DOI:
10.1039/C8RA02442A
(Paper)
RSC Adv., 2018,
8, 17551-17557
Preparation of thermo-responsive drug-loaded nanofibrous films created by electrospinning
Received
20th March 2018
, Accepted 6th May 2018
First published on 15th May 2018
Abstract
We prepared thermosensitive and biocompatible drug-loaded nanofibrous films by an electrospinning technique using a block copolymer, poly(N-isopropylacrylamide)-b-poly(L-lactide) (PNLA), and poly(L-lactide) (PLLA). The copolymer PNLA was synthesized by the radical polymerization of N-isopropylacrylamide (NIPAAm), followed by the ring-opening polymerization of L-lactide. The properties of PNIPAAm and PNLA were selectively discussed based on the results of NMR, FT-IR, GPC, and CA analyses. Because of the low molecular weight of PNIPAAm and PNLA and the hydrolysis of PNLA resulting from its hydrophilicity, these copolymers were inappropriate for electrospinning separately. Hence, a mixture of PNLA and PLLA was used to prepare electrospun nanofibrous films. SEM images of the PNLA/PLLA electrospun films showed that homogeneous fibres with smooth surfaces were obtained. In vitro release studies indicated that the drug-release rate of the PNLA/PLLA electrospun nanofibrous films can be adjusted by the content and molecular weight of PNLA and by the environmental temperature. The results demonstrate that electrospinning is a promising way to create stimuli-responsive fibrous films with potential applications in the design of controllable drug delivery systems.
1. Introduction
Electrospinning is a unique processing technique that can be used to fabricate ultra-fine fibres with diameters in the sub-micrometre down to the nanometre range, a size range that is otherwise difficult to access by conventional ultra-fine fibre fabrication techniques.1–3 These fibres exhibit an extremely high surface area-to-mass or volume ratio, small inter-fibrous pore size with high porosity and malleability to conform to a wide variety of sizes and shapes.4–6 Due to these advantages, many notable applications of electrospinning have become increasingly attractive in the biomedical field, including filtration5,7 and dialysis membranes, tissue engineering,8–10 immobilized enzymes and catalysts,11–13 wound dressings,14 artificial blood vessels15 and controlled drug/gene delivery.16–19 A typical example is its use in the biomedical field combined with PLA, which is a particularly interesting biocompatible and biodegradable material with potential environmental and biomedical applications. On the one hand, PLA has a relatively high breaking strength and initial modulus compared with other polymer materials.20 On the other hand, the poor hydrophilic properties of PLA are well known for causing a lack of bioactivity and cell affinity, and the degradation rate of PLA is relatively slow.21 For successful application in the biomedical field, many techniques to modify PLA materials have been created in the past years. Among these techniques, an important method is to modify the electrospinning fabrication process and setup. For example, the structure and morphology of the fibre can be controlled by different parameters,14 such as the electric field strength, distance between the spinneret and the collecting plate, temperature and humidity, or by modification of the setup, including side-by-side electrospinning systems,22,23 multi-jet electrospinning24,25 and co-axial electrospinning.26 In addition to focusing on the parameters and setup, adjusting the material compositions also plays an important role in the improvement and refinement of electrospun fibres, allowing manipulation of the properties and structure. Among these modified formulations, using rational polymer material designs, including copolymers27 and polymer mixtures with organic28,29 and inorganic components,8,30–32 and adjusting the component ratio have been considered efficient schemes to generate desirable materials with hydrophilicity,28,33 antibacterial properties,34 biodegradability,33 spinning ability,35 and mechanical integrity.15
In this study, we provided a new pathway incorporating biodegradable and thermosensitive poly(N-isopropylacrylamide) (generally abbreviated PNIPAAm) with PLLA as an electrospinning solution formulation to fabricate an excellent fibrous film that overcomes the previously reported deficiencies. PNIPAAm is a temperature-responsive polymer, the lower critical solution temperature (LCST) of which is at a temperature near that of the human body.
In general, PNIPAAm-modified PLLA materials can be obtained through either copolymerization of PNIPAAm and PLA27 or mixtures of PNIPAAm and PLA36 (as we have previously reported). The problem is that, as reported by Dai et al.27 and Hu et al.,37 the copolymer is difficult to electrospin into fibres when the parameters are changed. This could be attributed to the low molecular weight of PNIPAAm-b-PLLA. Additionally, the films electrospun from these mixtures did not show a desirable increase in the rate of drug release with an increase in temperature, which was ascribed to the blended, not encapsulated, drug-loaded models resulting from the incompatibility between PNIPAAm and PLA.
In this study, to avoid the problems mentioned above, PNIPAAm was added to PLA in the form of the copolymer PNIPAAm-b-PLA and electrospun to produce a biodegradable and thermo-responsive fibrous film. PNIPAAm-b-PLA was synthesized by radical polymerization and ring-opening polymerization. Through blending with PLA, the electrospinnability of PNIPAAm-b-PLA improved, and the diameter of fibres can be well controlled by adjusting the proportion of PNIPAAm blocks in PNIPAAm-b-PLA and the amount of PLA in the mixture. The thermo-sensitivity of the fibrous films resulting from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H groups in the PNIPAAm blocks can also be manipulated by adjusting the proportion of PLA. The PLA/PNLA electrospun fibrous film prepared in this manner was used as a stimuli-responsive carrier of rifampicin in a drug-release system to prevent the formation of Gram-positive and Gram-negative microbial communities on prosthetic surfaces after major orthopaedic surgeries.38 A patient may undergo multiple surgeries and prolonged antibiotic treatment following the current standard of care. Thus, use of the prepared system in wound dressing would be of clinical significance.
O and N–H groups in the PNIPAAm blocks can also be manipulated by adjusting the proportion of PLA. The PLA/PNLA electrospun fibrous film prepared in this manner was used as a stimuli-responsive carrier of rifampicin in a drug-release system to prevent the formation of Gram-positive and Gram-negative microbial communities on prosthetic surfaces after major orthopaedic surgeries.38 A patient may undergo multiple surgeries and prolonged antibiotic treatment following the current standard of care. Thus, use of the prepared system in wound dressing would be of clinical significance.
2. Experimental
2.1. Materials
N-Isopropylacrylamide (NIPAAm), recrystallized twice with toluene/cyclohexane (v/v, 40/60) solvents, was purchased from TCI (Tokyo). N,N′-Azobisisobutyronitrile (AIBN), recrystallized twice with ethanol, was provided by Shanghai Chemical Reagent Co., Ltd. 2-Mercaptoethanol (98%) and stannous octoate (96%) were purchased from Alfa Aesar. L-Lactide was purified by recrystallization from toluene three times and dried in a vacuum at room temperature. Poly(L-lactide) (PLLA) with an average molecular weight of 180![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and dried in vacuum at 60 °C for 12 h was obtained from Shanghai TJL Co., Ltd. (China). Other reagents and solvents were purchased from Shanghai Chemical Reagent Co., Ltd. (China) and used without further processing.
000 and dried in vacuum at 60 °C for 12 h was obtained from Shanghai TJL Co., Ltd. (China). Other reagents and solvents were purchased from Shanghai Chemical Reagent Co., Ltd. (China) and used without further processing.
2.2. Synthesis of PNIPAAm
PNIPAAm was synthesized via radical polymerization with 2-mercaptoethanol as the chain-transfer agent and AIBN as the initiator. The typical detailed preparation procedures were as follows. NIPAAm (8.23 g/72.8 mmol), AIBN (6.9 mg/0.042 mmol, 0.06 mol%) and 2-mercaptopropanol (6.88 mg, 0.06 mol%) were dissolved in DMF (30 mL) first, then nitrogen was bubbled through the solution for 30 min to remove oxygen from the solvent before polymerization, and the solution was polymerized at 70 °C for 20 h under nitrogen atmosphere. Afterward, the white solid was obtained through spin-evaporation condensing under reduced pressure, repeatedly precipitating three times in excess ether and suction filtering. Then, the product was washed twice with ether and dried in a vacuum oven for 24 h. A white powder was finally obtained.
2.3. Synthesis of PNIPAAm-b-PLLA(PNLA)
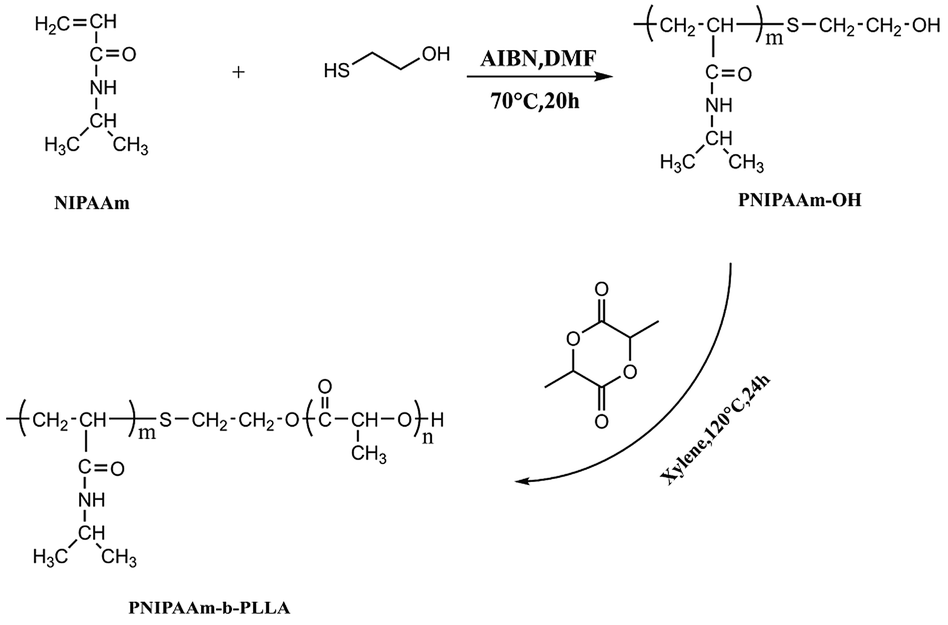
With the terminal hydroxyl of PNIPAAm as the initiation point, PNLA was synthesized from L-lactide by ring-opening polymerization, and the synthesis route is shown in Scheme 1. The typical detailed preparation procedure was as follows. First, the macromolecule initiator PNIPAAm (1 g) and L-lactide (5 g/34.7 mmol) were added to dry xylene (3 mL), then the initiator stannous octoate (0.1 mol% of the monomer) was added after stirring to dissolve. Then, nitrogen was charged three times after evacuation to remove the air from the reaction flask, and the reactants were polymerized at 120 °C for 24 h afterward. Subsequently, the products were dissolved in a small amount of THF and then precipitated in excess diethyl ether three times. The white powder PNLA was finally obtained after suction filtering, washing twice with ethyl ether and drying in a vacuum oven.
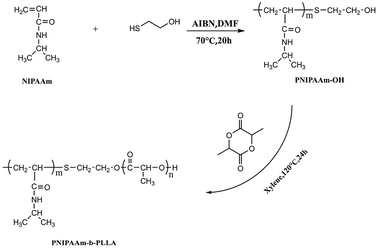 |
| | Scheme 1 Synthesis route of the amphiphilic block copolymer PNIPAAm-b-PLLA. | |
2.4. Nuclear magnetic resonance spectroscopy (NMR)
Nuclear magnetic resonance (NMR) spectra of the samples were obtained on a Bruker DMX 500 NMR spectrometer operating at 400 MHz using deuterated chloroform (CDCl3) as solvent, and the chemical shifts were evaluated relative to tetramethylsilane (TMS).
2.5. Attenuated total reflection Fourier-transform infrared (ATR FT-IR) spectra
Attenuated total reflection Fourier-transform infrared (ATR FT-IR) spectra of the powders, after being ground and compressed with KBr, were recorded on a magna FT-IR 550 FTIR spectrometer (Nicole, America) under the following parameter: measuring range 4000–400 cm−1.
2.6. Gel permeation chromatography (GPC)
The average molecular weight and polydispersity of the samples were measured on a gel permeation chromatograph instrument (GPC, WATERS-150C) at the flow rate of 1.0 mL min−1 using tetrahydrofuran (THF) as the eluent and polystyrene as the standard.
2.7. Preparation of blank fibrous films
The PLLA was dissolved in a mixed solvent composed of chloroform, dichloromethane and DMF (v/v/v: 7/2/1) first, and a concentration of 0.1 g mL−1 solution was obtained after magnetically stirring for 12 h at room temperature. Then, the solution was divided into two parts, one of which was used for electrostatic spinning directly, and the rest of the PLLA solution was added to PNLA powder slowly with further magnetic stirring for 12 h to formulate a solution of the desired concentration. Next, each of the obtained spinning solutions was transferred into a 20 mL syringe with a truncated-tip needle (9#) for electrospinning, in which the spinning voltage was controlled at approximately 18 kV and the receiving distance was 18–20 cm, with the liquid feed rate adjusted by a micro injection pump at a flow rate of 2 mL h−1. After spinning for 4 h, the film was dried in a vacuum at room temperature for one day. The dried fibrous membrane was finally peeled off from the aluminium foil and encapsulated in a self-sealing bag for further experimentation. For comparison, a casting film of PLLA was made on a Petri dish.
2.8. Detecting the surface contact angle
The contact angle was measured on an OCA20 contact-angle analysis system (DataPhysics, Germany). The temperature was controlled using a super thermostat (Julabo F25, Germany). Water droplets (approximately 2 μL) were dropped carefully onto the samples. The average CA values were obtained by measuring five different positions on the same sample.
2.9. Preparation of drug-loaded fibres
Preparation of the PLLA/PNLA solution was as described above. Rifampicin (RFP) powder was added to the solution such that an 8% drug loading (RFP vs. PLLA) was obtained. The procedures of electrospinning were also as described above.
2.10. Scanning electron microscopy (SEM)
The surface morphology of the fibrous films was investigated on a S-2360N scanning electron microscope (SEM, HITACHI, Japan) at a 15 kV accelerating voltage after vacuum drying and gold plating. The average diameter and distribution of the ultra-fine fibres in the SEM images were obtained by measuring one hundred fibres via Sigma Scan Pro4.0.
2.11. In vitro release test
PBS solution was used to simulate a human body fluid environment for the test of controlled drug-release ability. Fibrous films were cut into small pieces (1 cm × 1 cm) and suspended into a wide jar containing PBS solution (300 mL, pH 7.4) that had been preheated to 25 °C or 40 °C. The absorption peak at λmax = 474 ± 1 nm was measured by a UV-Vis spectrophotometer every other time. The cumulative release Er (%) was calculated by the Lambert–Beer law.
3. Results and discussion
3.1. Molecular structure of polymers
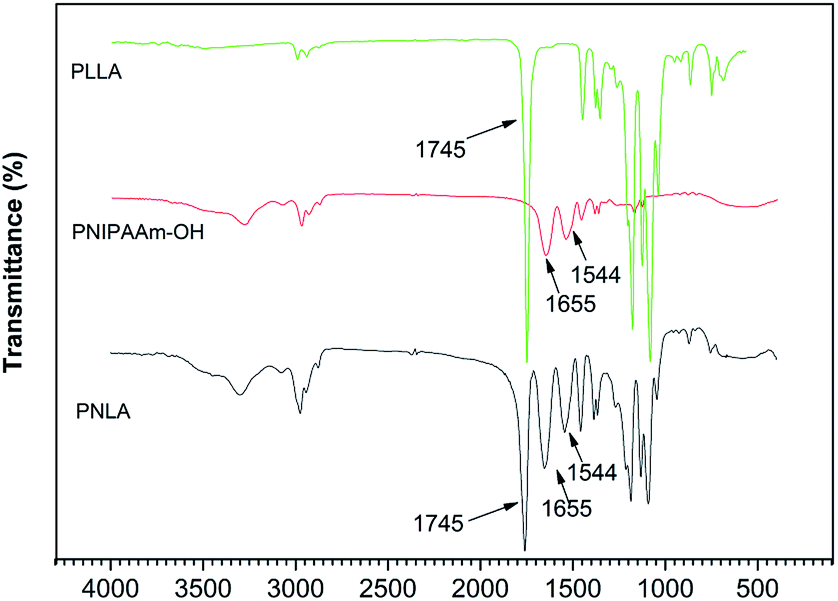
The microstructure of the polymers was measured by FT-IR. Fig. 1 shows the spectrum of the homopolymer PNIPAAm. The CO stretching vibration peak and N–H deformation vibration peak of the amide group are shown at 1655 cm−1 and 1544 cm−1, respectively. The peaks at 1386 cm−1 and 1367 cm−1 indicate the stretching vibration of the methyne groups on the pendant groups of PNIPAAm, and the peaks at 1332 cm−1and 1272 cm−1 represent the doublet of overlapping C–N and C–H. In addition, the CH3 scissor deformation (1459 cm−1), N–C stretching (1243 cm−1 and 1172 cm−1), C–C stretching (1130 cm−1), CH3 rocking (975 cm−1 and 925 cm−1), and CH3 twisting (883 cm−1 and 838 cm−1) of the pendant groups are also presented in the spectrum. Moreover, disappearance of the CC stretching vibration peak at 1600 cm−1 indicated the acquisition of PNIPAAm. The FT-IR spectrum of the copolymer PNLA is also shown in Fig. 1. The peak at 1745 cm−1 indicates the CO stretching in the ester bond of PLLA, the CO stretching (1655 cm−1) and N–H deformation (1544 cm−1) peaks, which are characteristic of PNIPAAm, did not disappear, supporting the conclusion that the copolymer PNLA was formed.
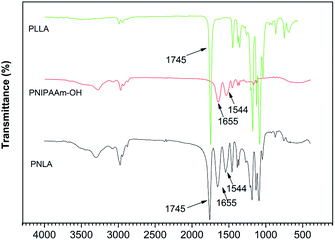 |
| | Fig. 1 FT-IR spectra of PLLA, PNIPAAm and PNLA. | |
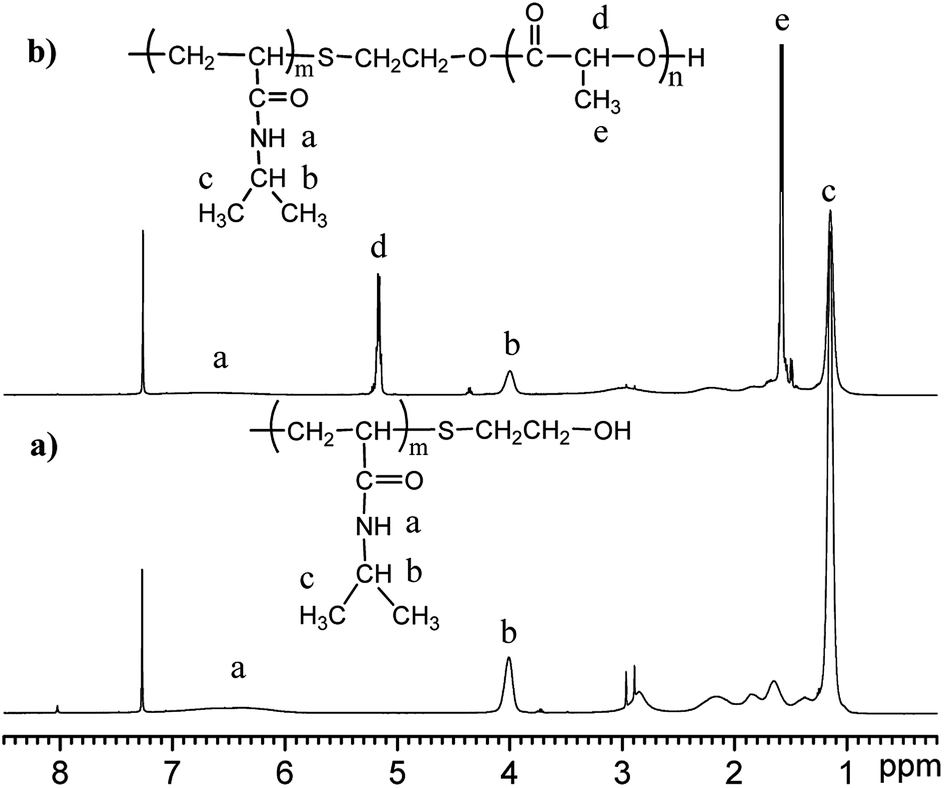
The molecular structure of the synthesized polymer was also analysed by nuclear magnetic resonance (NMR), and the spectrum is shown in Fig. 2. The spectrum of PNIPAAm (Fig. 2(a)) shows peaks at δ4.0 ppm and δ1.2 ppm that are attributed to the methyne and methyl protons of the –NH–CH(CH3)2 groups in the NIPAAm moieties, respectively. The peaks at δ1.5 ppm and δ2.0 ppm are attributed to the resonance of the protons of the methylene and methyne groups on the backbone of PNIPAAm, respectively. Since the hydrogen protons of –NH–CO– are active and its resonance peak is generally not obvious, it's hard to identify without using heavy water exchange. The resonance peak is approximately assigned to the broad peak at δ6.0–7.0 ppm. The spectrum of PNLA is also shown in Fig. 2(b), in which the characteristic proton resonance peak (δ1.6 ppm) of the methyne groups on the PLLA chain and the methyl peak (δ5.2 ppm) on the side chain increased apparently. At the same time, the characteristic proton resonance peaks (δ1.2 ppm, δ4.0 ppm) of the double methyl and methyne groups on the pendant groups of the PNIPAAm chain can be seen clearly. Combining the results of FT-IR and 1HNMR, all of the signals confirmed the successful preparation of PNIPAAm and the copolymer PNLA.
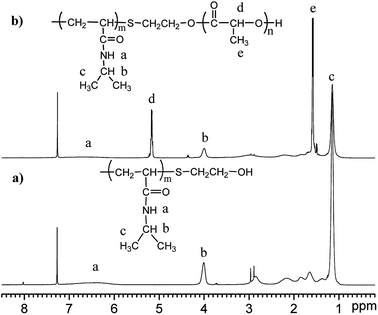 |
| | Fig. 2 The typical 1H-NMR spectra of (a) PNIPAAm and (b) PNLA in CDCl3. | |
3.2. Molecular weight and distribution of polymers
The molecular weights and distribution of the synthesized polymers are summarized in Table 1. The Mn and Mw/Mn values suggest that the molecular weights of both PNIPAAm and PNLA are too low to electrospin separately.39,40 Therefore, PNLA and PLLA were subsequently blended for electrospinning to obtain fibres with better structure.
Table 1 The characterization of PNIPAAm and PNLA block copolymers
| Samples |
PNIPAAm/LLA (wt) |
Mn |
Mw/Mn |
| PNIPAAm |
|
10500 |
1.65 |
| PNLA1 |
3:10 |
22600 |
1.59 |
| PNLA2 |
2:10 |
31600 |
1.31 |
| PNLA3 |
1:10 |
39700 |
1.45 |
3.3. The surface contact angle of the fibrous films
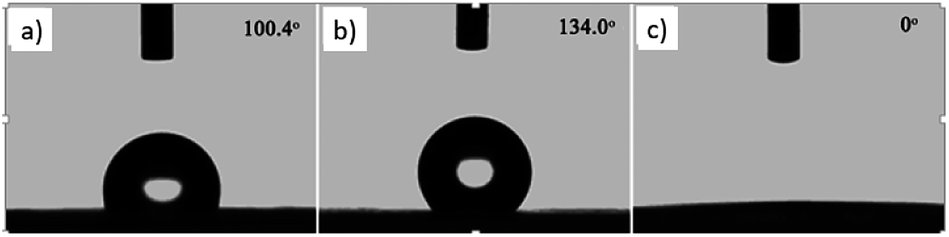
The wettability of the film was observed by water contact angle (CA) measurements. The coating film of pure PLLA [Fig. 3(a)] shows a water contact angle of 100.4°, and the electrospun film of pure PLLA [Fig. 3(b)] shows a CA of 134° at the same temperature (20 °C), which indicated an increase in the water contact angle resulting from the ultrafine fibrous film produced from the electrospinning process. The large water contact angle caused by strong hydrophobic interactions will slow down the release of drugs into aqueous fluids, enabling sustained release. A photograph of the water-droplet shape on the electrospun film of PNLA (bead-in-fibre) is shown in Fig. 3(c), which demonstrates that the suspended water droplets were quickly absorbed at 20 °C and that the water contact angle was nearly 0°. Obviously, the hydrophilicity of the PNLA electrospun film was caused by the PNIPAAm chain segments.
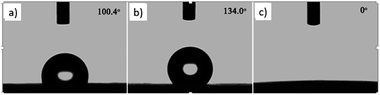 |
| | Fig. 3 Water contact angle of several samples: (a) casting film of PLLA, (b) electrospun film of PLLA, and (c) electrospun film of PNLA (bead-in-fibres). | |
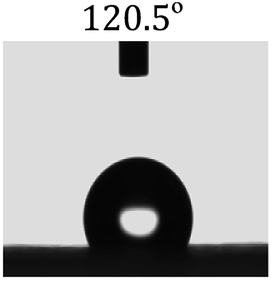
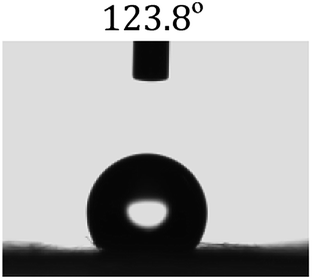
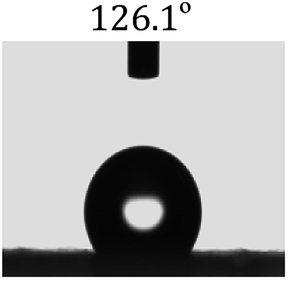
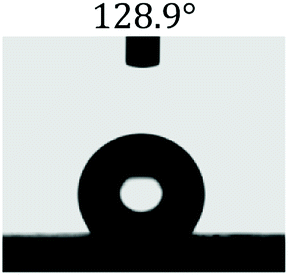
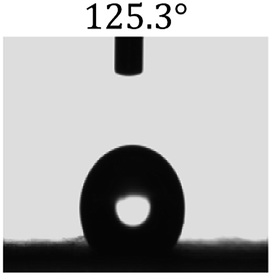
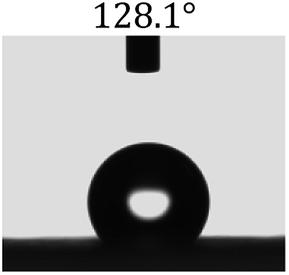
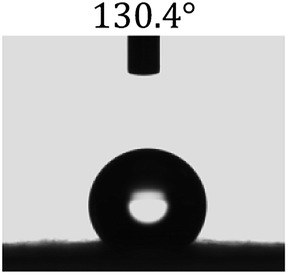
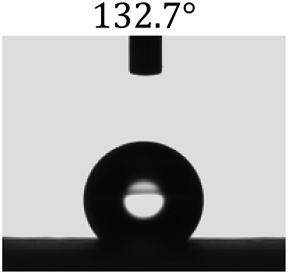
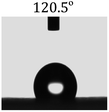
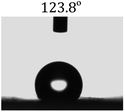
Table 2 shows the contact angles of the blend films with different proportions and at different temperatures. Combined with pure hydrophobic PLLA, the blended films showed more hydrophobicity. Among them, the PNLA1/PLLA (3:10) electrospun film showed the lowest contact angle of 120.5° when the temperature was 25 °C. Apparently, the contact angles of the PNLA/PLLA electrospun films were gradually enhanced with the increase in the hydrophobic PLLA proportion of the copolymer PNLA and the PNLA/PLLA blends. Additionally, the contact angles of the electrospun films also increased to some extent when the testing temperature increased from 25 °C to 40 °C. This phenomenon was due to competition between the intermolecular and intramolecular hydrogen bonds of PNIPAAm as the temperature hovered around the LCST (approximately 32 °C). When the temperature was lower than the LCST, despite the hydrophobic PLLA, the hydrophilic groups N–H and CO of the amide bond on the PNIPAAm molecular chain tended to form intermolecular hydrogen bonds with the water molecules. Therefore, the PNLA/PLLA electrospun film was more hydrophilic. However, when the films were heated, intramolecular hydrogen bonds tended to form between the CO and N–H groups, and the PNIPAAm molecular chain curled, which made it difficult for water molecules to interact with these hydrophilic groups. Therefore, the materials transformed to show more obvious hydrophobicity.
Table 2 Water contact angles of films of different proportions at 25 °C and 40 °C
| T/(°C) |
PNLA1:PLLA = 3:10 |
PNLA2:PLLA = 3:10 |
PNLA3:PLLA = 3:10 |
PNLA1:PLLA = 1:10 |
| 25 °C |
 |
 |
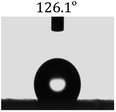 |
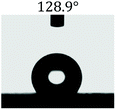 |
| 40 °C |
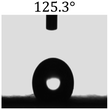 |
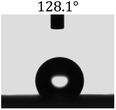 |
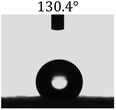 |
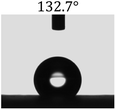 |
3.4. Micro-morphology of the electrospun films
As shown in Fig. 4(a) and (b), the morphology of the pure PNLA electrospun film was a microsphere structure instead of a fibre structure, which indicated that pure PNLA could not be electrospun into ultra-fine fibres. It is reasonable to speculate that the hydrophilicity of pure PNLA is excellent (CA = 0° after spinning). In contrast, PLLA solution (8% w/v) could be spun into uniform fibres [Fig. 4(d)], and the fibres spun from PLLA solution (10% w/v) presented a “microbead/nanofibre” structure [Fig. 4(c)]. Obviously, PLLA has a great spinnability and will make up for the spinning deficiencies of PNLA. Therefore, combining PNLA with PLLA for blend electrospinning will provide better results to a certain extent.
 |
| | Fig. 4 SEM photographs of electrospun films: (a) and (b) PNIPAAm-b-PLLA(PNLA), (c) pure PLLA (10%), (d) pure PLLA (8%). | |
3.5. Micro-morphology and diameter distribution statistics of the drug-loaded PNLA/PLLA electrospun films
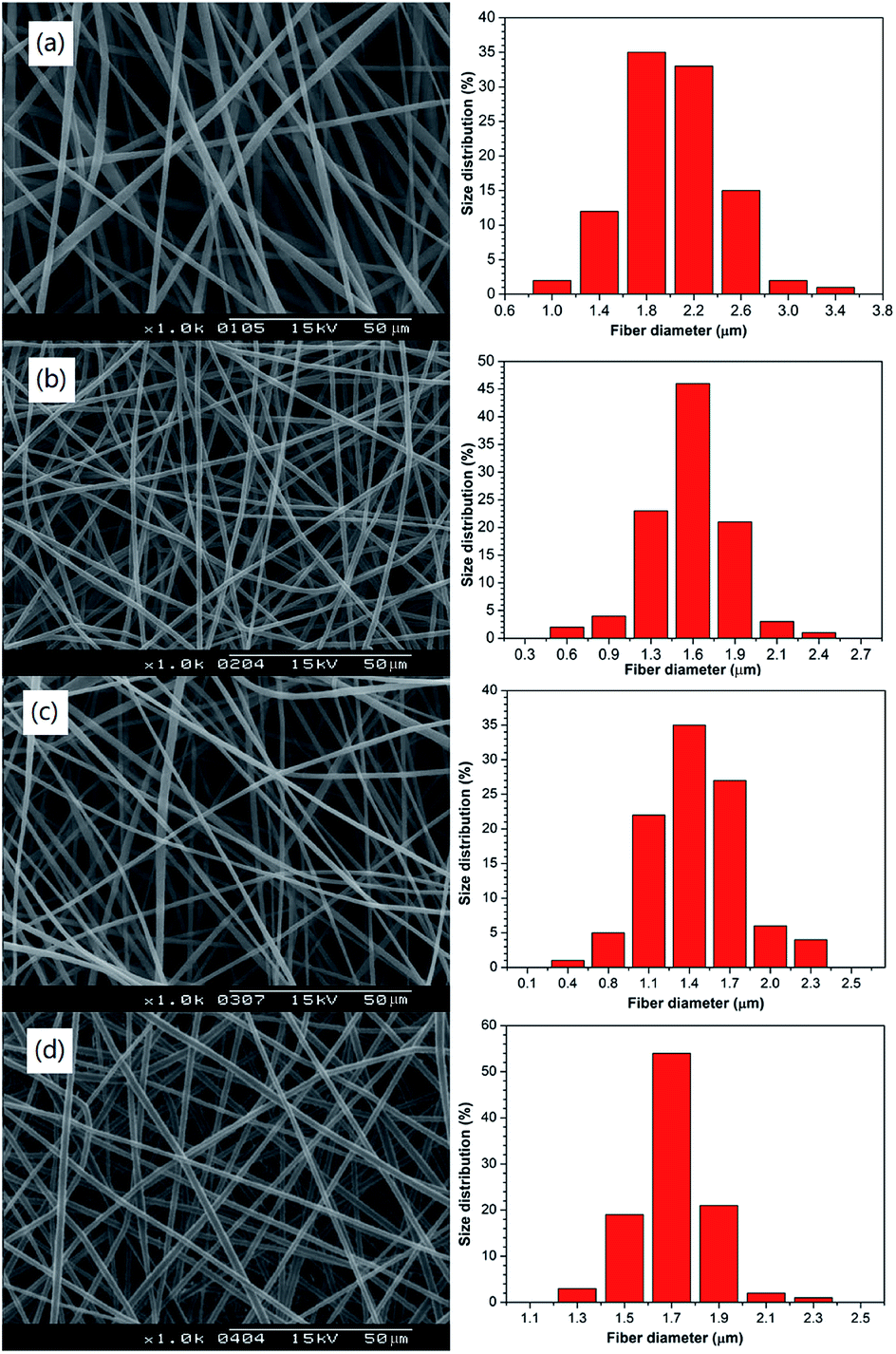
Fig. 5 shows that the fibre diameter of PNLA/PLLA was very uniform and that there was no bead formation. Comparing Fig. 5(a) with 5(d), the average diameter of the ultra-fine fibres was proportional to the PNLA mass fraction, which was consistent with the previous analysis. Furthermore, with the increase in the molecular weight of PNLA, the average diameter of the fibres tended to decrease.
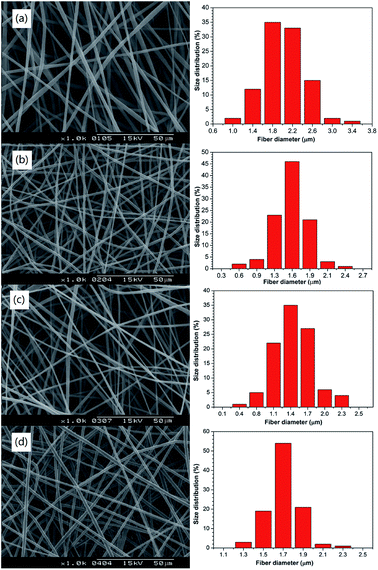 |
| | Fig. 5 SEM images and diameter distribution of the electrospun nanofibrous films of (a) PNLA1/PLLA = 3:10, (b) PNLA2/PLLA = 3:10, (c) PNLA3/PLLA = 3:10, and (d) PNLA1/PLLA = 1:10. | |
This effect was because as the molecular weight of PNLA increased, it became closer to the molecular weight of PLLA, and the spinning behaviour became much more similar to that of PLLA. Table 3 shows the average fibre diameters of PNLA/PLLA films (a to d), and the results are in line with Fig. 5.
Table 3 The average diameters of PNLA/PLLA drug-loaded nanofibres
| |
Samples |
Average fibre diameter (μm) |
| a# |
PNLA1 (22600)/PLLA = 3:10 |
1.95 |
| b# |
PNLA2 (31600)/PLLA = 3:10 |
1.57 |
| c# |
PNLA3 (39700)/PLLA = 3:10 |
1.49 |
| d# |
PNLA1 (22600)/PLLA = 1:10 |
1.73 |
3.6. In vitro drug-release behaviour of the drug-loaded PNLA/PLLA electrospun nanofibrous films
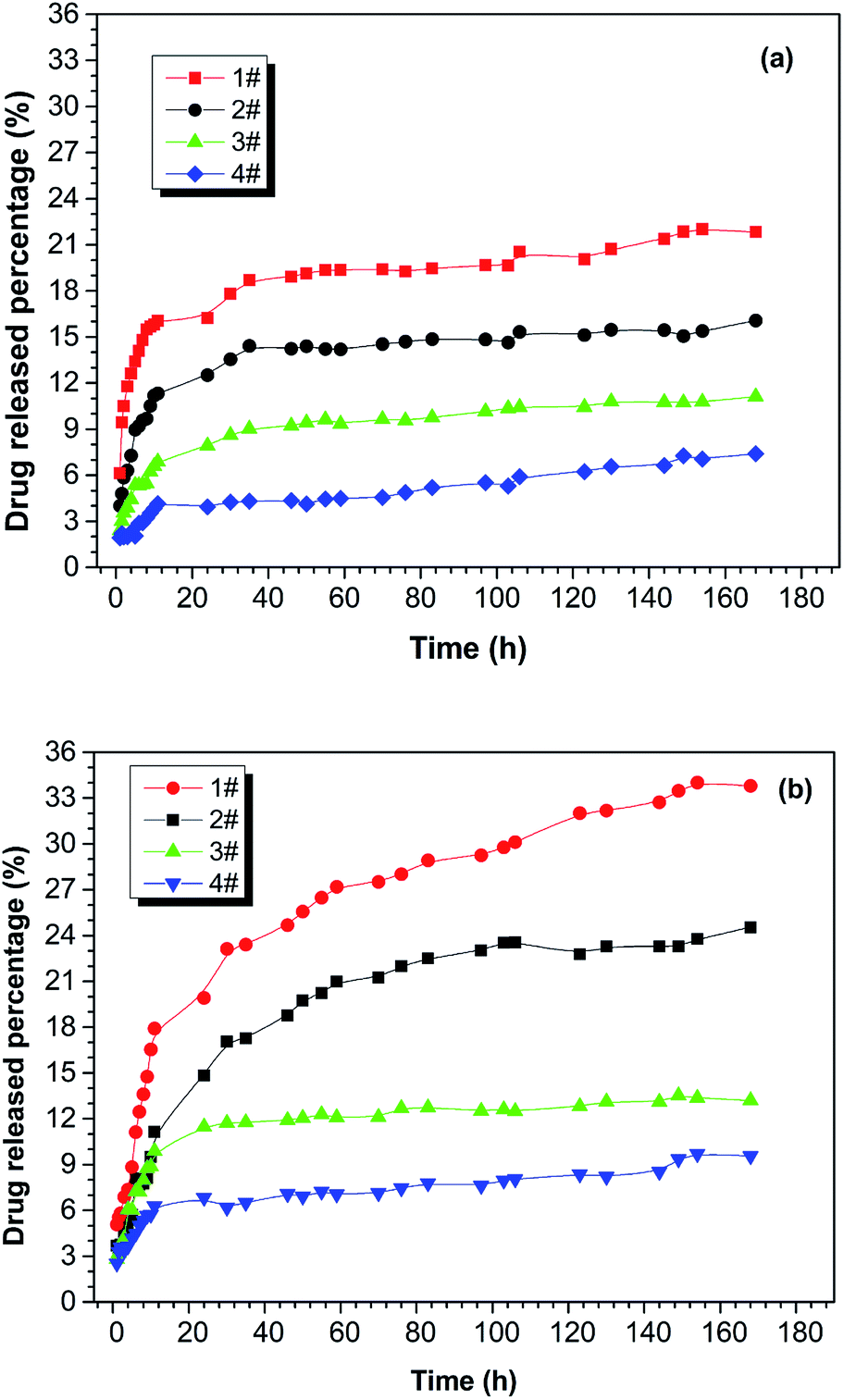
The drug-release behaviour of the drug-loaded electrospun nanofibrous films was studied using a hydrophobic drug that was physically loaded and stabilized in the ultra-fine fibres of PNLA/PLLA electrospun films. Rifampicin (RFP), an anti-pneumonia drug with very low solubility in water, was chosen as the model drug. Fig. 6 demonstrates the in vitro release curves of drug-loaded PNLA/PLLA electrospun films at different temperatures. By comparing Fig. 6(a) and (b), it can be seen that the electrospun nanofibrous films had higher drug release rates at 40 °C than at 25 °C which indicated that the prepared electrospun films possessed thermosensitive drug-release behaviour. This was because at higher temperature, PNIPAAm tends to form intramolecular hydrogen bonds, causing curling of the molecular chains and release of the contained drug.41,42 From the 1#, 2#, and 3# curves in Fig. 6, it could be found that the drug release rate of the electrospun films became significantly faster at the same temperature with an increase in the molecular weight of PNLA. The copolymer PNLA with the lower molecular weight contains more PNIPAAm structural units, which could lead to more thermosensitive amide bonds in electrospun nanofibrous films. Additionally, the drug release rate of the PNLA1/PLLA (3:10) electrospun film was dramatically higher than that of the PNLA1/PLLA (1:10) electrospun film. The significant increase in the drug release rate is believed to be attributable to the blending of more PNIPAAm chains into the PNLA/PLLA electrospun films. Therefore, the drug-release rate of the PNLA/PLLA electrospun nanofibrous films can be adjusted by the content and molecular weight of PNLA and by the environmental temperature.
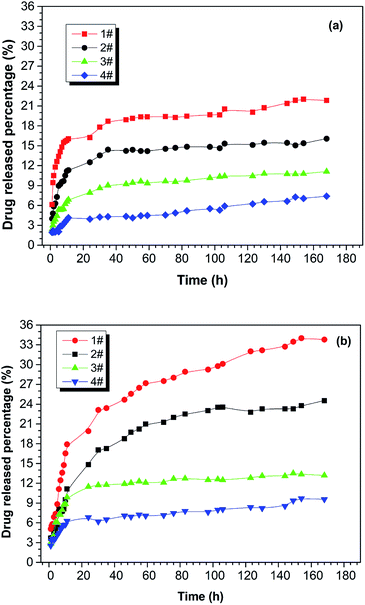 |
| | Fig. 6 Drug release curves of the drug-loaded electrospun nanofibrous films of 1# PNLA1/PLLA = 3:10, 2# PNLA2/PLLA = 3:10, 3# PNLA3/PLLA = 3:10, 4# PNLA1/PLLA = 1:10 at (a) 25 °C and (b) 40 °C. | |
4. Conclusions
Thermosensitive and biocompatible drug-loaded nanofibrous films were produced by an electrospinning technique using a PNLA/PLLA blend. The copolymer PNLA was synthesized by the radical polymerization of N-isopropylacrylamide, followed by the ring-opening polymerization of L-lactide. The molecular structures of PNIPAAm and PNLA were verified by the results of NMR, FT-IR, GPC, and CA analyses. Because of the low molecular weight and hydrophilicity of PNIPAAm and PNLA, they are unsuitable for application in electrospinning. Hence, a mixture of PNLA and PLLA was used to prepare electrospun nanofibrous films with different morphologies and diameters. Finally, the results indicated that the drug-release rate of the PNLA/PLLA electrospun nanofibrous films could be adjusted by the content and molecular weight of PNLA and by the environmental temperature. The results demonstrate that electrospinning is a promising way to create stimuli-responsive fibrous films with potential applications in the design and application of controllable drug delivery systems.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National High-Tech R&D Program of China (No. 2013AA032202), the National Natural Science Foundation of China (No. 51203118), the Fundamental Research Funds for the Central Universities and the Open Funds for Characterization of Tongji University.
Notes and references
- F. Anton, US pat., 1,975,504, 1934.
- J. M. Deitzel, J. D. Kleinmeyer, J. K. Hirvonen and N. C. B. Tan, Polymer, 2001, 42, 8163–8170 CrossRef.
- P. Ke, X.-N. Jiao, X.-H. Ge, W.-M. Xiao and B. Yu, RSC Adv., 2014, 4, 39704–39724 RSC.
- Y.-L. Cheng, J.-T. Chen, X.-B. Yan, Z.-M. Zheng and Q.-J. Xue, RSC Adv., 2013, 3, 20606–20612 RSC.
- S. S. Ray, S.-S. Chen, C.-W. Li, N. C. Nguyen and H. T. Nguyen, RSC Adv., 2016, 6, 85495–85514 RSC.
- C.-Q. Chen, Y. Yu, W. Li, C.-Y. Cao, P. Li, Z.-F. Dou and W. G. Song, J. Mater. Chem., 2011, 21, 12836–12841 RSC.
- W.-J. Ma, Q.-L. Zhang, D.-W. Hua, R.-H. Xiong, J.-T. Zhao, W.-D. Rao, S.-L. Huang, X.-X. Zhan, F. Chen and C.-B. Huang, RSC Adv., 2016, 6, 12868–12884 RSC.
- S.-J. Shao, S.-B. Zhou, L. Li, J.-R. Li, C. Luo, J.-X. Wang, X.-H. Li and J. Weng, Biomaterials, 2011, 32, 2821–2833 CrossRef PubMed.
- F. Mohabatpour, A. Karkhaneh and A. M. Sharifi, RSC Adv., 2016, 6, 83135–83145 RSC.
- J. Zhang, H. Liu, J.-X. Ding, X.-L. Zhuang, X.-S. Chen and Z.-M. Li, RSC Adv., 2015, 5, 32604–32608 RSC.
- L.-L. Wu, X.-Y. Yuan and J. Sheng, J. Membr. Sci., 2005, 250, 167–173 CrossRef.
- X.-H. Ma, S.-W. Gu and Z.-L. Xu, Catal. Today, 2016, 276, 133–138 CrossRef.
- C.-Q. Chen, W. Li, C.-Y. Cao and W.-G. Song, J. Mater. Chem., 2010, 20, 6968–6974 RSC.
- L.-H. Zhang, X.-P. Duan, X. Yan, M. Yu, X. Ning, Y. Zhao and Y.-Z. Long, RSC Adv., 2016, 6, 53400–53414 RSC.
- C. M. Vaz, S. V. Tuijl, C. V. C. Bouten and F. P. Baaijens, Acta Biomater., 2005, 1, 575–582 CrossRef PubMed.
- J.-W. Qin, Y.-Y. Jiang, J.-J. Fu, Y.-Q. Wan, R.-H. Yang, W.-D. Gao and H.-B. Wang, Iran. Polym. J., 2013, 22, 729–737 CrossRef.
- A. Balaji, M. V. Vellayappan, A. A. John, A. P. Subramanian, S. K. Jaganathan, E. Supriyanto and S. I. A. Razak, RSC Adv., 2015, 5, 57984–58004 RSC.
- M. V. Vellayappan, J. R. Venugopal, S. Ramakrishna, S. Ray, A. F. Ismail, M. Mandal, A. Manikandan, S. Seal and S. K. Jaganathan, RSC Adv., 2016, 6, 83638–83655 RSC.
- C.-T. Yeh and C.-Y. Chen, RSC Adv., 2017, 7, 6023–6030 RSC.
- J. Ren, Biodegradable Poly (Lactic Acid): Synthesis, Modification, Processing and Applications, Springer Press, 2011 Search PubMed.
- T. Yu, J. Ren, S.-M. Li, H. Yuan and Y. Li, Composites, Part A, 2010, 41, 499–505 CrossRef.
- H. Nie, B. W. Soh, Y.-C. Fu and C.-H. Wang, Biotechnol. Bioeng., 2008, 99, 223–234 CrossRef PubMed.
- C.L. Li, C. Liu, W. Wang, J. Bell, Z. Mutlu, K. Ahmed, R. Ye, M. Ozkan and C. S. Ozkan, Chem. Commun., 2016, 52, 11398–11401 RSC.
- W.-G. Cui, X.-H. Li, S.-B. Zhou and J. Weng, J. Biomed. Mater. Res., Part A, 2007, 82, 831–841 CrossRef PubMed.
- W. Li, C.-Y. Cao, C.-Q. Chen, Y. Zhao, W.-G. Song and L. Jiang, Chem. Commun., 2011, 47, 3619–3621 RSC.
- B. Sun, B. Duan and X.-Y. Yuan, J. Appl. Polym. Sci., 2006, 102, 39–45 CrossRef.
- X.-H. Dai, H. Jin, M.-H. Cai, H. Wang, Z.-P. Zhou, J.-M. Pan, X.-H. Wang, Y.-S. Yan, D.-M. Liu and L. Sun, React. Funct. Polym., 2015, 89, 9–17 CrossRef.
- X.-Q. Shan, F.-Q. Li, C.-S. Liu and Q. Gao, J. Appl. Polym. Sci., 2014, 131, 41098 CrossRef.
- P. Sarazin, X. Roy and B. D. Favis, Biomaterials, 2004, 25, 5965–5978 CrossRef PubMed.
- Z.-Q. Su, J.-W. Ding and G. Wei, RSC Adv., 2014, 4, 52598–52610 RSC.
- A. Lazaro, S. Haddad, S. Sacca, C. Orellana-Tavra, D. Fairen-Jimenez and R. S. Forgan, Chem, 2017, 2, 561–578 Search PubMed.
- M. S. Kwon, J. H. Jordahl, A. W. Phillips, K. Chung, S. Lee, J. Gierschner, J. Lahann and J. Kim, Chem. Sci., 2016, 7, 2359–2363 RSC.
- E.-R. Kenawy, F. I. Abdel-Hay, M. H. El-Newehy and G. E. Wnek, Mater. Sci. Eng., A, 2007, 459, 390–396 CrossRef.
- X.-Y. Xu, Q.-B. Yang, Y.-Z. Wang, H.-J. Yu, X.-S. Chen and X.-B. Jing, Eur. Polym. J., 2006, 42, 2081–2087 CrossRef.
- W. Tomaszewski, W. Swieszkowski, M. Szadkowski, M. Kudra and D. Ciechanska, J. Appl. Polym. Sci., 2012, 125, 4261–4266 CrossRef.
- S.-Y. Gu, Z.-M. Wang, J.-B. Li and J. Ren, Macromol. Mater. Eng., 2010, 295, 32–36 CrossRef.
- Y.-F. Hu, V. Darcos, S. Monge and S.-M. Li, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3274–3283 CrossRef.
- H. V. D. Belt, D. Neut, W. Schenk, J. R. V. Horn, H. C. V. D. Mei and H. J. Busscher, Acta Orthop. Scand., 2001, 72, 557–571 CrossRef PubMed.
- H. Lee, H. Y. Kim, H. J. Bang, Y. H. Jung and S. G. Lee, Polymer, 2003, 44, 4029–4034 CrossRef.
- K. T. Tan and S. K. Obendorf, J. Membr. Sci., 2007, 305, 287–298 CrossRef.
- C.-L. Lo, K.-M. Lin and G.-H. Hsiue, J. Controlled Release, 2005, 104, 477–488 CrossRef PubMed.
- C.-Y. Choi, S. Y. Chae and J.-W. Nah, Polymer, 2006, 47, 4571–4580 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Chengwei Peng,
Zhimei Wang and
Jie Ren
*,
Chengwei Peng,
Zhimei Wang and
Jie Ren