
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence(Ba,Sr)TiO3:RE perovskite phosphors (RE = Dy, Eu): nitrate pyrolysis synthesis, enhanced photoluminescence, and reversible emission against heating
Wanlu Liu abc,
Qian Liu*ab,
Jia Niabc,
Zhenzhen Zhou*abc and
Guanghui Liuab
abc,
Qian Liu*ab,
Jia Niabc,
Zhenzhen Zhou*abc and
Guanghui Liuab
aThe State Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, 1295 Dingxi Road, Shanghai 200050, China. E-mail: qianliu@mail.sic.ac.cn
bShanghai Institute of Materials Genome, 99 Shangda Road, Shanghai 200444, China
cUniversity of Chinese Academy of Sciences, 19A Yuquan Road, Beijing 100049, China
First published on 6th June 2018
Abstract
A series of (Ba,Sr)TiO3 phosphors singly doped with Eu3+ and Dy3+ were successfully synthesized using the nitrate pyrolysis method at 750 °C. Eu3+ or Dy3+ single-doped BaTiO3 retained the tetragonal crystal structure of the host, while the Sr2+-substituted (Ba,Sr)TiO3:RE3+ (RE3+ = Eu3+ or Dy3+) experienced a phase transformation from tetragonal to cubic phase with a unit cell shrinkage. For Eu3+ doped phosphors, BaTiO3:xEu3+ (x = 0.02–0.10) exhibited red photoluminescence and the highest intensity of emission belonged to the optimal-doped BaTiO3:xEu3+ (x = 8 mol%). Moreover, the substitution of 30 mol% Sr2+ for Ba2+ (that is Ba0.7Sr0.3TiO3:xEu3+, x = 8 mol%) further enhanced the emission intensity of BaTiO3:xEu3+ (x = 8 mol%). For Dy3+ doped phosphors, BaTiO3:xDy3+ (x = 0.02–0.10) showed yellow photoluminescence and the highest light intensity was from the optimal-doped BaTiO3:xDy3+ (x = 4 mol%). In addition, the substitution of 20 mol% Sr2+ for Ba2+ (the phosphor Ba0.8Sr0.2TiO3:xDy3+, x = 4 mol%) induced further increase in emission intensity of BaTiO3:xDy3+ (x = 4 mol%). The emission intensities at higher temperature of 100 °C retained about 70% and 90% of the initial values at room temperature (RT) for the optimal BaTiO3:xEu3+ (x = 8 mol%) and BaTiO3:xDy3+ (x = 4 mol%) phosphors, respectively, while the emission intensities at the temperature of 100 °C retained about 60% and 80% of the initial intensities at RT for the optimal Sr2+-substituted Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) and Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%) phosphors, respectively. It is worth noting that on cooling down to RT again from 210 °C, the BaTiO3:xDy3+ (x = 4 mol%) phosphor exhibited excellent luminescent thermal stability (with a high activation energy of 0.387 eV) and the strongest recovery (∼95%) of PL emission among the series of phosphors. The as-prepared phosphors with optimal compositions would be good candidates for the applications in lighting, display, and related fields.
1. Introduction
In recent years, trivalent rare-earth (RE) ions doped phosphors have been developed rapidly because of their gradually widespread applications in many fields, such as field emission displays (FEDs), cathode-ray tubes (CRTs), light-emitting diodes (LEDs), plasma display panels (PDPs), colour televisions, and optical fiber telecommunication.1–4 Currently, LEDs are considered as one of the most important solid-state illumination sources owing to their environment-friendly nature, low power consumption, as well as promising applications.5 In the use of LED devices, heat generates in phosphor materials coated on the LED chip, resulting in the increase in phosphor layer temperature to 120–150 °C. Generally, the luminescence thermal quenching of phosphors occurs at such temperatures, which further decrease the performance of LED devices.6 It is well known that thermal stability of phosphors is one of the important parameters for evaluating the performance of LEDs, which influences lighting efficiency and colour performance.7 Thus, the detailed investigation of phosphors' thermal stability is of great significance for understanding their practical properties and exploring their potential in various applications.8 Furthermore, more efforts should be made to develop and research some promising phosphors that maintain their emission intensities and colour coordinates at higher temperatures for applications in lighting.The selection of appropriate host materials as well as dopants (for example rare earth ions), is a crucial for obtaining phosphors with improved photoluminescence properties.9 Perovskite structured titanate is a traditional material that has been widely used in functional materials fields for its excellent dielectric and ferroelectric properties. Moreover, increasing number of researchers have recently began to pay more attention to the optical properties of perovskite structured titanate due to its low phonon energy and stable physical and chemical properties.10,11 The preparation conditions of titanate are not very harsh and hence, titanate phosphors have always been the subject of attention. Concerning dopants, Eu3+ is usually selected because of its specific energy level and high radiative recombination rate. Dy3+ is also an important dopant in many luminescent materials owing to its various transitions between different energy levels, leading to three special emission bands of blue (480 nm), yellow (575 nm) and red (625 nm). Numerous research reports have exhibited that Dy3+ has been applied in white LEDs as well as in mercury-free luminescence lamps, and the white emission mainly comes from two dominant emission bands: blue and yellow.12,13
In the past decades, several reports have been published on the luminescent properties of lanthanide (Ln3+ or RE3+) doped BaTiO3 and SrTiO3.9,14–19 Recently, RE3+ (Gd3+,Dy3+,Tb3+, Lu3+)-doped BaTiO3-based phosphors with tetragonal structure were synthesized by a modified solid-state reaction at 1000 °C, and their photoluminescence properties have been studied. It has been found that Gd3+ ion doped BaTiO3 phosphor showed the strongest yellow emission intensity compared to the other phosphors.20 The preparation of bulk BaTiO3:Pr3+ by a solid-state reaction and nanocrystalline BaTiO3:Pr3+ by a solvothermal method were also studied. It was found that the photoluminescence (PL) intensity of the bulk material was stronger than that of the nanocrystalline BaTiO3:Pr3+, which was attributed to more surface defects on nanocrystal powder.21 Furthermore, SrTiO3:Pr3+ phosphors with different morphologies have been synthesized by three different methods: high-temperature solid-state reaction, solvothermal synthesis, and sol–gel synthesis. The red-emitting phosphors possess cubic phase, and the PL intensity of SrTiO3:Pr3+ synthesized by the solid-state method was the strongest compared to the phosphors synthesized by the other two methods.19 However, to the best of our knowledge, there are few reports on the luminescence intensity, thermal stability, and emission recovery against heating/cooling of Ba1−xSrxTiO3-based phosphors concerning the effects of Sr2+ ion substitution and rare earth activators.
In this prospective study, we designed and prepared a series of comparable Eu3+ and Dy3+ singly-doped (Ba,Sr)TiO3 phosphors for the first time, and the luminescence thermal stability of phosphors was evaluated systematically. The resultant phosphors exhibited enhanced emission via the substitution of Sr2+ for Ba2+ in (Ba,Sr)TiO3:RE3+ (RE3+ = Eu3+ or Dy3+). Herein, excellent luminescence stability against heating in Dy3+ doped BaTiO3 phosphors was found, presenting a promising phosphor candidate for lighting applications.
2. Experimental
A series of Eu3+ and Dy3+ singly-doped BaTiO3:xEu3+ (x = 0.02–0.10), BaTiO3:xDy3+ (x = 0.02–0.10), Ba1−ySryTiO3:xEu3+ (y = 0–1.0) and Ba1−ySryTiO3:xDy3+ (y = 0–1.0) phosphors were designed and synthesized using the nitrate pyrolysis method. Herein, it has to be emphasized that the doping sites of rare-earth Eu3+ and Dy3+ in the BaTiO3 host depended on many factors, including the ionic radius, dopant concentration, and electronegativity. In fact, large lanthanide ions such as La3+ and Ce3+ replaced the Ba2+ sites in the BaTiO3 lattice, whereas small lanthanide ions such as Dy3+, Ho3+, and Er3+ showed amphoteric behaviour in the BaTiO3 lattice and could get incorporated at both Ba2+ and Ti4+ sites in the lattice with comparable probabilities.22,23 In our experiments, the starting raw materials were barium nitrate (Ba(NO3)2, 99.9% purity), strontium nitrate (Sr(NO3)2, 99.9% purity), tetrabutyl titanate (C16H36O4Ti, 98% purity), europium(III) oxide (Eu2O3, 99.9% purity), dysprosium(III) oxide (Dy2O3, 99.9% purity), acetylacetone (C5H8O2), and absolute ethyl alcohol (CH3CH2OH). During the preparation, titanium solution was obtained by adding appropriate amounts of C5H8O2 and CH3CH2OH to tetrabutyl titanate (the molar ratio of tetrabutyl titanate, CH3CH2OH, and C5H8O2 was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:0.2). Typically, Eu2O3 and Dy2O3 were dissolved in diluted nitric acid (HNO3) solutions with constant stirring and heating to obtain the nitrate solutions of Eu(NO3)3 (0.03 mol L−1) and Dy(NO3)3 (0.03 mol L−1). Ba(NO3)2 (0.04 mol) and Sr(NO3)2 (0.04 mol) were separately dissolved in deionized water (200 mL) for dilution. Ba(NO3)2 and Sr(NO3)2 aqueous solutions were first mixed and then added to the titanium solution to obtain a mixed solution. Eventually, Eu(NO3)3 (0.03 mol L−1) or Dy(NO3)3 (0.03 mol L−1) was added to the above mixed solution. The amount of each solution added was calculated based on the stoichiometric ratio of the corresponding chemical formula. The solution mixture was then stirred on a magnetic stirrer for 4 hours, followed by drying in a box furnace at 160 °C for 6 h. Following this, the as-obtained mixture was preheated at 600 °C for 4 h in air to decompose the nitrates, and then sintered at certain temperatures for some time in air to derive the final powder products. We tried to optimize the crystallization conditions, including sintering temperature and time, for the purpose of fabricating (Ba,Sr)TiO3 phosphors at a fixed Eu3+ or Dy3+ doping concentration. Based on the phase purity and luminescence intensity analyses of samples, the optimal sintering condition was considered as 750 °C for 4 hours to obtain the designed phosphors. As a result, throughout this article, we focussed on the phosphors prepared under the above optimal sintering conditions.
:4:0.2). Typically, Eu2O3 and Dy2O3 were dissolved in diluted nitric acid (HNO3) solutions with constant stirring and heating to obtain the nitrate solutions of Eu(NO3)3 (0.03 mol L−1) and Dy(NO3)3 (0.03 mol L−1). Ba(NO3)2 (0.04 mol) and Sr(NO3)2 (0.04 mol) were separately dissolved in deionized water (200 mL) for dilution. Ba(NO3)2 and Sr(NO3)2 aqueous solutions were first mixed and then added to the titanium solution to obtain a mixed solution. Eventually, Eu(NO3)3 (0.03 mol L−1) or Dy(NO3)3 (0.03 mol L−1) was added to the above mixed solution. The amount of each solution added was calculated based on the stoichiometric ratio of the corresponding chemical formula. The solution mixture was then stirred on a magnetic stirrer for 4 hours, followed by drying in a box furnace at 160 °C for 6 h. Following this, the as-obtained mixture was preheated at 600 °C for 4 h in air to decompose the nitrates, and then sintered at certain temperatures for some time in air to derive the final powder products. We tried to optimize the crystallization conditions, including sintering temperature and time, for the purpose of fabricating (Ba,Sr)TiO3 phosphors at a fixed Eu3+ or Dy3+ doping concentration. Based on the phase purity and luminescence intensity analyses of samples, the optimal sintering condition was considered as 750 °C for 4 hours to obtain the designed phosphors. As a result, throughout this article, we focussed on the phosphors prepared under the above optimal sintering conditions.
The X-ray diffraction (XRD) data were collected by a Bruker D8 Advance diffractometer (Cu Kα radiation, λ = 1.5405 Å) in the 2θ range from 20° to 80° to characterize the crystal phase. The phosphors used for the XRD analysis were pressed into a thin disc on a quartz slide before testing. The morphology of the powder samples was observed using a S-4800 Field Emission Scanning Electron Microscope (SEM) (Hitachi, Tokyo, Japan). For SEM observation, the phosphors were processed by ultrasonically dispersing in ethanol and then putting the drops on an aluminium substrate. Photoluminescence (PL), PL excitation (PLE) spectra, and temperature-dependent luminescence properties (from room temperature to 210 °C) were measured by a Hitachi F-4600 fluorescence spectrophotometer, equipped with a 150 W Xe lamp as the excitation source. The scan speed was fixed at 240 nm min−1, and the excitation slit as well as the emission slit width were set as 2.5 nm. In PL spectrum tests, the size of the powder container was kept constant in the Hitachi F-4600 fluorescence spectrophotometer. Hence, the amount of phosphor powder put into the container was the same which ensured that the test results were comparable. Also, the comparison of luminescence intensities of different samples was carried out by calculating the integral area under the emission peak of each phosphor. The decay times of Eu3+-doped phosphors (monitored at 615 nm with the 465 nm excitation) and Dy3+-doped phosphors (monitored at 573 nm with the 452 nm excitation) were measured with a FLS 980 fluorescence spectrofluorometer equipped with photonics μF2 (Edinburgh Instruments, UK).
3. Results and discussion
3.1 Morphology, crystal structure, and photoluminescence of BaTiO3:RE phosphors
The representative SEM morphology images of undoped BaTiO3, BaTiO3:xEu3+ (x = 8 mol%), and Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) phosphors indicated the irregular morphology of particles consisted of agglomerated short-rod-like sub-particles, as shown in Fig. 1. The average particle sizes of BaTiO3, BaTiO3:xEu3+ (x = 8 mol%), and Ba0.7Sr0.3TiO3:xEu3+ (x 0 = 8 mol%) phosphors were almost the same with the agglomerated irregular morphology. | ||
| Fig. 1 Representative SEM images of as-prepared powders (a) undoped BaTiO3, (b) BaTiO3:xEu3+ (x = 8 mol%), and (c) Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%), prepared at 750 °C for 4 hours. | ||
The XRD patterns of Eu3+ and Dy3+ singly-doped BaTiO3 samples with different doping concentrations are shown in Fig. 2a and b, respectively. In all the samples doped with different Eu3+ concentrations, a tetragonal main phase of BaTiO3 (JCPDF #79-2264, with lattice constants a = b = 3.9998 Å, c = 4.018 Å) was observed due to the appearance of all the standard XRD peaks of the tetragonal phase. The diffraction patterns of Dy3+ doped BaTiO3 phosphors also agreed well with the literature values of tetragonal BaTiO3 (JCPDF #79-2264, with lattice constants a = b = 3.9998 Å, c = 4.018 Å), and there was no detectable impurity or shifting of XRD peaks after doping rare-earth ions into the BaTiO3 host.
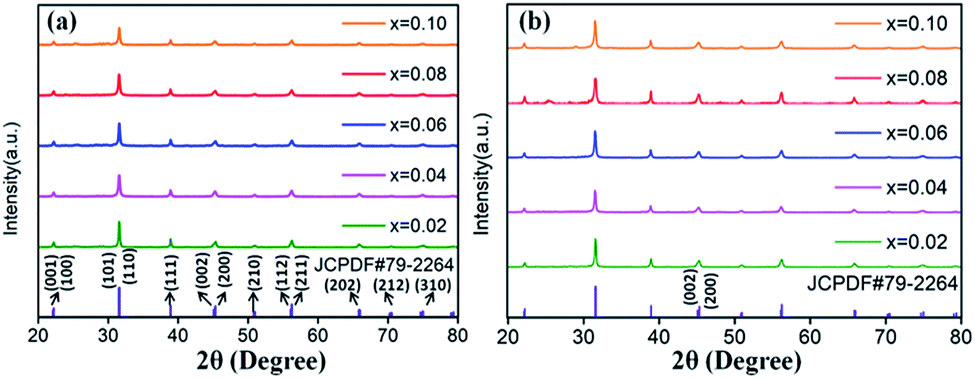 | ||
| Fig. 2 XRD patterns of (a) BaTiO3:xEu3+; (b) BaTiO3:xDy3+, x = 0.02–0.10, prepared at 750 °C for 4 hours. | ||
Room temperature PL and PLE spectra of BaTiO3:xEu3+ (x = 0.02–0.10) phosphors are depicted in Fig. 3. As shown in the figure, the PL spectra of BaTiO3:xEu3+ under excitation at 394 nm are indicated in Fig. 3a. With the increase in Eu3+ content up to 8 mol%, the emission intensity increased to a maximum and then decreased with the further increase in concentration of Eu3+, which was attributed to the concentration quenching effect. At the same time, the dependence of the emission intensity on the concentration of Eu3+ is shown in the inset of Fig. 3a. As shown in Fig. 3a, in the spectral range from 560 nm to 680 nm, there are four peaks centred at 580, 593, 615 and 655 nm, which were assigned to the transitions from the higher excited state (5D0) to the lower states (7F0, 7F1, 7F2, 7F3) of Eu3+ owing to the splitting of the 4f energy level of Eu3+.14,24 In general, the strongest peak at 615 nm (red emission) corresponds to the electric dipole transition 5D0 → 7F2 and its intensity is strongly influenced by the ligand ions around Eu3+ in the host. The second strong emission peak at 593 nm belongs to the magnetic dipole transition 5D0 → 7F1. Interestingly, the intensity ratio of (5D0 → 7F2)/(5D0 → 7F1) is usually regarded as a measurement of the site symmetry around Eu3+ in the host.25,26 If Eu3+ occupies an inversion symmetry, a dominant red-orange emission will be obtained according to the magnetic dipole transition 5D0 → 7F1. In contrast, red emission will predominate in the spectra due to the electric dipole transition 5D0 → 7F2. In this study, we can see that the (5D0 → 7F2)/(5D0 → 7F1) ratio increases with the increase in x value up to 0.08 and then decreases, as shown in Fig. 4. When more Eu3+ ions were doped, the distortion of the ligand ions around Eu3+ was severe, indicating that more Eu3+ ions occupied the non-inversion symmetry sites in the host. The intensity of the electric dipole transition 5D0 → 7F2 was more sensitive to the forthcoming distortion than the magnetic dipole transition 5D0 → 7F1. Therefore, the increase in the (5D0 → 7F2)/(5D0 → 7F1) ratio could be witnessed with the increasing x value up to 0.08. Furthermore, the PL intensity started to decrease on 8 mol% Eu3+ doping. The reduction in intensity is mainly attributed to the non-radiative transition, which contributed to the concentration quenching. The PLE spectra (monitored at 615 nm) are shown in Fig. 3b. A strong excitation peak is centred at 465 nm, which corresponds to the 7F0 → 5D2 (f–f) transition of Eu3+. The other excitation peaks belonged to different f–f transitions: 7F0 → 5D4 (360 nm), 7F0 → 5L6 (394 nm), 7F0 → 5D3 (412 nm), and 7F0 → 5D1 (532 nm). The PLE spectrum consists of two regions of charge transfer transitions (CTT): (i) between the host and Eu3+ ions and (ii) f–f intra-transition in Eu3+. However, in the present research, the charge transfer (O2−–Eu3+) was not found explicitly in the region of 230–320 nm, which was possible due to the weak covalent bonding between the 2p orbitals of O2− and the partially filled f orbitals of Eu3+ in the perovskite BaTiO3.27 From the viewpoint of the PL and PLE spectral intensity, the optimal doping concentration of Eu3+ was 8 mol% in the BaTiO3 host.
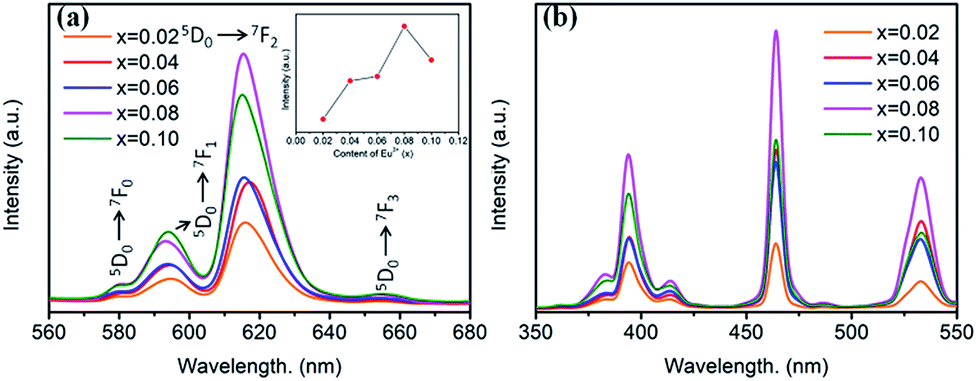 | ||
| Fig. 3 (a) PL spectra (λex = 394 nm), inset is the dependence of the emission intensity on the Eu3+ content. (b) PLE spectra (λem = 615 nm) of BaTiO3:xEu3+ (x = 0.02–0.10) phosphors. | ||
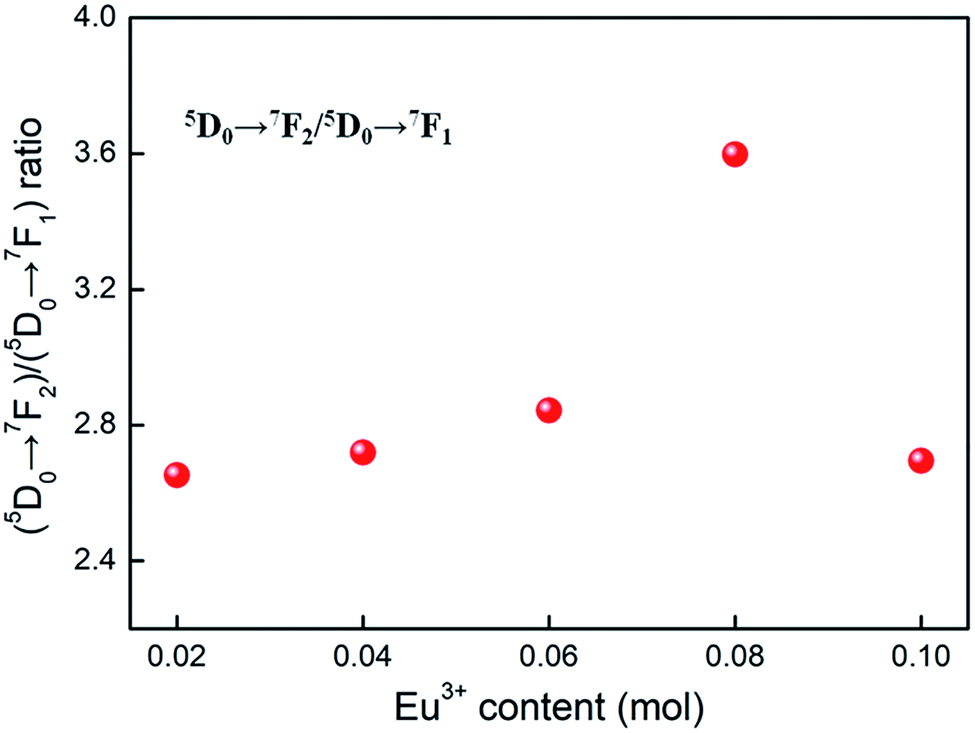 | ||
| Fig. 4 The dependence of (5D0 → 7F2)/(5D0 → 7F1) ratio on the Eu3+ content, derived from the PL spectra (λex = 394 nm) of BaTiO3:xEu3+ (x = 0.02–0.10) phosphors. | ||
Fig. 5 shows the PL and PLE spectra of BaTiO3:xDy3+ (x = 0.02–0.10) phosphors. In Fig. 5a, a broad peak can be seen in the yellow light emission region centred at 573 nm, corresponding to the 4F9/2 → 6H13/2 transition of Dy3+. This is a forced electric dipole transition allowed only at low symmetries without no inversion centres in the host crystal structure, indicating dominant occupation of Dy3+ at Ba2+ sites.16 As shown in Fig. 5b, the PLE spectra of BaTiO3:xDy3+ phosphors monitored at 573 nm consist of several strong peaks centred at 380, 428, 453 and 475 nm in the wide wavelength region, which were ascribed to the f–f transitions of Dy3+. The dependence of the emission intensity on the content of Dy3+ is illustrated in the inset of Fig. 5a. It is clearly observed that the PL intensities increased when x ≤ 0.04, and then suddenly decreased when the content of Dy3+ is more than 4 mol% due to concentration quenching, as mentioned for the BaTiO3:xEu3+ phosphors. Thus, the optimal doping concentration of Dy3+ in the BaTiO3 host to gain the strongest PL emission intensity was obtained, viz., 4 mol%.
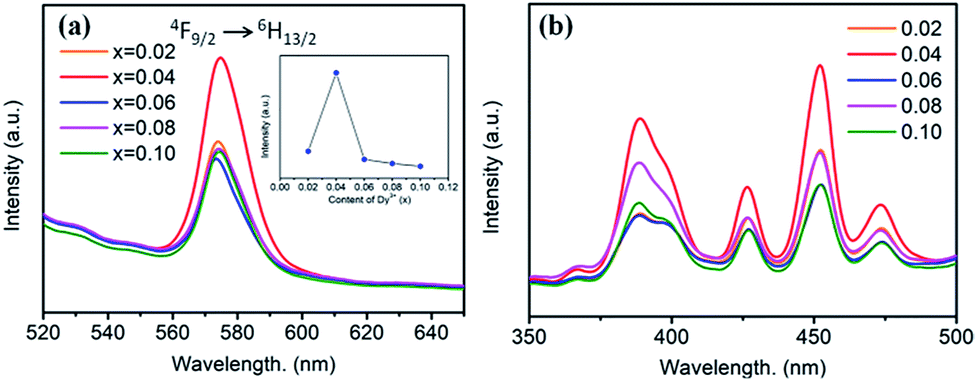 | ||
| Fig. 5 (a) PL spectra (λex = 453 nm), inset is the dependence of the emission intensity on the Dy3+ content. (b) PLE spectra (λem = 573 nm) of BaTiO3:xDy3+ phosphors (x = 0.02–0.10). | ||
3.2 Crystal structure and photoluminescence of Sr2+-substituted (Ba,Sr)TiO3:RE phosphors
Based on the fixed optimal doping content of activator Eu3+ (8 mol%) or Dy3+ (4 mol%) in BaTiO3 (mentioned in the Section 3.1), phosphors with smaller Sr2+ ions incorporated into the BaTiO3 host were designed and prepared to cause crystal field changes to enhance the emission.The XRD patterns of the as-prepared Ba1−ySryTiO3:xEu3+ (x = 8 mol%) and Ba1−ySryTiO3:xDy3+ (x = 4 mol%) (y is the substitution value of Sr2+ for Ba2+, y = 0.0–1.0) phosphors with different Sr2+ contents are presented in Fig. 6a and 7a, respectively. In addition, Fig. 6b and 7b show the magnified diffraction peaks, in which the value of 2θ is 45–49° for all the samples. From Fig. 6b and 7b, it is clear that there exists a clear diffraction peak shifting towards the high-angle direction, which suggests that the lattice parameter gradually decreased with the increase in substitution of Sr2+ for the Sr-substituted (Ba,Sr)TiO3:RE3+ (RE3+ = Eu3+, Dy3+) phosphors. This also suggested that the substitution of Sr2+ was achieved in BaTiO3:RE3+ phosphors. From Fig. 6b and 7b, it could be seen that the (002) peak and the (200) peak of the tetragonal phase at around 2θ = 46° gradually converted into a single (200) peak of the cubic phase, illustrating that the crystal structure transformed from the tetragonal phase (indexed as JCPDF #79-2264, with lattice constants a = b = 3.9998 Å, c = 4.018 Å) to the cubic phase (JCPDF #74-1296, with lattice constants a = b = c = 3.905 Å) as the concentration of Sr2+ increased for either Ba1−ySryTiO3:xEu3+ (x = 8 mol%) or Ba1−ySryTiO3:xDy3+ (x = 4 mol%) powder. The main reason for the above phenomena is the replacement of larger Ba2+ ions (r = 1.35 Å) by smaller Sr2+ ions (r = 1.18 Å), since both the BaTiO3:xEu3+ (x = 8 mol%) and BaTiO3:xDy3+ (x = 4 mol%) powder retained the tetragonal crystal structure without Sr2+ substitution, as indicated in Fig. 2. In order to clearly understand the phase transformation from tetragonal to cubic phase, the calculation of fractions of cubic and tetragonal phases was performed and the proportions of the cubic phase were 32.92% and 19.70% for the compositions of Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) and Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%), respectively. The compositions of both Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) and Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%) were nearly at the critical line of the phase transformation, as shown in Fig. 6b and 7b.
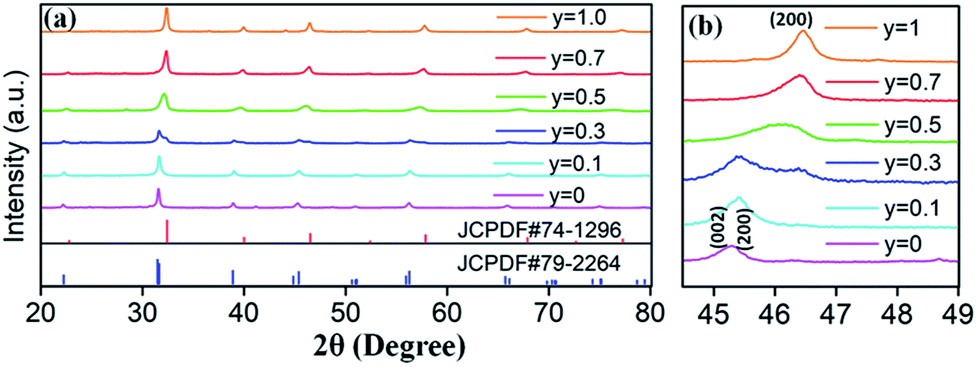 | ||
| Fig. 6 (a) XRD patterns of Ba1−ySryTiO3:xEu3+ (x = 8 mol%) phosphors (y = 0–1), with the standard XRD patterns of tetragonal phase BaTiO3 (JCPDF #79-2264) and cubic phase SrTiO3 (JCPDF #74-1296). (b) Magnified 2θ diffraction peaks at around 45–49° for all Ba1−ySryTiO3:xEu3+ (x = 8 mol%) phosphors, prepared at 750 °C for 4 hours. | ||
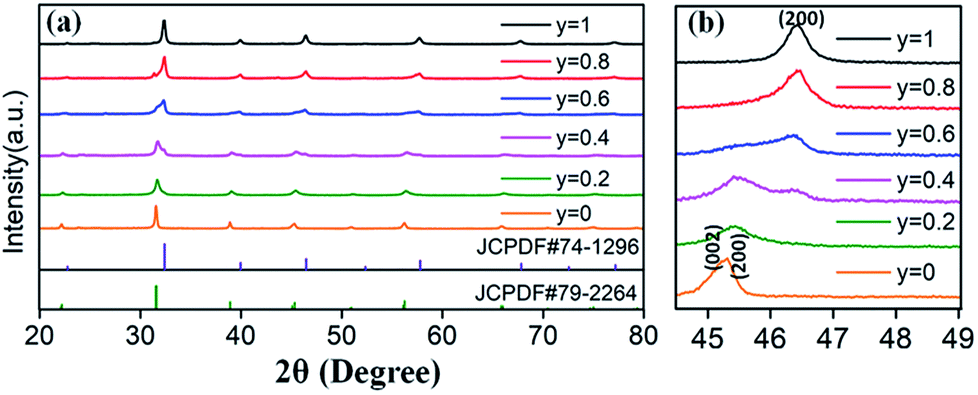 | ||
| Fig. 7 (a) XRD patterns of Ba1−ySryTiO3:xDy3+ (x = 4 mol%) phosphors (y = 0–1.0), with the standard XRD patterns of tetragonal phase BaTiO3 (JCPDF #79-2264) and cubic phase SrTiO3 (JCPDF #74-1296). (b) Magnified 2θ diffraction peaks at around 45–49° for all Ba1−ySryTiO3:xDy3+ (x = 4 mol%) phosphors, prepared at 750 °C for 4 hours. | ||
The substitution of Sr2+/Ba2+ in hosts can change the crystal field and the covalence portion of the host.28 Therefore, the photoluminescence of BaTiO3-based phosphors could be fine-tuned by the partial substitution of the host lattice cation Ba2+ by Sr2+ ions. To discern the luminescent properties of Ba1−ySryTiO3:xEu3+ (x = 8 mol%) phosphors, the PL and PLE spectra are shown in Fig. 8. As shown in Fig. 8a, the profiles of the PL spectra of the phosphors with different Sr2+ contents are similar, and the emission peaks remained at the same positions as those of the BaTiO3:xEu3+ (x = 8 mol%) phosphor without Sr2+ (Fig. 3). A previous study showed that the crystal structure transition only affects the PL/PLE intensity in BaTiO3 based phosphors doped by Li+ and Er3+; thus, the position of PL/PLE remains unchanged.29 Furthermore, the PL intensity increased until the Sr2+ content reached y = 0.3 and then, an abrupt decreasing trend could be observed on continually increasing the substitution concentration of Sr2+, as shown in the inset of Fig. 8a. Generally, the phase transition of (Ba,Sr)TiO3 occurs when the substitution of Sr2+ is about 40%.30 With the structure variation from tetragonal to cubic, the structure symmetry increases, inducing higher site symmetry surrounding Eu3+ ions. Hence, the probability of f–f (5D0 → 7FJ) transitions decreases, which further reduces the emission intensity because the electric dipole transition between f–f levels is forbidden when a rare-earth ion is located at a high centro-symmetric site, according to the quantum mechanical selection rules. When y ≤ 0.3, the probability of f–f transitions increases due to the lower portion of cubic phase and low local point symmetry, which improves the luminescence intensity of Ba1−ySryTiO3:xEu3+ (x = 8 mol%). To sum up, the intensity of luminescence mainly depends on the concentration of Eu3+ and the proportion of tetragonal phase in phosphors with Sr2+ substitution. Clearly, the emission intensity was much stronger than the other Ba1−ySryTiO3:xEu3+ (x = 8 mol%) when the substituting concentration of Sr2+ was at ∼30 mol%, and the crystal structure of the Ba1−ySryTiO3:xEu3+ (x = 8 mol%) phosphors with y ≤ 0.3 was dominated by the tetragonal phase. Therefore, the optimum substitution concentration of Sr2+ in the series of (Ba,Sr)TiO3:xEu3+ (x = 8 mol%) phosphors was considered to be 30 mol%. Fig. 8b shows the Sr2+-content-dependent PLE spectra of Ba1−ySryTiO3:xEu3+ (x = 8 mol%) phosphors. As shown in the figure, the excitation intensity of the Sr2+-totally-substituted SrTiO3:xEu3+ (x = 8 mol%) phosphor, particularly the broadband peaks at around 380 nm, is the highest among others, due to the band gap absorption of the SrTiO3 cubic phase host.14
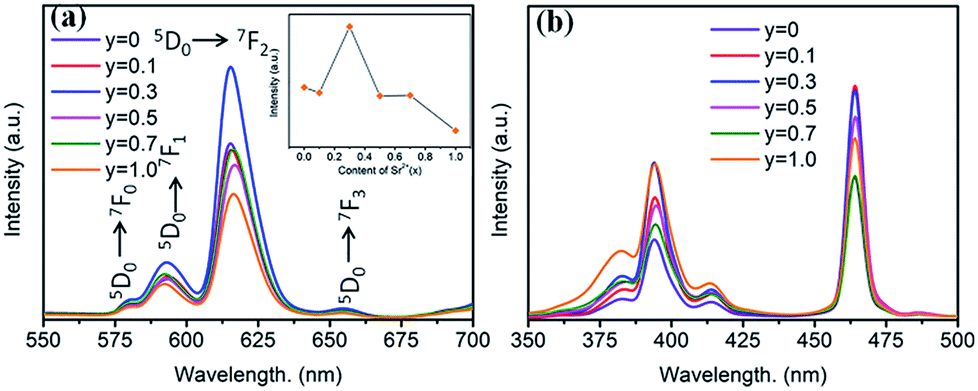 | ||
| Fig. 8 (a) PL spectra (λex = 465 nm), inset is the dependence of the emission intensity on the Sr2+ content. (b) PLE spectra (λem = 615 nm) of Ba1−ySryTiO3:xEu3+ (x = 8 mol%) phosphors (y = 0–1.0). | ||
Fig. 9 shows the Sr2+-content-dependent PL and PLE spectra of Ba1−ySryTiO3:xDy3+ (x = 4 mol%) phosphors. Clearly, all the PL and PLE spectra had the same profiles. From the inset in Fig. 9a, we could see that the optimal substitution content of Sr2+ was y = 0.2 (tetragonal phase dominated) for obtaining the strongest emission intensities among the series of Ba1−ySryTiO3:xDy3+ (x = 4 mol%) phosphors. Fig. 9b shows the Sr2+-content-dependent PLE spectra of Ba1−ySryTiO3:xDy3+ (x = 4 mol%) samples. There are several typical Dy3+ excitations, and all the PLE spectra present similar shapes with the increase in Sr2+ content. Given the above information, moderate substitution of Ba2+ by Sr2+ in the BaTiO3 host can improve the luminescence intensity of Eu3+ and Dy3+ singly-doped (Ba,Sr)TiO3 phosphors because of the partial formation of cubic phases in the tetragonal BaTiO3 crystal structure. As mentioned in case of BaTiO3:xEu3+ (x = 8 mol%), enhanced electric dipole transition (5D0 → 7F2) relates to the degree of distortion or the non-centrosymmetry of Eu3+'s local symmetry groups in the BaTiO3 host.25,26 In other words, when the partial formation of highly symmetrical cubic phases occurs in the Sr-substituted tetragonal (Ba,Sr)TiO3 host crystal structure, the distortion of the Eu3+'s local site symmetry groups aggravates because of the mixed structure with a co-existing bi-phase and then, the emission intensity increases accordingly. The situation is the same as that in the 4F9/2 → 6H13/2 electric dipole transition in (Ba,Sr)TiO3:xDy3+ (x = 4 mol%) phosphors after substituting larger Ba2+ by smaller Sr2+. With the further addition of Sr2+, the phase transformation from tetragonal to cubic phase completes, the degree of distortion or the non-centrosymmetry of RE3+'s local symmetry groups in (Ba,Sr)TiO3:RE3+ reduces and then, the emission intensity from electric dipole transition declines for both 5D0 → 7F2 of Eu3+ and 4F9/2 → 6H13/2 of Dy3+. Therefore, the optimal Sr2+ substitution content was considered to be 30 mol% for (Ba,Sr)TiO3:xEu3+ (x = 8 mol%) and 20 mol% for (Ba,Sr)TiO3:xDy3+ (x = 4 mol%) phosphors, respectively.
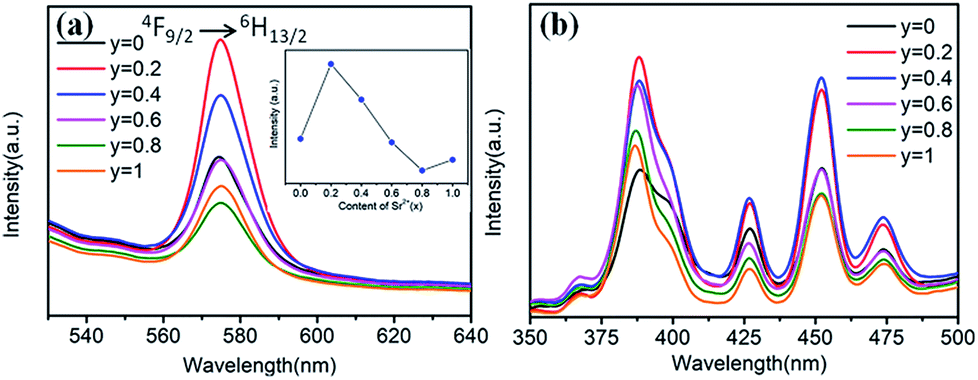 | ||
| Fig. 9 (a) PL spectra (λex = 452 nm), inset is the dependence of the emission intensity on the Sr2+ content. (b) PLE spectra (λem = 573 nm) of Ba1−ySryTiO3:xDy3+ (x = 4 mol%) phosphors (y = 0–1.0). | ||
3.3 Temperature-dependent luminescence and reversible emission against heating in the series of phosphors
Generally, in the process of the application of phosphors in LED, the phosphor's emission intensities are reduced against the temperature as the result of the thermal phonon-assisted relaxation between the excited states and the ground states.31,32 In fact, the temperature of the phosphor can be increased to about 150 °C during the operation of a LED.33 Therefore, the thermal quenching properties are of great significance to phosphors. For example, it has considerable effects on the output of light and the colour rendering index. To evaluate the effect of temperature on luminescence, the temperature dependent emission spectra for our four different samples were measured and are shown as follows.Fig. 10a and c show the temperature-dependent PL spectra of BaTiO3:xEu3+ (x = 8 mol%) and BaTiO3:xDy3+ (x = 4 mol%), respectively. As shown in the figures, the PL profiles of the Eu3+-optimal-doped BaTiO3:xEu3+ (x = 8 mol%) as well as Dy3+-optimal-doped BaTiO3:xDy3+ (x = 4 mol%) phosphors are similar at different temperatures. The PL intensities of both phosphors decreased with the increase in temperature from RT to 210 °C. In addition, the CIE chromaticity coordinates of the optimal BaTiO3:xEu3+ (x = 8 mol%) phosphors gradually moved from (0.624, 0.370) to (0.567, 0.419), while the colour coordinates of the optimal BaTiO3:xDy3+ (x = 4 mol%) phosphor were stable at (0.390 ± 0.01, 0.571 ± 0.007) with the increase in temperature, which are also clearly shown in Fig. 11 and Table 1. The curves of relative PL intensities of these two optimal phosphors as a function of temperature are illustrated in Fig. 10b and d. While heating the BaTiO3:xEu3+ (x = 8 mol%) phosphor up to 100 °C and 150 °C, it retained about 70% and 50% of its initial PL intensity at RT, respectively, because of the non-radiative transitions from the excited state to the ground state under heating. While cooling down to RT again from 210 °C, the phosphor retained 85% of its initial PL intensity, and is known as thermal degradation.34 However, while heating the BaTiO3:xDy3+ (x = 4 mol%) phosphor up to 100 °C and 150 °C, it retained about 90% and 60% of its initial PL intensity at RT, respectively. While cooling down to the room temperature again from 210 °C, the PL intensity of the BaTiO3:xDy3+ (x = 4 mol%) phosphor could be reserved at ∼95% of its initial intensity. The above results indicated that the BaTiO3:xDy3+ (x = 4 mol%) phosphor possessed superior thermal stability than the BaTiO3:xEu3+ (x = 8 mol%) phosphor, and it could be a good candidate for applications in lighting.
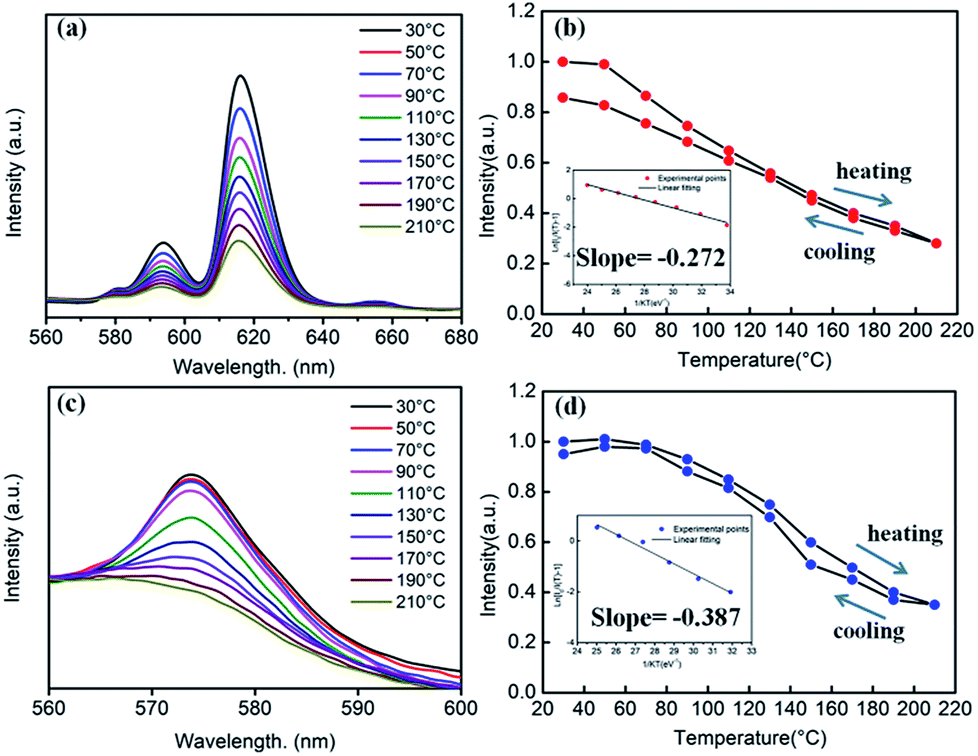 | ||
| Fig. 10 (a) Temperature-dependent PL spectra of the BaTiO3:xEu3+ (x = 8 mol%) phosphor (λex = 465 nm). (b) The relative PL intensity of BaTiO3:xEu3+ (x = 8 mol%) as a function of temperature. Inset is the linear fitting of the calculated activation energy for thermal quenching. (c) Temperature-dependent PL spectra of the BaTiO3:xDy3+ (x = 4 mol%) phosphor (λex = 452 nm). (d) The relative PL intensity of BaTiO3:xDy3+ (x = 4 mol%) as a function of temperature. Inset is the linear fitting of the calculated activation energy for thermal quenching. | ||
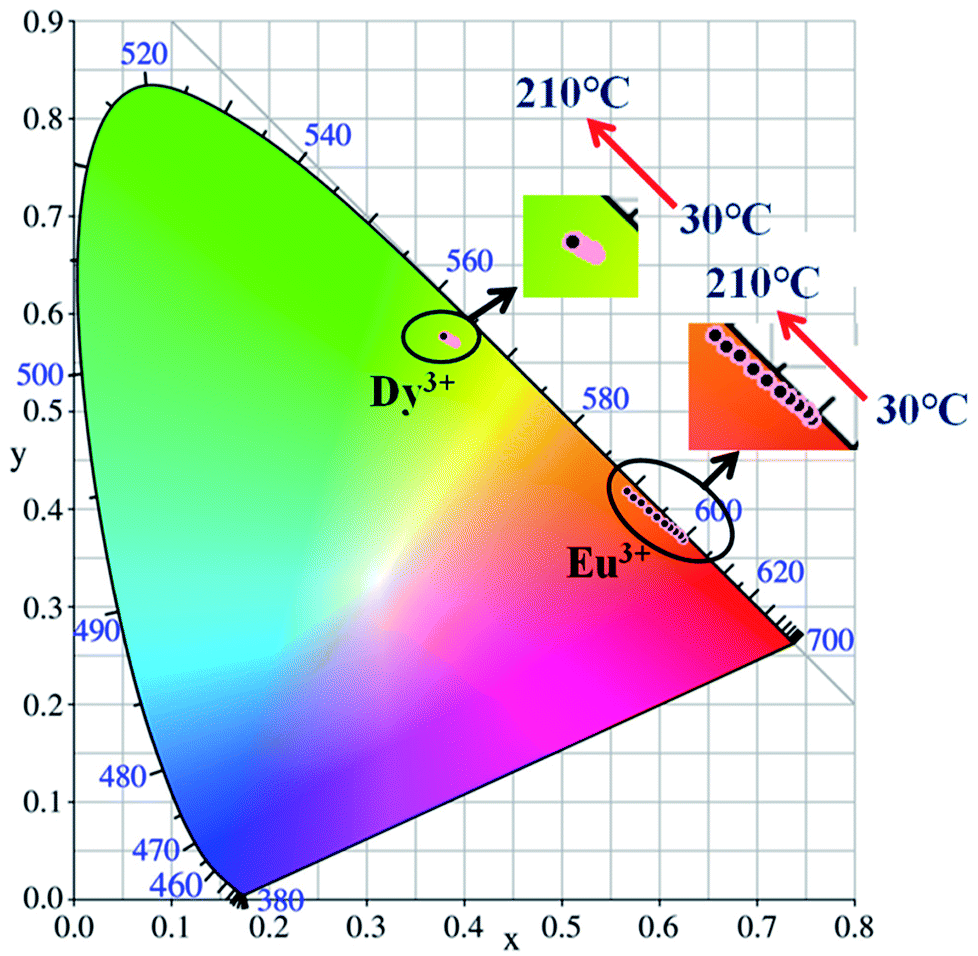 | ||
| Fig. 11 CIE chromaticity diagram of BaTiO3:xEu3+ (x = 8 mol%) and BaTiO3:xDy3+ (x = 4 mol%) phosphors at elevated temperatures. | ||
| Temperature, (°C) | BaTiO3:xEu3+, (x = 8 mol%) | BaTiO3:xDy3+, (x = 4 mol%) | ||
|---|---|---|---|---|
| X | Y | X | Y | |
| 30 | 0.624 | 0.370 | 0.390 | 0.571 |
| 50 | 0.621 | 0.373 | 0.390 | 0.571 |
| 70 | 0.616 | 0.377 | 0.389 | 0.572 |
| 90 | 0.611 | 0.381 | 0.388 | 0.572 |
| 110 | 0.605 | 0.386 | 0.386 | 0.573 |
| 130 | 0.598 | 0.392 | 0.384 | 0.575 |
| 150 | 0.590 | 0.399 | 0.382 | 0.576 |
| 170 | 0.582 | 0.406 | 0.382 | 0.576 |
| 190 | 0.574 | 0.412 | 0.380 | 0.577 |
| 210 | 0.567 | 0.419 | 0.380 | 0.578 |
In principle, the effect of luminescence thermal quenching is put down to the non-radiative relaxation between the excited and ground state through the crossing points of configuration coordinate diagram of RE3+ electrons in host.35 The most important factor affecting non-radiative transition is the temperature. The relationship between the emission intensity and the temperature can be described by the following Arrhenius equation:36
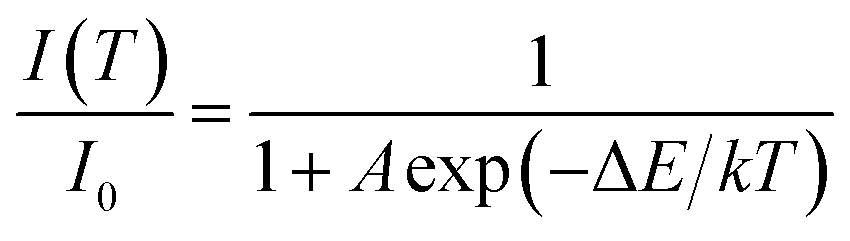 | (1) |
Generally, the thermal stability of nitride phosphors with strong covalent bonds is higher than that of oxide phosphors, while the thermal stability of oxide phosphors is higher than that of sulphide phosphors. Luminescence thermal stability is also related to the unavoidable defects in/on imperfect phosphors (such as dangling bonds, and oxygen defects, introducing extra defect energy levels), which can induce non-radiative transitions during the heating or cooling process. Although the unavoidable defects in/on the imperfect phosphors are difficult to be detected at present, these defects would affect the luminescence properties. Singh et al. reported that the ΔE of Eu3+–K+ co-doped Ba(1−x−y)TiO3:xEu3+,yK+ was only 0.088 eV,9 which indicated that the BaTiO3:xDy3+ (x = 4 mol%) phosphor prepared by our group possesses much higher activation energy and better thermal stability.
With the replacement of Ba2+ by Sr2+ for phosphors (Ba,Sr)TiO3:RE3+ (RE = Eu, Dy), there were some differences in thermal stabilities compared to the initial unsubstituted BaTiO3:RE3+ (RE = Eu, Dy). Clearly, the PL intensities of both Sr2+-substituted phosphors Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) and Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%) decreased with the increase in temperature and there was no evident shift in the emission wavelengths of these two phosphors at higher temperatures (Fig. 12a and c). Furthermore, as shown Fig. 12b and d, when the temperature of the phosphors increases to 100 °C and 150 °C, the PL intensity drops to about 60% and 40%, and 80% and 50% of its initial intensity for Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) and Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%), respectively. While cooling down to the RT again, the PL intensities of these two phosphors were irreversible, indicating thermal degradation. The measured PL intensities after cooling to RT from 210 °C were approximately 85% of the values at the RT before heating up. As shown in the insets of Fig. 12b and d, the calculated activation energy of Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) was 0.266 eV, which was lower than that of Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%) (0.322 eV), calculated according to the Arrhenius equation.
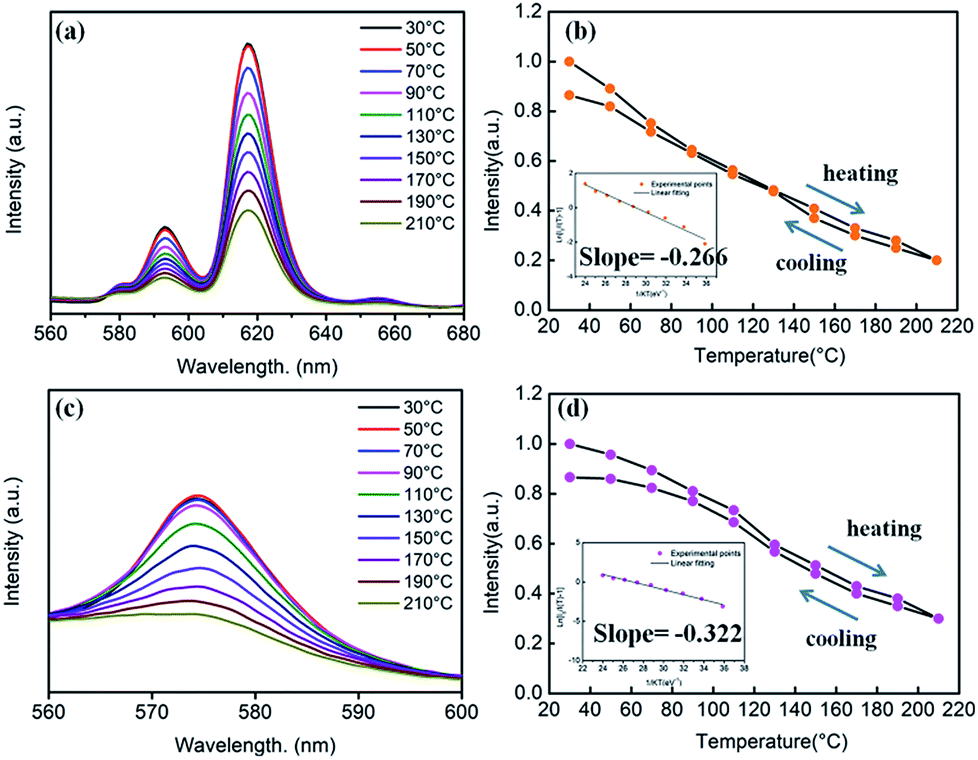 | ||
| Fig. 12 (a) Temperature-dependent PL spectra of the Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) phosphor (λex = 465 nm). (b) Relative PL intensity of the phosphor as a function of temperature. Inset: the linear fitting of the calculated activation energy for thermal quenching of the Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) phosphor. (c) Temperature-dependent PL spectra of the Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%) phosphor (λex = 452 nm). (d) Relative PL intensity of the phosphor as a function of temperature. Inset: the linear fitting of the calculated activation energy for thermal quenching of the Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%) phosphor. | ||
The thermal stability of Sr2+-substituted Ba1−ySryTiO3:RE3+ (RE3+ = Eu3+, Dy3+) phosphors decreased compared to the unsubstituted phosphors (BaTiO3:RE3+, RE3+ = Eu3+, Dy3+), and the results are summarized in Table 2. Upon the comparison of two phosphors BaTiO3:xEu3+ (x = 8 mol%) and Ba0.7Sr0.3TiO3:xEu3+(x = 8 mol%), reduced thermal stability could be noticed in the latter. For the other two phosphors BaTiO3:xDy3+ (x = 4 mol%) and Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%), there a similar trend was observed. The decreased thermal stability of Sr2+-substituted Ba1−ySryTiO3:RE3+ could be mainly attributed to the fact that a part of larger Ba2+ ions were replaced by smaller Sr2+, resulting in shrinkage of the unit cell and an increase in lattice defects. It has to be emphasised that the partial phase transformation of the tetragonal phase of Sr2+-substituted Ba1−ySryTiO3:RE3+ may also contribute to the reduced thermal stability because the optimal Sr-substitution contents of both Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) and Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%) were around the critical line where the phase transformation occurs (seen in Fig. 6b and 7b). Lattice defects or lattice mismatch induced by phase transformation may influence the thermal stability to some extent by introducing additional defect energy levels.
| Sample | Retained emission intensity at 100 °C (100% at RT) | Activation energy ΔE (eV) |
|---|---|---|
| BaTiO3:xEu3+ (x = 8 mol%) | 70% | 0.272 |
| BaTiO3:xDy3+ (x = 4 mol%) | 90% | 0.387 |
| Ba0.7Sr0.3TiO3:xEu3+ (x = 8 mol%) | 60% | 0.266 |
| Ba0.8Sr0.2TiO3:xDy3+ (x = 4 mol%) | 80% | 0.322 |
3.4 Decay times of the series of phosphors
The decay times of the series of phosphors were measured and are shown in Table 3. Additionally, the obtained decay curves were fitted by the double exponential equation:|
I = I0 + A1exp(−t/τ1) + A2exp(−t/τ2)
| (2) |
| τav = (A1τ12 + A2τ22)/(A1τ1 + A2τ2) | (3) |
| Sample | τ1/μs | τ2/μs | τav/μs |
|---|---|---|---|
| BaTiO3:xEu3+ (x = 8 mol%) | 53.41 | 452.78 | 414.32 |
| BaTiO3:xDy3+ (x = 4 mol%) | 1.01 | 12.25 | 5.178 |
| Ba0.7Sr0.3TiO3:xEu3+, (x = 8 mol%) | 61.11 | 465.09 | 433.95 |
| Ba0.8Sr0.2TiO3:xDy3+, (x = 4 mol%) | 1.06 | 13.607 | 5.567 |
As shown in Table 3, the decay time of Dy3+-doped (Ba,Sr)TiO3 phosphors was much shorter than that of Eu3+-doped (Ba,Sr)TiO3 phosphors. The replacement of Ba2+ by Sr2+ slightly increased the decay time of phosphors. The detailed investigations on the decay behaviours of our as-prepared phosphors are in progress.
4. Conclusions
In summary, a series of (Ba,Sr)TiO3 phosphors singly-doped with Eu3+ and Dy3+ were successfully prepared by the nitrate pyrolysis method at 750 °C. The XRD patterns illustrated that BaTiO3:xEu3+ (x = 0.02–0.10), BaTiO3:xDy3+ (x = 0.02–0.10), Ba1−ySryTiO3:xEu3+ (x = 0.08, y ≤ 0.3), and Ba1−ySryTiO3:xDy3+ (x = 0.04, y ≤ 0.2) phosphors primarily showed tetragonal phase structure. When the Sr2+ substitution content was higher than the optimal value (y > 0.3 for Ba1−ySryTiO3:0.08Eu3+ and y > 0.2 for Ba1−ySryTiO3:0.04Dy3+), a remarkable phase transformation from tetragonal to cubic phase occurred. In comparison, the photoluminescence intensities of RE3+-optimally-doped BaTiO3:0.08Eu3+ and BaTiO3:0.04Dy3+ phosphors were the highest in the serial phosphors of BaTiO3:xEu3+ and BaTiO3:xDy3+ (x = 0.02–0.10), respectively. Interestingly, the substitution of 30 mol% Sr2+ (Ba0.7Sr0.3TiO3:0.08Eu3+) further enhanced the photoluminescence intensity of the BaTiO3:0.08Eu3+ phosphor, while the substitution of 20 mol% Sr2+ (Ba0.8Sr0.2TiO3:0.04Dy3+) further enhanced the photoluminescence intensity of the BaTiO3:0.04Dy3+ phosphor. Moreover, the appropriate Sr/Ba isomorphic substitution improved the luminescence intensity of the compounds in various degrees, as compared to the luminescence thermal stabilities of BaTiO3:RE3+ phosphors without Sr2+ substitution. According to curves of the temperature-dependent PL intensity, the calculated activation energies ΔE were about 0.272 and 0.387 eV for BaTiO3:0.08Eu3+ and BaTiO3:0.04Dy3+, while they were around 0.266 and 0.322 eV for Ba0.7Sr0.3TiO3:0.08Eu3+ and Ba0.8Sr0.2TiO3:0.04Dy3+, indicating that the best luminescence thermal stability was obtained in the BaTiO3:0.04Dy3+ phosphor. As a result, about 70% and 85% of the emission intensities at 100 °C compared to those measured at room temperature is retained for BaTiO3:0.08Eu3+ and BaTiO3:0.04Dy3+, respectively, while about 60% and 80% is retained for the Sr2+-substituted Ba0.7Sr0.3TiO3:0.08Eu3+ and Ba0.8Sr0.2TiO3:0.04Dy3+, respectively. It has to be emphasized that while cooling from 210 °C to room temperature, the BaTiO3:0.04Dy3+ phosphor showed the strongest recovery of PL emission (∼95% of the intensity measured at room temperature) compared to other phosphors. Thus, BaTiO3:0.04Dy3+ phosphor with excellent luminescent stability is a good candidate for applications in lighting, display, and other related fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the financial supports from the National Key R&D Program of China (Grant No. 2016YFB0700204 and 2016YFB0701004), the National Natural Science Foundation of China (Grant No. 51502329 and 51702343), and the Research Program of Shanghai Sciences and Technology Commission Foundation (Grant No. 14DZ2261203 and 16ZR1441100).Notes and references
- X. Li, Y. Zhang, D. Geng, J. Lian, G. Zhang, Z. Hou and J. Lin, J. Mater. Chem. C, 2014, 2, 9924–9933 RSC.
- X. Wu, Y. Liang, S. Liu, Y. Zhu, R. Xu, M. Tong and K. Li, Spectrosc. Lett., 2017, 50, 48–54 CrossRef.
- C. S. Lewis, H. Liu, J. Han, L. Wang, S. Yue, N. A. Brennan and S. S. Wong, Nanoscale, 2016, 8, 2129–2142 RSC.
- L. L. Noto, S. K. K. Shaat, D. Poelman, M. S. Dhlamini, B. M. Mothudi and H. C. Swart, Ceram. Int., 2016, 42, 9779–9784 CrossRef.
- J. Zhong, D. Chen, H. Xu, W. Zhao, J. Sun and Z. Ji, J. Alloys Compd., 2017, 695, 311–318 CrossRef.
- H. Ji, L. Wang, M. S. Molokeev, N. Hirosaki, Z. Huang, Z. Xia, O. M. ten Kate, L. Liu and R. Xie, J. Mater. Chem. C, 2016, 4, 2359–2366 RSC.
- K.-H. Chen, M.-H. Weng, C.-T. Pan and R.-Y. Yang, Powder Technol., 2016, 288, 117–122 CrossRef.
- H. Ji, Z. Huang, Z. Xia, M. S. Molokeev, M. Chen, V. V. Atuchin, M. Fang, Y. g. Liu, X. Wu and J. A. Varela, J. Am. Ceram. Soc., 2015, 98, 3280–3284 CrossRef.
- D. Kumar Singh, K. Mondal and J. Manam, Ceram. Int., 2017, 43, 13602–13611 CrossRef.
- G. Nag Bhargavi and A. Khare, Opt. Spectrosc., 2015, 118, 902–917 CrossRef.
- G. K. Ribeiro, F. S. Vicente, M. I. B. Bernardi and A. Mesquita, J. Alloys Compd., 2016, 688, 497–503 CrossRef.
- Y. Kamiyanagi, M. Kitaura and M. Kaneyoshi, J. Lumin., 2007, 122, 509–511 CrossRef.
- B. Liu, L. Kong and C. Shi, J. Lumin., 2007, 122–123, 121–124 CrossRef.
- D. Chen, W. Xu, Y. Zhou and Y. Chen, J. Alloys Compd., 2016, 676, 215–223 CrossRef.
- L. Zhang, H. Pan, H. Liu, B. Zhang, L. Jin, M. Zhu and W. Yang, J. Alloys Compd., 2015, 643, 247–252 CrossRef.
- D. P. Dutta, A. Ballal, J. Nuwad and A. K. Tyagi, J. Lumin., 2014, 148, 230–237 CrossRef.
- S.-Y. Kang, Y. H. Kim, J. Moon, K. S. Suh, D. J. Lee and S.-G. Kang, Jpn. J. Appl. Phys., 2009, 48, 052301 CrossRef.
- J. Li and M. Kuwabara, Sci. Technol. Adv. Mater., 2003, 4, 143–148 CrossRef.
- Z. Fu, W. Li, H. Kyoung Yang, B. Kee Moon, B. Chun Choi and J. Hyun Jeong, Phys. Scr., 2010, T139, 014015 CrossRef.
- E. Korkmaz and N. O. Kalaycioglu, Bull. Mater. Sci., 2012, 35, 1011–1017 CrossRef.
- Z. Fu, B. K. Moon, H. K. Yang and J. H. Jeong, J. Phys. Chem. C, 2008, 112, 5724–5728 Search PubMed.
- M. Buscaglia, V. Buscaglia, M. Viviani, P. Nanni and M. Hanuskova, J. Eur. Ceram. Soc., 2000, 20, 1997–2007 CrossRef.
- D. K. Patel, B. Vishwanadh, V. Sudarsan, R. K. Vatsa and S. K. Kulshreshtha, J. Am. Ceram. Soc., 2011, 94, 482–487 CrossRef.
- W. E. I. Yun-Ge, Y. Qian and L. I. Gui-Fang, Journal of Inorganic Materials, 2017, 32, 936 CrossRef.
- Y. Cao, G. Zhu and Y. Wang, RSC Adv., 2015, 5, 65710–65718 RSC.
- G. Zhu, Z. Ci, Y. Shi, M. Que, Q. Wang and Y. Wang, J. Mater. Chem. C, 2013, 1, 5960 RSC.
- X. Zhang, Y. Chen, S. Zeng, L. Zhou, J. Shi and M. Gong, Ceram. Int., 2014, 40, 14537–14541 CrossRef.
- X. Chen, J. Zhao, L. Yu, C. Rong, C. Li and S. Lian, J. Lumin., 2011, 131, 2697–2702 CrossRef.
- L. Chen and X. H. Wei, Integr. Ferroelectr., 2012, 140, 187–194 CrossRef.
- Z. Lei, L. Hui, X. Ting-xian and T. Jin, Piezoelectr. Acoustoopt., 2002, 24, 469–472 Search PubMed.
- Z. Ci, Q. Sun, S. Qin, M. Sun, X. Jiang, X. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2014, 16, 11597–11602 RSC.
- R.-J. Xie, N. Hirosaki, T. Suehiro, F.-F. Xu and M. Mitomo, Chem. Mater., 2006, 5578–5583 CrossRef.
- H. Jing, C. Guo, G. Zhang, X. Su, Z. Yang and J. H. Jeong, J. Mater. Chem., 2012, 22, 13612 RSC.
- C. W. Yeh, W. T. Chen, R. S. Liu, S. F. Hu, H. S. Sheu, J. M. Chen and H. T. Hintzen, J. Am. Chem. Soc., 2012, 134, 14108–14117 CrossRef PubMed.
- V. Bachmann, C. Ronda, O. Oeckler, W. Schnick and A. Meijerink*, Chem. Mater., 2009, 21, 316–325 CrossRef.
- X. Zhang, L. Huang, F. Pan, M. Wu, J. Wang, Y. Chen and Q. Su, ACS Appl. Mater. Interfaces, 2014, 6, 2709–2717 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |