DOI:
10.1039/C8RA02658K
(Review Article)
RSC Adv., 2018,
8, 17535-17550
1,3,5-Triphenylbenzene: a versatile photoluminescent chemo-sensor platform and supramolecular building block
Received
27th March 2018
, Accepted 26th April 2018
First published on 14th May 2018
Abstract
Fluorescence chemo-sensors for species of environmental and biological significance have emerged as a major research area in recent years. In this account, we describe fluorescence quenching as well as enhancement-based chemo-sensors obtained by employing C3-symmetric 1,3,5-triphenylbenzene (1,3,5-TPB) as the fluorescence signalling unit. 1,3,5-TPB is a thermally and photochemically stable fluorescent platform with π-electron-rich characteristics. Starting from this platform, supramolecular, discrete, triphenylbenzene-carbazole, covalent-organic framework, covalent-organic polymer and conjugated polymer based sensors have been developed for the selective detection of polynitroaromatic compounds, trinitrotoluene (TNT), dinitrotoluene (DNT) and picric acid (PA). Tris-salicylaldimine Schiff bases have been synthesized for the selective sensing of fluoride ions through a fluorescence turn-on mechanism. It is likely that it should be possible to develop other highly selective and sensitive chemo-sensors by incorporating 1,3,5-TPB as the fluorophore unit.
 Pratap Vishnoi | P. Vishnoi is presently a Post-Doctoral Fellow with Professor C. N. R. Rao (FRS) at the Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Bangalore. He was born in Bukaram sotan (Nagaur), Rajasthan. He obtained his BSc from Maharaja Ganga Singh University, Bikaner (formerly Bikaner University) and his MSc from the Department of Chemistry, Rajasthan University, Jaipur. He received his PhD from the Department of Chemistry, Indian Institute of Technology Bombay (IITB), Mumbai, under the supervision of Professor R. Murugavel. His research interests are in the field of photoactive materials, chemo-sensors and nanomaterials. |
 Dhananjayan Kaleeswaran | D. Kaleeswaran is presently a Research Associate at the Department of Chemistry, Indian Institute of Technology Bombay (IITB), Mumbai. He was born in Sattur, Tamil Nadu. He obtained his BSc from Sri Ramasamy Naidu Memorial College, Sattur, and his MSc. in chemistry from VHNSN College, Virudhunagar. He received his PhD from the Department of Chemistry, IITB, Mumbai, under the supervision of Prof. R. Murugavel. His research interests are in the area of porous and photoactive materials. |
 Ramaswamy Murugavel | R. Murugavel is presently the Biswas Palepu Distinguished Chair Professor and J. C. Bose National Fellow at the Department of Chemistry, IIT-Bombay. He is a fellow of the Indian Academy of Sciences, the Indian National Science Academy, and Royal Society of Chemistry. He is a recipient of the Swarnajayanti Fellowship, DAE-SRC Outstanding Investigator Award, C. N. R. Rao National Prize in Chemical Sciences, and S. C. Bhattacharya Award for Excellence in Basic Sciences. His current research interest lies in developing new generations of molecular magnets, porous materials, phosphate-based ceramic materials and catalysts for energy-related applications. |
1. Introduction
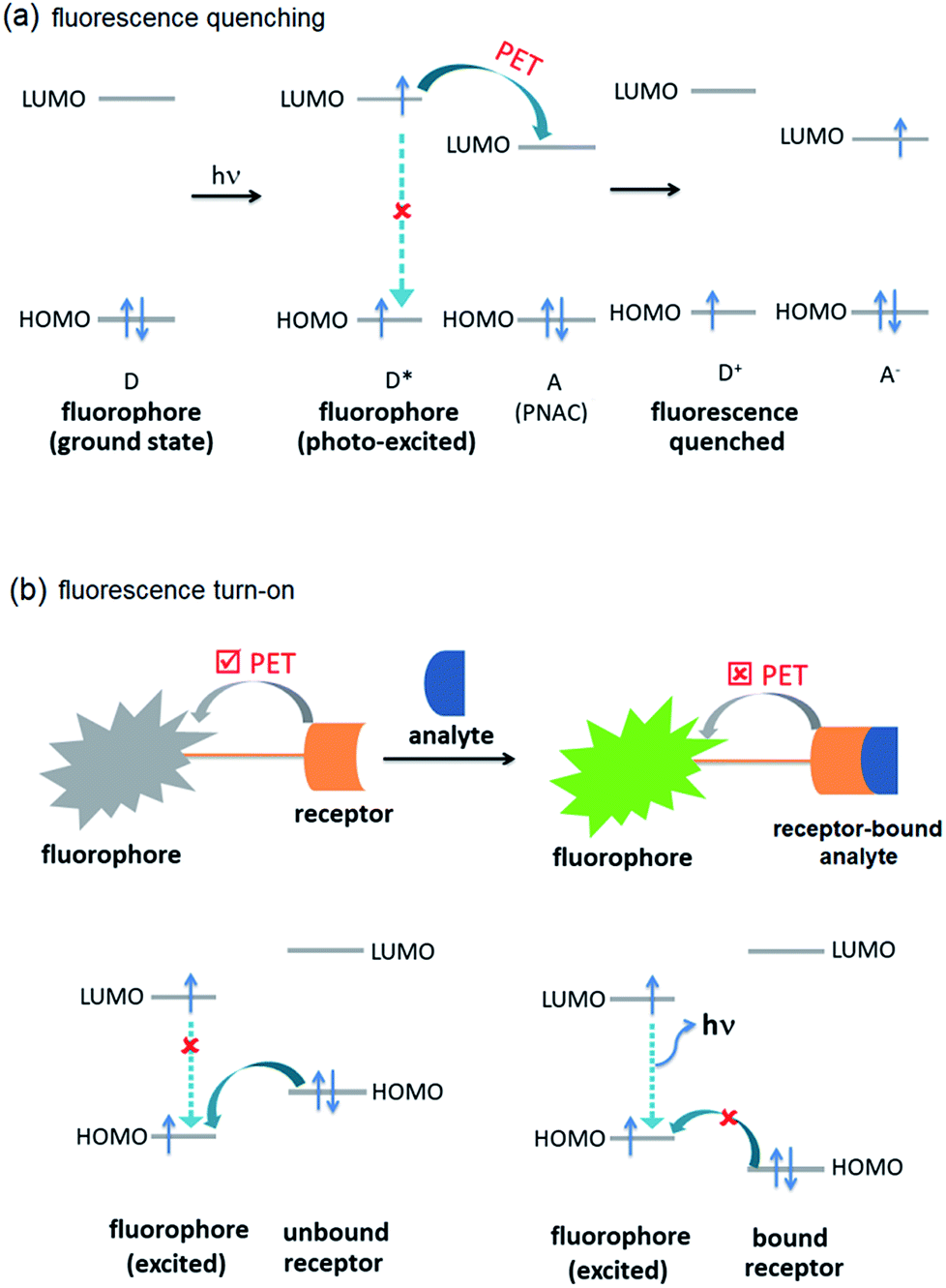
The selective and efficient detection of environmentally and biologically significant analytes has become a major area of research.1 One of the most common classes of such analytes is polynitroaromatic compounds (PNACs) that include trinitrotoluene (TNT), dinitrotoluene (DNT), picric acid (PA), etc. PNACs are high energy density materials and explode on applying external stimuli, and therefore cause security threats.2–5 Some of these PNACs are non-biodegradable environmental pollutants as well. For instance, TNT causes eye, liver and neurological damages as well as other diseases such as anaemia and skin diseases.6,7 Picric acid causes skin irritation and allergy, cancer and respiratory disorder and liver damage.7,8 PNACs are chemically non-reactive and charge-neutral species and, therefore, their chemo-sensing is challenging.2 The presence of electron-withdrawing –NO2 groups, however, makes them good electron-acceptors. The commonest approach to develop a sensor for electron-acceptors includes electron-donor systems that can effectively form donor–acceptor (D–A) interactions. Fluorescence spectroscopy based sensing methods have received much attention because they are fast and efficient, and can easily be incorporated into portable microelectronic devices.1–5 Typically, the fluorescence intensity of the sensor is attenuated by PNACs through intermolecular photo-induced electron transfer (PET) (Fig. 1a). In an intermolecular PET, a fluorophore (donor) is likely to donate the excited electron to the PNAC (acceptor) to form a D–A complex which returns to the ground state without photon emission. In some cases, ground state D–A complexation also contributes to the fluorescence attenuation. Several fluorescent organic π-conjugated polymers have been reported to show fluorescence quenching on interaction with PNACs. Typical of these sensors are poly(phenyleneethynylene),9 poly(phenylenevinylene),10 polysilanes,11,12 and polymetalloles.11,13 Small organic molecules, such as pyrene,14 anthracene,15 and coumarin,16 have been shown to be efficient chemo-sensors.
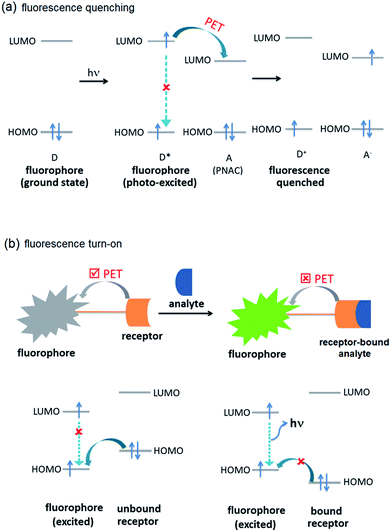 |
| | Fig. 1 Molecular orbital schematic presentation of PET between (a) fluorophore (D) and quencher (A), and (b) fluorophore and receptor. | |
Another development in the area of chemo-sensing is the case of fluorescence turn-on sensors.17,18 Typically, fluorescence turn-on sensors consist of fluorophore and guest-receptor units, connected to each other by a covalent linkage. The pristine sensors exhibit very weak fluorescence due to intramolecular PET (Fig. 1b). On binding with analytes, the PET becomes unfavourable and, therefore, the sensors exhibit fluorescence turn-on/or enhancement. These sensors are more sensitive compared to those based on fluorescence quenching. They have been successfully applied in the detection of inorganic ions, such as Al3+, Zn2+, Cu2+, CN−, F− and others, with a high sensitivity and selectivity.19–23
Given such useful properties associated with π-electron-rich small organic molecules and π-conjugated organic polymers, we intend to explore the photoluminescence of C3-symmetric 1,3,5-triphenylbenzene (1,3,5-TPB) systems and their application in chemo-sensors of PNACs and other analytes. 1,3,5-TPB is a thermally and photochemically stable fluorophore with π-electron-rich characteristics.24,25 The study of its physical properties is of current interest. With different substitutions at the peripheral phenyl rings, 1,3,5-TPB has emerged as one of the most useful building blocks for a wide variety of organic light emitting,26,27 discotic liquid crystal28,29 and high-porosity materials.30,31 For example, carboxylic acid substituted 1,3,5-TPB is a rigid building block for metal–organic frameworks (MOFs).32–35 Similarly, amine (–NH2),31,35 aldehyde (–CHO),31,36 boronic acid (–B(OH)2),37 and cyano (–CN)38 substituted 1,3,5-TPBs have been employed for the preparation of covalent-organic frameworks (COFs).
In this account, we present our recent results on the synthesis and chemo-sensing properties of systems that have been derived from the 1,3,5-TPB fluorophore. Firstly, sensors based on supramolecules of 1,3,5-TPB and the role of non-covalent interactions on the sensing efficiency will be discussed. Then, sensors based on covalently linked 1,3,5-TPB units will be presented. Finally, the development of fluorescence turn-on sensors and their fluoride sensing ability will be summarized. A few other materials based on 1,3,5-TPB fluorophores, developed by other research groups, will also be presented.
2. Polynitroaromatic compound (PNAC) sensing
2.1. Supramolecular 1,3,5-tris(4′-aminophenyl)benzene
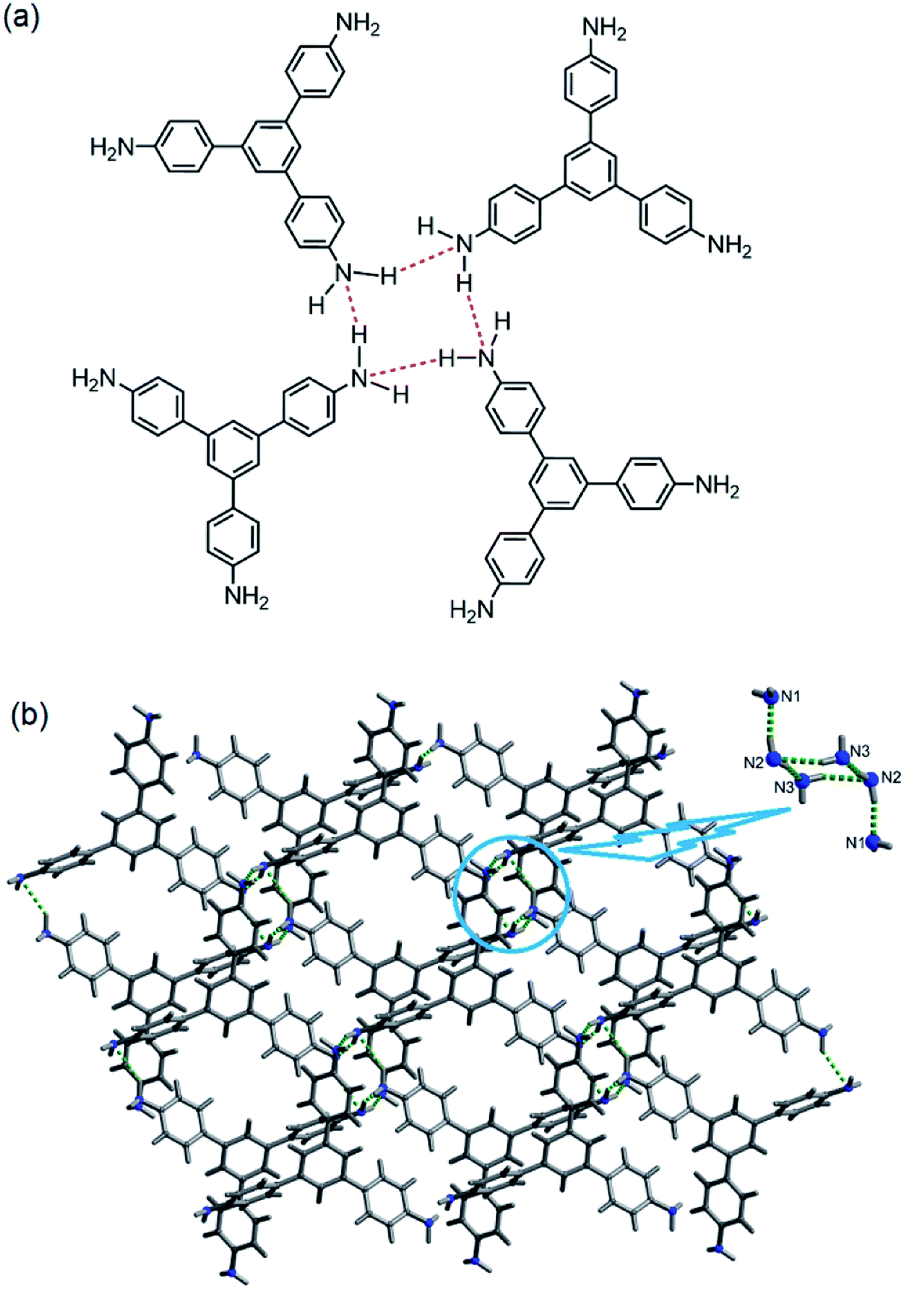
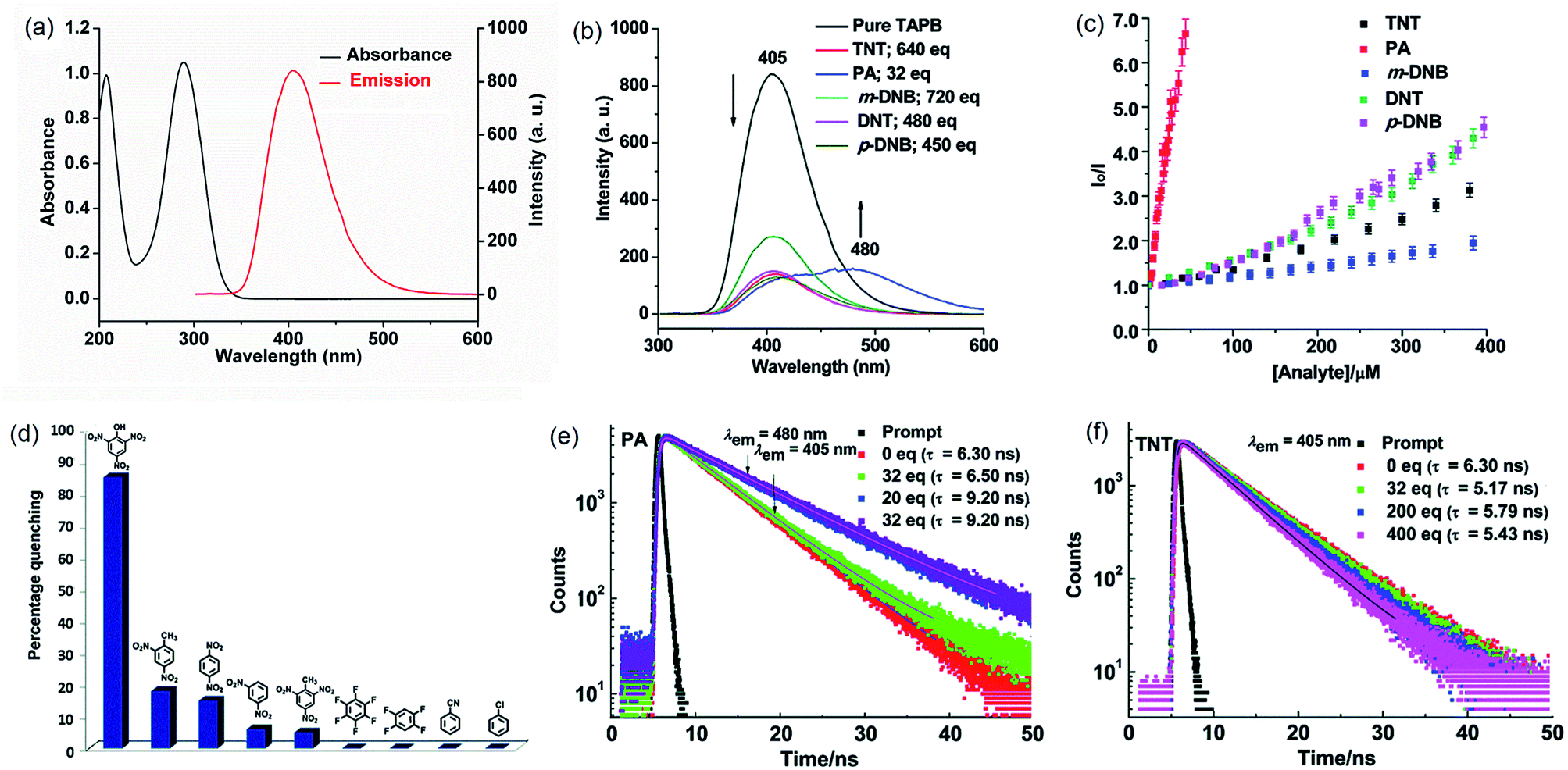
In 2014, we demonstrated that the tris-amine 1,3,5-tris(4′-aminophenyl)benzene (1) is an efficient supramolecular chemo-sensor for PNACs.39 Fig. 2 shows that the 1,3,5-TPB units are connected with each other through N–H⋯N hydrogen bonds between the –NH2 groups. The extended 3D network helps in the easy diffusion of analytes through the bulk and provides more surface for host–guest interactions.39,40 Due to the presence of the π-electron-rich 1,3,5-TPB core and –NH2 groups, 1 forms intermolecular π–π interactions as well as hydrogen bonds with PNACs. Fig. 3a shows the UV-visible absorption and photoluminescence (PL) spectra of 1 in acetonitrile. The PL spectrum shows a band at 405 nm on excitation at 290 nm. The PL intensity quenches progressively on increasing the concentration of PA, TNT, DNT, p-DNB and m-DNB (Fig. 3b). The quenching efficiencies in terms of Stern–Volmer constants (Ksv) have been found to be 1.2 × 105, 2.6 × 103, 8.0 × 103, 8.3 × 103 and 4.0 × 103 M−1 for PA, m-DNB, p-DNB, DNT and TNT respectively (Table 1). The quenching effect of PA is more significant than those by other analytes (Fig. 3c and d) which could be attributed to the more electronegative and acidic nature of PA. From single crystal X-ray diffraction studies and 1H NMR spectroscopy, it has been concluded that PA transfers –OH proton to –NH2 groups of 1. Strong N–H⋯O hydrogen bonds are formed between picrate and –NH3+ moieties. The static nature of the fluorescence quenching has been established from time-correlated single-photon counting (TCSPC), where the lifetime of the excited state of 1 remains unchanged in the presence of PA, while the lifetime changes slightly with TNT and other PNACs (Fig. 3e and f). It is remarkable to note that photoluminescence of a thin film of 1 on a quartz plate is quenched efficiently in the presence of DNT and TNT vapour. The film lost nearly 60% of its initial photoluminescence intensity on exposure to DNT and TNT vapour within 20 minutes.
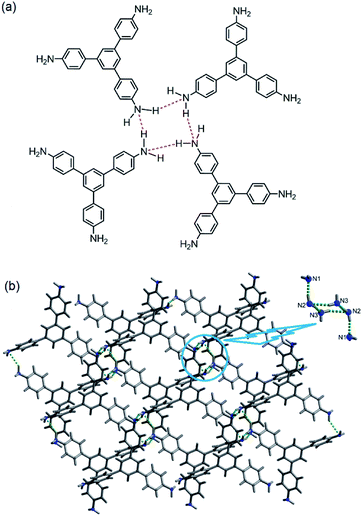 |
| | Fig. 2 Supramolecular structure of 1, (a) depiction of intermolecular N–H⋯N hydrogen bonds (red dotted bonds) and (b) crystal structure (inset shows a cluster of six –NH2 groups, connected by hydrogen bonds). Reproduced from ref. 40 with permission. Copyright© 2014, American Chemical Society. | |
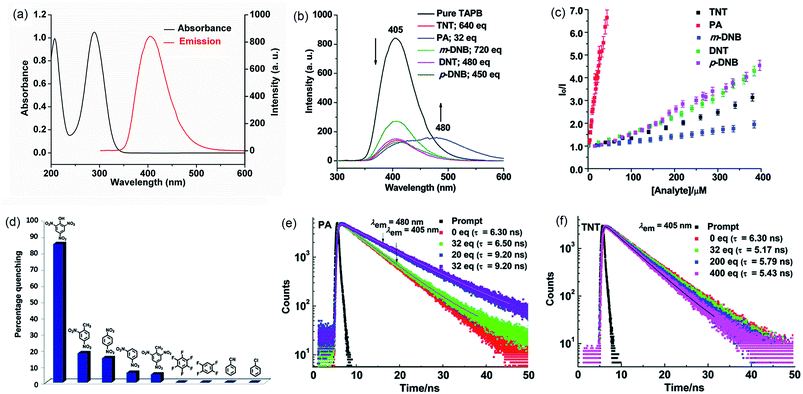 |
| | Fig. 3 (a) UV-Vis absorption spectrum (1.6 × 10−6 M, ε1 = 6.18 × 104 M−1 cm−1, ε2 = 6.59 × 104 M−1 cm−1) and photoluminescence spectrum (1.0 × 10−6 M, λex = 290 nm) of 1 in acetonitrile, (b) photoluminescence spectra of 1 with TNT (640 equivalents), PA (32 equivalents), m-DNB (720 equivalents), DNT (480 equivalents) and p-DNB (450 equivalents), (c) Stern–Volmer plots, (d) relative PL quenching on the addition of 32.0 equivalents of each PNAC and other electron-deficient analytes, and (e and f) time-resolved photoluminescence spectra before and after the additions of multiple concentrations of PA and TNT, respectively (λex = 295 nm). Reproduced from ref. 39 with permission. Copyright© 2014, Royal Society of Chemistry. | |
Table 1 Stern–Volmer constants (Ksv) of 1,3,5-TPB sensors for PNAC analytesa
| Fluorophores |
PA |
m-DNB |
p-DNB |
DNT |
TNT |
NA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Data not available. :Data not available. |
| 1 |
1.2 × 105 |
2.6 × 103 |
8.0 × 103 |
8.3 × 103 |
4.0 × 103 |
| 2 |
2.8 × 105 |
1.7 × 103 |
7.0 × 103 |
4.4 × 103 |
1.0 × 103 |
| 3 |
3.8 × 105 |
3.0 × 103 |
5.4 × 103 |
1.2 × 103 |
1.9 × 103 |
| 4 |
1.3 × 105 |
1.8 × 103 |
8.1 × 103 |
6.9 × 103 |
2.4 × 103 |
| 5 |
2.4 × 104 |
4.2 × 103 |
1.0 × 104 |
9.5 × 103 |
NA |
| 6 |
2.0 × 104 |
1.6 × 103 |
7.9 × 103 |
9.7 × 103 |
NA |
| 7 |
2.5 × 104 |
3.1 × 102 |
6.2 × 103 |
9.2 × 103 |
NA |
| 8 |
2.4 × 104 |
4.2 × 103 |
1.0 × 104 |
9.5 × 103 |
NA |
| 9 |
3.7 × 104 |
3.5 × 103 |
9.3 × 103 |
8.0 × 103 |
NA |
| 10 |
3.9 × 104 |
1.0 × 103 |
2.0 × 103 |
1.5 × 103 |
NA |
| 11 |
2.0 × 104 |
1.8 × 103 |
6.7 × 103 |
4.7 × 103 |
NA |
| 12 |
2.5 × 104 |
3.2 × 103 |
9.5 × 103 |
6.0 × 103 |
NA |
| 19 |
5.9 × 104 |
3.3 × 103 |
7.6 × 103 |
8.7 × 103 |
NA |
| 20 |
3.0 × 104 |
1.7 × 103 |
3.1 × 103 |
1.2 × 103 |
NA |
| 21 |
3.2 × 104 |
2.8 × 103 |
9.6 × 103 |
8.7 × 103 |
NA |
| 22 |
1.8 × 104 |
4.2 × 103 |
1.1 × 103 |
8.8 × 103 |
NA |
| 23 |
1.1 × 104 |
8.9 × 101 |
4.3 × 103 |
1.7 × 103 |
NA |
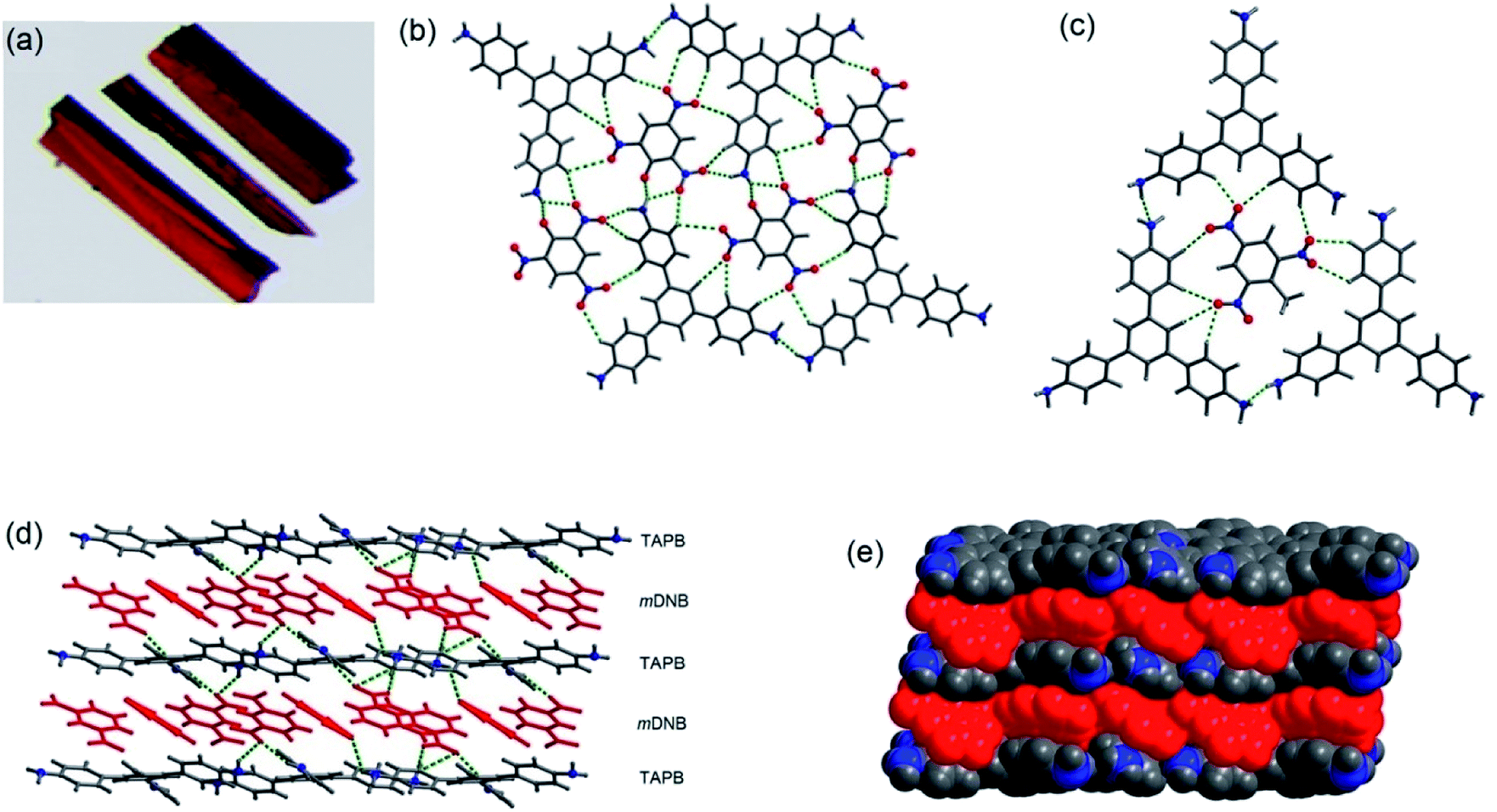
The co-crystals of 1 with TNT, PA and m-DNB have been obtained in methanol or ethanol solvent by a slow evaporation method at room temperature.40 Fig. 4 shows the crystal structures of co-crystals, indicating the simultaneous presence of π–π and hydrogen bonds between 1 and PNAC analytes. While TNT and PA form π-interactions with the central benzene ring of 1, m-DNB forms π-interactions with all the three peripheral benzene rings. The N–H⋯N hydrogen bonds maintain the supramolecular architecture of 1 in the co-crystals. In addition, the acidic –OH proton of PA migrates to one of the three –NH2 groups of 1, forming a 1:1 –NH3–picrate complex through N–H⋯O hydrogen bonds and electrostatic interactions. The distances between the centroids of 1 and the PNACs are 3.58–3.64, 3.74–3.81, and 3.57–4.11 Å for 1–TNT, 1–PA and 1–m-DNB respectively. These distances are indicative of the formation of donor–acceptor π–π interactions.41 On forming co-crystals, physical properties, such as melting points of the PNACs, improved significantly. From differential scanning calorimetry (DSC), the melting temperatures (mp) were determined to be 170, 243, and 121 °C for 1–TNT, 1–PA, and 1–m-DNB co-crystals, respectively. These melting temperatures are higher than those of PNACs (mp; 122.5, 89.6 and 80.8 °C for PA, m-DNB and TNT, respectively) and lower than that of 1 (mp; 250–252 °C).
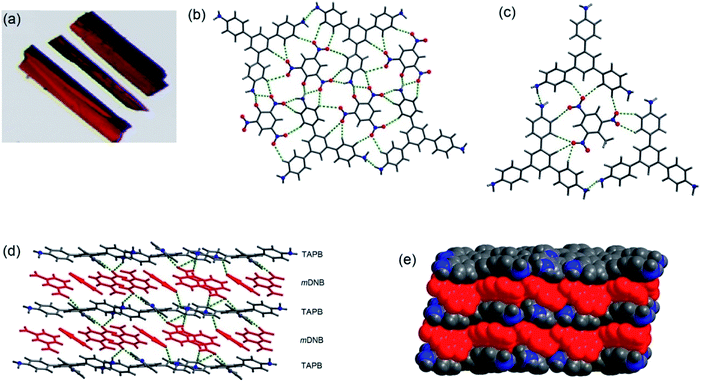 |
| | Fig. 4 (a) Photographs of co-crystals of 1–PA, (b) showing hydrogen bonds in 1–PA, (c) showing hydrogen bonds in 1–TNT, (d) showing alternate layers in 1–m-DNB, connected by N–H⋯O hydrogen bonds, and (e) space-filling model of co-crystals of 1–m-DNB in which the atoms are shown by spheres of van der Waals radii. Reproduced from ref. 40 with permission. Copyright© 2014, American Chemical Society. | |
2.2. Discrete 1,3,5-tris(4′-aminophenyl)benzenes
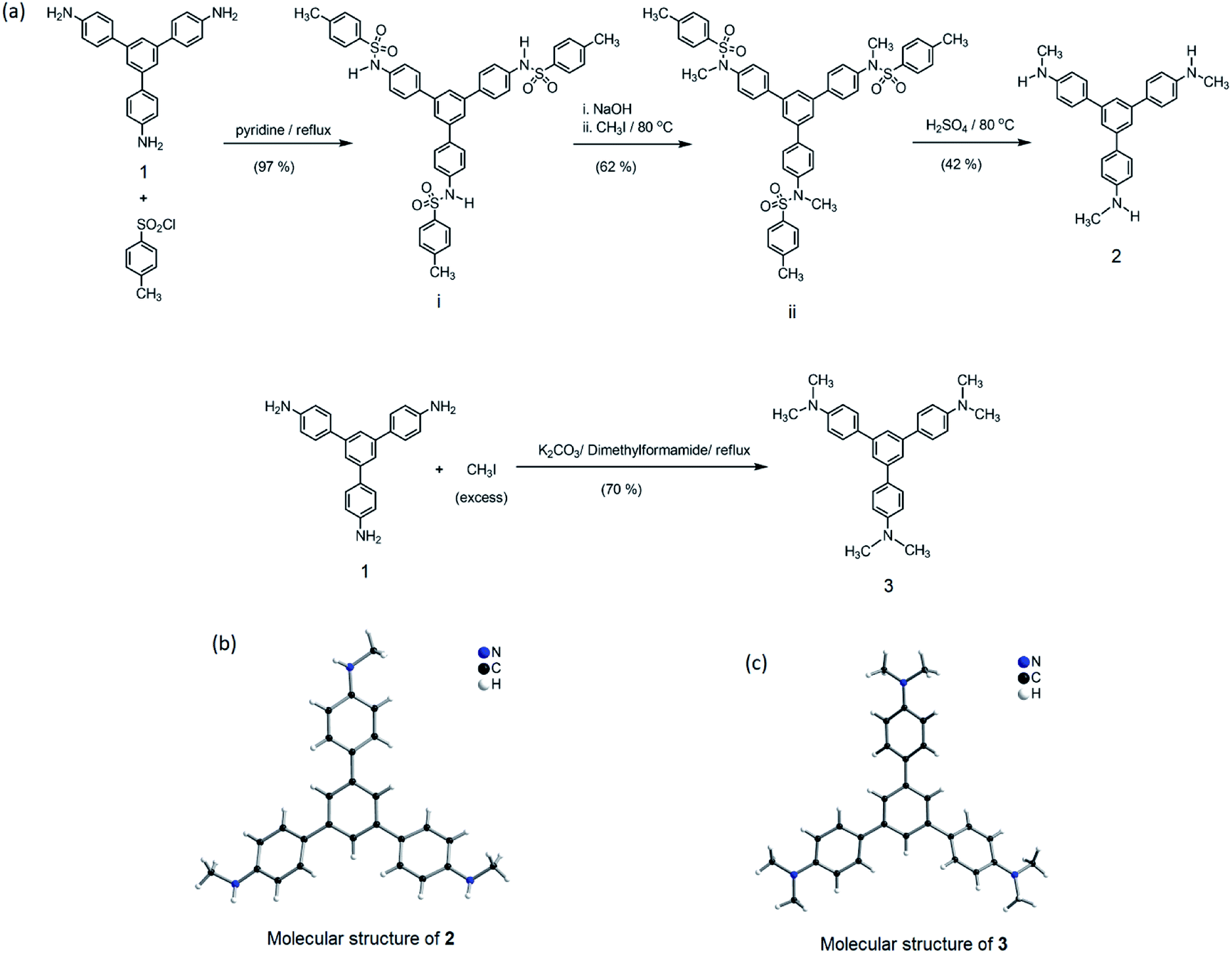
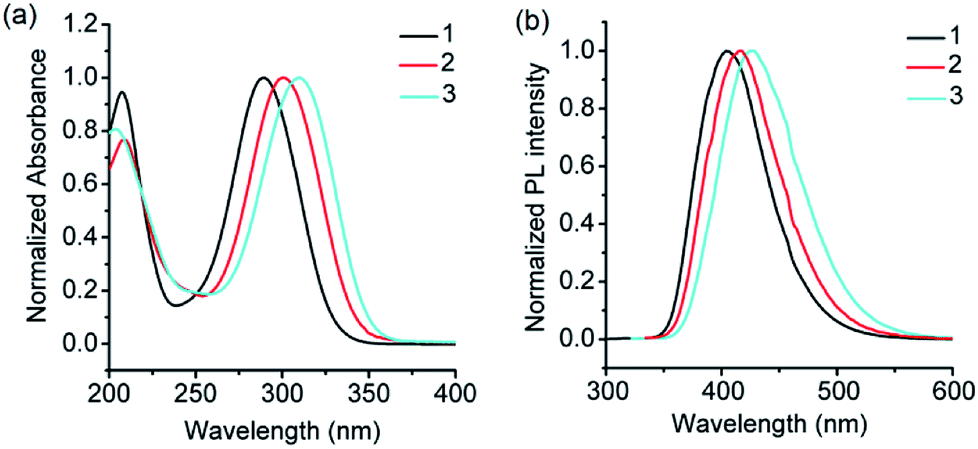
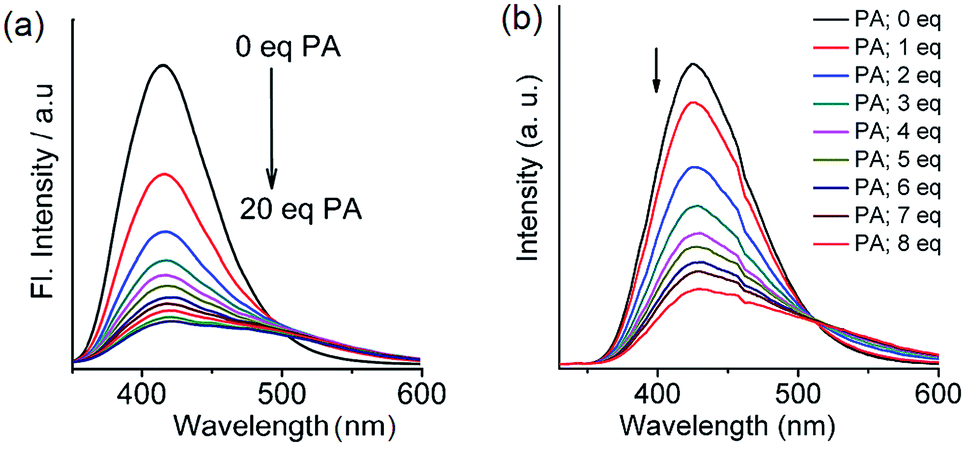
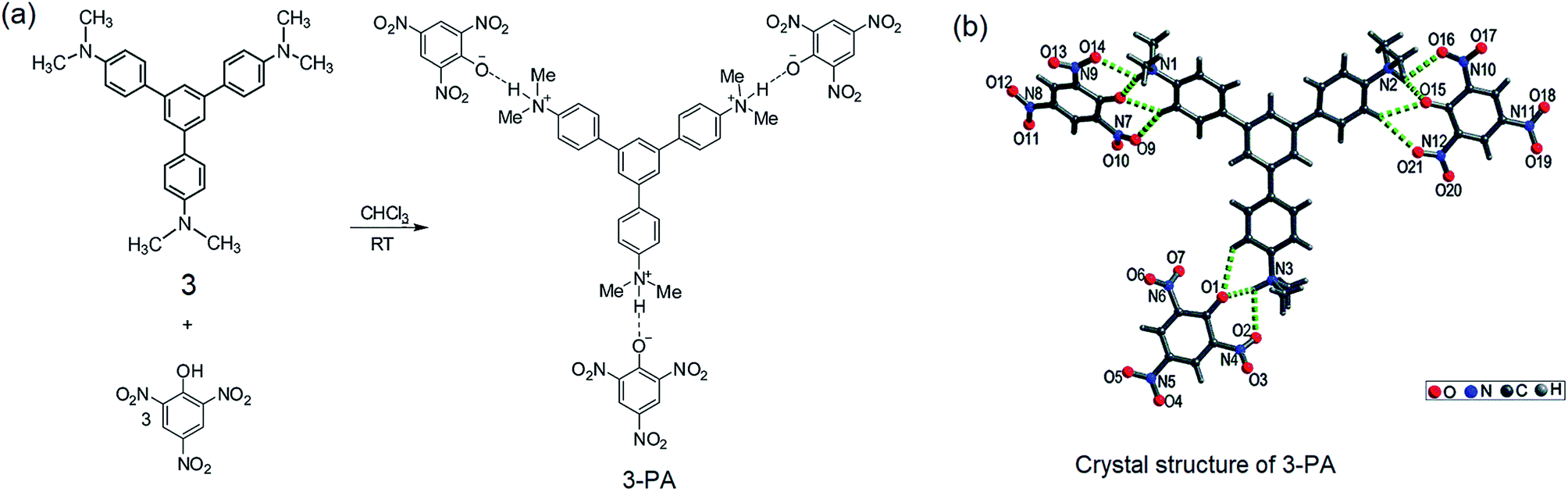
From the structural analysis of the co-crystals of 1 formed with TNT, PA and m-DNB, it can be perceived that hydrogen bonds help in the formation of donor–acceptor complexes, which, in turn, enhances the sensing efficiency of 1. We substituted the –NH2 protons of 1 with methyl groups (hydrogen bond inactive groups) in order to study the effect of hydrogen bonds on fluorescence quenching.8 Scheme 1 shows the synthesis of partially and fully N-methyl substituted fluorophores 2 and 3 respectively. To synthesize 2, each –NH2 group of 1 was first protected with 4-tosyl group to obtain an intermediate compound (i). Fluorophore 2 has been obtained by the reaction of this intermediate compound with methyl iodide in a basic medium followed by de-protection in an acidic medium. The fully N-methyl substituted fluorophore 3 has been synthesized by the reaction of 1 with excess methyl iodide in basic medium.42 Fluorophores 2 and 3 do not form the supramolecular structures, as confirmed from their single crystal structures (Scheme 1). It is important to note that the substitution of N–H groups with N-methyl causes a sizeable red-shift to the absorption and photoluminescence bands of 1 (Fig. 5). It is expected that the photoluminescence of 1, 2 and 3 would behave differently with PNACs. Indeed, we found marked changes in the photoluminescence quenching constants of these sensors for each analyte (Table 1). These fluorophores exhibit a parts per million (ppm) level of detection of PA. Fig. 6 shows the photoluminescence spectra of 2 and 3 with increasing concentrations of PA. Nearly 85–90% of the initial PL intensities of 2 and 3 were quenched by PA on the addition of 20 and 8 equivalents, respectively. Close to 85% of the initial photoluminescence of 1 is quenched by a mere 32 equivalents of PA. The Stern–Volmer constants (Ksv) of 1–3 for PA increase in the order 1 < 2 < 3, suggesting an enhanced interaction between fluorophore and PA. For other analytes, the Ksv values are not only small but also vary in the reverse order. PA also showed sharp visual changes in the fluorophores. These results show that PA binds more strongly with –N(CH3)2 compared to the –NH(CH3) and –NH2 groups. Therefore, PA quenches the photoluminescence of 3 the most efficiently among 1–3. We obtained red single crystals and subsequently determined the structure of a 1:3 complex of 3 with PA (Scheme 2). The structure reveals the transfer of the –OH proton of PA to the –N(CH3)2 groups of 3, transforming them into –NH(CH3)2+. The –NH(CH3)2+ and picrate groups efficiently form N–H⋯O and C–H⋯O hydrogen bonds with each other. The interaction between 3 and PA has been further established by 1H NMR spectroscopy. The aromatic protons of 3 showed a downfield-shift due to the deshielding caused by picrate ions.
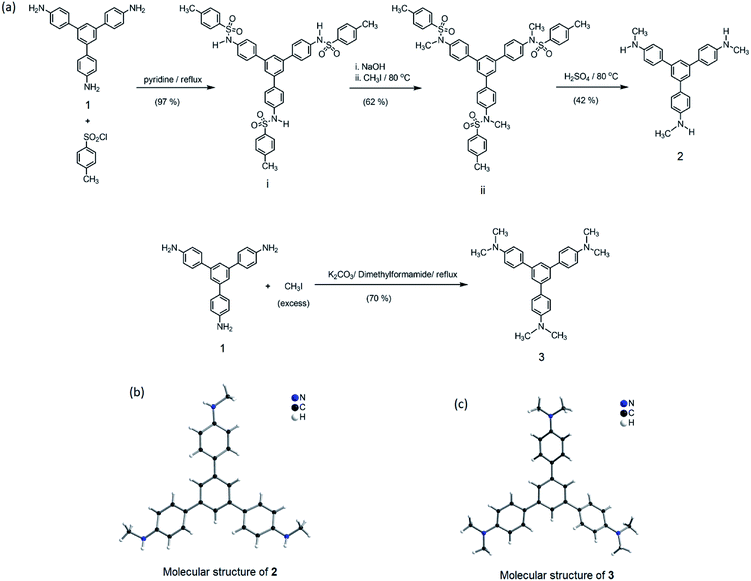 |
| | Scheme 1 (a) Synthesis of 2 and 3 from 1. (b) Molecular structure of 2, drawn using CIF obtained from ref. 42. Copyright© 2017, Springer Nature. (c) Molecular structure of 3 drawn using CIF obtained from ref. 8. Copyright© 2015, Royal Society of Chemistry and the Centre National de la Recherche Scientifique. | |
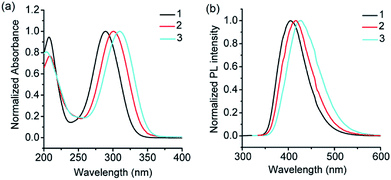 |
| | Fig. 5 (a) UV-visible absorption spectra of 1, 2 and 3 (λmax for the n–π* transition = 289, 300 and 309 nm), and (b) photoluminescence spectra of 1, 2 and 3 (λem = 405, 415 and 425 nm) in acetonitrile (see Scheme 1 for the structures of 1–3). | |
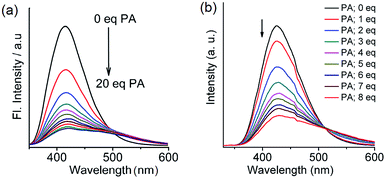 |
| | Fig. 6 Photoluminescence spectra of (a) 2 (1.0 × 10−6 M in acetonitrile, excitation wavelength = 300 nm) with PA (0.0 to 20.0 equivalents). Reproduced from ref. 42 with permission. Copyright© 2017, Springer Nature. (b) 3 (1.0 × 10−6 M in acetonitrile, excitation wavelength = 310 nm) with PA (0.0 to 8.0 equivalents). Reproduced from ref. 8 with permission. Copyright© 2015, Royal Society of Chemistry and the Centre National de la Recherche Scientifique. | |
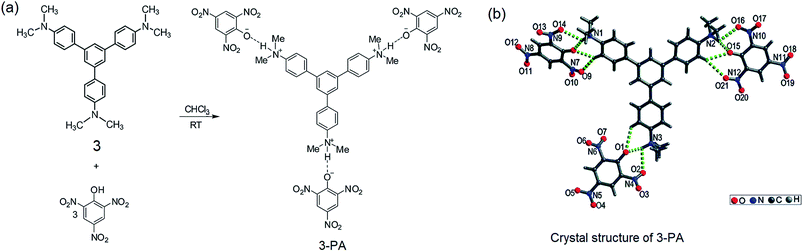 |
| | Scheme 2 (a) Preparation of the 3–PA complex in a 1:3 stoichiometry. (b) Crystal structure of 3–PA; hydrogen bonds are indicated by green dotted bonds. Reproduced from ref. 8 with permission. Copyright© 2015, Royal Society of Chemistry and the Centre National de la Recherche Scientifique. | |
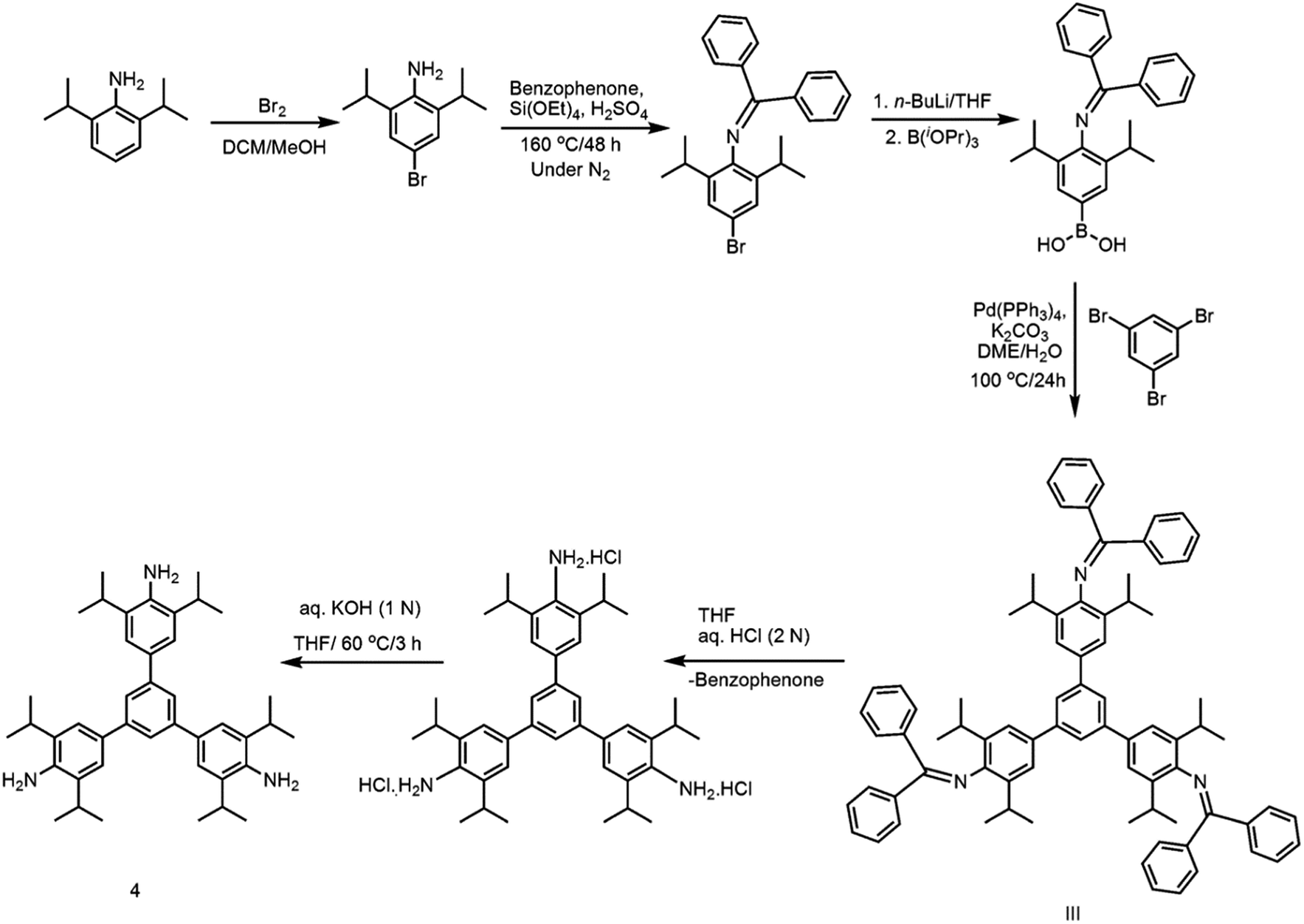
Closely related to 1, but much more sterically encumbered, 1,3,5-tris(4′-amino-3′,5′-di-isopropylphenyl)benzene (4) has been subsequently synthesized through a multistep synthetic protocol (Scheme 3).31 The isopropyl groups in 4 are placed on the sides of the –NH2 groups of peripheral benzene rings. On excitation at 293 nm, this per-alkylated 4 in acetonitrile exhibited an emission band at 412 nm. Surprisingly, the PNAC sensing ability of 4 is comparable to that of 1, suggesting the importance of –NH2 groups in chemo-sensing over the bulky alkyl groups present on the peripheral rings.
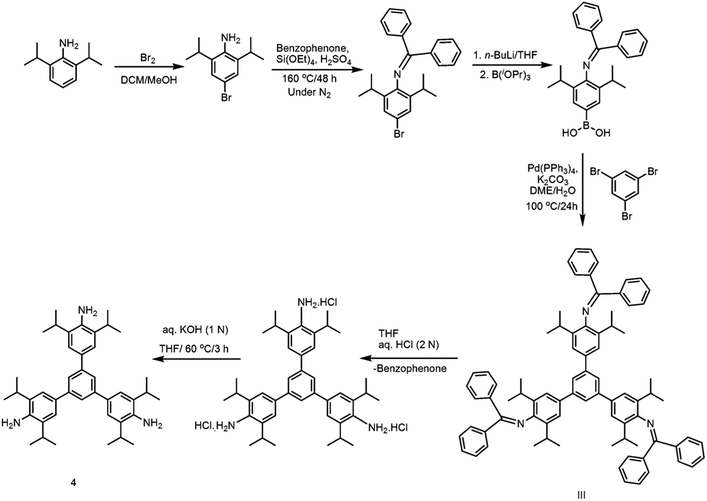 |
| | Scheme 3 Synthesis of 4. | |
2.3. 1,3,5-Tris(4′-hydroxyphenyl)benzene and its carbazole derivatives
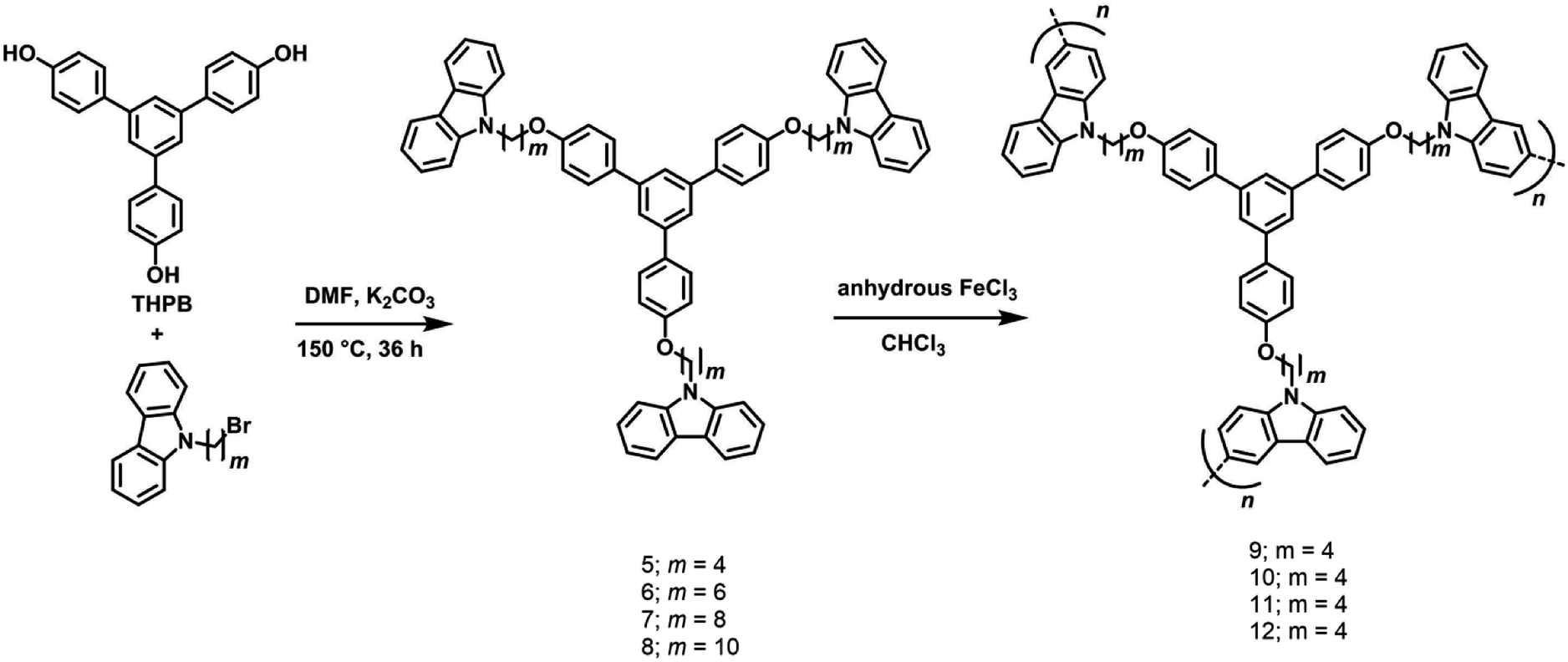
Building on the chemistry described in Section 2.2, with the objective to evaluate the effect of non-conjugated long alkyl chain spacers on the net absorption and emission properties of a range of 1,3,5-TPB-carbazoles, four monomeric triphenylbenzene-carbazoles, 5–8 and four polymeric triphenylbenzene-carbazoles, 9–12 have been synthesized and characterized (Scheme 4).43 The monomers 5–8 have been synthesized from the reaction of 1,3,5-tris(4′-hydroxyphenyl)benzene with 9-(4-bromobutyl)-9H-carbazole, 9-(6-bromobutyl)-9H-carbazole, 9-(8-bromobutyl)-9H-carbazole or 9-(10-bromobutyl)-9H-carbazole, respectively, in anhydrous DMF solvent and K2CO3. The 3- and 6- positions of the N-alkylcarbazole units of the monomers are reactive and can easily be coupled with other monomer units to form polymers. The polymers 9–12 have been obtained in the presence of an FeCl3 catalyst as well as under electrochemical conditions through a highly stable 3,6-bicarbazylium cation-radical intermediate. The amorphous nature of the polycarbazoles has been established by DSC measurements.
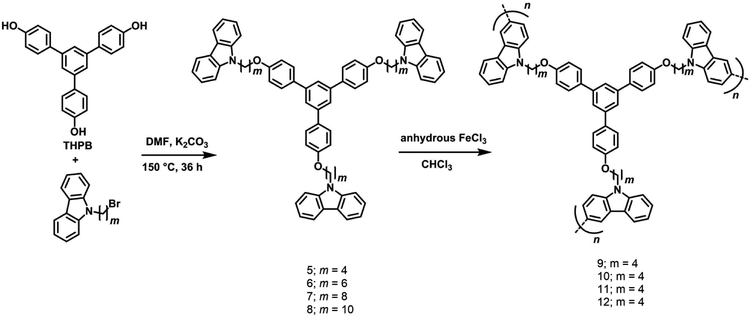 |
| | Scheme 4 Synthesis of monomers 5–8 and polymers carbazoles 9–12. Reproduced from ref. 43 with permission. Copyright© 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
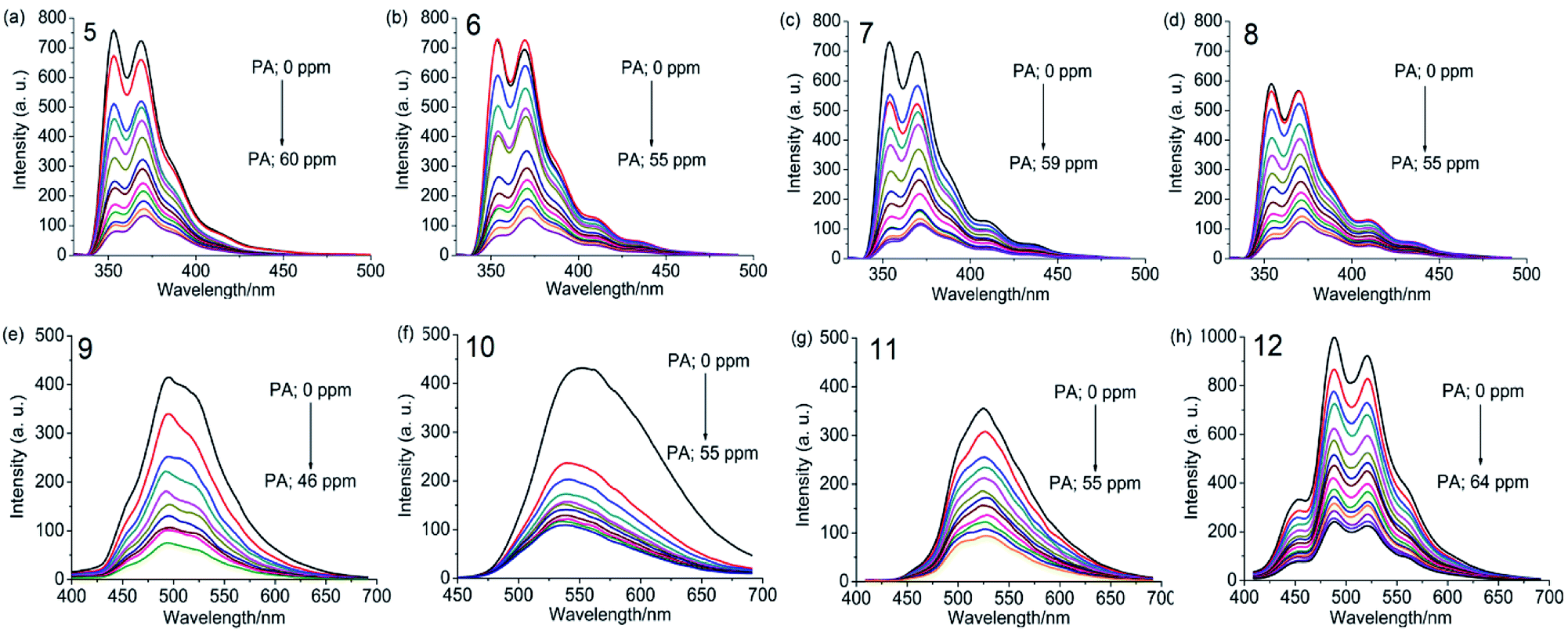
UV-visible absorption spectra of 5–8 as a dichloromethane solution or as a thin film show bands at 238, 265, 294, 332 and 346 nm due to the π–π* and n–π* transitions of carbazole and 1,3,5-TPB units. The photoluminescence (PL) spectrum of 5 shows two intense bands at 353 and 369 nm in dichloromethane. Besides, two additional weak bands at 410 and 435 nm have been observed in the PL spectra of 6–8, which are more intense in the case of thin films. These additional bands could arise due to excimers formed between carbazole and 1,3,5-TPB units. Increased lengths of alkyl chains in 6–8 facilitate the formation of these excimers. Due to the extended π-conjugation, the intense PL bands are positioned at 495 and 520 nm and are red-shifted in the case of polymers compared to the corresponding monomers. The electrochemical studies unveiled the electron-donating ability of carbazole fluorophores to the acceptor PNACs. PA, DNT, p-DNB and m-DNB quench the PL bands of the monomers as well as the polymers in acetonitrile, with the most marked quenching being observed with PA (∼90% of the initial PL intensity quenched on the addition of ∼55 equivalents of PA) (Fig. 7). The Ksv values of all these carbazoles are listed in Table 1. The PL of all four monomers showed a fast response to DNT vapours. Nearly 80% of the initial PL intensity of the monomers, spin-coated on quartz plates, is quenched on exposure to DNT vapour for 15 minutes.
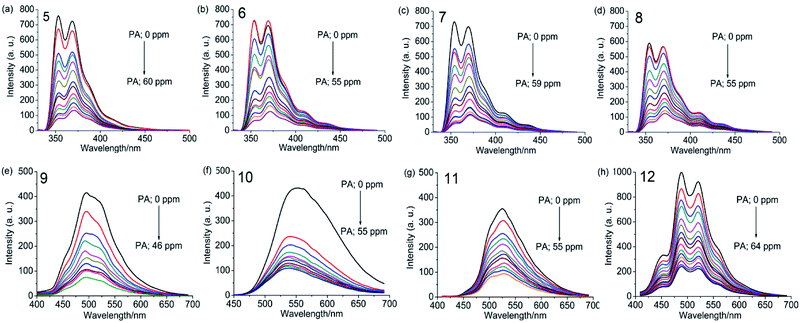 |
| | Fig. 7 Photoluminescence spectra of 5–8 in dichloromethane (1.0 × 10−6 M, λex = 295 nm) and 9–12 in acetonitrile (0.2 mg mL−1, λex = 300 nm), with progressive additions of PA. Reproduced from ref. 43 with permission. Copyright© 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
2.4. Covalent-organic frameworks (COFs) and polymers (COPs)
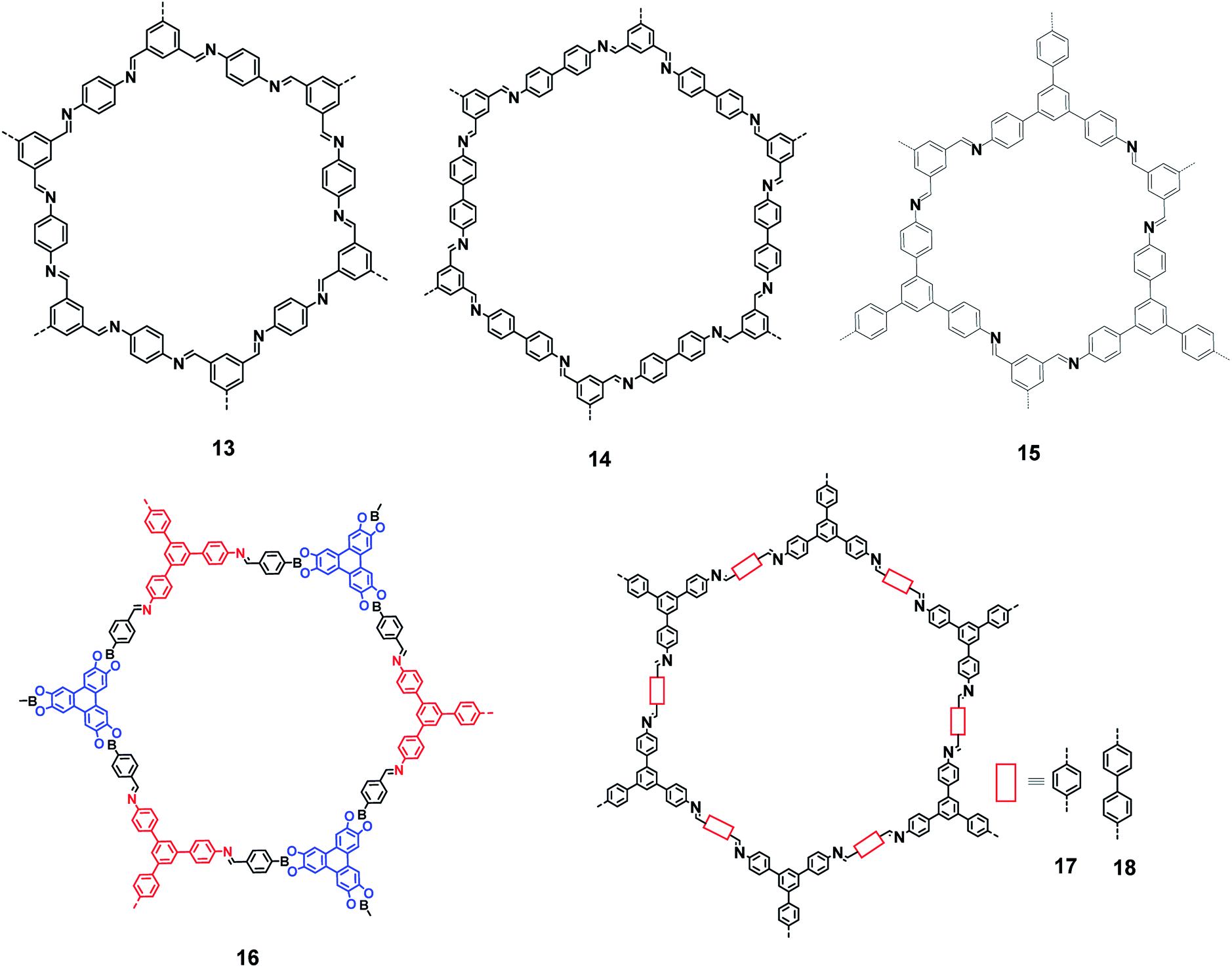
In 2005, Yaghi et al. demonstrated the condensation of –B(OH)2 groups to form covalent organic framework materials exhibiting a high surface area and crystallinity.44 These studies led to the search for newer types of COFs with potential applications. Several COFs with exciting gas storage and separation, catalysis, energy storage and proton conduction properties have been reported over the last decade.45–48 Condensation between an amino group and a carbonyl group (of an aldehyde and a ketone) has been extensively exploited for preparing COFs with imine linkages. Fig. 8 shows some of the imine linkage-based COFs (13–18) that have been realized by the condensation of amine and aldehyde building blocks.49–55 The photoluminescence properties of these COFs have not been investigated, although some of these frameworks contain 1,3,5-TPB units.
 |
| | Fig. 8 Various covalent-organic frameworks with imine linkages. | |
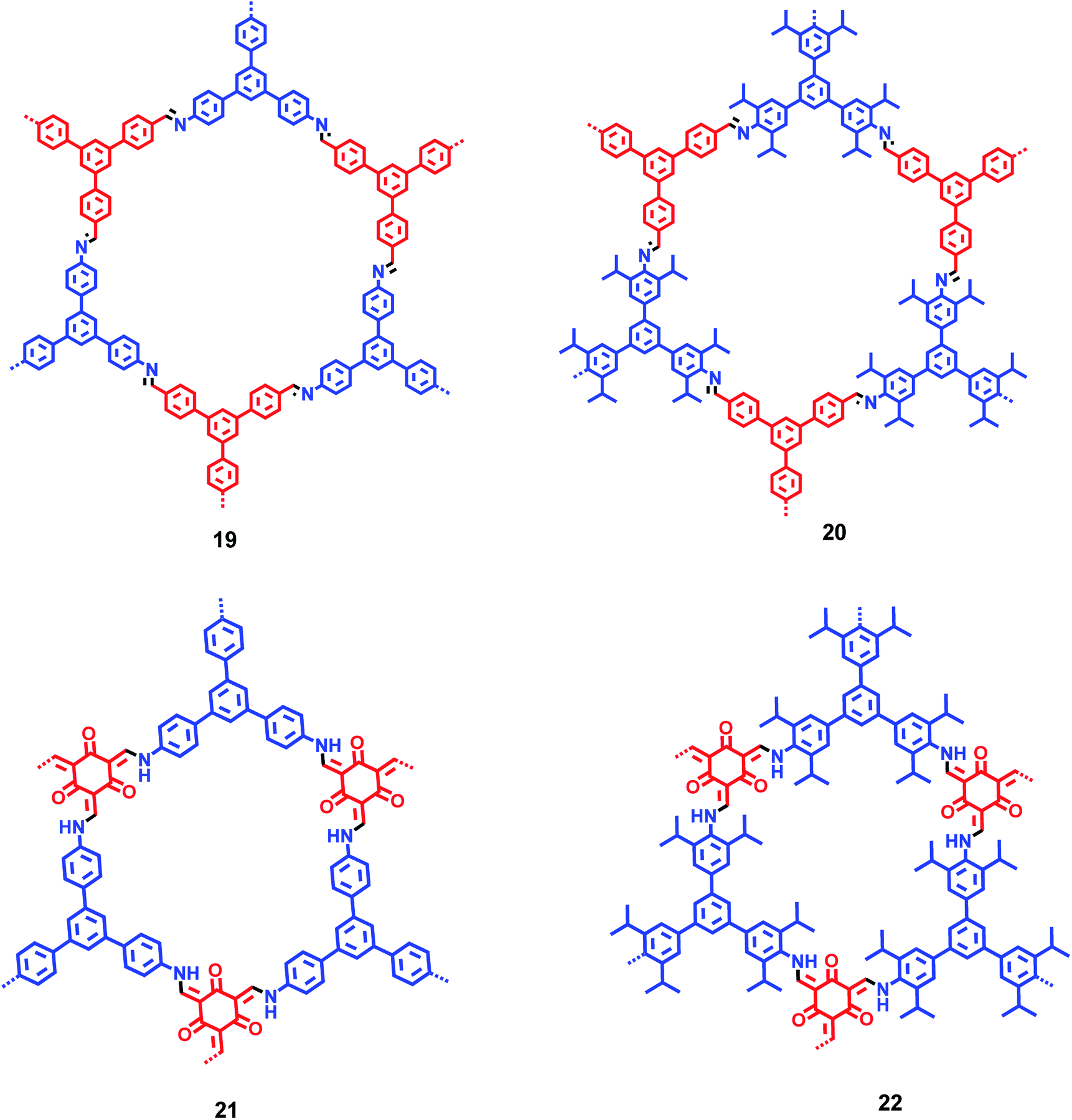
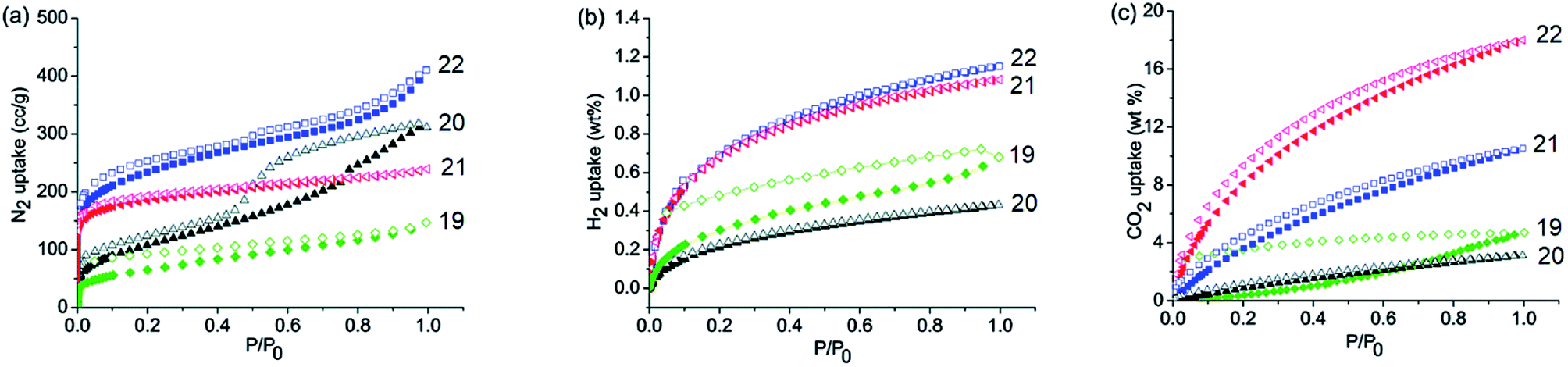
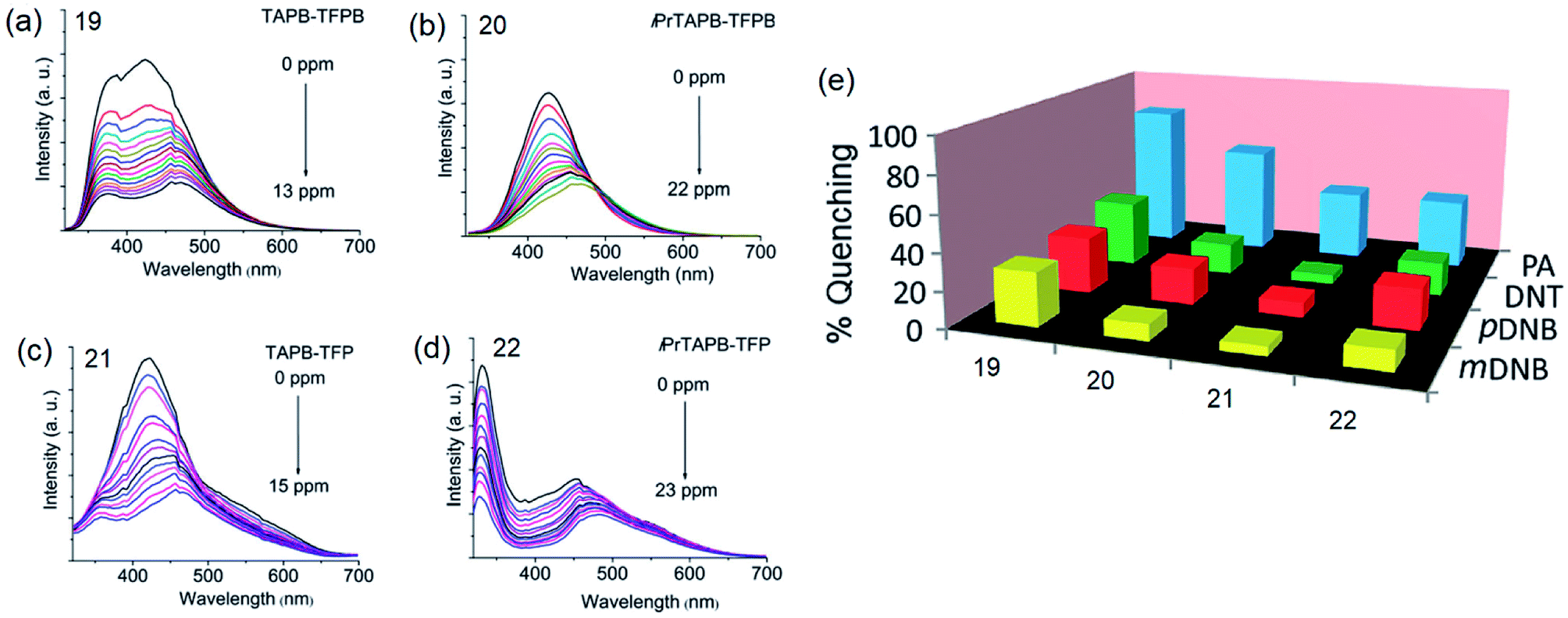
By incorporating 1,3,5-tris(4′-aminophenyl)benzene (TAPB; 1) and 1,3,5-tris(4′-amino-3′,5′-di-isopropylphenyl)benzene (iPrTAPB; 4) as the fluorophore units, we had synthesized four new covalent organic frameworks, TAPB-TFPB (19), iPrTAPB-TFPB (20), TAPB-TFP (21) and iPrTAPB-TFP (22) in dry 1,4-dioxane and aqueous acetic acid (as a catalyst) under solvothermal conditions (Fig. 9).31 The –NH2 groups containing building blocks 1 and 4 have been condensed with –CHO groups containing 1,3,5-tris(4′-formylphenyl)benzene (TFPB) or 1,3,5-triformylphluroglucinol (TFP) building blocks. The formation of COFs takes place via the formation of new imine linkages (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) between the –NH2 and –CHO groups through reversible dehydration with the elimination of water. These COFs have been characterized by infrared (IR) and 13C nuclear magnetic resonance (NMR) spectroscopy. The porosity and crystallinity of these COFs have been established by BET surface area and PXRD measurements, respectively. Fig. 10 shows the N2, H2 and CO2 sorption isotherms. The BET surface areas of these COFs are 229.4 (19), 390.6 (20), 567.0 (21) and 756.0 m2 g−1 (22) at 77 K and the corresponding pore sizes are 40, 50, 26 and 34 Å. These COFs showed a high CO2 uptake of 40.1 mg g−1 (4 wt%, 19), 31.2 mg g−1 (3.1 wt%, 20), 180 mg g−1 (18 wt%, 21) and 105.2 mg g−1 (10.5 wt%, 22) at a 1 bar pressure and a 273 K temperature. Due to the high surface area of the extended structures and π-conjugated 1,3,5-TPB units, 19 and 20 showed strong photoluminescence. The PL spectra of acetonitrile suspensions of 19–21 showed broad bands at 425 nm, while in the case of 22, two bands were observed at 285 and 450 nm. The photoluminescence is more effectively quenched by PA, compared to DNT, p-DNB and m-DNB. Fig. 11 shows the quenching of PL spectra of the COFs on the addition of PA. The efficiency of PL quenching with PA decreases as 19 > 20 > 21 > 22. The time-resolved photoluminescence spectrum of 19 shows two components with lifetimes of 1.7 and 6.8 ns. With PA, lifetimes remain almost unchanged, indicating the static mechanism of quenching. In contrast, other PNACs showed a decrease in the lifetimes due to a possible involvement of dynamic quenching.
N–) between the –NH2 and –CHO groups through reversible dehydration with the elimination of water. These COFs have been characterized by infrared (IR) and 13C nuclear magnetic resonance (NMR) spectroscopy. The porosity and crystallinity of these COFs have been established by BET surface area and PXRD measurements, respectively. Fig. 10 shows the N2, H2 and CO2 sorption isotherms. The BET surface areas of these COFs are 229.4 (19), 390.6 (20), 567.0 (21) and 756.0 m2 g−1 (22) at 77 K and the corresponding pore sizes are 40, 50, 26 and 34 Å. These COFs showed a high CO2 uptake of 40.1 mg g−1 (4 wt%, 19), 31.2 mg g−1 (3.1 wt%, 20), 180 mg g−1 (18 wt%, 21) and 105.2 mg g−1 (10.5 wt%, 22) at a 1 bar pressure and a 273 K temperature. Due to the high surface area of the extended structures and π-conjugated 1,3,5-TPB units, 19 and 20 showed strong photoluminescence. The PL spectra of acetonitrile suspensions of 19–21 showed broad bands at 425 nm, while in the case of 22, two bands were observed at 285 and 450 nm. The photoluminescence is more effectively quenched by PA, compared to DNT, p-DNB and m-DNB. Fig. 11 shows the quenching of PL spectra of the COFs on the addition of PA. The efficiency of PL quenching with PA decreases as 19 > 20 > 21 > 22. The time-resolved photoluminescence spectrum of 19 shows two components with lifetimes of 1.7 and 6.8 ns. With PA, lifetimes remain almost unchanged, indicating the static mechanism of quenching. In contrast, other PNACs showed a decrease in the lifetimes due to a possible involvement of dynamic quenching.
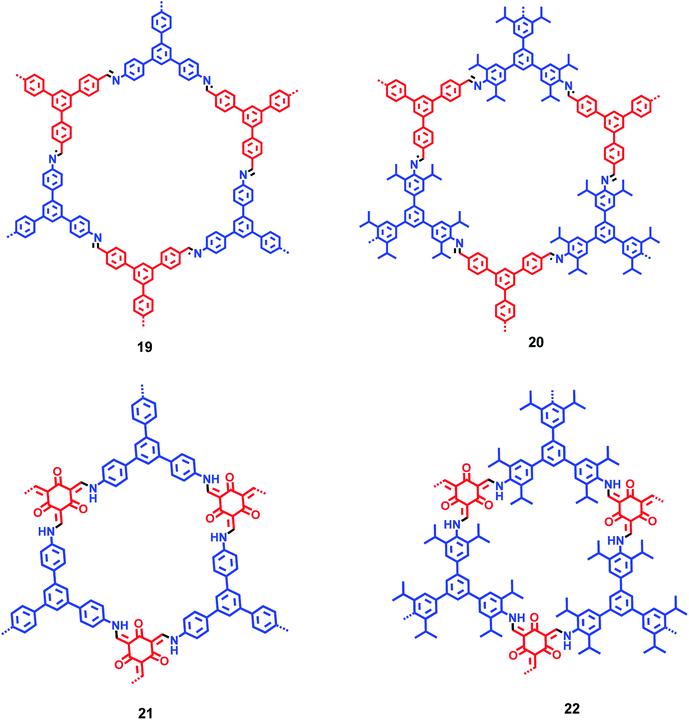 |
| | Fig. 9 Plausible structures of COFs 19–22. Different colours (blue and red) represent different building blocks. | |
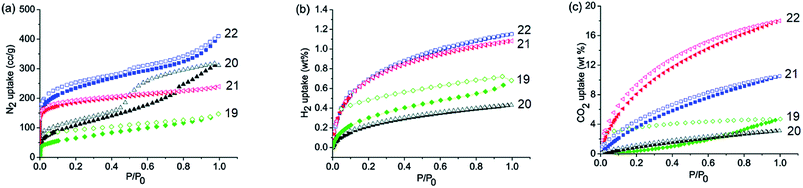 |
| | Fig. 10 Gas absorption isotherms of 19–22. (a) N2 adsorption isotherms at 77 K, (b) H2 adsorption isotherms at 77 K and (c) CO2 adsorption isotherms at 273 K and 1 bar. Reproduced from ref. 31 with permission. Copyright© 2015, Royal Society of Chemistry. | |
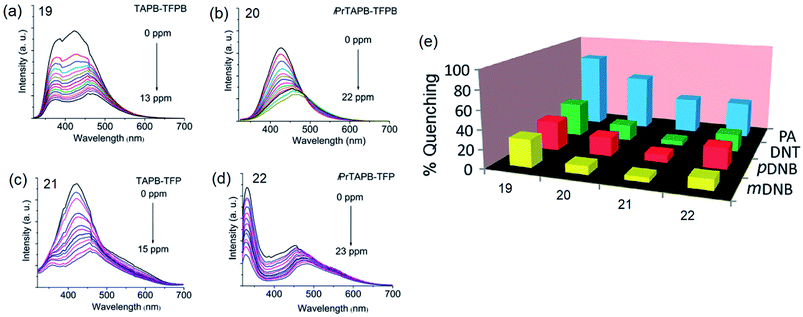 |
| | Fig. 11 Photoluminescence spectra of acetonitrile dispersions (0.2 mg mL−1) of (a) 19 (λex = 285 nm), (b) 20 (λex = 300 nm), (c) 21 (λex = 285 nm) and (d) 22 (λex = 290 nm), showing progressive quenching of photoluminescence on the addition of PA, and (e) the degree of PL quenching of COFs (%) with 13.0 ppm of PA, DNT, m-DNB and p-DNB. Reproduced from ref. 31 with permission. Copyright© 2015, Royal Society of Chemistry. | |
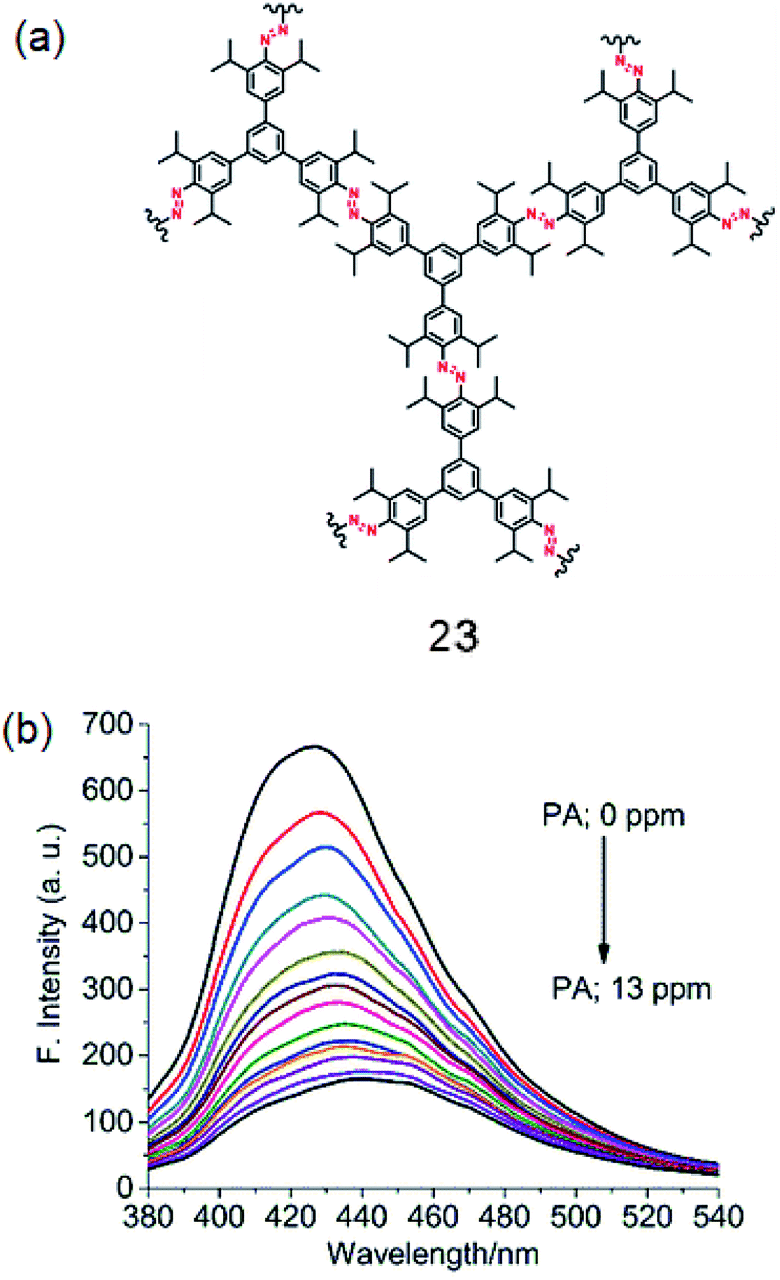
By using the Cu(I) catalyzed homocoupling reaction (amine–amine) of 1,3,5-tris(4′-amino-3′,5′-di-isopropylphenyl)benzene (iPrTAPB; 4), the fluorescent azo-linked covalent organic polymer (COP) 23 has been synthesized (Fig. 12a).56 Owing to the bulky isopropyl groups, the BET surface area of this polymer is 395 m2 g−1, which is smaller than that reported for the unsubstituted variant synthesized from 1.57 The COP 23 showed a photoluminescence band at 428 nm on excitation at 300 nm. PA quenches the PL intensity efficiently with a slight red-shift (Fig. 12b). Other PNACs quench the PL much less efficiently and the quenching efficiency decreases in the order of PA ≫ p-DNB > DNT ≫ m-DNB. Owing to the presence of microspores as well as the CO2-philic azo (–NN–) sites, 23 uptakes 6.5 and 19.4 wt% CO2 at 1 and 30 bar, respectively, at 273 K.
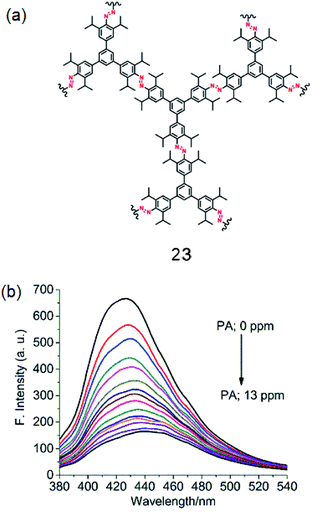 |
| | Fig. 12 (a) Structure of 23 and (b) photoluminescence spectra of an acetonitrile suspension of 23 (0.2 mg mL−1; λex = 300 nm) in the presence of PA (0.0 to 13.0 ppm). Reproduced from ref. 56 with permission. Copyright© 2018, Springer Nature. | |
2.5. π-Conjugated polymers
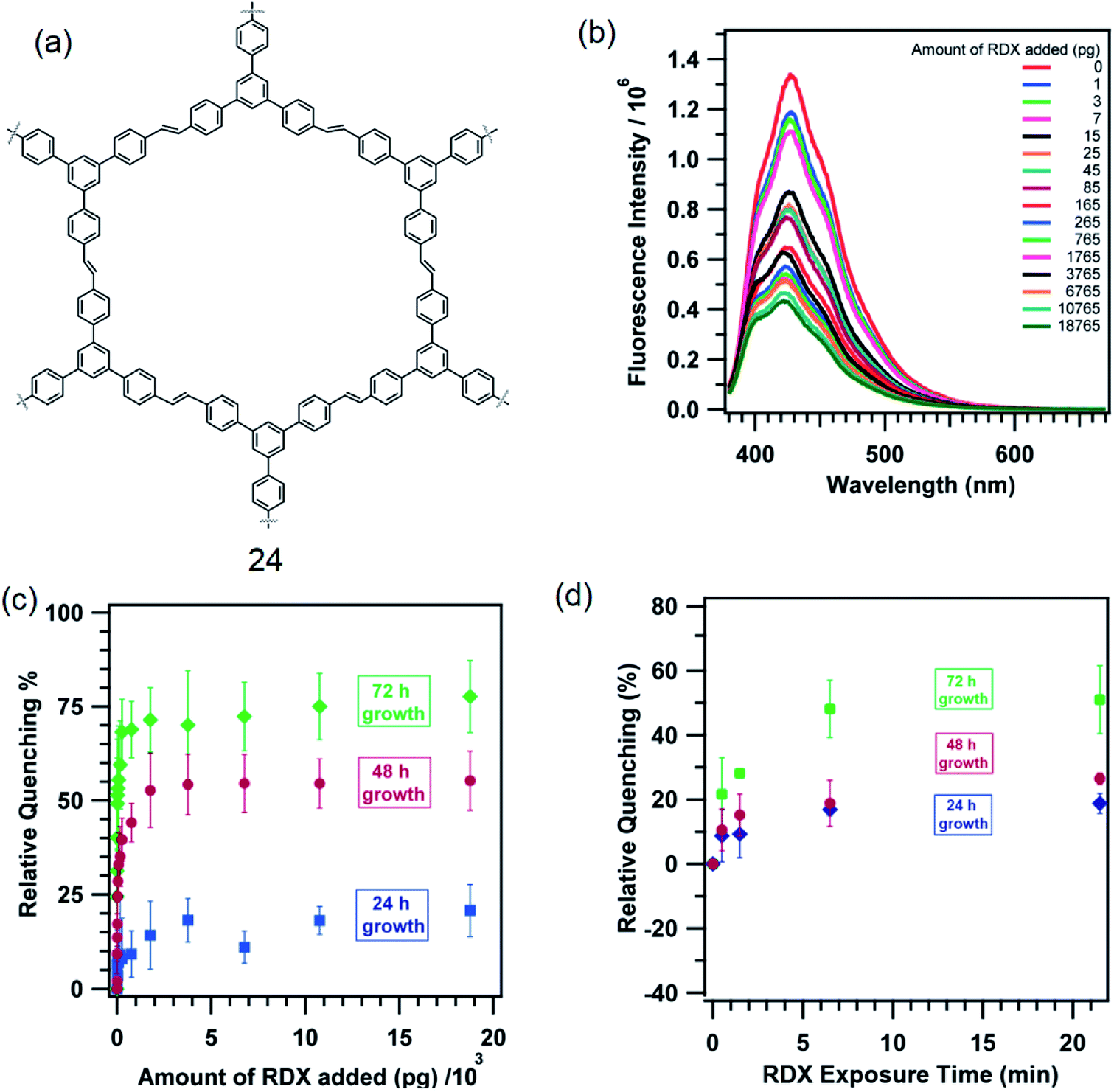
Fluorescent π-conjugated organic polymers provide an efficient transport media for the electronic excited state (or exciton). This leads to an amplification of the sensing ability of such polymers. Typical of these polymers are poly(phenyleneethynylene) and poly(phenylenevinylene). Dichtel and co-workers grew thin films of a 1,3,5-TPB containing polymer, tris(phenylene)vinylene (24), on fused SiO2 substrates (Fig. 13a).58 The polymer 24 itself has been synthesized from a metathesis reaction of trifunctional styrene under solvothermal conditions in CH2Cl2 at 45 °C in the presence of 6.0 mol% of a Grubbs second-generation olefin metathesis catalyst. The thin films of 24 grown for 24, 48 and 72 h detect 1,3,5-trinitroperhydro-1,3,5-triazine (RDX) at the pictogram level. The fluorescence of these thin films is effectively quenched on exposure to RDX (Fig. 13b). The films exhibited an increased quenching response as a function of their reaction time to a RDX solution in a mixture of CH3CN:MeOH (Fig. 13c). The film grown for 72 h showed a 51 ± 15% quenching on exposure to 25 pg of RDX and saturated at 71 ± 9% on exposure to larger doses. The 48 h films showed 24 ± 6% quenching on exposure to 25 pg of RDX and saturated at 53 ± 10%, while the 24 h films showed only 9 ± 4% when exposed to 25 pg of RDX and saturated at 14 ± 9%. On exposure to RDX vapour, films grown for 72 h showed 22 ± 11% quenching within 30 s, which saturated at 50 ± 11% on longer exposure times (Fig. 13d). The 48 and 24 h films showed a qualitatively similar quenching behaviour, but with a reduced response.
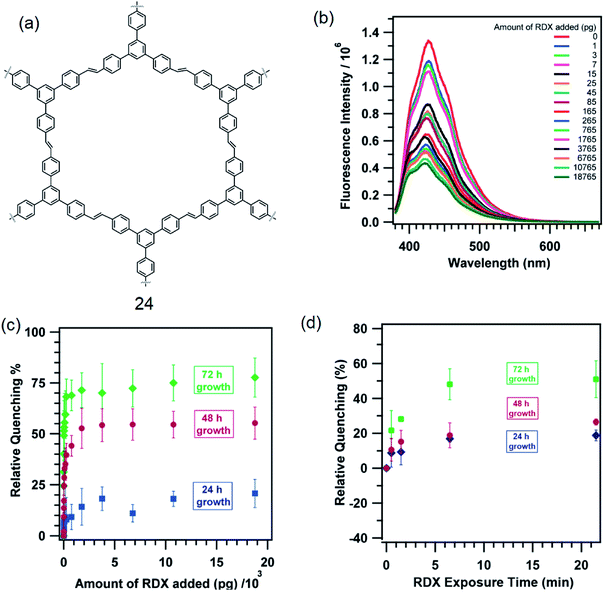 |
| | Fig. 13 (a) Structure of 24, (b) quenching of PL of a film grown for 72 h on exposure to RDX solution prepared in a mixture of CH3CN:MeOH (1:1 v/v), (c) relative PL quenching (%) of films on exposure to RDX in solution with different reaction time, and (d) PL quenching (%) of films on exposure to RDX vapour at different times. Reproduced from ref. 58 with permission. Copyright© 2013, American Chemical Society. | |
3. Ion sensing
3.1. Fluoride sensing
The fluoride ion plays an important role in preventing tooth decay and osteoporosis.23 Excess intake of fluoride, however, causes fluorosis, urolithiasis and other diseases. There have been remarkable efforts in developing sensors for the trace level detection of fluoride in biological systems as well as in drinking water sources. Owing to the high selectivity, sensitivity and ease of handling, fluorescence spectroscopy has become a powerful tool for the detection and quantification of the fluoride ion.59–62
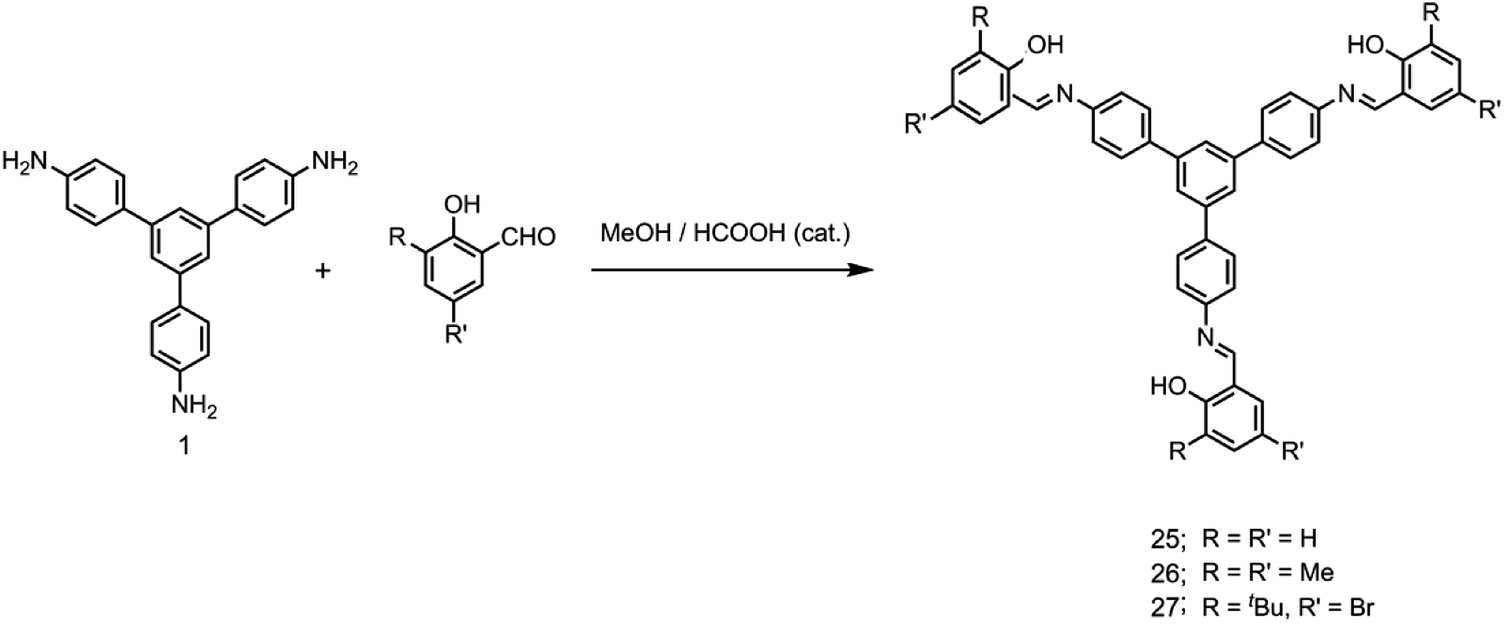
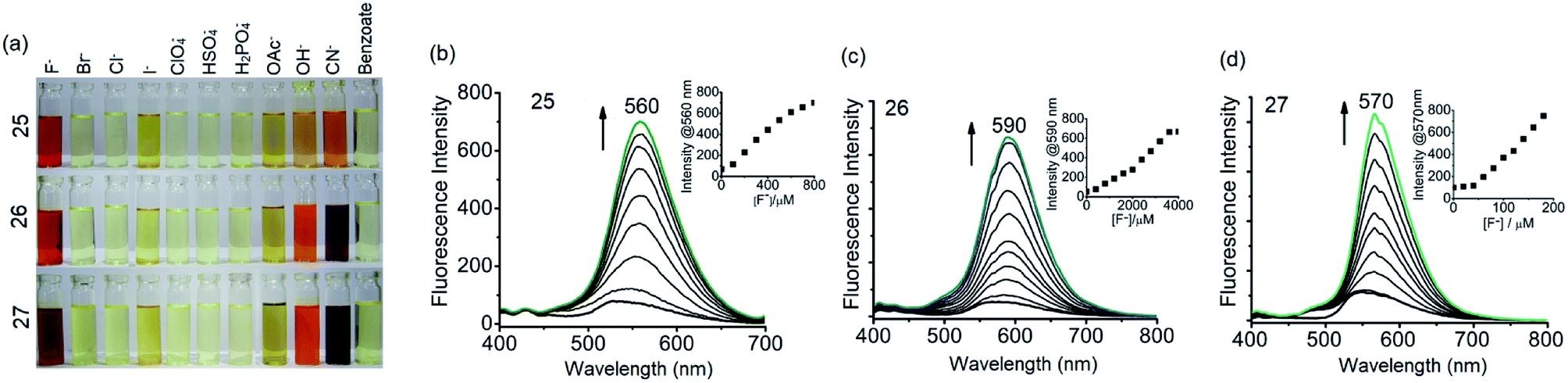
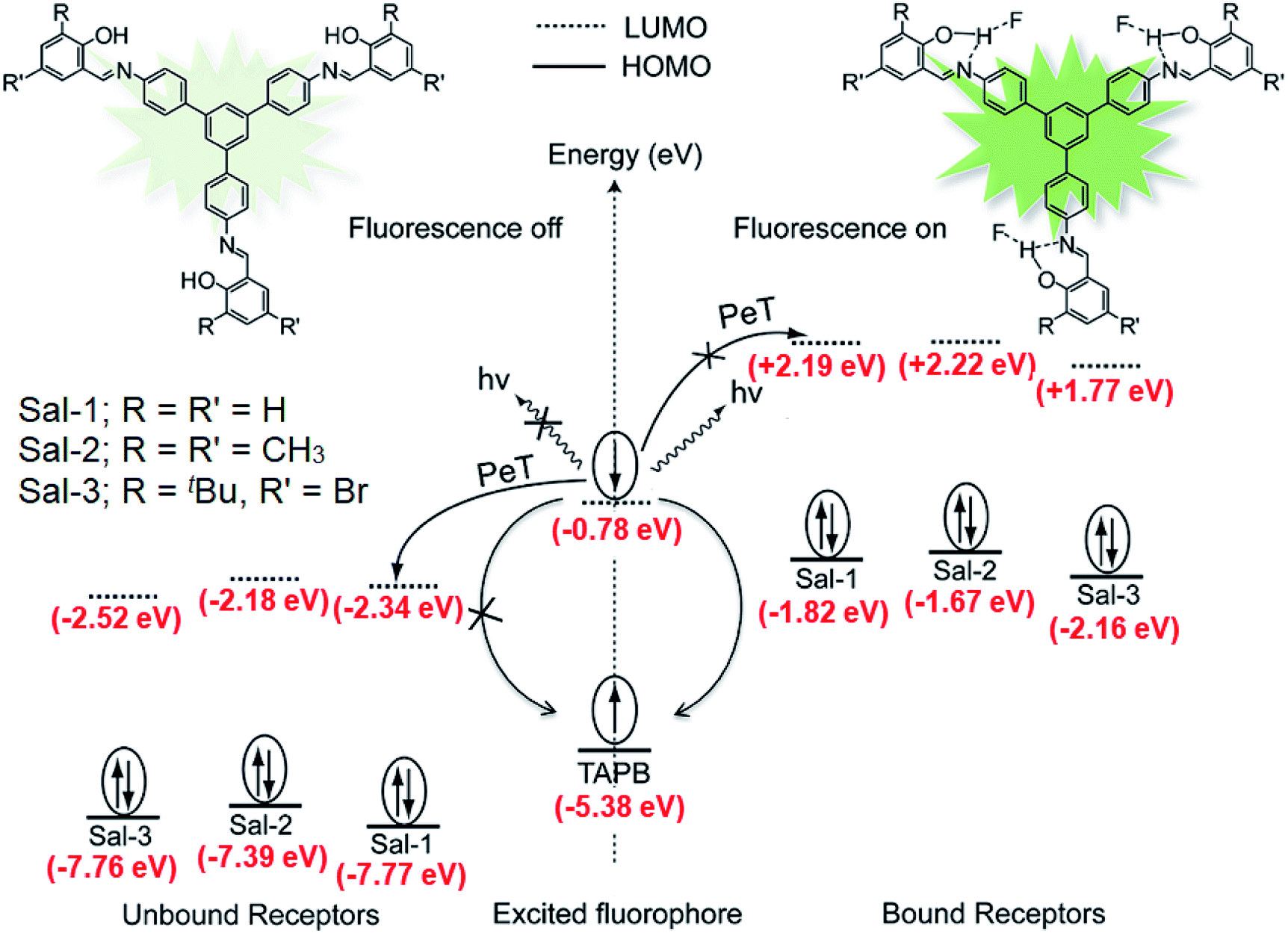
It has been shown that fluoride binds with the –OH proton of the salicylaldimine Schiff base through hydrogen bonds.59,63–67 A sensor molecule that comprises a salicylaldimine receptor connected to a fluorophore unit can effectively be used as a fluorescence turn-on sensor for fluoride. We have synthesized three different Schiff bases, 25–27, from a reaction of 1 with three different salicylaldehydes (Scheme 5).63 These Schiff bases contain three salicylaldimine groups and a 1,3,5-TPB core to serve as the anion receptor and fluorophore, respectively. Due to the rapid isomerization of –CN– linkages and intramolecular PET, these Schiff bases are very weakly fluorescent in their pristine state. The fluoride ion binds with the –OH group through hydrogen bond interactions, and its binding hinders –CN– isomerization and PET, leading to a dramatic change in the photoluminescence as well as in the colour. The light yellow solution of pristine Schiff bases turned deep red on exposure to fluoride and cyanide ions (Fig. 14a). On the addition of tetra-n-butylammonium fluoride (TBAF), the Schiff bases 25, 26 and 27 showed a photoluminescence turn-on at 560, 590 and 570 nm, respectively. The emission spectra showed a band enhancement on increasing the concentration of fluoride ions (Fig. 14b–d). The fluoride ion detection limits are 0.75, 3.0 and 0.17 ppm for 25, 26 and 27, respectively. Compound 27 shows the lowest detection limit (highest sensitivity) due to the presence of a more electronegative bromide substituent close to the –OH group. Energies of the frontier molecular orbitals of the receptors and the Schiff bases have been calculated. Fig. 15 shows the mechanism of fluorescence off-on deduced from first-principles calculations. The lowest unoccupied molecular orbitals (LUMOs) of the unbound salicylaldimine receptors lie at a lower energy compared to that of the LUMO of the 1,3,5-TPB unit. The excited electrons from the LUMO of 1,3,5-TPB can easily transfer to the LUMO of the receptor units. Therefore, the photoluminescence of the Schiff bases remains off in the pristine state. On binding with fluoride, the energies of the LUMOs of the receptors increase significantly. This leads to an energetically unfavourable PET from the 1,3,5-TPB core to the salicylaldimine receptors. Therefore, the excited electron relaxes back to the ground state through radiative decay with photon emission. The selective binding of the fluoride with the Schiff bases has been further studied by 1H NMR spectroscopy. The 1H NMR signal of the –OH protons diminishes on the addition of TBAF. The signal for [F⋯H⋯F]− has not been observed, which suggests that the fluoride forms hydrogen bonding rather than causes deprotonation.
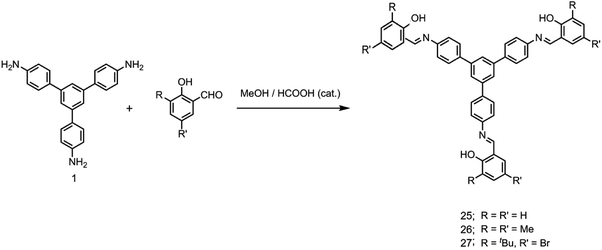 |
| | Scheme 5 Synthesis of 25–27. | |
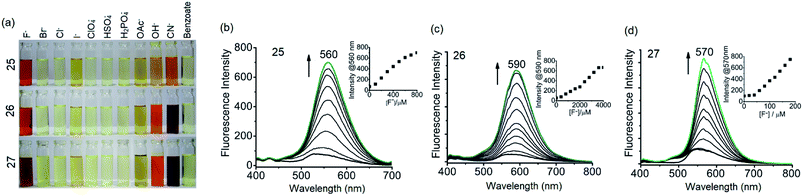 |
| | Fig. 14 (a) Chromogenic response of 25–27 (10.0 × 10−3 M in dry tetrahydrofuran) in the presence of 20 equivalents of various anions, (b) PL spectra of 25 with TBAF (0 to 40 equivalents), (c) PL spectra of 26 with TBAF (0 to 200 equivalents) and (d) PL spectra of 27 with TBAF (0 to 10 equivalents). The insets in b–d show emission intensity versus concentration of TBAF. Reproduced from ref. 63 with permission. Copyright© 2016, Springer Nature. | |
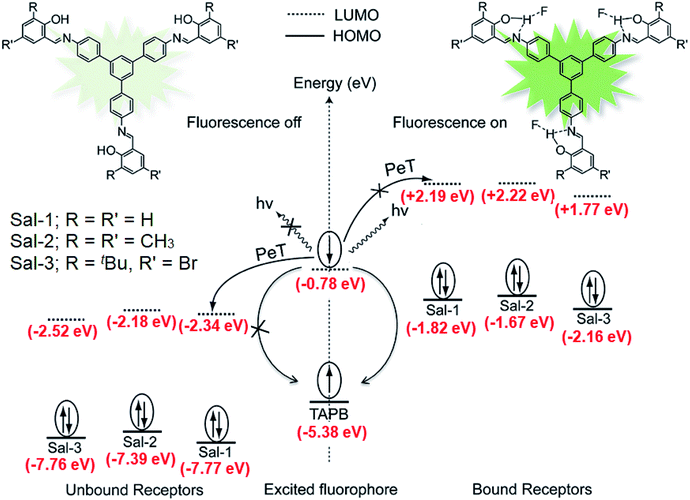 |
| | Fig. 15 Schematic of the intramolecular photo-induced electron transfer (PET) mechanism of photoluminescence “off-on” for tris-salicylaldimine Schiff bases in the absence and presence of fluoride ions. Sal-1, Sal-2 and Sal-3 indicate the receptors formed by salicylaldehyde, 3,5-dimethyl-2-hydroxy benzaldehyde and 5-bromo-2-hydroxy-3-tert-butylbenzaldehyde, respectively. Reproduced from ref. 63 with permission. Copyright© 2016, Springer Nature. | |
3.2. α-Hydroxycarboxylic acids sensing
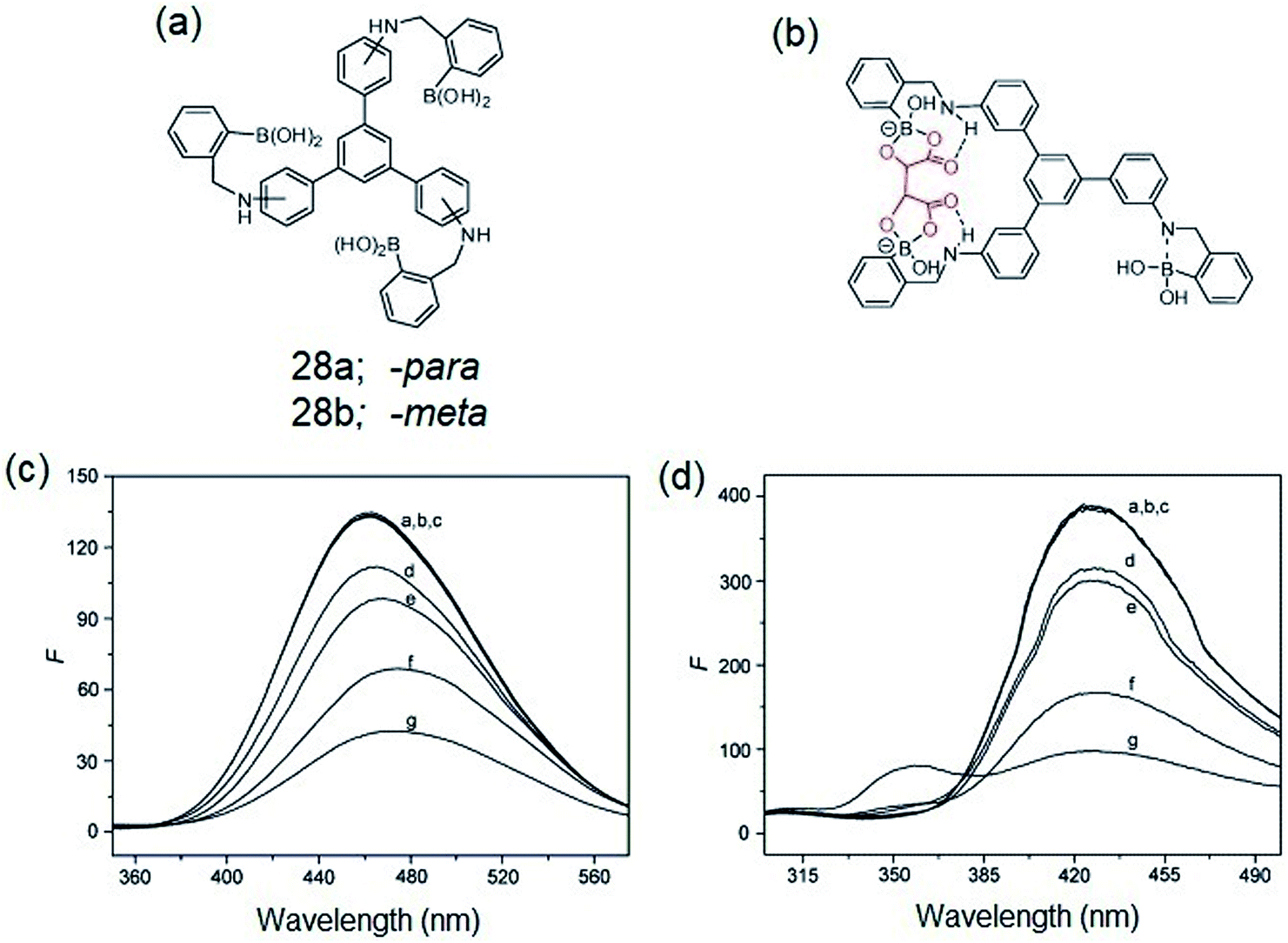
Zheng et al. reported on the synthesis of -para (28a) and -meta (28b) isomers of C3-symmetric tris-boronic acids containing –B(OH)2 and –NH binding sites and a 1,3,5-TPB fluorophore (Fig. 16a).68 The PL spectra of these fluorophores show bands at 460 and 358 nm on excitation at 297 and 257 nm, respectively, in a pH 8.71 buffer of methanol–water. The PL of both the fluorophores quenches selectively with α-hydroxycarboxylic and sugar acids over other saccharides. It is believed that intramolecular charge-transfer (ICT) facilitates the quenching by the formation of stable complexes with the hydroxyl and carboxyl groups of α-hydroxycarboxylic and sugar acids (Fig. 16b). Fig. 16c and d show the photoluminescence spectra of 28a and 28b after the addition of multiple concentrations of α-hydroxycarboxylic acids and saccharides. The PL is quenched selectively by α-hydroxycarboxylic acids with the selectivity order: tartaric acid > malic acid > lactic acid > galacturonic acid. Tartaric acid forms a bidentate complex with the sensors due to the presence of two α-hydroxycarboxylic groups.
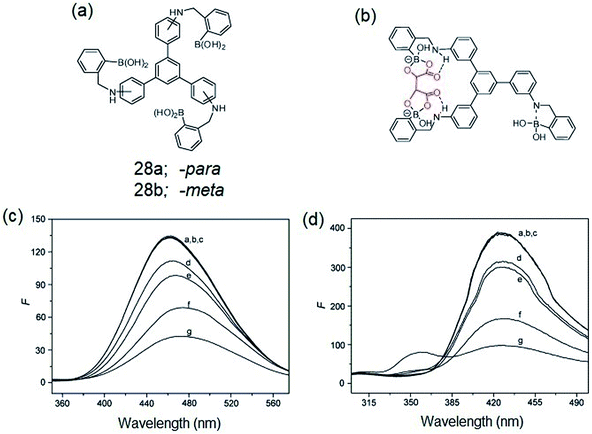 |
| | Fig. 16 (a) C3-symmetric tris-boronic acids, (b) proposed complex of α-hydroxycarboxylic acid with the sensor molecule, (c) PL spectra of 28a (1.4 × 10−2 mM, λex = 297 nm) and (d) 28b (6 × 10−2 mM, λex = 257 nm) in the presence of 5.0 mM of different analytes (a) free, (b) D-glucose, (c) D-galactose, (d) galacturonic acid, (e): lactic acid, (f): malic acid, (g): tartaric acid. Reproduced from ref. 68 with permission. Copyright© 2008, Elsevier Ltd. | |
3.3. Cu(II) sensing
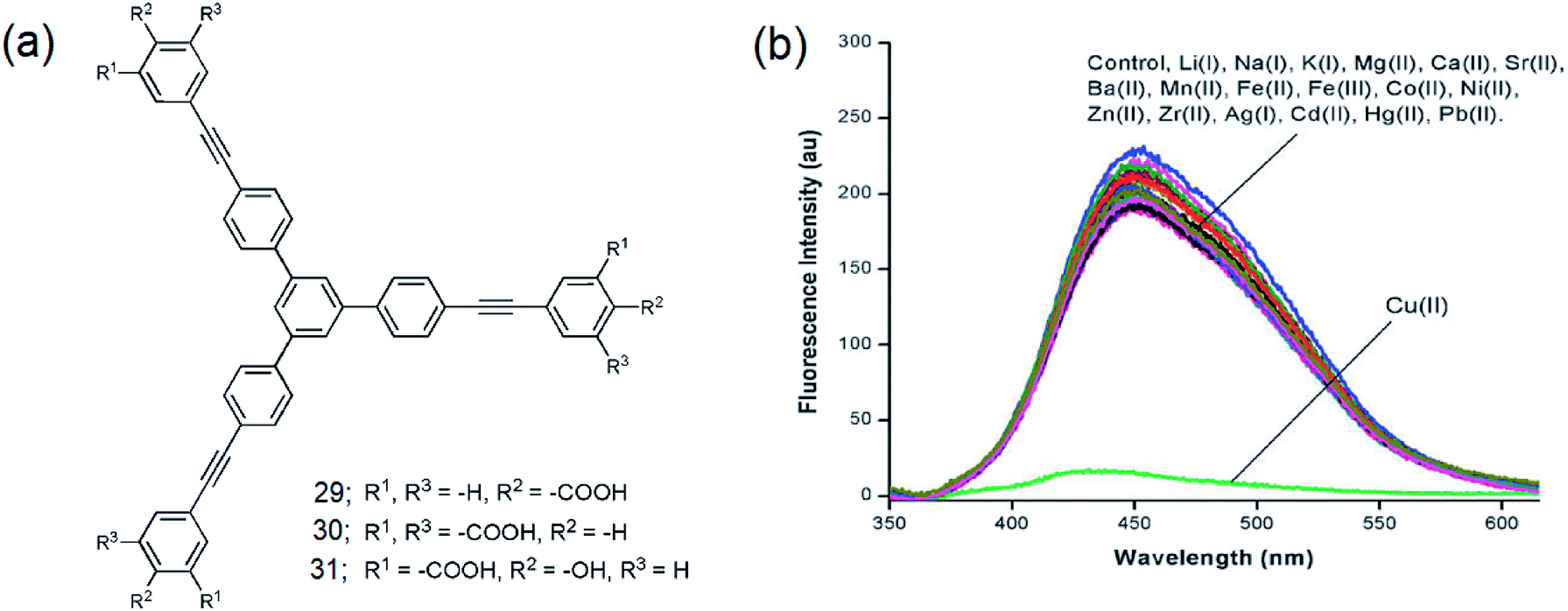
Sirilaksanapong et al. synthesized the water-soluble 1,3,5-TPB fluorophores 29 and 30 by Sonogashira coupling between 1,3,5-tri-(4′-iodophenyl)benzene and the peripheral units ethyl-4-ethynylbenzoate and diethyl 5-ethynyl iso-phthalate (Fig. 17a).24 Fluorophore 31 has been obtained by Sonogashira coupling between 1,3,5-tris-(4-ethynylphenyl)-benzene and ethyl-5-iodo-salicylate. The fluorophores 29–31 show photoluminescence bands at 426, 365, and 465 nm, respectively. The photoluminescence of 31 is selectively quenched on binding with Cu(II) (Fig. 17b). This could be attributed to the highly selective binding of peripheral salicylate groups with Cu(II) ions. The photoluminescence of 29 and 30 did not show any changes in the presence of various metal ions, including Cu(II) ions. Further, it was observed that Triton X-100 improves the quantum yields as well as the detection limit for Cu(II) from 6.49 to 0.19 ppb. Increases in the temperature of the sensor from 30 (room temperature) to 50 °C decreases the Stern–Volmer constant from 1.62 × 106 to 0.68 × 106 M−1. This indicates that Cu(II) binding with the sensor molecule is reversible and the mechanism of the PL quenching is static in nature. The addition of EDTA (ethylenediaminetetraacetic acid) to the mixture of 31 and Cu(II) leads to the complete restoration of the photoluminescence signal.
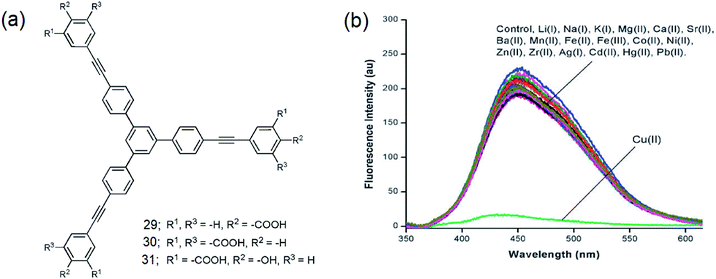 |
| | Fig. 17 (a) Structures of 29–31 and (b) photoluminescence spectra of 31 (1.0 × 10−3 M) in the presence of various metal ions (10.0 × 10−3 M). Cu(II) selectively quenches the photoluminescence. Reproduced from ref. 24 with permission. Copyright© 2011, Royal Society of Chemistry. | |
4. Conclusions and outlook
In this account, we have presented the recent studies on photoluminescent chemo-sensory materials based on a π-electron-rich 1,3,5-triphenylbenzene (1,3,5-TPB) platform. We have discussed the inherent properties of 1,3,5-TPB which make it a suitable fluorophore. The synthesis and sensing efficiency of supramolecular, discrete triphenylbenzene-carbazole, covalent-organic framework, covalent-organic polymer and π-conjugated polymer sensors have been presented in detail. 1,3,5-TPB is an easily synthesizable and stable material with modifiable spectroscopic and analyte binding properties. Fluorophores with attractive optical properties can be developed by changing the substituents on peripheral benzene rings, which can selectively recognize a wide variety of analytes. For instance, picric acid binds strongly with amino substituted 1,3,5-TPB fluorophores. The binding ability of PA with various amino groups decreases as –N(CH3)2 > –NH(CH3) and –NH2. Similarly, the photoluminescence of 1,3,5-TPB can be turned off by substituting with salicylaldimine groups and turned on by fluoride ion binding with the salicylaldimine receptors. As indicated by analyte binding constants (Table 1), 1,3,5-TPB based sensors can be placed among the best-known sensors for picric acid and other PNAC analytes.8,69,70 Many of the covalent-organic frameworks synthesized from 1,3,5-TPB exhibit appreciable photoluminescence properties, which can be of use for chemo-sensing and other related applications.50–52 Looking forward, there is substantial scope for the development of new materials using this fluorophore, some of which might find applications in sensing real-time samples.
Conflicts of interest
The authors declare that there is no conflict of interests regarding the publication of this paper.
Acknowledgements
P. V. and D. K. thank CSIR-New Delhi and UGC-New Delhi for research fellowships. R. M. acknowledges SERB-New Delhi for a J. C. Bose National Fellowship. The authors are also thankful to DST-Nano Mission, SERB-New Delhi and DAE-Mumbai for financial support.
Notes and references
- J. L. Kolanowski, F. Liu and E. J. New, Chem. Soc. Rev., 2018, 47, 195–208 RSC.
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019–8061 RSC.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef PubMed.
- X. Ma, F. Tao, Y. Zhang, T. Li, F. M. Raymo and Y. Cui, J. Mater. Chem. A, 2017, 5, 14343–14354 Search PubMed.
- S. Shanmugaraju and P. S. Mukherjee, Chem. Commun., 2015, 51, 16014–16032 RSC.
- A. Chowdhury and P. S. Mukherjee, J. Org. Chem., 2015, 80, 4064–4075 CrossRef PubMed.
- P. Vishnoi, S. Sen, G. N. Patwari and R. Murugavel, New J. Chem., 2015, 39, 886–892 RSC.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 5321–5322 CrossRef.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef PubMed.
- W. Chen, N. B. Zuckerman, J. P. Konopelski and S. Chen, Anal. Chem., 2010, 82, 461–465 CrossRef PubMed.
- S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871–2883 RSC.
- H. Sohn, M. J. Sailor, D. Magde and W. C. Trogler, J. Am. Chem. Soc., 2003, 125, 3821–3830 CrossRef PubMed.
- S. Shanmugaraju, H. Jadhav, R. Karthik and P. S. Mukherjee, RSC Adv., 2013, 3, 4940–4950 RSC.
- B. Gole, S. Shanmugaraju, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 10046–10048 RSC.
- J. Li, C.-F. Zhang, S.-H. Yang, W.-C. Yang and G.-F. Yang, Anal. Chem., 2014, 86, 3037–3042 CrossRef PubMed.
- H. S. Jung, K. C. Ko, J. H. Lee, S. H. Kim, S. Bhuniya, J. Y. Lee, Y. Kim, S. J. Kim and J. S. Kim, Inorg. Chem., 2010, 49, 8552–8557 CrossRef PubMed.
- B. Sui, X. Liu, M. Wang and K. D. Belfield, Chem.–Eur. J., 2016, 22, 10351–10354 CrossRef PubMed.
- A. S. Gupta, K. Paul and V. Luxami, Anal. Methods, 2018, 10, 983–990 RSC.
- V. Bhalla, Roopa and M. Kumar, Org. Lett., 2012, 14, 2802–2805 CrossRef PubMed.
- C.-H. Chen, D.-J. Liao, C.-F. Wan and A.-T. Wu, Analyst, 2013, 138, 2527–2530 RSC.
- A. Gupta and N. Kumar, RSC Adv., 2016, 6, 106413–106434 RSC.
- B. Sui, B. Kim, Y. Zhang, A. Frazer and K. D. Belfield, ACS Appl. Mater. Interfaces, 2013, 5, 2920–2923 Search PubMed.
- S. Sirilaksanapong, M. Sukwattanasinitt and P. Rashatasakhon, Chem. Commun., 2012, 48, 293–295 RSC.
- J. Pang, E. J. P. Marcotte, C. Seward, R. S. Brown and S. Wang, Angew. Chem., Int. Ed., 2001, 40, 4042–4045 CrossRef PubMed.
- K. Deng, Q.-Y. Huai, Z.-L. Shen, H.-J. Li, C. Liu and Y.-C. Wu, Org. Lett., 2015, 17, 1473–1476 CrossRef PubMed.
- B. P. Dash, R. Satapathy, E. R. Gaillard, J. A. Maguire and N. S. Hosmane, J. Am. Chem. Soc., 2010, 132, 6578–6587 CrossRef PubMed.
- T. Wohrle, S. J. Beardsworth, C. Schilling, A. Baro, F. Giesselmann and S. Laschat, Soft Matter, 2016, 12, 3730–3736 RSC.
- C. Bao, R. Lu, M. Jin, P. Xue, C. Tan, T. Xu, G. Liu and Y. Zhao, Chem.–Eur. J., 2006, 12, 3287–3294 CrossRef PubMed.
- H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. Go, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef PubMed.
- D. Kaleeswaran, P. Vishnoi and R. Murugavel, J. Mater. Chem. C, 2015, 3, 7159–7171 RSC.
- Y.-L. Li, Y. Zhao, Y.-S. Kang, X.-H. Liu and W.-Y. Sun, Cryst. Growth Des., 2016, 16, 7112–7123 Search PubMed.
- W. Salomon, G. Paille, M. Gomez-Mingot, P. Mialane, J. Marrot, C. Roch-Marchal, G. Nocton, C. Mellot-Draznieks, M. Fontecave and A. Dolbecq, Cryst. Growth Des., 2017, 17, 1600–1609 Search PubMed.
- H. Furukawa, Y. B. Go, N. Ko, Y. K. Park, F. J. Uribe-Romo, J. Kim, M. O'Keeffe and O. M. Yaghi, Inorg. Chem., 2011, 50, 9147–9152 CrossRef PubMed.
- Z. Wang, B. Zhang, H. Yu, G. Li and Y. Bao, Soft Matter, 2011, 7, 5723–5730 RSC.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef PubMed.
- A. P. Cote, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914–12915 CrossRef PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef PubMed.
- P. Vishnoi, M. G. Walawalkar, S. Sen, A. Datta, G. N. Patwari and R. Murugavel, Phys. Chem. Chem. Phys., 2014, 16, 10651–10658 RSC.
- P. Vishnoi, M. G. Walawalkar and R. Murugavel, Cryst. Growth Des., 2014, 14, 5668–5673 Search PubMed.
- V. G. Saraswatula, D. Sharada and B. K. Saha, Cryst. Growth Des., 2018, 18, 52–56 Search PubMed.
- S. Nagendran, P. Vishnoi and R. Murugavel, J. Fluoresc., 2017, 27, 1299–1305 CrossRef PubMed.
- D. Kaleeswaran, P. Vishnoi, S. Kumar, S. Chithiravel, M. G. Walawalkar, K. Krishnamoorthy and R. Murugavel, ChemistrySelect, 2016, 1, 6649–6657 CrossRef.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef PubMed.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- D. Kaleeswaran, R. Antony, A. Sharma, A. Malani and R. Murugavel, ChemPlusChem, 2017, 82, 1253–1265 CrossRef.
- S. K. Gupta, D. Kaleeswaran, S. Nandi, R. Vaidhyanathan and R. Murugavel, ACS Omega, 2017, 2, 3572–3582 CrossRef.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef PubMed.
- E. Vitaku and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 12911–12914 CrossRef PubMed.
- A. de la Peña Ruigómez, D. Rodríguez-San-Miguel, K. C. Stylianou, M. Cavallini, D. Gentili, F. Liscio, S. Milita, O. M. Roscioni, M. L. Ruiz-González, C. Carbonell, D. Maspoch, R. Mas-Ballesté, J. L. Segura and F. Zamora, Chem.–Eur. J., 2015, 21, 10666–10670 CrossRef PubMed.
- Y. Zeng, R. Zou, Z. Luo, H. Zhang, X. Yao, X. Ma, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 1020–1023 CrossRef PubMed.
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 4999–5002 CrossRef PubMed.
- J. L. Segura, M. J. Mancheno and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- R. Jangir, A. C. Kalita, D. Kaleeswaran, S. K. Gupta and R. Murugavel, Chem.–Eur. J., 24, 6178–6190 CrossRef PubMed.
- D. Kaleeswaran and R. Murugavel, J. Chem. Sci., 2018, 130(1) DOI:10.1007/s12039-017-1403-2.
- P. Arab, M. G. Rabbani, A. K. Sekizkardes, T. İslamoğlu and H. M. El-Kaderi, Chem. Mater., 2014, 26, 1385–1392 CrossRef.
- D. Gopalakrishnan and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 8357–8362 CrossRef PubMed.
- J.-S. Wu, W.-M. Liu, X.-Q. Zhuang, F. Wang, P.-F. Wang, S.-L. Tao, X.-H. Zhang, S.-K. Wu and S.-T. Lee, Org. Lett., 2007, 9, 33–36 CrossRef PubMed.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- C. A. Swamy P, R. N. Priyanka, S. Mukherjee and P. Thilagar, Eur. J. Inorg. Chem., 2015, 2015, 2338–2344 CrossRef.
- C. A. Swamy P and P. Thilagar, Inorg. Chem., 2014, 53, 2776–2786 CrossRef PubMed.
- P. Vishnoi, S. Sen, G. N. Patwari and R. Murugavel, J. Fluoresc., 2016, 26, 997–1005 CrossRef PubMed.
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571 CrossRef PubMed.
- M. E. Moragues, R. Martinez-Manez and F. Sancenon, Chem. Soc. Rev., 2011, 40, 2593–2643 RSC.
- L. E. Santos-Figueroa, M. E. Moragues, E. Climent, A. Agostini, R. Martinez-Manez and F. Sancenon, Chem. Soc. Rev., 2013, 42, 3489–3613 RSC.
- M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- W.-Z. Xu, Z.-T. Huang and Q.-Y. Zheng, Tetrahedron Lett., 2008, 49, 4918–4921 CrossRef.
- S. Sanda, S. Parshamoni, S. Biswas and S. Konar, Chem. Commun., 2015, 51, 6576–6579 RSC.
- V. Vij, V. Bhalla and M. Kumar, ACS Appl. Mater. Interfaces, 2013, 5, 5373–5380 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Dhananjayan Kaleeswaran
*ab,
Dhananjayan Kaleeswaran