
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNotoginsenoside R1 prevents H9c2 cardiomyocytes apoptosis against hypoxia/reoxygenation via the ERs/PI3K/Akt pathway
Guang Li abc,
Xiaoyan Xing*ac,
Yun Luoac,
Xuehong Dengac,
Shan Luac,
Shimin Tangd,
Guibo Sunac and
Xiaobo Sun*ac
abc,
Xiaoyan Xing*ac,
Yun Luoac,
Xuehong Dengac,
Shan Luac,
Shimin Tangd,
Guibo Sunac and
Xiaobo Sun*ac
aThe Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Beijing 100093, China. E-mail: sun_xiaobo163@163.com; lilyclxy@163.com; Fax: +86-10-57833013; Tel: +86-10-57833013
bYunnan Branch, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Jing Hong 666100, China
cBeijing Key Laboratory of Innovative Drug Discovery of Traditional Chinese Medicine (Natural Medicine) and Translational Medicine, Institute of Medicinal Plant Development, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing 100193, China
dChangchun University of Chinese Medicine, 130117, Changchun, China
First published on 13th April 2018
Abstract
Notoginsenoside R1 (NGR1) is separate from Panax notoginsenosides (PNS), and plays a role similar to phytoestrogen in preventing and treating cardiovascular diseases. However, the protective mechanism of NGR1 in the myocardial ischemia/reperfusion injury via the estrogen receptor (ER) pathway remains unclear, which hinder its application. This study aimed to study the preventive mechanisms of NGR1 in the apoptosis of H9c2 cardiomyocytes after hypoxia/reoxygenation (H/R). NGR1 did not affect the expression of ERα and ERβ proteins in normal H9c2 cardiomyocytes. However, NGR1 could upregulate the ERα and G protein-coupled receptor 30 (GPR30) proteins in H9c2 cardiomyocytes after H/R without affecting ERβ levels. Moreover, it significantly affected the expression levels of PI3K and its downstream apoptosis proteins such as Bcl-2 Associated X Protein (Bax), B cell lymphoma/lewkmia-2 (Bcl-2), caspase-3, and so forth. Whereas, after adding the PI3K protein antagonist, the modulatory expression levels of PI3K and its downstream apoptosis proteins were remarkably abolished. After adding ERα and GPR30 antagonists, NGR1 had no significant effect on the expression of PI3K and its downstream Akt protein in the model group. The data of flow cytometry showed that after adding the ERα, GPR30 and PI3K antagonists, the apoptotic rate of cardiomyocytes had no significant changes compared with the model group. This study demonstrated that NGR1 protected H9c2 cardiomyocytes from the injury after H/R by affecting ERα and GPR30 to regulate the expression levels of PI3K and its downstream apoptosis proteins.
1. Introduction
Coronary heart disease is one of the main causes of death in humans.1 In most cases, reperfusion therapy after myocardial ischemia could restore heart function and repair damaged structures. However, sometimes, reperfusion after ischemia not only is unable to restore organ functions, but also causes myocardial energy metabolism disorder, ultrastructural changes, and no vascular reflow, aggravating functional disorder and structural damage. This phenomenon is termed as myocardial ischemia-reperfusion injury (MIRI).2 At present, MIRI is the major obstacle in obtaining the optimal therapeutic effect from reperfusion of ischemic myocardium. Prevention and mitigation of a reperfusion injury have become the major focus for researchers nowadays.Clinical studies have shown that estrogen plays a role in preventing ischemic heart disease.3 In 1997, Kolodgie et al. proved that 17β-estradiol could effectively reduce the myocardial infarct size after ischemia-reperfusion in a female rat model with bilateral ovarian resection. Kang et al. found that estrogen (E2) significantly reduced the myocardial oxidative stress injury and dilated the coronary artery.4,5 Li et al. found that estrogen could significantly reduce heart rate and blood pressure in normal and propylepinephrine-treated hearts and myocardial energy consumption and prevent Ca2+ overload.6 However, the underlying mechanisms of these effects remain unclear. Moreover, estrogen could also protect myocardial cells through anti-inflammatory and anti-apoptotic responses.7
It is expensive and has obvious adverse effects for treating menopausal syndrome and cardiovascular disease using estrogen replacement therapy (ERT). The long-term use of ERT could increase the incidence of a series of diseases, such as uterine and breast cancers,8 endometrial hyperplasia and hypercoagulable state,9–13 venous thrombosis,14 gallbladder disease and stroke.15,16 Drug withdrawal after ERT could easily result in repeated illness.17 On the contrary, oral estrogen had a severe first-pass effect on liver,18 but subcutaneous injection or changing the dosage form virtually increases costs and difficulty in treatment. Considering the aforementioned disadvantages of ERT, estrogen replacements are urgently needed. It was hoped that these substitutes could preserve the protective effects of estrogen on the cardiovascular system while avoiding the adverse effects. Phytoestrogen is one of them.19
Pseudo-ginseng is the subterraneous root of Panax notoginseng (Burk) F. H. Chen (Araliaceae, Panax), which was used for promoting blood circulation and removing blood stasis. It has obvious controlling effects on cardio-cerebrovascular diseases,20 and has attracted much attention for an effective MIRI control drug in the worldwide.21 Notoginsenoside R1 (NGR1, Fig. 1a), one of the main components, accounts for about 5% of the total saponin. Wang et al. proved that NGR1 exerted phytoestrogen-like effects by activating the phosphorylation of phosphorylated estrogen response element (pERE).22 Our group found that NGR1 could protect cardiomyocytes from H/R–induced injuries by regulating the expression of endoplasmic reticulum stress-related proteins, thereby promoting the expression of apoptosis proteins.23 Moreover, we proved that NGR1 could inhibit SH-SY5Y cell apoptosis via the mitogen-activated protein kinase 1 and 2-extracellular signal-regulated kinases 1 and 2 (MEK1/2-ERK1/2) pathway.24
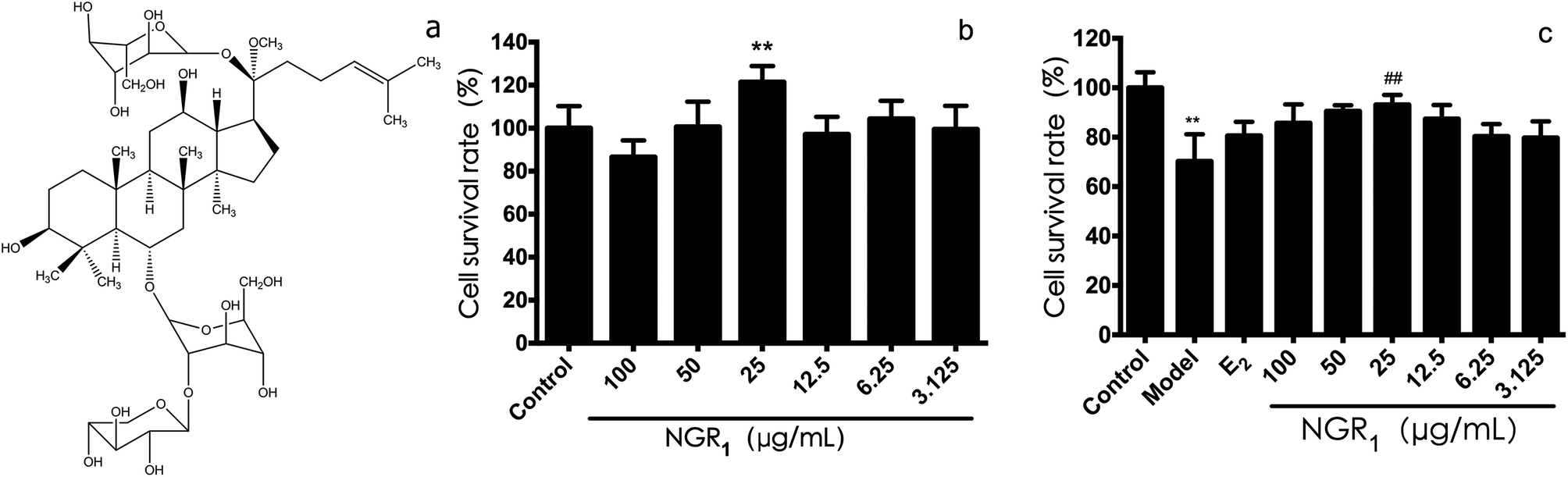 | ||
| Fig. 1 Protective effects of NGR1 against H/R-induced H9c2 cell injury. (a) Structural formula of NGR1. (b) Effects of NGR1 on H9c2 cell viability. Cells were incubated with different doses of NGR1 for 24 h. Cell viability was measured by MTT assay (c) effects of NGR1 on H/R-induced H9c2 cell viability. H9c2 cells were pretreated with indicated doses of NGR1 and E2 for 24 h before exposure to H/R for 12 h. Cell viability was measured by MTT assay. (B and C, expressed as the percentage of control). All results are expressed as the mean ± SD of three independent experiments. **P < 0.01 vs. control group; ##P < 0.01 vs. H/R-treated cells. | ||
However, the mechanisms underlying its estrogen-like effects and its role in preventing MIRI remain unclear. Using ER and PI3K pathway antagonists, this study proved that NGR1 could antagonize the apoptosis of cardiomyocytes by regulating PI3K and its downstream pathways via the ER pathway, highlighting the mechanism of MIRI prevention and treatment by NGR1.
2. Materials and methods
2.1 Drug and reagents
NGR1 (molecular weight: 933.15, purity ≥ 98%) was obtained from the Shanghai Winherb Medical S & T Development Co. Ltd (Shanghai, China). ERα, ERβ, and GPR30 receptor agonists and antagonists were procured from Cayman Chemical Inc (Michigan, USA). ERβ, GPR30, and Bax antibodies, as well as secondary antibodies, including goat anti-rabbit immunoglobulin G (IgG) and goat anti-mouse IgG, were purchased from Abcam (Cambridge, England). PI3K, Akt, caspase-3, Bcl-2, ERα, and β-actin antibodies were purchased from Millipore Corporation (CA, USA). Dulbecco's modified Eagle's medium (DMEM) was purchased from Hyclone (Utah, USA). Foetal bovine serum (FBS) was purchased from Gibco (NY, USA). Double antibiotics were purchased from Gibco (NY, USA). The Alexa Fluor 488 Annexin V/propidium iodide (PI) Apoptosis Kit was procured from Life Technologies (CA, USA). The protein extraction and separation reagent were obtained from CW Biotech (Beijing, China). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).2.2 Cell culture and treatment
H9c2 cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), cultured in Dulbecco's modified Eagle's medium with high glucose, supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 of penicillin, 100 mg mL−1 of streptomycin, and maintained in a humidified incubator at 37 °C with 5% CO2. The H/R model was established according to previous study25 The anaerobic incubator (Coy Laboratory, Michigan, USA) was used to establish the model at 37 °C. First, the complete medium was replaced with the FBS-free and sugar-free medium to culture cells at 37 °C with 5% CO2, 5% H2, and 90% N2 for 6 h. The medium was replaced with the FBS-free high-glucose medium to continue cell culture with 5% CO2 for another 12 h. The control group was cultured under the same conditions in the carbon dioxide incubator with 5% CO2.2.3 Cell viability
The cell viability was measured using the MTT assay. The H9c2 cells were cultured in a 96-well plate (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well), and NGR1 was added at doses of 100, 50, 25, 12.5, 6.25, and 3.125 μg mL−1 for 24 h. MTT solution (1 mg mL−1) was added after administrating NGR1, and the cells were cultured continuously for another 4 h at 37 °C. By the end of cell incubation, dimethyl sulfoxide (100 μL per well) was added for dissolution. Optical density (OD) values were measured at 570 nm using the automatic multifunctional microplate reader (Model i3, Molecular Devices, CA, USA) for calculating cell survival rates.
000 cells per well), and NGR1 was added at doses of 100, 50, 25, 12.5, 6.25, and 3.125 μg mL−1 for 24 h. MTT solution (1 mg mL−1) was added after administrating NGR1, and the cells were cultured continuously for another 4 h at 37 °C. By the end of cell incubation, dimethyl sulfoxide (100 μL per well) was added for dissolution. Optical density (OD) values were measured at 570 nm using the automatic multifunctional microplate reader (Model i3, Molecular Devices, CA, USA) for calculating cell survival rates.
2.4 Flow cytometry
Cell apoptosis was detected by flow cytometry using the Annexin V/PI kit. The cell culture medium in each group was collected after treatment. The cells were trypsinized, and culture solutions were collected and combined with centrifugation at 1000 rpm for 10 min. The cells were then washed with cold phosphate-buffered saline and centrifuged at 1000 rpm for 10 min. The cells in each group were treated following the manufacturer's instructions for detection. The flow cytometer was BD Accuri C6 (Accuri Cytometers, Inc. Michigan, USA).2.5 Detection of protein expression using western blot
The cells were cultured in 25 cm2 flasks. Molding was performed 24 h after drug administration, and then the proteins were extracted and purified using the mammalian protein extraction kit (CW Biotech, Beijing, China). Then, the proteins were quantified by the BCA protein quantification kit (CW Biotech, Beijing, China). Protein extracts were diluted to the same concentration and added into the loading buffer (CW Biotech, Beijing, China) at a ratio of 1:4 for 10 min in a boiling water bath. The protein samples were separated from the sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto the polyvinylidene difluoride (PVDF) membrane using a semidry blotter. The resulting PVDF membrane was blocked for 2 h using the blocking buffer [5% milk in Tris-buffered saline with Tween 20 (TBST)]. Then, primary antibodies were prepared and incubated overnight at 4 °C. The membrane was washed three times using TBST. The membrane was incubated with secondary antibodies in the blocking buffer for 2 h and then washed three times using TBST. After adding the enhanced chemiluminescence, the membrane was incubated in the dark for 5 min. The band intensities were quantified using the Photo-Image System (Molecular Dynamics, Sweden).
2.6 Statistical analysis of data
All data were repeated three times. Three to five wells for each group were placed on the 96-well plate. Data were presented as mean ± SD. The statistical analysis was performed by one-way ANOVA using the SPSS version 13.0 (SPSS, IL, USA). Comparisons were tested by one-way ANOVA and least significant difference. A P value less than 0.05 was considered to be statistically significant.3. Results
3.1 Effects of NGR1 on the survival rate of H9c2 cells after normal and H/R treatment
First of all, we evaluated the cell viability effects of various concentration of NGR1. As shown in Fig. 1b, the survival rate of H9c2 cells at the dose of 25 μg mL−1 was the highest (121.4 ± 7.5%), which was significantly different from that in the control group. Next, the protective effects of NGR1 on H/R induced H9c2 cell injury were investigated. As shown in Fig. 1c, after 6 h of hypoxia and 12 h of reoxygenation, the cell survival rate (70 ± 10%) decreased obviously compared with the control group. However, 25 μg mL−1 of NGR1 significantly increased the cell survival rate (86.53 ± 7.77%) compared with the model group, suggesting that 25 μg mL−1 of NGR1 could obviously inhibit cell apoptosis induced by H/R. Therefore, 25 μg mL−1 was considered to be the concentration of drug administration for NGR1.3.2 Effects of NGR1 on the expression of ER proteins
ER protein plays an important role in ischemia reperfusion injury.26–28 Then, we accessed the effects of NGR1 on the expression of ER proteins in H/R treated H9c2 cells. Different doses of ER antagonists were added for different times. Fig. 2a and b showed that a single administration of ERα antagonist MPP significantly decreased the expression of ERα. If NGR1 was added, the expression of ERα did not change significantly. A single administration of NGR1 had no effect on the expression of ERα and ERβ in normal H9c2 cardiomyocytes. Fig. 2a and c indicated that a single administration of ERβ antagonist HPTPP significantly decreased the expression of ERβ. When NGR1 was added, the expression of ERβ did not change significantly. A single administration of GPR30 receptor antagonist G-15 significantly decreased the expression of GPR30. If NGR1 was added, the expression of GPR30 increased. A single administration of NGR1 also increased the expression of GPR30 (Fig. 2a and d). Thus, when ER antagonists were added, they directly inhibited the expression of ERα and ERβ. NGR1 upregulated the expression of GPR30 in normal H9c2 cardiomyocytes irrespective of the addition of G-15. These data together suggested that the mechanism underlying the regulatory effect of NGR1 on the expression of ERα and ERβ was different from that in the case of GPR30. The aforementioned findings also showed that NGR1 had no effect on the expression of ERα and ERβ protein in normal H9c2 cardiomyocytes, but had a significant stimulatory effect on the expression of GPR30.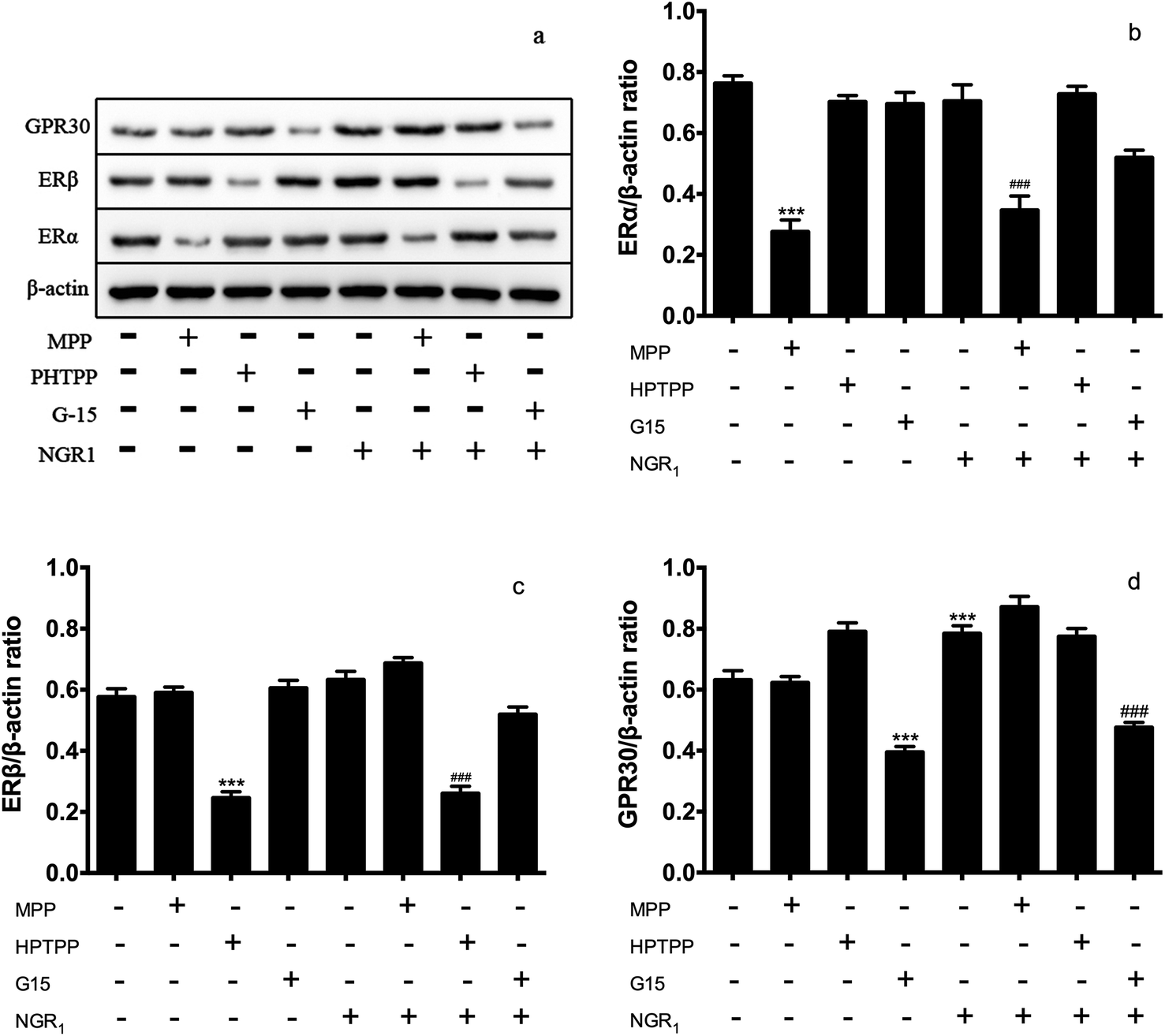 | ||
| Fig. 2 Effects of ER antagonists on the expression of ER α/β in normal H9c2 cardiomyocytes. H9c2 cells were pretreated with ERα antagonist MPP (100 nM) for 30 h, ERβ antagonist PHTPP (1 μM) for 24 h and GPR30 receptor antagonist G-15 (1 μM) for 24 h, then cells were exposed to NGR1 (25 μg mL−1) for 24 h. Expression levels of GRP30, ERβ, ERα, and β-actin were investigated by western blot. (a) Representative images of GRP30, ERβ, ERα, and β-actin (b–d) statistical results of GRP30, ERβ, ERα relative to control group. ***p < 0.001 vs. control; ###p < 0.001 vs. H/R-treated cells. | ||
3.3 Effects of NGR1 on the PI3K/Akt pathway and the expression level of downstream proteins
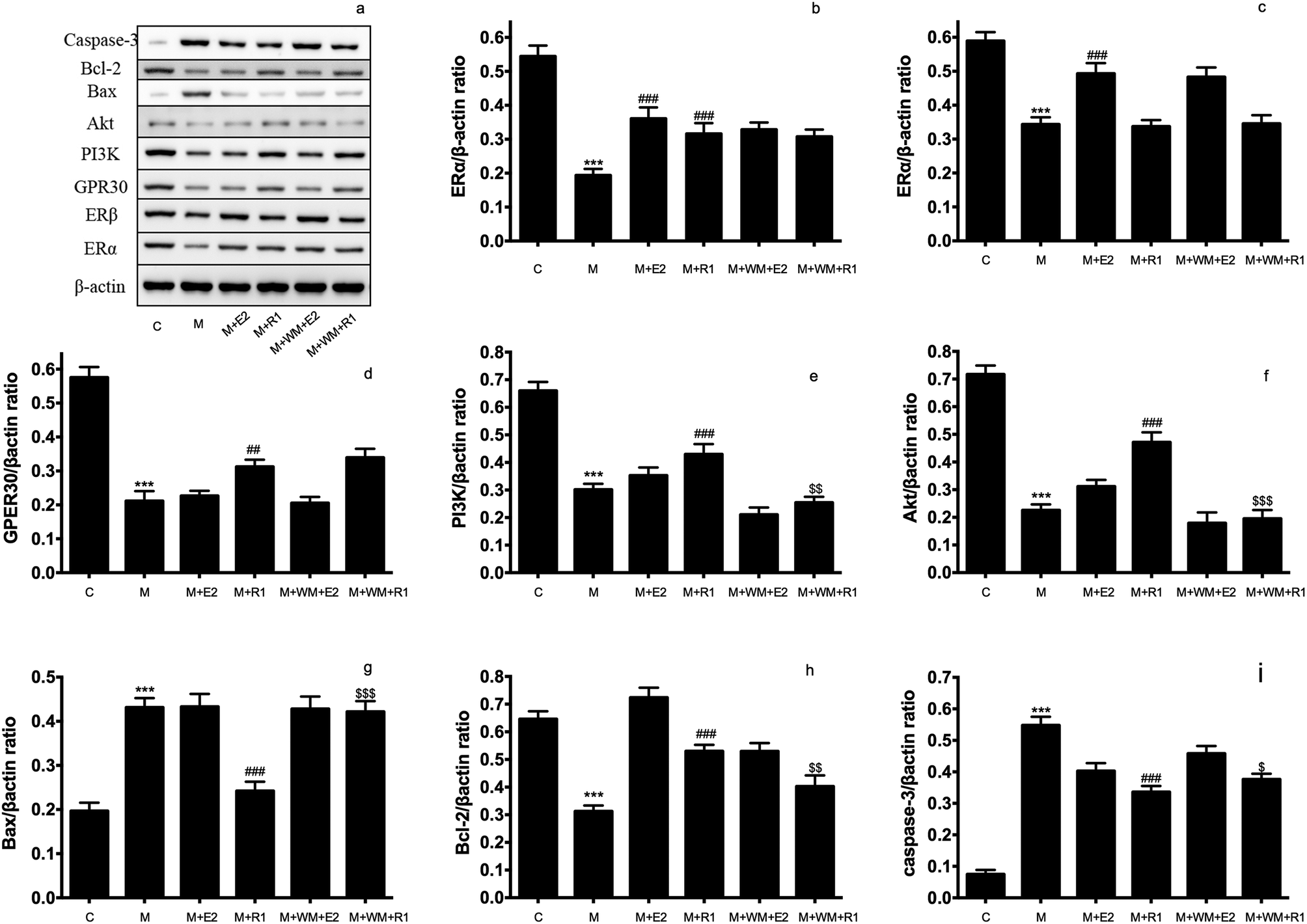 | ||
| Fig. 3 Effects of PI3K inhibitor on the protein expression in H/R-induced H9c2 cardiomyocytes. H9c2 cells were pretreated with PI3K inhibitor WM (100 nM) for 24 h, then cells were exposed to NGR1 (25 μg mL−1) for 24 h. Expression levels of Caspase-3, Bcl-2, Bax, Akt, PI3K, GRP30, ERβ, ERα and β-actin were investigated by western blot. (a) Representative images of Caspase-3, Bcl-2, Bax, Akt, PI3K, GRP30, ERβ, ERα and β-actin (b–i) Statistical results of ERβ, ERα, GRP30, PI3K, Akt, Bax, Bcl-2, Caspase-3 relative to control group. ***p < 0.001 vs. control; ###p < 0.001 vs. H/R-treated cells; $p < 0.05, $$p < 0.01 vs. model + NGR1 group. | ||
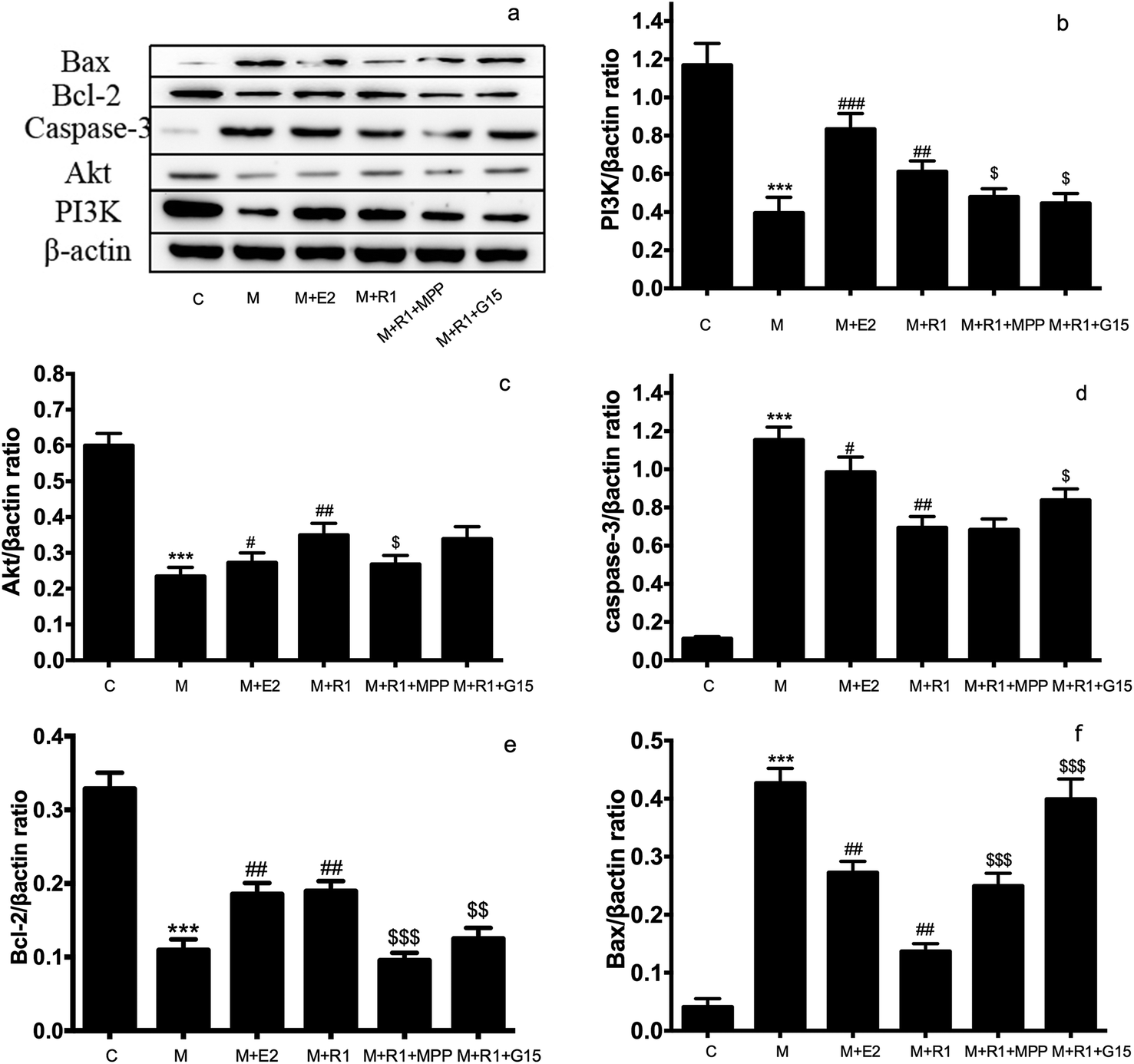 | ||
| Fig. 4 Effects of ER inhibitor on the expression of PI3K/Akt pathway and apoptosis-related proteins in H/R-induced H9c2 cardiomyocytes. H9c2 cells were pretreated with ERα antagonist MPP (100 nM) for 30 h and GPR30 receptor antagonist G-15 (1 μM) for 24 h, then cells were exposed to NGR1 (25 μg mL−1) for 24 h. Expression levels of Bax, Bcl-2, Caspase-3, Akt PI3K and β-actin were investigated by western blot. (a) Representative images of Bax, Bcl-2, Caspase-3, Akt PI3K and β-actin. (b–f) Statistical results of Bax, Bcl-2, Caspase-3, Akt PI3K relative to control group. ***p < 0.001 vs. control; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R-treated cells; $p < 0.05, $$p < 0.01, $$$p < 0.001 vs. model + NGR1 group. | ||
3.4 Effects of ER and PI3K inhibitors on the apoptosis of H9c2 cardiomyocytes
Cell apoptosis and necrosis were identified by the Annexin V-FITC/PI assay. The cell apoptosis rate increased significantly after H/R treatment compared with the normal control group. The addition of E2 and NGR1 significantly decreased cell apoptosis. After adding the antagonists, the cell apoptosis rate increased significantly, suggesting that NGR1 could inhibit the apoptosis of H9c2 cardiomyocytes via the ERα, GPR30, and PI3K pathways (Fig. 5).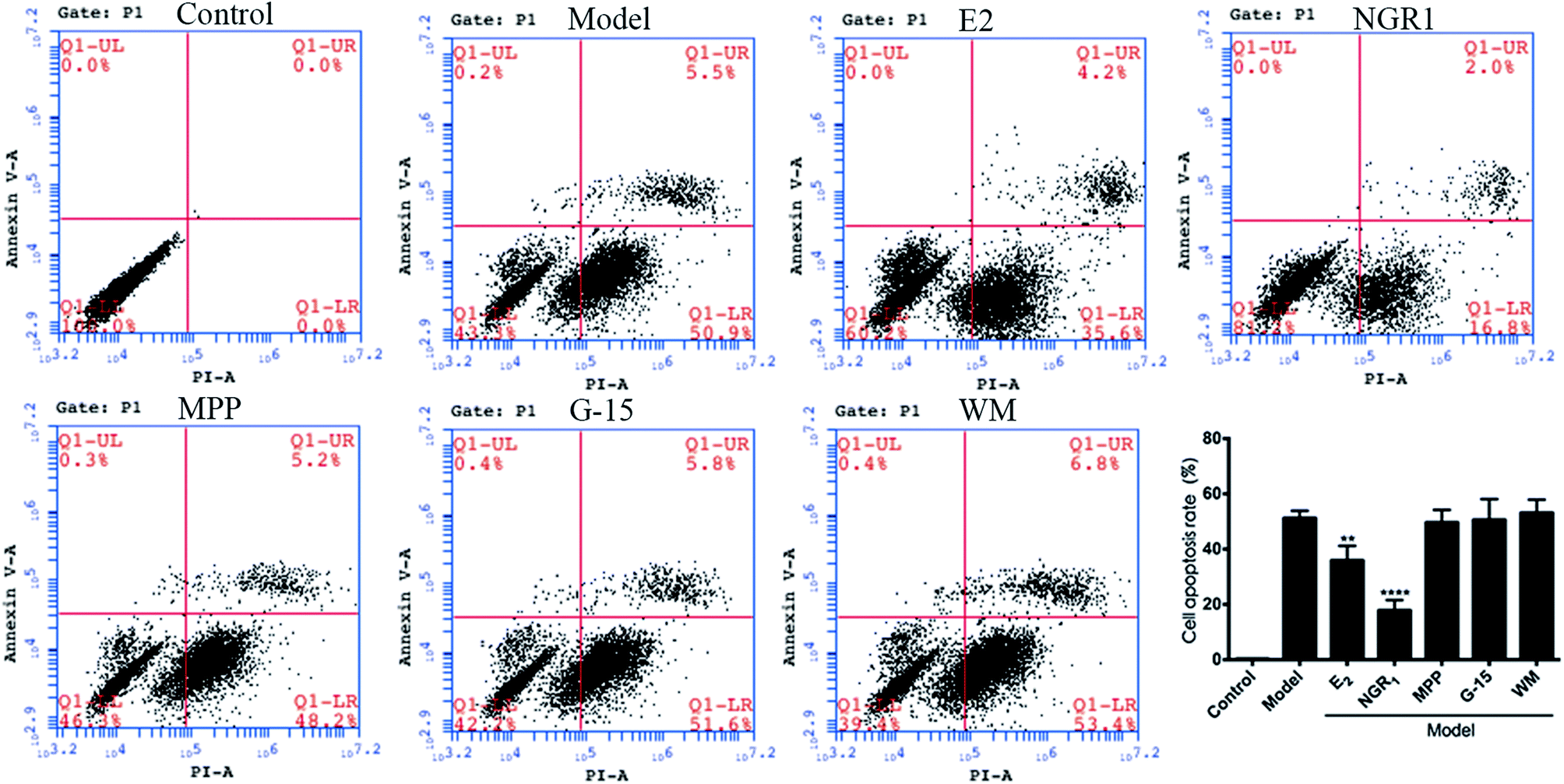 | ||
| Fig. 5 Effects of NGR1 on H/R-induced apoptosis in H9c2 cardiomyocytes. H9c2 cells were pretreatment with ERα antagonist MPP (100 nM) for 30 h, PI3K inhibitor WM (100 nM) for 24 h and GPR30 receptor antagonist G-15 (1 μM) for 24 h, then cells were exposed to NGR1 (25 μg mL−1) for 24 h. Annexin V-FITC/PI staining in H9c2 cells was detected by flow cytometry. ##p < 0.01, ###p < 0.001 vs. H/R-treated cells. | ||
4. Discussion
NGR1 is one of the major components of PNS. The therapeutic effects of NGR1 on organ ischemia-reperfusion injury has gained the attention of researchers in recent years. NGR1 plays a therapeutic role through anti-inflammatory, anti-oxidative stress, and anti-apoptosis pathways. It has been proved that NGR1 could be used for treating a neonatal cerebral hypoxic-ischemic injury by affecting endoplasmic reticulum stress through ERs.30 NGR1 could decrease the inflammatory damage induced by oxidized low-density lipoprotein in vascular endothelial cells by activating the nuclear transcription factor peroxisome proliferator-activated receptor γ.31 At the same time, NGR1 could improve the intestinal metabolism, and thereby exerted a protective effect on the intestinal ischemia-reperfusion injury. Moreover, NRG1 played a protective role in lung functions through the transforming growth factor β1/TGF β activated kinase 1 (TGF-β1/TAK1) signaling pathway in the pulmonary ischemic model.32 NGR1 affected cardiac function by inhibiting a myocardial ischemia-reperfusion injury through the tumor suppressor protein vitamin D3 upregulated protein 1/Nuclear factor-κB (NF-κB) pathway.33 For H9c2 myocardial cell apoptosis induced by endotoxins, NGR1 played an anti-oxygenation and anti-apoptosis role through ERα.34 Chen proved that PNS upregulated the expression of PI3K and Akt proteins in heart and H9c2 cardiomyocytes after hypoxia stimulation.35 It was found in a recent study that NGR1 could inhibit the apoptosis of H9c2 cardiomyocytes by affecting the expression of endoplasmic reticulum stress-related proteins such as GRP78, p-PERK, ATF6, and IRE.23H9c2 cardiomyocytes treated with H/R is the major in vitro model of myocardial ischemia-reperfusion injury.36–39 This study focused on the roles of NGR1 in the ER and PI3K pathways during the myocardial H/R injury. We found that NGR1 could not directly stimulate the expression of ERα and ERβ proteins in normal H9c2 cardiomyocytes. However, NGR1 could increase the expression of GPR30 in normal H9c2 cells, and increase the expression of ERα and GPR30 in H9c2 cardiomyocytes after H/R treatment, suggesting that although NGR1 could be used as a phytoestrogen, its mechanism of preventing the myocardial ischemia-reperfusion injury was not completely consistent with that of estradiol.40 NGR1 could regulate the expression of PI3K and its downstream protein in the model, thereby controlling the expression of apoptosis proteins such as caspase-3 and Bax to inhibit the apoptosis of cardiomyocytes. The inhibition of apoptosis of cardiomyocytes disappeared on adding PI3K inhibitors, suggesting that NGR1 protects cells from the ischemic-reperfusion injury through the PI3K pathway. ERα inhibitor and MPP and GPR30 inhibitor G-15 were used to evaluate the expression of PI3K and its downstream proteins to further explore effects of ERα and GPR30 on the PI3K pathway. Interestingly, although NGR1 upregulated the expression of GPR30, the addition of GPR30 inhibitor G-15 could also increase the expression of PI3K and its downstream protein Akt, thereby promoting the apoptosis of cardiomyocytes. This is likely to be one of the mechanisms of action of estrogen with bidirectional regulation.
The role of ER in preventing and treating cardiovascular diseases has long been the interest of many researchers. Simoncini et al. found that estradiol could reduce the ischemia-reperfusion injury by increasing endothelial nitric oxide synthase and PI3K activities.29 The role of the nonnuclear ER signaling pathway between ERα and PI3K was elucidated. GPR30 agonist could also regulate the expression of PI3K and its downstream proteins to prevent the myocardial ischemia-reperfusion injury.41 We have proved that NGR1 could protect the myocardium of mice via the ER pathway,42 suggesting an estrogen-like effect of NGR1. The present study found that NGR1 had no effect on the expression of ER in normal H9c2 cardiomyocytes, probably because NRG1 could not activate the estrogen response elements in normal H9c2 cardiomyocytes but activated the estrogen response elements after H/R treatment.22
This study proved that one of the mechanisms of NGR1 in preventing the myocardial H/R injury is by controlling the expression of PI3K and its downstream apoptosis proteins. However, some mechanisms remain unclear: first, the mechanism how NGR1 increases the expression of proteins in the model group, although it has no effect on the expression of ERα in normal H9c2 cardiomyocytes; second, the mechanism for GPR30 to regulate the expression of Akt and the downstream apoptosis proteins through non-PI3K pathways. These would be the focus of the in-depth studies in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Natural Science Foundation of China (Grant No supported this study. 81503289, 81303257), the Central Public–interest Scientific Research Found of China, Peking Union Medical College (Grant No. 2016ZX350064), the Applied basic Research Foundation of Yunnan Province, China (Grant No. 2016FB143), Beijing Natural Science Foundation (Grant No. 7172138).References
- S. Negi and A. Anand, Cardiovasc. Hematol. Disord.: Drug Targets, 2010, 10, 257–261 CrossRef CAS.
- A. L. Moens, M. J. Claeys, J. P. Timmermans and C. J. Vrints, Int. J. Cardiol., 2005, 100, 179–190 CrossRef CAS PubMed.
- D. J. Lerner and W. B. Kannel, Am. Heart J., 1986, 111, 383–390 CrossRef CAS PubMed.
- L. S. Kang, B. Chen, R. A. Reyes, A. J. LeBlanc, B. Teng, S. J. Mustafa and J. M. Muller-Delp, Am. J. Physiol., 2011, 300, H2105–H2115 CrossRef CAS PubMed.
- P. I. Moreira, J. B. A. Cust dio, E. Nunes, P. J. Oliveira, A. N. Moreno, R. Sei a, C. R. Oliveira and M. S. Santos, J. Steroid Biochem. Mol. Biol., 2011, 123, 8–16 CrossRef CAS PubMed.
- H. Y. Li, J. S. Bian, Y. W. Kwan and T. M. Wong, J. Pharmacol. Exp. Ther., 2000, 293, 592–598 CAS.
- M. Wang, B. M. Tsai, K. M. Reiger, J. W. Brown and D. R. Meldrum, J. Mol. Cell. Cardiol., 2006, 40, 205–212 CrossRef CAS PubMed.
- S. A. Narod, Nat. Rev. Clin. Oncol., 2011, 8, 669–676 CrossRef CAS PubMed.
- G. F. T. W. Writing, JAMA, 2002, 288, 321–333 CrossRef.
- R. T. Chlebowski, S. L. Hendrix, R. D. Langer and A. Et, JAMA, 2003, 289, 3243–3253 CrossRef CAS PubMed.
- S. Yusuf and S. Anand, Can. Med. Assoc. J., 2002, 167, 357–359 Search PubMed.
- B. C. T. C. Early, Lancet, 2011, 378, 771–784 CrossRef.
- R. Saxena and A. Dwivedi, Med. Res. Rev., 2012, 32, 166–215 CrossRef CAS PubMed.
- J. E. Rossouw, G. L. Anderson, R. L. Prentice, A. Z. LaCroix, C. Kooperberg, M. L. Stefanick, R. D. Jackson, S. A. Beresford, B. V. Howard, K. C. Johnson, J. M. Kotchen and J. Ockene, JAMA, 2002, 288, 321–333 CrossRef CAS PubMed.
- M. L'Hermite, Climacteric, 2013, 16(suppl. 1), 44–53 CrossRef PubMed.
- J. E. Manson, R. T. Chlebowski, M. L. Stefanick, A. K. Aragaki, J. E. Rossouw, R. L. Prentice, G. Anderson, B. V. Howard, C. A. Thomson, A. Z. LaCroix, J. Wactawski-Wende, R. D. Jackson, M. Limacher, K. L. Margolis, S. Wassertheil-Smoller, S. A. Beresford, J. A. Cauley, C. B. Eaton, M. Gass, J. Hsia, K. C. Johnson, C. Kooperberg, L. H. Kuller, C. E. Lewis, S. Liu, L. W. Martin, J. K. Ockene, M. J. O'Sullivan, L. H. Powell, M. S. Simon, L. Van Horn, M. Z. Vitolins and R. B. Wallace, JAMA, 2013, 310, 1353–1368 CrossRef CAS PubMed.
- G. Perrone, O. Capri, P. Galoppi, F. R. Patacchioli, E. Bevilacqua, M. G. de Stefano and R. Brunelli, Gynecol. Obstet. Invest., 2013, 76, 38–43 CrossRef CAS PubMed.
- M. Canonico, E. Oger, G. Plu-Bureau, J. Conard, G. Meyer, H. Levesque, N. Trillot, M. T. Barrellier, D. Wahl, J. Emmerich and P. Y. Scarabin, Circulation, 2007, 115, 840–845 CrossRef CAS PubMed.
- T. Usui, Endocr. J., 2006, 53, 7–20 CrossRef CAS PubMed.
- T. B. Ng, J. Pharm. Pharmacol., 2006, 58, 1007–1019 CrossRef CAS PubMed.
- P. Chan, G. N. Thomas and B. Tomlinson, Acta Pharmacol. Sin., 2002, 23, 1157–1162 CAS.
- T. Wang, D. Wan, L. Shao, J. Dai and C. Jiang, Biochem. Biophys. Res. Commun., 2015, 466, 232–239 CrossRef CAS PubMed.
- Y. Yu, G. Sun, Y. Luo, M. Wang, R. Chen, J. Zhang, Q. Ai, N. Xing and X. Sun, Sci. Rep., 2016, 6, 21730 CrossRef CAS PubMed.
- X. Meng, G. Sun, M. Wang, J. Sun, M. Qin and X. Sun, J. Evidence-Based Complementary Altern. Med., 2013, 2013, 971712 Search PubMed.
- J. Sun, G. Sun, X. Meng, H. Wang, M. Wang, M. Qin, B. Ma, Y. Luo, Y. Yu, R. Chen, Q. Ai and X. Sun, J. Evidence-Based Complementary Altern. Med., 2013, 2013, 690190 Search PubMed.
- J. Santollo and L. A. Eckel, Physiol. Behav., 2009, 97, 193–198 CrossRef CAS PubMed.
- J. Santollo, B. S. Katzenellenbogen, J. A. Katzenellenbogen and L. A. Eckel, Horm. Behav., 2010, 58, 872–877 CrossRef CAS PubMed.
- D. R. Compton, S. Sheng, K. E. Carlson, N. A. Rebacz, I. Y. Lee, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Med. Chem., 2004, 47, 5872–5893 CrossRef CAS PubMed.
- T. Simoncini, A. Hafezi-Moghadam, D. P. Brazil, K. Ley, W. W. Chin and J. K. Liao, NATURE, 2000, 407, 538–541 CrossRef CAS PubMed.
- Y. Wang, L. Tu, Y. Li, D. Chen and S. Wang, J. Pharmacol. Exp. Ther., 2016, 357, 591 CrossRef CAS PubMed.
- P. Su, S. Du, H. Li, Z. Li, W. Xin and W. Zhang, Eur. J. Pharmacol., 2016, 770, 9–15 CrossRef CAS PubMed.
- Z. R. Ge, M. C. Xu, Y. Huang, C. J. Zhang, J. Lin and C. W. Ruan, Exp. Ther. Med., 2016, 11, 2341–2348 CrossRef CAS PubMed.
- K. Xia, H. Cao and C. Shao, J. Pharm. Sci., 2015, 70, 740–744 CAS.
- L. Zhong, X. L. Zhou, Y. S. Liu, Y. M. Wang, F. Ma, B. L. Guo, Z. Q. Yan and Q. Y. Zhang, Mol. Med. Rep., 2015, 12, 119–126 CrossRef CAS PubMed.
- S. Chen, J. Liu, X. Liu, Y. Fu, M. Zhang, Q. Lin, J. Zhu, L. Mai, Z. Shan, X. Yu, M. Yang and S. Lin, J. Ethnopharmacol., 2011, 137, 263–270 CrossRef CAS PubMed.
- H. J. Ranki, G. R. Budas, R. M. Crawford, A. M. Davies and A. Jovanovi, J. Am. Coll. Cardiol., 2002, 40, 367–374 CrossRef CAS PubMed.
- Y. Urata, Y. Ihara, H. Murata, S. Goto, T. Koji, J. Yodoi, S. Inoue and T. Kondo, J. Biol. Chem., 2006, 281, 13092–13102 CrossRef CAS PubMed.
- T. Ballantyne, Q. Du, S. Jovanovi, A. Neemo, R. Holmes, S. Sinha and A. Jovanovi, Int. J. Biochem. Cell Biol., 2012, 45, 283–291 CrossRef PubMed.
- M. Wang, X. Meng, Y. Yu, G. Sun, X. Xu, X. Zhang, X. Dong, J. Ye, H. Xu and Y. Sun, Apoptosis, 2014, 19, 1727–1735 CrossRef CAS PubMed.
- J. P. Stice, L. Chen, S. Kim, J. S. Jung, A. L. Tran, T. T. Liu and A. A. Knowlton, Endocrinology, 2011, 152, 1589–1598 CrossRef CAS PubMed.
- A. M. Deschamps and E. Murphy, Am. J. Physiol., 2009, 297, H1806–H1813 CrossRef CAS PubMed.
- B. Sun, J. Xiao, X. B. Sun and Y. Wu, Br. J. Pharmacol., 2013, 168, 1758–1770 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |