | 0.062 | 3.52 ± 0.17 | 7.35 ± 0.36 | 5.63 ± 0.28 | 4.59 ± 0.23 | 0.41 |
| PPG1 | 0.066 | 4.68 ± 0.23 | 5.52 ± 0.28 | 3.35 ± 0.17 | 5.77 ± 0.29 | 0.44 |
| PPG2 | 0.075 | 2.29 ± 0.11 | 11.30 ± 0.56 | 4.48 ± 0.22 | 5.95 ± 0.29 | 0.49 |
| PPG3 | 0.088 | 0.71 ± 0.03 | 36.20 ± 1.81 | 3.26 ± 0.16 | 7.92 ± 0.39 | 0.58 |
| PPG4 | 0.103 | 0.07 ± 0.01 | 361.81 ± 18.09 | 3.14 ± 0.16 | 8.23 ± 0.41 | 0.68 |
The electrocatalytic behaviors of these electrodes were further characterized with symmetric dummy cells that were tested with EIS and linear scan voltammetry (LSV), in which the Co(bpy)32+/3+ electrolyte solution was sandwiched between two identical CEs. The Nyquist plots obtained from the Pt, GnP, PP, and PPG4 dummy cells for the Co(bpy)32+/3+ redox couple are shown in Fig. 3b. All Nyquist plots showed two distinct semicircles corresponding to the electron transfer resistance (Rct) at the CE/electrolyte interface and the Nernst diffusion process in the high and low-frequency regions, respectively. The EIS spectra of PPGx (x = 1–3) dummy cells are presented in Fig. S7b.† The numerical data obtained from the EIS spectra of all dummy cells are summarized in Table 1. These values were obtained by fitting the EIS spectra with an appropriate equivalent circuit (EC) model (inset of Fig. 3b). The Rct at the CE/electrolyte interface is a vital parameter in evaluating the catalytic behavior of CE materials in DSSCs. Generally, a low Rct corresponds to a decrease in the total internal resistance, effective reduction of a redox couple, and efficient regeneration of dye, which concurrently enhances the photocurrent (Jsc) and fill factor (FF) in a DSSC.36
The PPG4-CE exhibited a significantly low Rct value (0.07 Ω cm2) compared to the Pt (1.19 Ω cm2), GnP (1.29 Ω cm2), PP (3.52 Ω cm2), and PPGx (x = 1–3) (4.68, 2.29, 0.71 Ω cm2, respectively) electrodes. The relationship between Rct and the exchange current density (J0) can be described by the following equation.37–39
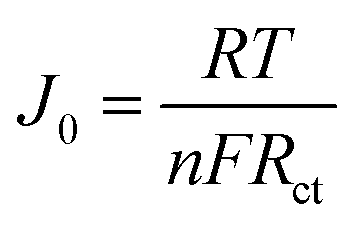 | (1) |
Here, R is the gas constant, T is the absolute temperature, n is the number of electrons, and F is the Faraday constant. Using eqn (1), the exchange current density from EIS (J0,EIS) in the Pt, GnP, PP, and PPG4 CE-based dummy cells were determined to be 21.7, 20, 7.35, and 361.81 mA cm−2, respectively, whereas it was 5.52, 11.30, and 36.20 mA cm−2 for PPGx (x = 1–3), respectively (Table 1). This clearly suggested that the PPG4-CE had the highest electron transfer kinetics at the CE/electrolyte interface.
Fig. 3c shows the LSV plots of the Pt, GnP, PP, and PPG4 CE-based dummy cells. The LSV plots of other PPGx (x = 1–3) CE-based dummy cells are shown in Fig. S6c.† The inverse slope of the LSV at ∼0 V was determined to characterize the catalytic activity of the CE materials.35 The overall cell resistance (RLSV) of the dummy cells can be determined at low current densities and these are summarized in Table 1. The LSV plot of the PPG4 dummy cell exhibits a steeper slope than all other CEs, indicating that the RLSV of PPG4 (3.14 Ω cm2) is much lower than that of Pt (4.77 Ω cm2) and other CEs. The RLSV values of GnPs, PP, and PPGx (x = 1–3) were 5.63, 3.35, 4.48, 4.35, and 3.26 Ω cm2, respectively. Using eqn (1) and the RLSV values of all CE-based dummy cells, the exchange current densities (J0,LSV) for Pt, GnPs, PP, and PPG4 were determined to be 5.42, 7.7, 4.59, and 8.23 mA cm−2, respectively, whereas it was 5.77, 5.95, and 7.92 mA cm−2 for PPGx (x = 1–3), respectively (Table 1). This trend is in good agreement with that obtained from EIS, and clearly suggests that the highest electron transfer kinetics occurs at the PPG4 electrode.
Chronoamperometry (CA) measurements were performed at room temperature to investigate the mass transport properties of Co(bpy)32+/3+ at the Pt, GnP, PP, and PPGx (x = 1–4) CEs. Fig. 3d shows the CA plots for the Pt, GnP, PP, and PPG4 CE dummy cells. The current follows semi-infinite Cottrell-like decay after a certain potential range.40 The current decays linearly with t−1/2 until the current profiles of each CE merge to form a single linear profile and attain a steady state value, which is equal to the limiting current (Jlim). The extrapolations of both linear components of the CA plots intersect at a point, termed as the transition time (ttrans), which was used to measure the diffusion coefficient (D) of the Co(bpy)32+/3+ redox mediator according to eqn (2) and (3).40
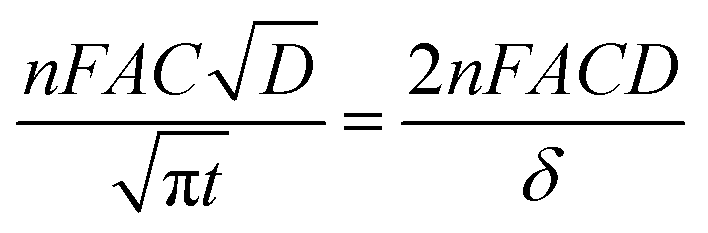 | (2) |
 | (3) |
From the data and eqn (3), the value of D for PPG4 was determined to be approximately 9.30 × 10−6 cm2 s−1, which was higher than that of Pt (approximately 7.09 × 10−6 cm2 s−1) and all other CEs. This strongly suggested the higher mass transport capacity of PPG4 among the CEs. Additionally, Jlim of the PPG4-CE was much higher than that of Pt and its other counterparts (Fig. 3d), which was due to lower Rct of PPG4 (Fig. 3b). The catalytic activities of CEs depend significantly on the surface roughness and surface area between the CEs and the redox couples.36,37 In addition, the effective surface area values of all the CEs calculated according to the Randles–Sevcik equation (see ESI: eqn (S1)†)41,42 are summarized in Table 1. As can be seen, the estimated effective surface area of the Pt and PPG4 electrodes were 0.58 and 0.68 cm2, respectively, which were in good agreement with the aforementioned AFM measurements (Fig. 2c), indicating that the intrinsic catalytic activity of the PPG4 electrode is higher than that of the Pt electrode.
To examine the electrochemical stability of the CEs, freshly assembled dummy cells were first investigated with CV at room temperature and then subjected to EIS cycles, followed by additional CV measurements (Fig. 4a and S8†). Fig. 4a shows the EIS plots of Pt and PPG4 dummy cells. The EIS plots of other dummy cells are shown in Fig. S8.† The CV cycling does not induce any significant change in the semicircles in both high- and low-frequency regions of the EIS spectrum for all samples, which demonstrated that the Co(bpy)32+/3+ redox couples were invariant to the electrode surface conditions.11,14,43 Nevertheless, the Pt electrode showed significantly high difference in Rct in the EIS after CV cycling than all other CEs. This can be ascribed to a substantial change in the surface morphology of the Pt CE after CV cycling because of its poor stability in the Co(bpy)32+/3+ electrolyte, as depicted in some other reports.11,14,43 The Rct at the CE/electrolyte interface of the PPG4-CE was 0.1 Ω cm2 in the first cycle, which increased to 0.30 Ω cm2 after the 10th cycle, whereas its Pt counterpart exhibited Rct of 3.17 Ω cm2 in the first cycle and 7.38 Ω cm2 in the 10th cycle (Fig. 4b and Table S1†). Thus, it can be concluded that the PPG4 film in the Co(bpy)32+/3+ redox medium has better electrochemical stability under prolonged potential cycling.
 | ||
| Fig. 4 (a) Nyquist plots of the Pt and PPG4 CEs-based dummy cells under potential cycling in an acetonitrile solution of Co(bpy)32+/3+. The sequence of the measurements was as follows: 100 × CV scans (from 0 V → 1 V → −1 V → 0 V→ 1 V; scan rate 50 mV s−1); 30 s relaxation at 0 V; EIS measurements at 0 V from 106 to 0.1 Hz. This sequence of electrochemical stability testing was repeated 10 times. (b) Rct changes of the Pt- and PPG4-CEs versus the EIS scan number. | ||
After the in-depth evaluation of the Pt, GnP, PP, and PPGx (x = 1–4) CEs, their electrocatalytic activity in actual DSSCs were examined with the Co(bpy)32+/3+ redox couple by using Y123 sensitizer with a CDCA co-adsorbent. The current–voltage (J–V) characteristics of the Pt, GnP, PP, and PPG4 CE-based DSSCs are shown in Fig. 5a. The J–V characteristics of other PPGx (x = 1–3) CE-based DSSCs are shown in Fig. S9a.† The photovoltaic parameters (PV) parameters of the DSSCs are summarized in Table 2. The PPG4 CE-based DSSCs exhibited Jsc, Voc, and FF of 13.64 mA cm−2, 0.92 V, and 67.01%, respectively, with PCE of 8.33%, whereas the Pt CE-based DSSCs exhibited Jsc, Voc, and FF of 13.27 mA cm−2, 0.92 V, and 65.52%, respectively, with PCE of 7.99%. The PPG4-CE showed net enhancement in PCE of approximately 4.25% compared to Pt CE, mostly due to the higher Jsc and FF, which can be attributed to the lower Rct at the PPG4/electrolyte interface. Meanwhile, the GnP CE exhibited decreased Jsc (12.74 mA cm−2) compared to PPG4 and Pt-CEs, and had PCE of 7.61%. On the other hand, the PP-CE showed significantly low FF and Jsc compared to PPG4, Pt, and GnP CE-based DSSCs. This was attributed to the fact that the large size of the PSS chain prevented direct contact between the redox couples on the active sites of PEDOT,44–46 which concurrently increased the Rct at the PP/electrolyte interface. Meanwhile, the PPG1-CE showed the lowest PCE (6.46%) due to significantly low Jsc (11.31 mA cm−2) and FF (60.83%). EIS measurements were performed on the DSSCs to further elucidate the improved performance of the PPG4-CE compared to the Pt, GnP, PP, and PPGx (x = 1–3) CEs. The high-frequency regions of the Nyquist plots of the DSSCs with the Pt, GnP, PP, and PPG4 CEs are shown in Fig. 5b.47,48 The Nyquist plots of other CE-based DSSCs are shown in Fig. S9b.† The numerical data calculated with an EC (inset of Fig. S9b†) in the high-frequency region are summarized in Table S1.† The first semicircle of the DSSCs with the PPG4-CE is significantly smaller than that of all other CEs (Fig. 5c and Table S1†), which could enhance the DSSC performance, particularly with regard to Jsc and FF.
 | ||
| Fig. 5 (a) Current–voltage (J–V) characteristics of the DSSCs with the Pt, GnP, PP and PPG4 CEs under one sun illumination (AM 1.5G). The TiO2 film thickness and active area are 8.5 (5.5 + 3) μm and 0.16 cm2 with a black metal mask, respectively. (b) Nyquist plots of the same DSSCs obtained at a forward bias of −0.9 V under dark conditions. (c) Charge-transfer resistance (Rct) of the same DSSCs. | ||
| CEs | Jsc (mA cm−2) | Voc (V) | FF (%) | PCE (%) |
|---|---|---|---|---|
| Pt | 13.27 ± 0.09 | 0.92 ± 0.01 | 65.52 ± 0.30 | 7.99 ± 0.01 |
| GnPs | 12.74 ± 0.02 | 0.91 ± 0.01 | 65.69 ± 0.23 | 7.61 ± 0.04 |
| PP | 12.27 ± 0.07 | 0.94 ± 0.01 | 60.72 ± 0.28 | 7.03 ± 0.07 |
| PPG1 | 11.31 ± 0.01 | 0.94 ± 0.01 | 60.83 ± 0.30 | 6.46 ± 0.19 |
| PPG2 | 13.16 ± 0.42 | 0.93 ± 0.01 | 62.61 ± 0.25 | 7.66 ± 0.32 |
| PPG3 | 13.39 ± 0.07 | 0.93 ± 0.03 | 65.39 ± 0.66 | 8.12 ± 0.13 |
| PPG4 | 13.64 ± 0.31 | 0.92 ± 0.01 | 67.01 ± 0.90 | 8.33 ± 0.08 |
4. Conclusions
Graphene nanoplatelets (GnPs) and PEDOT:PSS (PP) composite films [PPGx, (x = 1–4)] were prepared by an electrospray method from the mixture solutions of different wt% of GnPs and PP on FTO/glass substrates. Among these, PPG4 film was found to be most suitable as Pt-free alternative CEs for the DSSCs with Co(bpy)32+/3+ redox couple. The conductivity and surface area of the PPG4 thin film was higher than that of Pt. The inter-stacking of the GnP layer between PEDOT and PSS was proved by XPS and Raman spectra. It could delocalize charges of the PEDOT back bone, and rapidly transfer electrons from the external circuit to Co(bpy)33+, thus reducing the charge-transfer resistance and improving the photovoltaic performance. The PPG4 film exhibited lowest charge-transfer resistance (Rct) at the PPG4/electrolyte interface. The PPG4-based DSSC exhibited a PCE of 8.33%, which was higher than that of Pt (7.99%). The improved PCE of the PPG4 CE-based DSSC is mainly caused by higher Jsc and FF compared to other DSSCs, which could also be ascribed to the low Rct at the PPG4/electrolyte interface, high conductivity, and high surface area. Additionally, the mechanical adhesion and stability of all PPG films on FTO was enhanced significantly while the film thickness is much decreased for more facile and practical device fabrication application. Most importantly, the electrochemical stability of the PPG4-CE for the Co(bpy)32+/3+ redox couple was better than that of Pt CE, indicating that PPG4 can be used as an effective Pt-free CE material in DSSCs with Co(bpy)32+/3+ redox couple in conjunction with an appropriate sensitizer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF-2015M1A2A2054996, NRF-2016R1A2B2012061). It was also supported by the Technology Development Program to Solve Climate Changes of the National Research Foundation (NRF), funded by the Ministry of Science, ICT & Future Planning (NRF-2016M1A2A2940912). This work was also supported by the Dongguk University Research Fund of 2016.Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–743 CrossRef.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, F. E. CurchodBasile, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, K. NazeeruddinMd and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef PubMed.
- R. Vogel, P. Hoyer and H. Weller, J. Phys. Chem., 1994, 98, 3183–3188 CrossRef.
- H. K. Jun, M. A. Careem and A. K. Arof, Renewable Sustainable Energy Rev., 2013, 22, 148–167 CrossRef.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S.-C. Ryu, J.-W. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef PubMed.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef PubMed.
- J.-H. Yum, E. Baranoff, F. Kessler, T. Moehl, S. Ahmad, T. Bessho, A. Marchioro, E. Ghadiri, J.-E. Moser, C. Yi, M. K. Nazeeruddin and M. Grätzel, Nat. Commun., 2012, 3, 631 CrossRef PubMed.
- M. J. Ju, I. T. Choi, M. Zhong, K. Lim, J. Ko, J. Mohin, M. Lamson, T. Kowalewski, K. Matyjaszewski and H. K. Kim, J. Mater. Chem. A, 2015, 3, 4413–4419 Search PubMed.
- M. Janani, P. Srikrishnarka, S. V. Nair and A. S. Nair, J. Mater. Chem. A, 2015, 3, 17914–17938 Search PubMed.
- L. Kavan, Top. Curr. Chem., 2014, 348, 53–94 CrossRef PubMed.
- S.-M. Jung, I. T. Choi, K. Lim, J. Ko, J. C. Kim, J.-J. Lee, M. J. Ju, H. K. Kim and J.-B. Baek, Chem. Mater., 2014, 26, 3586–3591 CrossRef.
- I.-Y. Jeon, H.-J. Choi, M. J. Ju, I. T. Choi, K. Lim, J. Ko, H. K. Kim, J. C. Kim, J.-J. Lee, D. Shin, S.-M. Jung, J.-M. Seo, M.-J. Kim, N. Park, L. Dai and J.-B. Baek, Sci. Rep., 2013, 3, 2260 CrossRef PubMed.
- M. J. Ju, J. C. Kim, H.-J. Choi, I. T. Choi, S. G. Kim, K. Lim, J. Ko, J.-J. Lee, I.-Y. Jeon, J.-B. Baek and H. K. Kim, ACS Nano, 2013, 7, 5243–5250 CrossRef PubMed.
- M. J. Ju, I.-Y. Jeon, J. C. Kim, K. Lim, H.-J. Choi, S.-M. Jung, I. T. Choi, Y. K. Eom, Y. J. Kwon, J. Ko, J.-J. Lee, H. K. Kim and J.-B. Baek, Adv. Mater., 2014, 26, 3055–3062 CrossRef PubMed.
- I.-Y. Jeon, M. J. Ju, J. Xu, H.-J. Choi, J.-M. Seo, M.-J. Kim, I. T. Choi, H. M. Kim, J. C. Kim, J.-J. Lee, H. K. Liu, H. K. Kim, S. Dou, L. Dai and J.-B. Baek, Adv. Funct. Mater., 2015, 25, 1170–1179 CrossRef.
- L. Kavan, J.-H. Yum and M. Grätzel, Nano Lett., 2011, 11, 5501–5506 CrossRef PubMed.
- L. Kavan, P. Liska, S. M. Zakeeruddin and M. Grätzel, Electrochim. Acta, 2016, 195, 34–42 CrossRef.
- L. Kavan, J.-H. Yum and M. Graetzel, Electrochim. Acta, 2014, 128, 349–359 CrossRef.
- I.-Y. Jeon, H. M. Kim, I. T. Choi, K. Lim, J. Ko, J. C. Kim, H.-J. Choi, M. J. Ju, J.-J. Lee, H. K. Kim and J.-B. Baek, Nano Energy, 2015, 13, 336–345 CrossRef.
- G. R. Li, J. Song, G. L. Pan and X. P. Gao, Energy Environ. Sci., 2011, 4, 1680–1683 Search PubMed.
- X. Xin, M. He, W. Han, J. Jung and Z. Lin, Angew. Chem., Int. Ed., 2011, 50, 11739–11742 CrossRef PubMed.
- J. M. Pringle, V. Armel and D. R. MacFarlane, Chem. Commun., 2010, 46, 5367–5369 RSC.
- D. K. Hwang, D. Song, S. S. Jeon, T. H. Han, Y. S. Kang and S. S. Im, J. Mater. Chem. A, 2014, 2, 859–865 Search PubMed.
- H. N. Tsao, J. Burschka, C. Yi, F. Kessler, M. K. Nazeeruddin and M. Grätzel, Energy Environ. Sci., 2011, 4, 4921–4924 Search PubMed.
- I.-Y. Jeon, H.-J. Choi, M. Choi, J.-M. Seo, S.-M. Jung, M.-J. Kim, S. Zhang, L. Zhang, Z. Xia, L. Dai, N. Park and J.-B. Baek, Sci. Rep., 2013, 3, 1810 CrossRef PubMed.
- W. Hong, Y. Xu, G. Lu, C. Li and G. Shi, Electrochem. Commun., 2008, 10, 1555–1558 CrossRef.
- H. N. Tsao, C. Yi, T. Moehl, J.-H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, ChemSusChem, 2011, 4, 591–594 CrossRef PubMed.
- D. Yoo, J. Kim and J. H. Kim, Nano Res., 2014, 7, 717–730 CrossRef.
- G. H. Kim, D. H. Hwang and S. I. Woo, Phys. Chem. Chem. Phys., 2012, 4, 3530–3536 RSC.
- H. Han, H. Kim, D. Y. Kim, S. M. Jo and S. Y. Jang, ACS Nano, 2010, 4, 3503–3509 CrossRef PubMed.
- Y. S. Hsiao, W. T. Whang, C. P. Chen and Y. C. Chen, J. Mater. Chem., 2008, 18, 5948–5955 RSC.
- B. Q. Su, S. Pangm, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef.
- C.-C. Teng, C.-C. M. Ma, C.-H. Lu, S.-Y. Yang, S.-H. Lee, M.-C. Hsiao, M.-Y. Yen, K.-C. Chiou and T.-M. Lee, Carbon, 2011, 49, 5107–5116 CrossRef.
- J.-Y. Ouyang, Q. Xu, C.-W. Chu, Y. Yang, L. Gang and J. Shinar, Polymer, 2004, 45, 8443–8450 CrossRef.
- A. C. Ferrari1 and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef PubMed.
- M. Zhang, W. Yuan, B. Yao, C. Li and G. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 3587–3593 Search PubMed.
- L. Kavan, H. Krysova, P. Janda, H. Tarabkova, Y. Saygili, M. Freitag, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Electrochim. Acta, 2017, 251, 167–175 CrossRef.
- L. Kavan, Current Opinion in Electrochemistry, 2017, 2, 88–96 CrossRef.
- S. Das, P. Sudhagar, V. Verma, D. Song, E. Ito, S. Y. Lee, Y. S. Kang and W. Choi, Adv. Funct. Mater., 2011, 21, 3729–3736 CrossRef.
- S. Hou, X. Cai, H. Hongwei, X. Yu, M. Peng, K. Yan and D. Zou, Energy Environ. Sci., 2013, 6, 3356–3362 Search PubMed.
- M. Liberatore, A. Petrocco, F. Caprioli, C. La Mesa, F. Decker and C. A. Bignozzi, Electrochim. Acta, 2010, 35, 4025–4029 CrossRef.
- N. Papageorgiou, Coord. Chem. Rev., 2004, 248, 1421–1446 CrossRef.
- Y. Xue, J. Liu, H. Chen, R. Wang, D. Li, J. Qu and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 12124–12127 CrossRef PubMed.
- M. J. Ju, I.-Y. Jeon, K. Lim, J. C. Kim, H.-J. Choi, I. T. Choi, Y. K. Eom, Y. J. Kwon, J. Ko, J.-J. Lee, J.-B. Baek and H. K. Kim, Energy Environ. Sci., 2014, 7, 1044–1052 Search PubMed.
- Y. Saito, T. Kitamura, Y. Wada and S. Yanagida, Chem. Lett., 2002, 31, 1060–1061 CrossRef.
- M. M. Rahman, N. C. D. Nath and J.-J. Lee, Isr. J. Chem., 2015, 55, 990–1001 CrossRef.
- Y. Saito, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Photochem. Photobiol., A, 2004, 164, 153–157 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02668h |
| This journal is © The Royal Society of Chemistry 2018 |