
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA low-cost average valence detector for mixed electrolytes in vanadium flow batteries
Dongzhi Li *a,
Yunong Zhangb,
Zhuoyu Lib and
Le Liu*b
*a,
Yunong Zhangb,
Zhuoyu Lib and
Le Liu*b
aDepartment of Physics, School of Science, Shenyang University of Technology, Shenyang, China. E-mail: lidongzhi@tsinghua.org.cn
bInstitute of Green Chemistry and Energy, Graduate School at Shenzhen, Tsinghua University, Shenzhen, China. E-mail: liu.le@sz.tsinghua.edu.cn
First published on 6th June 2018
Abstract
The decay in capacity has hindered the applications of vanadium redox flow batteries (VFBs), which are promising energy storage devices with many benefits. Mixing positive and negative electrolytes can recover some of the lost capacity but is ineffective towards the increasing average valence of the mixed electrolyte caused by the side reactions. In this study, a low-cost optical average valence detector has been developed to monitor the average valence of the mixed electrolyte in VFBs. We demonstrate experimentally that with the aid of the average valence detector, the capacity of VFBs can be regularly recovered via electrolyte mixing and online electrolysis. This low-cost average valence detector has great potential for recovering the decayed capacities of VFBs in large-scale energy storage stations, which consist of thousands of VFBs.
1. Introduction
Nowadays, large-scale energy storage techniques are in demand to control the fluctuations of renewable energies including photovoltaics and wind power.1–3 Among all the energy storage devices, the vanadium redox flow battery (VFB, also known as VRB or VRFB), which was developed by Skyllas-Kazacos et al. in the 1980s,4–6 is regarded as one of the most promising energy storage devices due to its long cycle life, high safety, environmental friendliness and flexible design.7–12In VFB, positive and negative electrolytes with redox couples of VO2+/VO2+ and V3+/V2+ are stored in separate reservoirs and pumped into the battery where the redox reaction takes place.13,14 As both the active materials are the same elements in the positive and the negative electrolytes, the VFB is free from cross-contamination, which seriously affects other types of flow batteries.15–17 Therefore, VFBs have an infinite theoretical operation lifespan and reported cycle numbers of over ten thousand cycles,18 which are the most important advantages when compared with other energy storage devices. However, in their actual applications, the capacity of VFBs is found to decrease as a result of many factors including side reactions and transfer of water and other ions across the separator in the VFB.19,20 The traditional solution to this issue is to regularly replace both the positive and the negative electrolytes, which results in huge work quantity and renders the long cycle life of VFBs pointless. Thus, many studies have focused on the mechanism, the detection methods and the solution to the capacity decay in VFBs.
Theoretical and experimental studies show that there are two mechanisms resulting in the capacity decay of VFBs.19–28 One is the imbalance in the vanadium concentration and volume of the positive and the negative electrolytes, which is caused by water and vanadium ions transferring through the separator. The other is the imbalance in the vanadium valences of the positive and the negative electrolytes, which is due to the side reactions including gas evolution reactions and oxidation of V2+ by the oxygen in the air.
Based on the mechanism of the capacity decay of VFBs, methods to reduce this capacity decay have been proposed including altering the charging/discharging currents,29 controlling the hydraulic pressure of the electrolytes,30 balancing the osmotic pressure of the electrolytes,31 and using amphoteric membranes.32 Our group has also proposed a electrolyte-reflow method33 and an anolyte overhang strategy34 to effectively prolong the cycle numbers of VFBs. Although these methods can effectively reduce the decay rates of the VFB capacities, the capacity decay still exists and thus, methods to recover it are in demand.
Whitehead et al.35 proposed a concept to recover the capacity by reacting the evolved H2 with the positive electrolytes with the aid of catalysts; however, the use of an expensive catalyst can limit the industrial application of this method. Roznyatovskaya et al.36 recovered the imbalanced electrolytes by mixing them with extra electrolytes. However, this method needs the preparation of a lot more electrolytes, which can result in an increase in the work quantity of the capacity recovery process. Rudolph et al.37 discussed a method to recover the capacity of VFBs by mixing the electrolytes and by applying electrolysis, but their experiments only involved a dozen cycles, which did not fully explain the feasibility of the method. Our group has studied the benefits and limitations of electrolyte mixing in VFBs.38 We found that although the mix method can significantly regenerate the lost capacity by recovering the electrolyte imbalances in the vanadium concentration and volume, the effect of the mix method is limited after a few cycles due to electrolyte imbalances in the average valence caused by the side reactions. Therefore, methods such as online electrolysis are necessary to restore the average valence of the electrolytes. In addition, to accomplish online electrolysis, online detection techniques are required to monitor the variation in the electrolyte valence.
Conventional detection techniques such as potentiometric titration39 and mass spectrometry40 have disadvantages such as complicated operating procedures when they are used for the online detection of VFB electrolytes. Experimental and simulation studies have been devoted for detecting the state of charge (SOC) of VFBs by using their open circuit voltages (OCVs).41–45 There are also reports on the detection of SOC of the electrolyte using its conductivity,46 ultrasonic velocity,47 viscosity48 and density.49 Optical detection techniques are good choices for the online detection of chemical reactions.50–54 However, as a result of the interactions between VO2+ and VO2+ ions at high concentrations in VFBs, the absorbance of the positive electrolyte does not obey the Beer's law and is not a linear combination of the absorbance of its components; hence, traditional UV-vis spectroscopy methods cannot detect the electrolytes in VFBs.46 Our group proposed a transmittance spectroscopy detection method using a pre-prepared spectra database, and we successfully detected the SOC of the VFB electrolyte with an SOC resolution better than 0.5%.55–57 Many groups have focused on the spectroscopic detections of the VFB electrolytes,58–62 and our SOC detection resolution of 0.5% is the highest reported value. Recently, by changing the non-linear VO2+/VO2+ spectra from a negative factor into a positive factor for optical detection, we further improved the SOC detection resolution by 0.002% in certain experimental situations, and we enabled the fast detection of the positive side reactions in VFBs for the first time.63
In our previous study, we noticed that by mixing the positive and negative electrolytes, the imbalanced electrolyte volumes and concentrations can be recovered, and the total concentrations of the vanadium ions and sulphate ions can be restored as long as there is no leakage of the electrolytes. Only one factor of the electrolytes has changed: the average valence. Therefore, it is possible to detect the average valence of the electrolyte using a simplified optical detector; we can also recover the capacity of VFBs via online electrolysis with the help of the average valence detector.
In this paper, based on our previous studies on online detection techniques55–57,62 and capacity recovery methods,33,34,38 we have developed a low-cost optical detector, which can detect the average valence of the mixed electrolyte and enable the capacity recovery of VFBs via online electrolysis. This low-cost average valence detector has the potential to be used for recovering the decayed capacities of VFBs in large-scale energy storage stations, which consist of thousands of VFBs.
2. Experimental
2.1 System set-up of the average valence detector
In the VFB system, the initial electrolytes in the positive and negative reservoirs are aqueous solutions with equal amounts of VO2+ and V3+ ions. The average valence of the electrolytes in a VFB is 3.5. After several charge–discharge cycles, the average valence of the electrolytes in the entire VFB system is larger than 3.5 as a result of the side reactions, which occur in the negative side of the VFB. Therefore, the detection of the average valence of the electrolytes is very important for recovering the average valence using online electrolysis. Fortunately, unlike the correlated VO2+ and VO2+ ions in the positive electrolyte, the VO2+ and V3+ ions in the mixed electrolytes have no interactions, and their spectra obey the Beer's law. As the total concentrations of the vanadium ions and sulfate ions are constant in the mixed electrolyte, only one factor of the electrolytes is changed. Hence, the average valence can be detected using the absorption or transmission intensity of the electrolyte at one particular wavelength. Considering the absorption peaks of the V3+ ions at 400 nm and 605 nm and the absorption peaks of the VO2+ ions at 760 nm,60 a blue LED with the peak around 470 nm is chosen to be the light source in our average valence detector.Fig. 1 shows the schematic representation and the photograph of our homemade average valence detector, which consists of two modules: the optical detection module (ODM) and the acquisition and analyzing module (AAM).
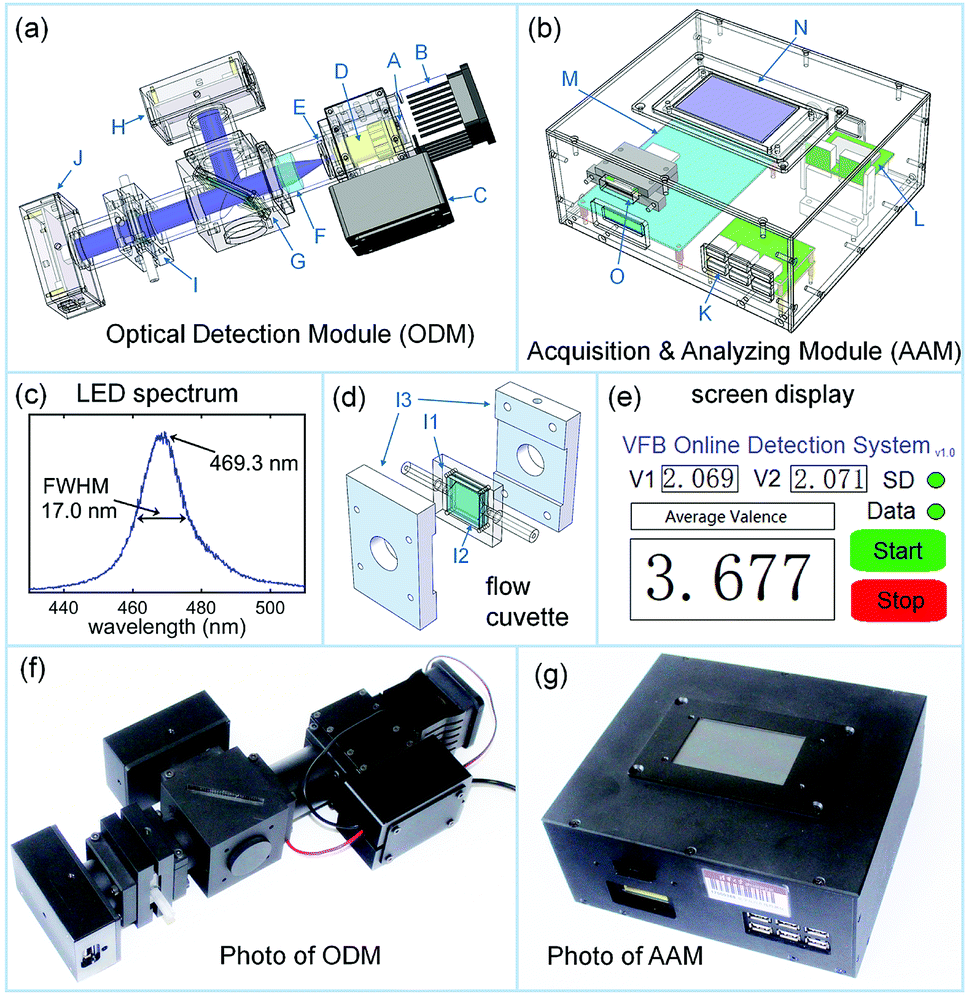 | ||
| Fig. 1 The schematic representation and the photograph of the average valence detector. (a) The schematic representation of the optical detection module (ODM). A, LED light source; B, heatsink with a fan; C, drive circuit of the LED; D, 25× objective; E, pinhole; F, achromatic convex lens; G, beam splitter; H, photodetector 1; I, flow cuvette; J, photodetector 2. (b) The schematic representation of the acquisition and analyzing module (AAM). K, USB interface; L, A/D converter; M, STM32 development board; N, 3.2 inch touch screen; O, SD card reader. (c) The spectrum of the LED in (a). (d) The structure of the flow cuvette in (a). I1, cuvette frame with an inlet and an outlet; I2, quartz window; I3, cuvette holder. (e) The content of the screen display in (b). (f) A photograph of the ODM. (g) A photograph of the AAM. | ||
Fig. 1a is a schematic representation of the ODM. The spectrum of the blue LED (A in Fig. 1a, Osram LB W5AP, central wavelength of 469.3 nm, bandwidth of 17.0 nm, electrical power of 5 W, and optical power of 75 lm) is shown in Fig. 1c; it was used as the light source. The LED (A in Fig. 1a) was adhered to a heat sink with a fan (B in Fig. 1a), and it was powered by an LED drive circuit (C in Fig. 1a). The light beam from the LED (A in Fig. 1a) was focused using a 25× objective (D in Fig. 1a) into a 0.5 mm pinhole (E in Fig. 1a) and then, it was collimated using an achromatic convex lens (F in Fig. 1a). The collimated beam was then split into two beams by a quartz plate placed at 45° (G in Fig. 1a). The reflected beam was recorded using a photodetector (H in Fig. 1a, OPT101, Texas Instruments) to monitor the fluctuation of the light source. The transmitted beam was passed through a homemade flow cuvette (I in Fig. 1a) and hit another photodetector (J in Fig. 1a, the same model as H). The structure of the homemade flow cuvette (I in Fig. 1a) is shown in Fig. 1d. Inside the cuvette holder (I3 in Fig. 1d), a polymethyl methacrylate (PMMA) frame (I1 in Fig. 1d) with an inlet and outlet had two quartz windows (I2 in Fig. 1d) attached on its both sides, forming a 0.5 mm thick flow cuvette. As the intensity of the transmitted beam was much larger than that of the reflected beam, the circuits of the two photodetectors (H and J in Fig. 1a) were optimized to maximize the detectors' dynamic range. The powers and the outputs of the photodetectors were designed with USB interfaces and thus, the detectors could be connected to AAM using ordinary USB cables.
Fig. 1b shows the schematic representation of AAM. A six channel USB interface (K in Fig. 1b) can power six photodetectors and collect their outputs. The outputs of the photodetectors are digitized using an analog to digital (A/D) converter (L in Fig. 1b, AD7606, Analog Devices), which are then entered in an embedded system (M in Fig. 1b, STM32F407, STMicroelectronics). The embedded system can work out the average valence of the measured electrolyte from the outputs of the two photodetectors (H and J in Fig. 1a). A LCD touch screen (N in Fig. 1b, TJC4024T032_11, 3.2 inch, 400 × 240 resolution, Taojingchi) can be used to control the AAM and display the real-time detection results, as shown in Fig. 1e. The detection results are also stored into an SD card using an SD storage board (O in Fig. 1b, Waveshare).
The photographs of the ODM and AAM prototypes corresponding to the schematics in Fig. 1a and b are shown in Fig. 1f and g. Fig. 2 shows the circuit diagram of our average valence detector including the ODM and AAM. The main chip in this embedded system is STM32 F407ZGT6, which can carry out the floating point calculation at a frequency of 168 MHz. The embedded system can acquire voltage signals using the parallel connected AD7606 converter at a sampling rate of 200 kHz. In this prototype, the outputs of the two photodetectors (H and J in Fig. 1a) are sampled every second (with 128 voltages averaged) and meanwhile, the average valence of the electrolyte is calculated, displayed and stored. Due to the use of embedded systems, the cost of the entire system of the average valence detector can be controlled within 200 U.S. dollars. Such low-cost gives it great potential to be used in large-scale energy storage stations, which consist of thousands of VFBs.
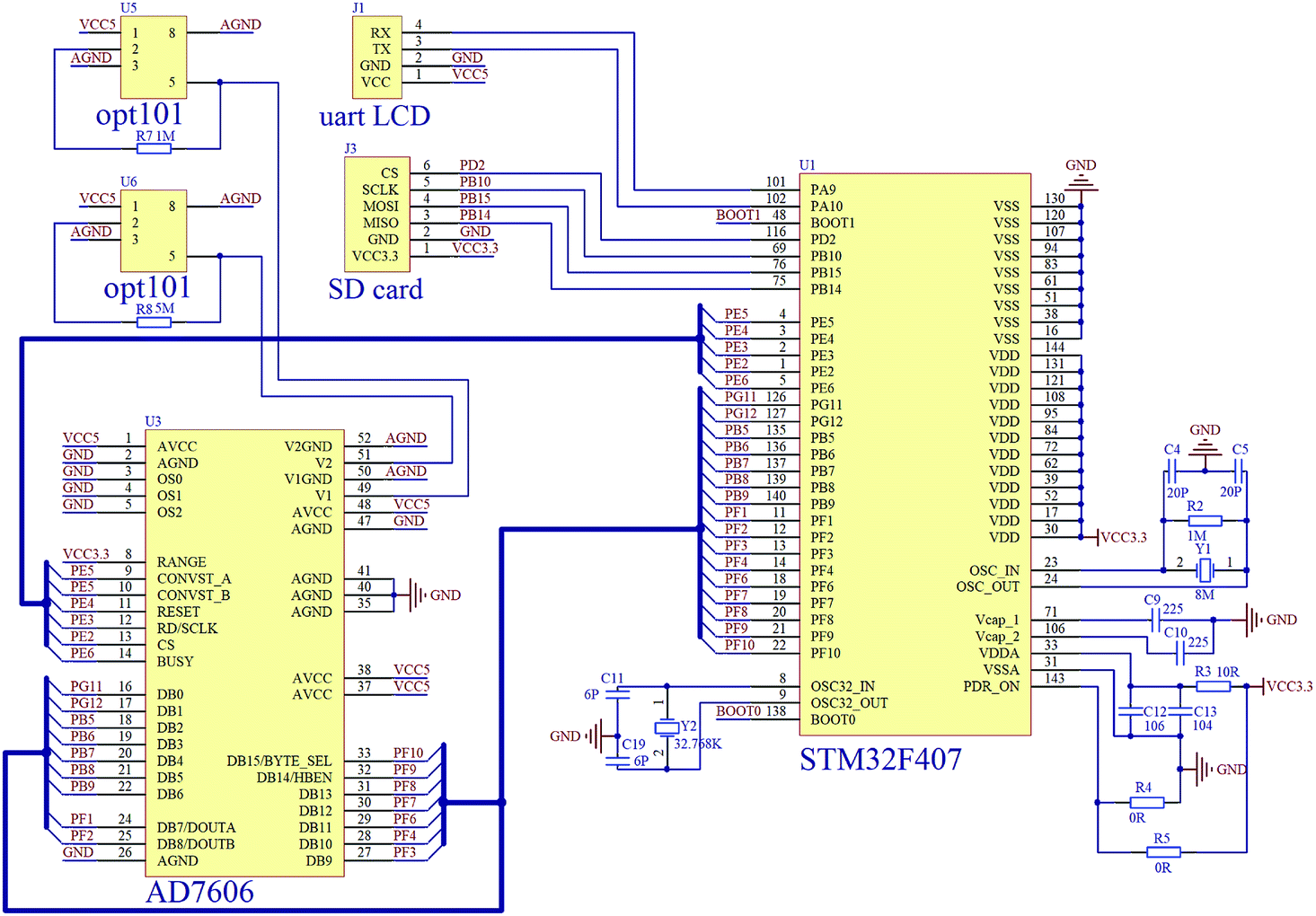 | ||
| Fig. 2 The circuit diagram of the average valence detector: opt101, photo detector; AD7607, A/D converter; STM32F407, embedded system; SD card, SD storage board and uart LCD, LCD touch screen. | ||
2.2 System set-up of the online electrolysis system
In this study, to demonstrate the application of our homemade average valence detector, an online electrolysis system is set-up to recover the capacity loss of an experimental VFB single cell with the aid of our average valence detector. The online electrolysis system, the photograph of which is shown in Fig. 3c, consists of an experimental VFB single cell, an electrolysis cell, two peristaltic pumps and our homemade average valence detector (ODM with a flow cuvette and AAM) as well as the corresponding reservoirs, tubes and flow controllers.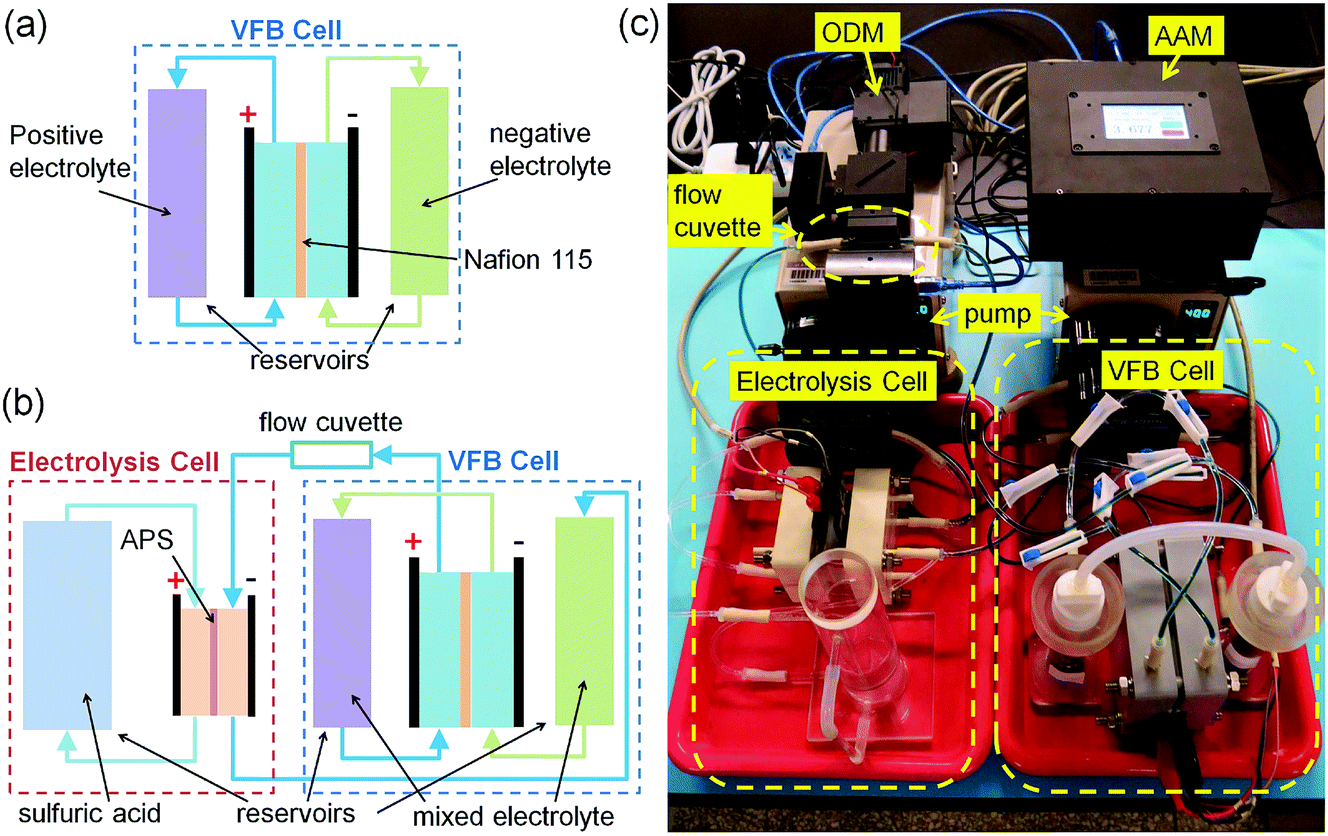 | ||
| Fig. 3 The schematic representation and the photograph of the VFB capacity recovery system based on online electrolysis with the aid of the average valence detector. (a) The schematic representation of the VFB cell in the normal working mode; (b) the schematic representation of the VFB cell in the capacity recovery mode; and (c) the photograph of the VFB capacity recovery system. | ||
The experimental VFB single cell was the same as that used in our previous study;64 a Nafion 115 membrane was used as the separator (Dupont, active area of 50 mm × 50 mm), carbon felts (5 mm thick, Gansu HaoShi Carbon Fiber Co., Ltd.) were used as the electrodes, and graphite plates (3 mm thick, Shanghai Hongfeng Industrial Co., Ltd.) were used as the current collectors. The Nafion 115 membrane in this VFB cell was reused several times to mimic the situation in real applications, in which the Nafion membranes are recycled due to their long life and high cost. After each use, the Nafion 115 membrane was treated by boiling it in 3 wt% H2O2 solution, deionized water, 1 M sulphuric acid, deionized water in sequence, for one hour each. The carbon felts were pre-treated at 420 °C in the air for 10 hours to improve the electrochemical activity. The two-channel peristaltic pumps (BT100M, Baoding Chuangrui Precision Pump Co., Ltd.) cyclically circulated the electrolytes through the corresponding half-cells at a flow rate of about 60 mL min−1. The initial positive and negative electrolytes were both 50 mL solutions of 1.5 M vanadium ions (V3+/VO2+ = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with 2.0 M free sulfuric acid (total 3.875 M SO42−). After a pre-charging process, it was found that the positive and the negative electrolytes consisted of VO2+ and V3+ ions, respectively.
:1) with 2.0 M free sulfuric acid (total 3.875 M SO42−). After a pre-charging process, it was found that the positive and the negative electrolytes consisted of VO2+ and V3+ ions, respectively.
The experimental VFB cell shown in Fig. 3c can be switched into two working modes (the normal working mode and the capacity recovery mode) using the flow controllers. When the VFB cell is in the normal working mode, as shown in Fig. 3a, the positive and negative electrolytes cycle separately from each of the reservoirs to the VFB half-cells and back to the reservoirs, and the redox reactions take place in the VFB cell during the charge/discharge process. When the VFB cell is in the capacity recovery mode, as shown in Fig. 3b, the positive and negative electrolytes are cycled in series for mixing, and they flow though the flow cuvette of the ODM in the average valence detector and the negative half of an electrolysis cell.
The electrolysis cell in Fig. 3b and c has a similar structure to the experimental VFB cell, but it has different membrane and electrolytes. The membrane in the electrolysis cell is an anion exchange membrane (APS, Selemion, active area of 30 mm × 30 mm), which is used to prevent the migration of vanadium ions. The positive electrolyte in the electrolysis cell is 100 mL 2.0 M sulfuric acid, and the negative electrolyte is the mixed electrolyte from the experimental VFB cell. After charging the electrolysis cell, the oxygen evolution reaction (OER) occurs at the positive electrode of the electrolysis cell, and the average valence of the negative electrode decreases. In this way, the average valence of the negative electrolytes (also the mixed electrolytes in the VFB cell) can be recovered using the electrolysis cell under the monitoring of the average valence detector (ODM and AAM).
3. Results and discussions
3.1 Calibration and performance of the average valence detector
In the average valence detector, one photodetector (J in Fig. 1a) detects the transmitted light intensity passing though the flow cuvette, and the other photodetector (H in Fig. 1a) monitors the fluctuation of the light source. Thus, the corrected voltage (CV) can be defined using the following equation:| CV = V1/(V2/V2(0)); | (1) |
To determine the relationship between the average valence of the electrolyte and the corrected voltage, a calibration experiment was conducted. By controlling the flow controllers in the system shown in Fig. 3, the experimental VFB cell was set in the normal working mode, whereas the positive electrolyte flowed through the flow cuvette of the ODM. The average valences of the initial positive and negative electrolytes were both 3.5 (V3+/VO2+ = 1:1). Then, the average valence of the electrolyte was varied by charging the VFB cell, and the average valence detector recorded the corrected voltage of the monitored electrolyte. The valence of the monitored electrolyte was first increased to over 4 by charging at a constant current density of 80 mA cm−2 and then, it was decreased to below 3 by reverse charging (reversing the positive and negative electrodes of the VFB cell) with the same constant current. From the absorption spectra of V2+, V3+ and VO2+, we inferred that the electrolyte with a valence of 4 (VO2+) had the largest transmittance in the wavelength range of ODM (around 470 nm), whereas the electrolyte with a valence of 3 (V3+) had the smallest transmittance in this wavelength range. Hence, the corrected voltages obtained upon changing the valence from 4 to 3 can be obtained from the detection results, as shown in Fig. 4a. The thick blue curve in Fig. 4a is the detected dependence of the average valence of the monitored electrolyte on the corrected voltage (eqn (1)) of the average valence detector. The thin red curve in Fig. 4a is the curve fitting result of the detection results, the equation of which is shown in the lower right of Fig. 4a. From this equation of the fitted curve, the average valence of the detected electrolyte can be calculated from the corrected voltages.
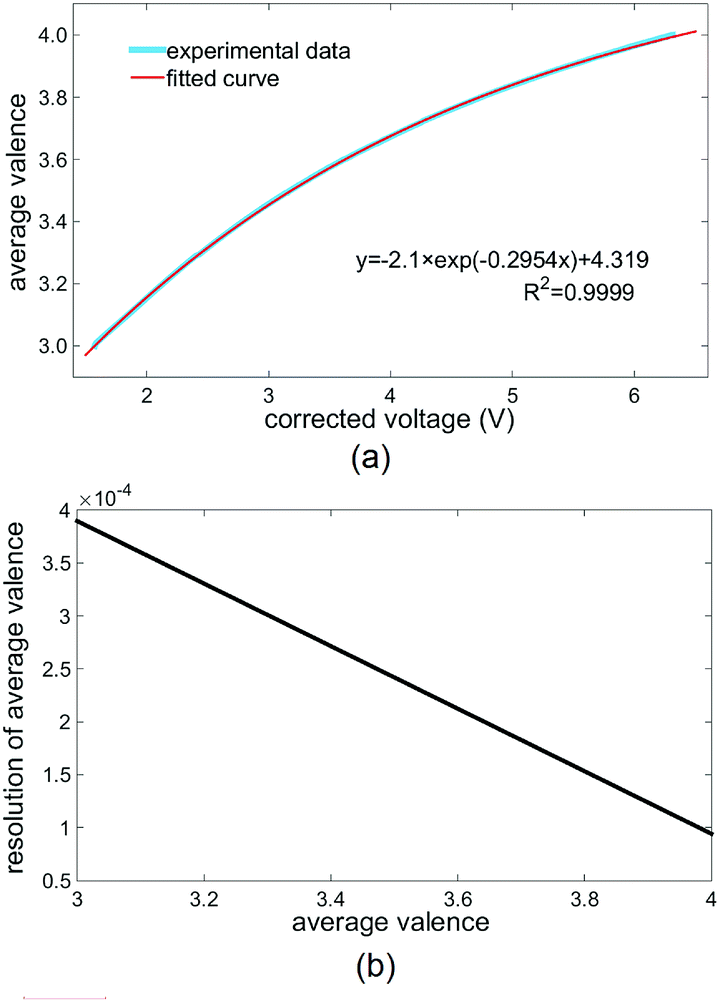 | ||
| Fig. 4 The calibration and resolution of the average valence detector. (a) The detected dependence of the average valence on the corrected voltage (thick blue curve) and the curve fitting result (thin red curve). (b) The detection resolution of the average valence detector at different average valences, calculated from (a) and the accuracy of the corrected voltage (0.001 V). | ||
In our average valence detector, both the output voltages of the photodetectors (J and H in Fig. 1a) are within 9.000 V with an accuracy of 0.001 V. Thus, the accuracy of the corrected voltage is also 0.001 V. Then, with the dependence of the average valence of the monitored electrolyte on the corrected voltage (Fig. 4a) and the accuracy of the corrected voltage, the detection resolution of our average valence detector at different average valences can be determined, as shown in Fig. 4b. We can conclude from Fig. 4b that the detection resolution is better at an average valence of 4, and the overall detection resolution of our average valence detector is more than 4 × 10−4. Although, in this study, the detected average valences of V3+ and VO2+ do not correspond to the state of charge (SOC) of the VFB, the resolution of our average valence detector can be converted to a nominal SOC resolution of more than 0.04%. This resolution is better than those of most existing techniques (which do not exceed 0.5%), and it is sufficient for assisting the recovery of the imbalanced electrolytes.
It should be discussed that this optical average valence detector is based on the assumption that the concentrations of vanadium ions and sulfate ions are constant in the mixed electrolyte. This assumption is true in most cases. However, in certain special situations of actual VFB applications, the concentrations of the vanadium ions and sulfate ions may change. In such cases, our optical average detector should be upgraded with more wavelengths to determine the average valence of the mixed electrolytes.
3.2 Validation experiment for the average valence detector
To validate the detection results of our optical average valence detector, a validation experiment was carried out. The average valences of the electrolytes during online electrolysis were measured using both potentiometric titration with a titrator (905 Titrando Metrohm) and online optical detection using our average valence detector. Column 1 and 2 in Table 1 show the detected average valences of the electrolytes by titration and our optical detector, respectively. Each uncertainty in Column 1 and 2 in Table 1 was calculated from five repeated measurements. Column 3 in Table 1 shows the difference between the two measured average valences. We can conclude from Table 1 that the detection results of our optical detector were consistent with those of the traditional potentiometric titration method (with differences of less than 1.3%), which validated the accuracy of our optical average valence detector. It should be noted that nearly all the average valences detected by titration were slightly higher than those obtained by using our optical detector. This may be due to the possible oxidation of V3+ by the oxygen in the air during the titration process. The absence of air-oxidation is another advantage of our optical online detector.| Average valence by titration | Average valence by our optical detector | Difference between two average valences |
|---|---|---|
| 3.067 ± 0.003 | 3.0280 ± 0.0004 | 1.28% |
| 3.246 ± 0.002 | 3.2110 ± 0.0003 | 1.08% |
| 3.419 ± 0.003 | 3.3940 ± 0.0002 | 0.73% |
| 3.612 ± 0.001 | 3.6000 ± 0.0002 | 0.34% |
| 3.800 ± 0.000 | 3.8000 ± 0.0001 | 0.00% |
| 3.994 ± 0.002 | 3.9640 ± 0.0001 | 0.77% |
3.3 Online electrolysis assisted by the average valence detector
To demonstrate the application of our average valence detector in VFB systems, a capacity recovery experiment using electrolyte mixing and online electrolysis assisted by our average valence detector was conducted. The experimental VFB cell shown in Fig. 3c was switched to the normal working mode (Fig. 3a), and charge/discharge cycles were conducted at a constant current density of 160 mA cm−2 in the voltage window of 1.65 V to 0.8 V. As was mentioned in Section 2.2, the Nafion 115 membrane in this VFB cell was reused several times to mimic the situation in real applications, in which the Nafion membrane is recycled due to its long life and high cost. Hence, the capacity of the VFB decayed faster when compared with the capacities of VFBs with new Nafion membranes, as reported in our previous studies.33,63Fig. 5a shows the discharge capacity of the VFB cell during the charge–discharge cycles. We can see from Fig. 5a that the discharge capacity of the VFB cell increased in the first few cycles and then rapidly decreased. When the discharge capacity decreased to approximately 75% of the initial value after the 78th cycle, the positive and negative electrolytes were mixed, and the average valence of the mixed electrolyte was detected to be 3.664, which was higher than that of the initial electrolyte (3.589, which was measured in advance). Then, the online electrolysis was conducted, and the average valence of the mixed electrolyte was monitored; it was found that the value decreased to 3.586 (Fig. 5b). Next, the charge–discharge cycle of VFB was continued after the pre-charging process. We can see from Fig. 5a that the discharge capacity of the VFB cell was significantly recovered after the 1st mix and online electrolysis.
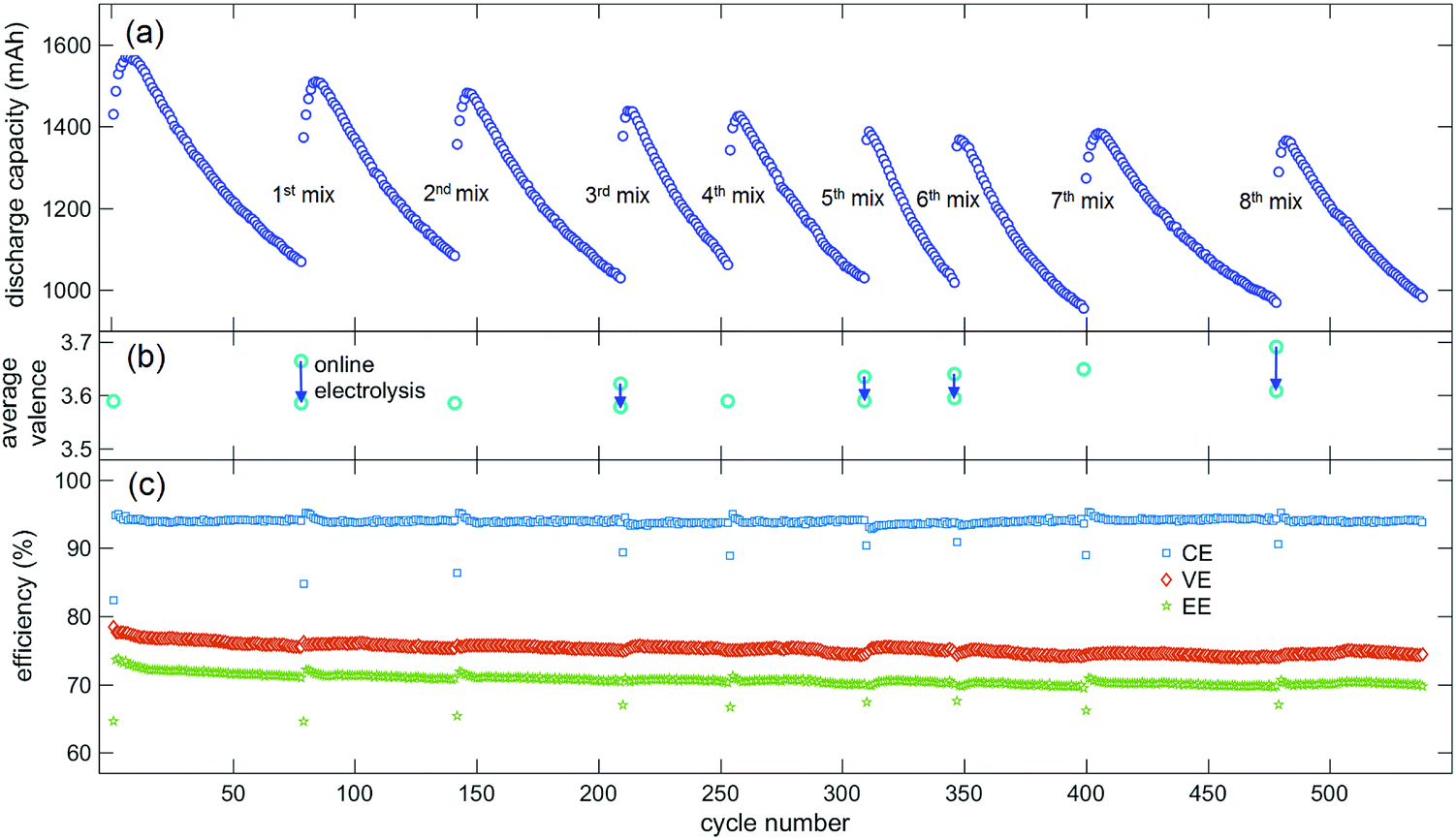 | ||
| Fig. 5 The data obtained for the experimental VFB cell during the charge–discharge cycles and eight capacity recovery processes via electrolyte mixing and online electrolysis. (a) The discharge capacity of the VFB cell; (b) the average valence of the mixed electrolytes and the electrolytes after online electrolysis; and (c) the coulomb efficiency (CE), voltage efficiency (VE) and energy efficiency (EE) of the VFB cell. | ||
The cycles were continued, and the mixing of electrolytes was conducted whenever the capacity decayed to about 75% of the initial value after the last mix. Thus, the mixing was conducted after the 78th, 141st, 209th, 253rd, 308th, 346th, 399th and 478th cycles (Fig. 5a). The online electrolysis was not conducted after each mixing; it was only conducted when the average valence of the mixed electrolyte was high enough. Thus, the average valence of the mixed electrolytes decreased during the online electrolysis from 3.622 to 3.578 after the 3rd mix, from 3.634 to 3.590 after the 5th mix, from 3.640 to 3.595 after the 6th mix, and from 3.690 to 3.608 after the 8th mix (Fig. 5b). Fig. 5c shows the efficiencies of the VFB cell during the cycling and capacity recovery processes. It can be seen from Fig. 5c that the coulomb efficiency and energy efficiency of each cycle after the mixing decreased due to the pre-charging process, and the coulomb efficiency of the VFB cell remained steady during the whole cycle and capacity recover processes; the voltage efficiency and energy efficiency slightly decreased (by about 2%) after more than 500 cycles. The decrease in voltage efficiency and recovered capacity of the VFB cell may be due to the reduction in the electrode activity during the charge–discharge cycles.
To sum up, when compared with only 2 or 3 times the recovery of the VFB capacity obtained by mixing the positive and negative electrolytes in our previous study,38 the electrolyte mixing and online electrolysis method can always effectively recover the lost VFB capacity. As a key device for electrolyte mixing and online electrolysis, our average valence detector has great potential for applications in large-scale energy storage stations in which thousands of VFBs have decayed capacities, which need to be recovered.
4. Conclusions
An optical detector has been developed to detect the average valence of the mixed electrolytes in VFBs. The prototype of this average valence detector can detect the average valences of the mixed electrolytes from 3 to 4 with a resolution of more than 0.04%; it has a low cost of less than 200 U.S. dollars due to the optical and embedded design. We have demonstrated experimentally that with the aid of our average valence detector, the electrolyte mixing and online electrolysis method can regularly and effectively recover the lost VFB capacity. As a key device of the electrolyte mixing and online electrolysis method, this low-cost average valence detector has great potential for applications in large-scale energy storage stations in which thousands of VFBs have decayed capacities, which need to be recovered.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was made possible with the financial support from the Doctoral Scientific Research Foundation of Liaoning province (20170520405), Natural Science Foundation of Guangdong Province (2015A030313794) and Basic Research Program of Shenzhen City (JCYJ20170307153749357, JCYJ20170412170756603).Notes and references
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928 CrossRef PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577 CrossRef PubMed.
- P. Leung, X. Li, C. P. de Leon, L. Berlouis, C. T. J. Low and F. C. Walsh, RSC Adv., 2012, 2, 10125 RSC.
- M. Skyllas-Kazacos, M. Rychcik, R. G. Robins, A. G. Fane and M. A. Green, J. Electrochem. Soc., 1986, 133(5), 1057 CrossRef.
- M. Ulaganathan, V. Aravindan, Q. Yan, S. Madhavi, M. Skyllas-Kazacos and T. M. Lim, Adv. Mater. Interfaces, 2016, 1500309 CrossRef.
- M. Skyllas-Kazacos, L. Cao, M. Kazacos, N. Kausar and A. Mousa, ChemSusChem, 2016, 9, 1 CrossRef PubMed.
- H. Zhang, Nature, 2014, 508, 319 CrossRef PubMed.
- K. Wang, Y. Zhang, L. Liu, J. Xi, Z. Wu and X. Qiu, Electrochim. Acta, 2018, 259, 11 CrossRef.
- J. Xi, W. Zhang, Z. Li, H. Zhou, L. Liu, Z. Wu and X. Qiu, Int. J. Electrochem. Sci., 2013, 8(4), 4700 Search PubMed.
- Z. Li, L. Liu, L. Yu, J. Xi, X. Qiu and L. Chen, J. Power Sources, 2014, 272, 427 CrossRef.
- J. Bo, L. Wu, L. Yu, X. Qiu and J. Xi, J. Membr. Sci., 2016, 510, 18 CrossRef.
- A. Tang, J. Bao and M. Skyllas-Kazacos, J. Power Sources, 2014, 248, 154 CrossRef.
- J. Noack, N. Roznyatovskaya, T. Herr and P. Fischer, Angew. Chem., Int. Ed., 2015, 54, 9776 CrossRef PubMed.
- G. L. Soloveichik, Chem. Rev., 2015, 115(20), 11533 CrossRef PubMed.
- C. Ding, H. Zhang, X. Li, T. Liu and F. Xing, J. Phys. Chem. Lett., 2013, 4, 1281 CrossRef PubMed.
- M. Park, J. Ryu, W. Wang and J. Cho, Nat. Rev. Mater., 2016, 16080 Search PubMed.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2016, 55, 2–28 CrossRef.
- Z. Yuan, Y. Duan, H. Zhang, X. Li, H. Zhang and I. Vankelecom, Energy Environ. Sci., 2016, 9, 441 Search PubMed.
- A. Tang, J. Bao and M. Skyllas-Kazacos, J. Power Sources, 2011, 196, 10737 CrossRef.
- X. G. Yang, Q. Ye, P. Cheng and T. S. Zhao, Appl. Energy, 2015, 145, 306 CrossRef.
- X. L. Zhou, T. S. Zhao, L. An, Y. K. Zeng and X. H. Yan, Appl. Energy, 2015, 158, 157 CrossRef.
- K. W. Knehr and E. C. Kumbur, Electrochem. Commun., 2012, 23, 76 CrossRef.
- E. Agar, K. W. Knehr, D. Chen, M. A. Hickner and E. C. Kumbur, Electrochim. Acta, 2013, 98, 66 CrossRef.
- P. A. Boettcher, E. Agar, C. R. Dennison and E. C. Kumbur, J. Electrochem. Soc., 2016, 163(1), A5244 CrossRef.
- M. Skyllas-Kazacos and M. Kazacos, J. Power Sources, 2011, 196, 8822 CrossRef.
- M. Skyllas-Kazacos and L. Goh, J. Membr. Sci., 2012, 399–400, 43 CrossRef.
- Q. Luo, L. Li, W. Wang, Z. Nie, X. Wei, B. Li, B. Chen, Z. Yang and V. Sprenkle, ChemSusChem, 2013, 6, 268 CrossRef PubMed.
- K. Oh, S. Won and H. Ju, Electrochim. Acta, 2015, 181, 238 CrossRef.
- E. Agar, A. Benjamin, C. R. Dennison, D. Chen, M. A. Hickner and E. C. Kumbur, J. Power Sources, 2014, 246, 767 CrossRef.
- B. Li, Q. Luo, X. Wei, Z. Nie, E. Thomsen, B. Chen, V. Sprenkle and W. Wang, ChemSusChem, 2014, 7, 577 CrossRef PubMed.
- L. Yan, D. Li, S. Li, Z. Xu, J. Dong, W. Jing and W. Xing, ACS Appl. Mater. Interfaces, 2016, 8, 35289 Search PubMed.
- F. J. Oldenburg, T. J. Schmidt and L. Gubler, J. Power Sources, 2017, 368, 68 CrossRef.
- K. Wang, L. Liu, J. Xi, Z. Wu and X. Qiu, J. Power Sources, 2017, 338, 17 CrossRef.
- D. Mu, Y. Zhao, L. Yu, L. Liu and J. Xi, Phys. Chem. Chem. Phys., 2017, 19, 29195 RSC.
- A. H. Whitehead and M. Harrer, J. Power Sources, 2013, 230, 271 CrossRef.
- N. Roznyatovskaya, T. Herr, M. Küttinger, M. Fühl, J. Noack, K. Pinkwart and J. Tübke, J. Power Sources, 2016, 302, 79 CrossRef.
- S. Rudolph, U. Schröder and I. M. Bayanov, J. Electroanal. Chem., 2013, 703, 29 CrossRef.
- Y. Zhang, L. Liu, J. Xi, Z. Wu and X. QIu, Appl. Energy, 2017, 204, 373 CrossRef.
- S. E. Quintar, J. P. Santagata and V. A. Cortinez, Talanta, 2005, 67, 843 CrossRef PubMed.
- H. Liu, Q. Xu and C. Yan, Electrochem. Commun., 2013, 28, 58 CrossRef.
- K. Ngamsai and A. Arpornwichanop, J. Power Sources, 2015, 282, 534 CrossRef.
- K. Ngamsai and A. Arpornwichanop, J. Power Sources, 2015, 298, 150 CrossRef.
- B. Xiong, J. Zhao, Z. Wei and M. Skyllas-Kazacos, J. Power Sources, 2014, 262, 50 CrossRef.
- B. Xiong, J. Zhao, Y. Su, Z. Wei and M. Skyllas-Kazacos, IEEE Trans. Sustainable Enery, 2016, 8(4), 1658 CrossRef.
- Z. Wei, K. J. Tseng, N. Wai, T. M. Lim and M. Skyllas-Kazacos, J. Power Sources, 2016, 332, 389 CrossRef.
- M. Skyllas-Kazacos and M. Kazacos, J. Power Sources, 2011, 196, 8822 CrossRef.
- Y. S. Chou, N. Y. Hsu, K. T. Jeng, K. H. Chen and S. C. Yen, Appl. Energy, 2016, 182, 253 CrossRef.
- X. Li, J. Xiong, A. Tang, Y. Qin, J. Liu and C. Yan, Appl. Energy, 2018, 211, 1050 CrossRef.
- S. Ressel, F. Bill, L. Holtz, N. Janshen, A. Chica, T. Flower, C. Weidlich and T. Struckmann, J. Power Sources, 2018, 378, 776 CrossRef.
- Z. Liu, S. Ma, Y. Ji, L. Liu, Z. Hu, J. Guo, H. Ma and Y. He, Anal. Chem., 2010, 82(18), 7752 CrossRef PubMed.
- L. Liu, S. Ma, Y. Ji, X. Chong, Z. Liu, Y. He and J. Guo, Rev. Sci. Instrum., 2011, 82(2), 023109 CrossRef PubMed.
- Y. Liu, L. Liu, Y. He, Q. He and H. Ma, Biosens. Bioelectron., 2016, 77, 886 CrossRef PubMed.
- R. Liao, N. Zeng, X. Y. Jiang, D. Z. Li, T. L. Liang, Y. H. He and H. Ma, J. Biomed. Opt., 2010, 15, 036014 CrossRef PubMed.
- S. G. Proskurin, Y. H. He and R. K. Wang, Phys. Med. Biol., 2004, 49, 1265 CrossRef PubMed.
- L. Liu, J. Xi, Z. Wu, W. Zhang, H. Zhou, W. Li and X. Qiu, J. Appl. Electrochem., 2012, 42, 1025 CrossRef.
- L. Liu, J. Xi, Z. Wu, W. Zhang, H. Zhou, W. Li and Y. He, J. Spectrosc., 2013, 453980 Search PubMed.
- W. Zhang, L. Liu and L. Liu, RSC Adv., 2015, 5, 100235 RSC.
- N. H. Choi, S. Kwon and H. Kim, J. Electrochem. Soc., 2013, 160, A973 CrossRef.
- S. Rudolph, U. Schröder, I. M. Bayanov, K. Blenke and D. Hage, J. Electroanal. Chem., 2013, 694, 17 CrossRef.
- D. N. Buckley, X. Gao, R. P. Lynch, N. Quill and M. J. Leahya, J. Electrochem. Soc., 2014, 161, A524 CrossRef.
- R. P. Brooker, C. J. Bell, L. J. Bonville, H. R. Kunz and J. M. Fenton, J. Electrochem. Soc., 2015, 162, A608 CrossRef.
- C. Petchsingh, N. Quill, J. T. Joyce, D. N. Eidhin, D. Oboroceanu, C. Lenihan, X. Gao, R. P. Lynch and D. N. Buckley, J. Electrochem. Soc., 2016, 163, A5068 CrossRef.
- L. Liu, Z. Li, J. Xi, H. Zhou, Z. Wu and X. Qiu, Appl. Energy, 2017, 185, 452 CrossRef.
- Z. Li, W. Dai, L. Yu, L. Liu, J. Xi, X. Qiu and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 18885 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |