
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the synthesis of fluorinated hydrazones
Rui Guo
 *ab and
Junting Chen
c
*ab and
Junting Chen
c
aInstitute of Environment and Health, Jianghan University, Wuhan 430056, P. R. China. E-mail: 13925825040@163.com
bState Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China
cMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany
First published on 10th May 2018
Abstract
Fluorinated compounds are widely used in pharmaceuticals, agrochemicals, and materials. As an important branch of fluorinated compounds, the introduction of fluorinated hydrazones remains unexplored due to the lack of convenient methods. However, as a powerful platform for generation of various fluorinated pyrazole species and diverse nitrogen-containing compounds, fluorinated hydrazones have been extensively studied in recent years. In this review, we highlight most of the important developments in fluorinated hydrazone synthesis.
 Rui Guo | Rui Guo was born in Hubei Province, P. R. of China in 1984. He received his BSc degree from Jiangxi Normal University in 2007 and MSc degree from Central China Normal University in 2010, under the supervision of Professor Shenghua Liu. Then as a project manager, he worked at HEC Pharm Co., Ltd. In 2013 he joined Professor Pingping Tang's research group at Nankai University to pursue his PhD degree. With interest in C–F bond formation reactions based on C–H activation, he obtained his PhD degree in 2016. After that, as an assistant researcher, he joined the Institute of Environment and Health at Jianghan University. In 2017, he also worked at the State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, as a post-doctoral researcher to continue studying fluorination reactions. |
 Junting Chen | Junting Chen was born in 1994, in Hunan, China. She started her bachelor research in the Tang Group in 2014 under the supervisor of Professor Tang at Nankai University. In 2015, she worked as a student intern in the Ritter Group, Harvard University for one year. After she graduated from Nankai University with a bachelor's degree, she joined Professor Ritter's research group at Max-Planck-Institut für Kohlenforschung to pursue her PhD degree. Her interests include the development of fluorination methods and transition metal catalysts. |
Introduction
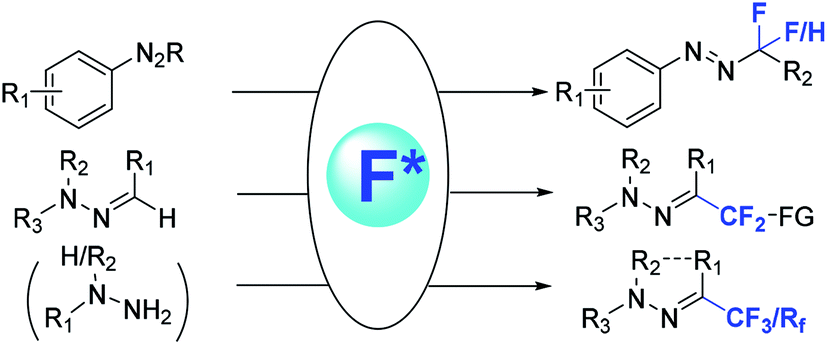
Organofluorine compounds possess unique physical and chemical properties because of fluorine's small size and high electronegativity, thus efficient construction of C–F bonds has been the subject of intensive research in the fields of synthetic and medicinal chemistry.1–11 Fluorinated hydrazone groups have received a lot of attention in pharmaceuticals and agrochemicals, also it is found in various biological active compounds (Fig. 1).12,13 The development of general methods for direct synthesis of fluorinated arylhydrazones is quite interesting and highly desirable. Generally, methods for direct synthesis of fluorinated arylhydrazones are comparatively scarce.14–16 But recent developments in organic and transition-metal catalysis have allowed new methods to prepare fluorinated hydrazones directly and efficiently. To the best of our knowledge, there is no review devoted to this emerging topic so far. A review highlighting the main advances in this area should be timely and desirable, which will attract more research interest to this field. Therefore, we highlight the recent advances on different kinds of fluorinated hydrazone reactions here, within which we will discuss different methods for the synthesis of fluorinated hydrazone species and their mechanisms reported during the previous decades (Fig. 2). | ||
| Fig. 1 Representative drug (Celecoxib) and active agrochemical ingredient (Bixafen) which contain fluorinated hydrazone functional groups. | ||
 | ||
| Fig. 2 Different methods for synthesis of fluorinated hydrazones. | ||
Water compatible monofluoro-hydrazones from aryl diazonium salts
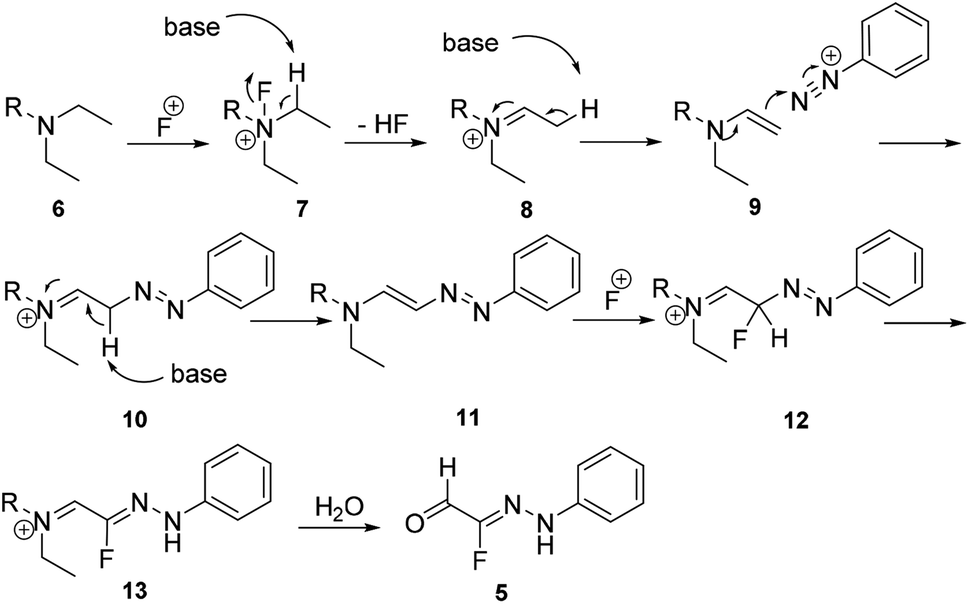
Traditionally, fluorinated arylhydrazones were prepared via the condensation of arylhydrazines17,18 or aryldiazonium salts (Japp–Klingemann reaction) with carbonyl compounds.19–22 For example, Reichardt has reported the coupling of fluorine-containing carbonyl compounds with aryldiazonium chlorides to generate fluorinated arylhydrazones (Scheme 1).23 As shown in Scheme 2, the dicarbonyl sodium salt 1 reacted with aryl diazonium salt 2 to form intermediate 3. With successive hydrolysis of intermediate 3, intermediate 4 might be prepared. Subsequent elimination of a formic acid from the intermediate 4 would provide the monofluorinated arylhydrazone product 5. Although the fluorination hydrazone product could be obtained in this manner with good yield, the drawback of this method is that the fluorinated dicarbonyl compound has to be prepared through several steps. Nevertheless, this study provides the first example of the preparation the monofluorinated arylhydrazones using the aryl diazonium salt in water.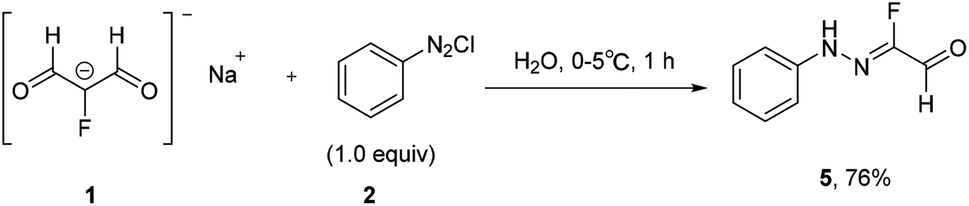 | ||
| Scheme 1 In water formation of fluorinated arylhydrazones via aryl diazonium salt. | ||
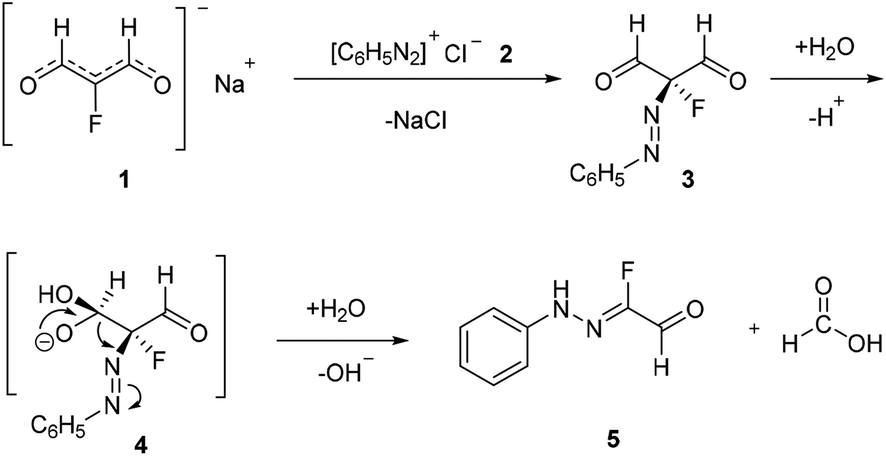 | ||
| Scheme 2 Proposed mechanism for the reaction in Scheme 1. | ||
Recently, we reported the first example of a mild and metal-free cascade reaction of aryl diazonium salts and trialkylamine in the presence of Selectfluor to prepare monofluorinated arylhydrazones.24 Some representative examples are shown in Scheme 3. The mild reaction conditions generally tolerate diverse functional groups, such as chloride, bromide, iodide, nitrile, nitro, and ester, on the aryl rings. Notably, the reaction was amenable to gram-scale synthesis, proving the practicality of our method. A plausible mechanism was proposed: trialkylamine was oxidized to the fluorinated quaternary ammonium salt 7 in the presence of Selectfluor, followed by elimination to generate the iminium ion 8, and the enamine 9 was formed after elimination. Subsequent treatment of the enamine 9 with aryldiazonium salts would generate the azo intermediate 10, which could produce 11 in the presence of base. The intermediate 11 could then react with Selectfluor to form the fluorinated azo intermediate 12. After then, 13 would react with H2O to generate the monofluorinated arylhydrazones product (Scheme 4).
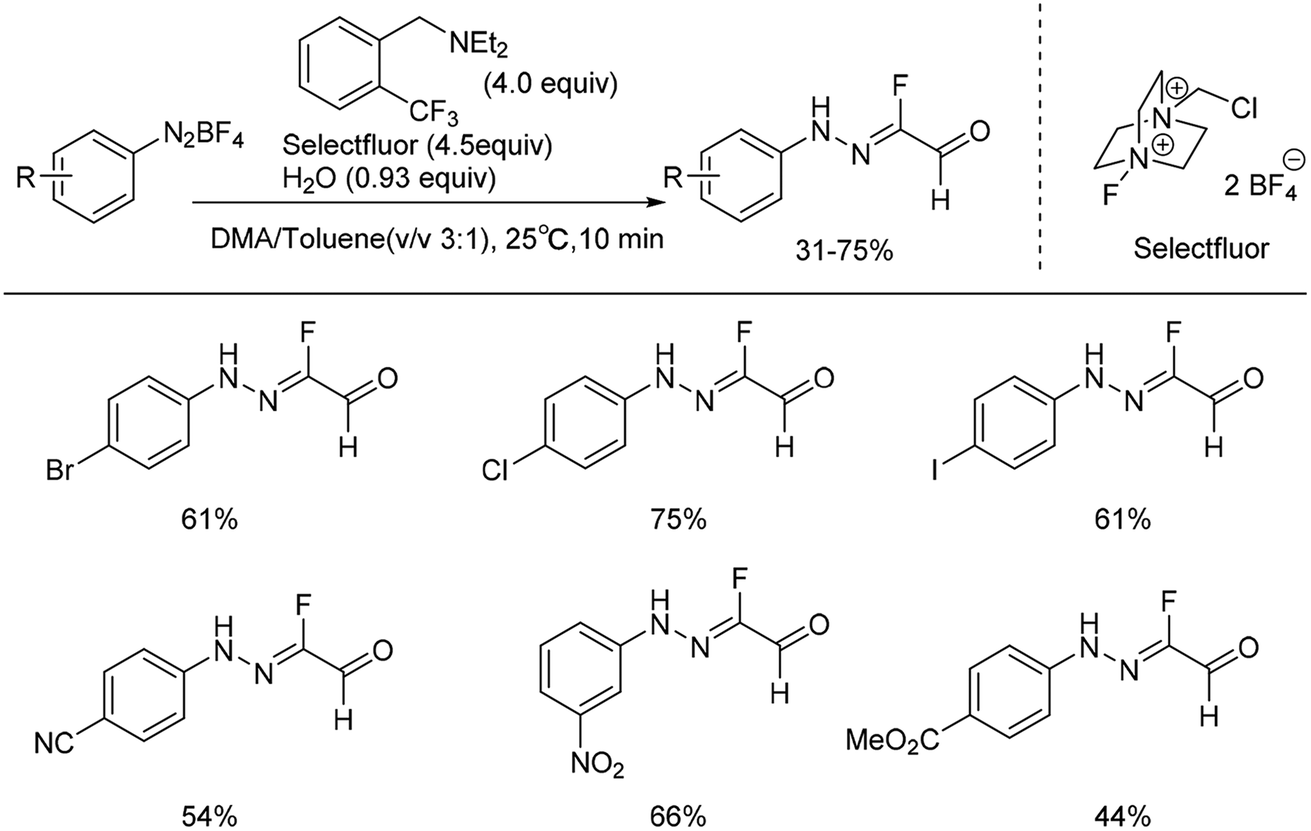 | ||
| Scheme 3 Formation of fluorinated arylhydrazones with Selectfluor and water. | ||
 | ||
| Scheme 4 Proposed mechanism for the reaction in Scheme 3. | ||
In addition, those fluorinated arylhydrazones were utilized to synthesise the fluorinated pyrazoles and other nitrogen-containing compound.
Various other amine compounds like triethylamine, diisopropylethylamine (DIPEA), diethylmethylamine and various N,N-diethylbenzenemethanamines, were also effective in generating the desired products. No desired products were obtained with tributylamine, dimethylethylamine, diethylamine, or N,N-diethylaniline. In addition, different fluorination reagents such as N-fluoropyridinium tetrafluoroborate, NFSI and Selectfluor were examined, and Selectfluor gave the highest yield. The reaction was proved to be operationally simple, and scalable under mild conditions.
Transition-metal catalysed difluoromethylation of hydrazones
Recently, palladium-catalysed coupling of hydrazones with fluorinated aryl halides has been reported for the preparation of fluorinated arylhydrazones.25–27 Monteiro and co-workers reported a palladium-catalysed C–H difluoromethylation of aldehyde-derived hydrazones (Scheme 5).28 This method proceeded with low catalyst loading, high regioselectivity, and excellent functional group compatibility. However, the presence of a vinyl substituent on the aryl moiety led to decomposition of the substrate. Importantly, several heterocyclic aldehyde-derived hydrazones (i.e. pyridinyl, quinolinyl, and pyrazolyl) were proved to be suitable substrates for the transformation with the heterocyclic ring being intact. Ethyl glyoxylate hydrazone participated substrates were also conducted with good yields.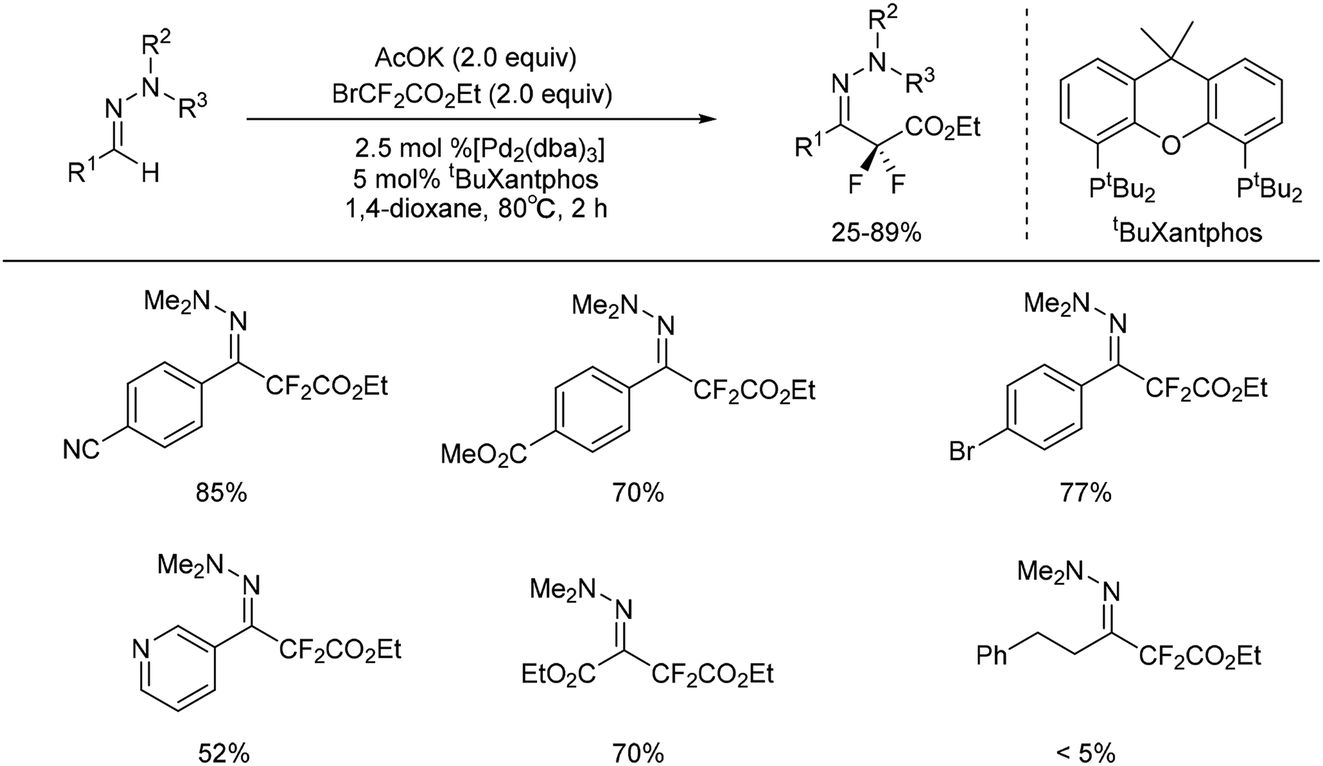 | ||
| Scheme 5 Formation of difluoromethylated hydrazones catalysed by palladium complex. | ||
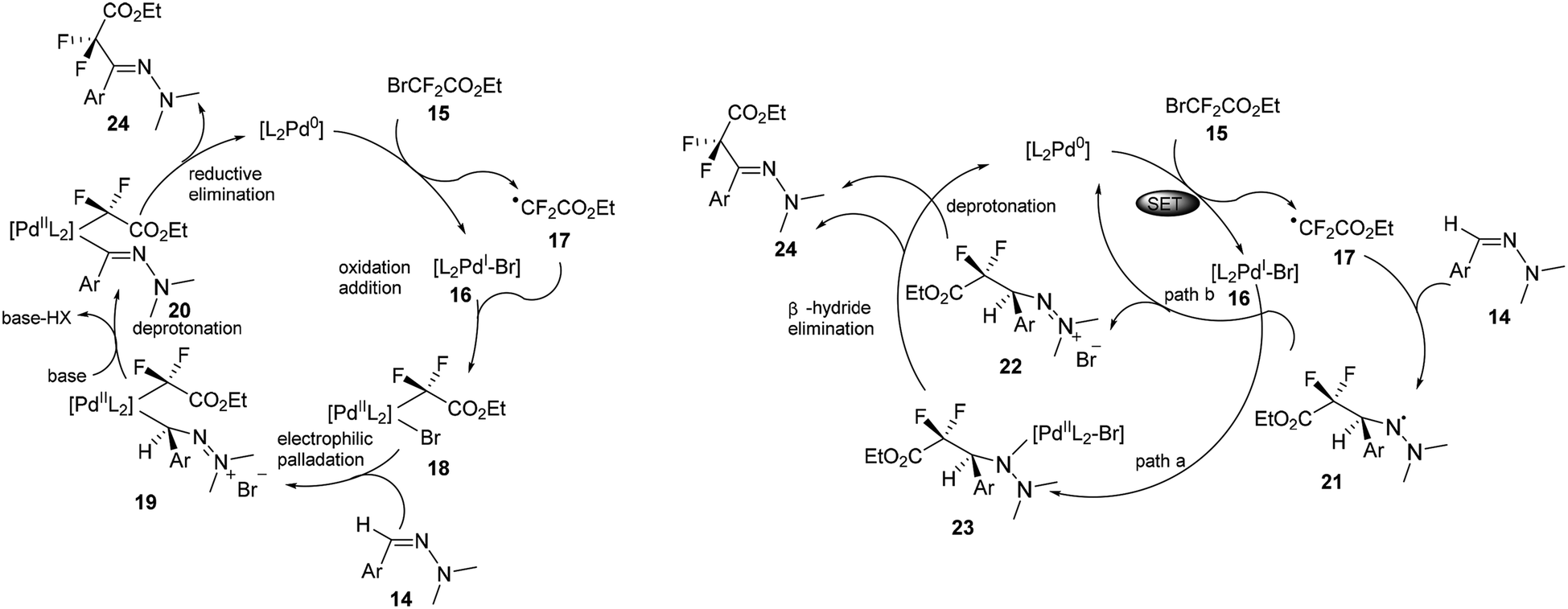
As shown in left of Scheme 6, a radical/SET pathway was proposed to initiate the coupling reaction. A palladium0/palladiumII catalytic cycle was suggested to be involved in this transformation. The suggested catalytic cycle would start with single-electron transfer from the Pd0 metal complex to the fluoroalkyl bromide 15 to form the PdIBr complex 16 and the difluoroalkyl radical intermediate 17. Subsequent recombination of these two species would give the PdII intermediate 18. Followed by electrophilic palladation of the hydrazone and deprotonation of the resulting cationic intermediate 19, the azomethinyl PdII complex 20 would be generated. Finally, reductive elimination would form the final product and recycle the Pd0 catalyst.
 | ||
| Scheme 6 Proposed mechanisms for palladium catalysed difluoromethyl hydrazones in Scheme 5. | ||
The authors also proposed an alternative electrophilic radical addition pathway, involving a key intermediate-aminyl radical 21, which could be formed from the addition of the difluoroalkyl radical 17 to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of the hydrazone. The following H-elimination of 21 was proposed to proceed via two possible pathways. One could be radical trapping of the radical 21 by PdI intermediate 16 to form the PdII intermediate 23, followed by β-hydride elimination to yield the final product. Another alternative pathway could be oxidation of the radical 21 by PdI intermediate 16 to regenerate the active Pd0 species and form the cationic intermediate 23, which could be subsequently deprotonated to restore the CN bond and yield the final hydrazone product.
N bond of the hydrazone. The following H-elimination of 21 was proposed to proceed via two possible pathways. One could be radical trapping of the radical 21 by PdI intermediate 16 to form the PdII intermediate 23, followed by β-hydride elimination to yield the final product. Another alternative pathway could be oxidation of the radical 21 by PdI intermediate 16 to regenerate the active Pd0 species and form the cationic intermediate 23, which could be subsequently deprotonated to restore the CN bond and yield the final hydrazone product.
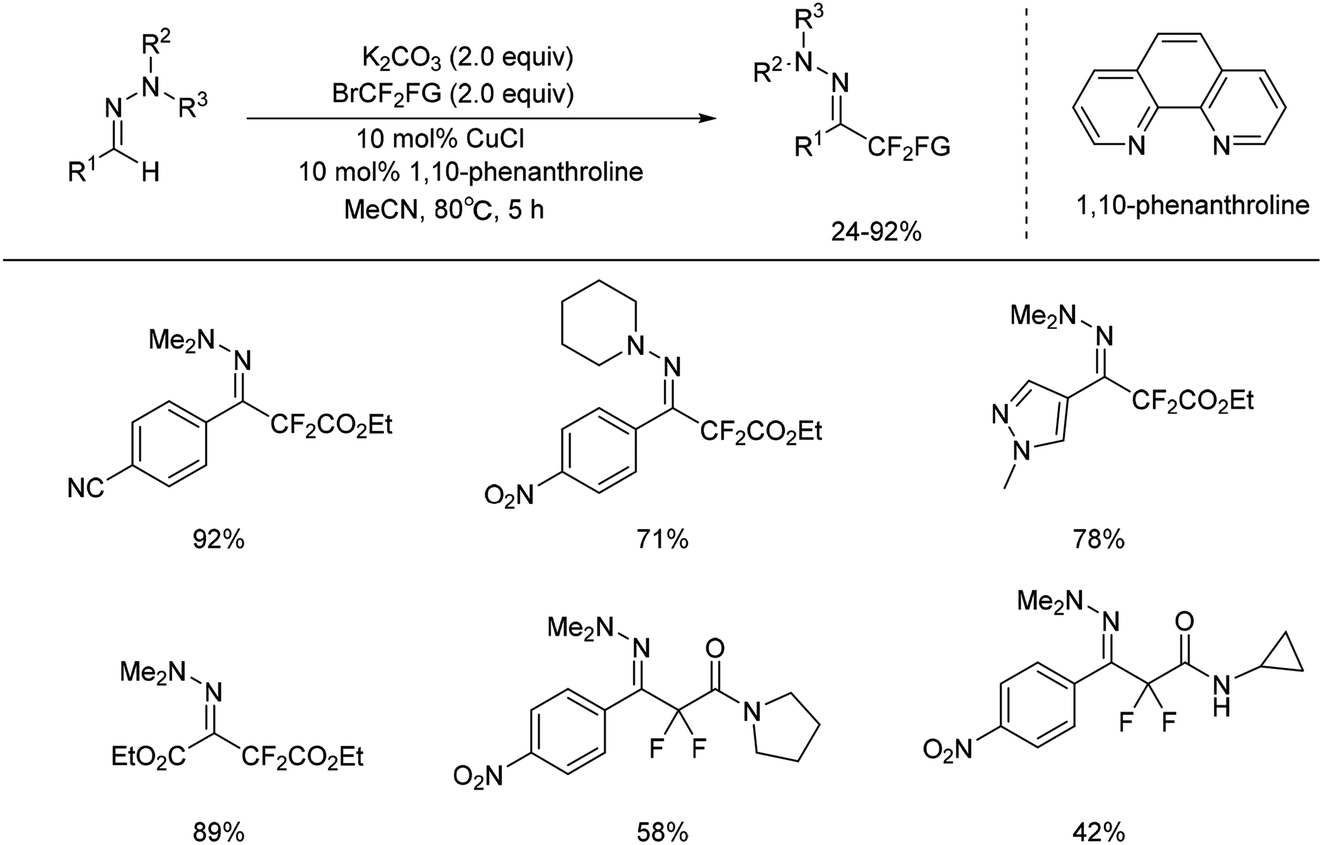
But aliphatic aldehyde-derived hydrazones were proved to be more challenging substrates for this reaction, and gave only small amounts of the desired product. Recently, the same authors found cuprous chloride could also catalyse the difluoromethylation of hydrazones from bromodifluoroacetate (Scheme 7).29 The mechanism studies indicated that a radical-chain or single-electron transfer (SET) mechanism could be involved in the transformation (Scheme 8). This mechanism could involve the formation of the electrophilic fluoroalkyl radical 27 from 25 via bromide abstraction by the CuI complex with concomitant generation of CuII, which would then be trapped by the hydrazone 26 to generate the difluoroalkylated aminyl radical intermediate 28. Oxidation of the intermediate 28 with CuII would lead to the cationic species 29, which would then undergo proton abstraction, thereby restoring the hydrazone functional group and recycling CuI. Such a CuI/CuII redox mechanism contrasts with radical-free CuI/CuIII catalytic cycles previously proposed for the difluoroalkylation of electron-rich alkenes and arenes proceeding via difluoroalkylcopper intermediates.30 As an efficient, scalable, and broad substrate scope protocol, the present method enables the preparation of functionalized difluoromethyl ketone hydrazones from aldehyde hydrazones and halodifluorinated reagents, it also holds promise for application to the β-difluoromethylation of α,β-unsaturated hydrazones.
 | ||
| Scheme 7 Copper catalysed difluoromethylation of hydrazones. | ||
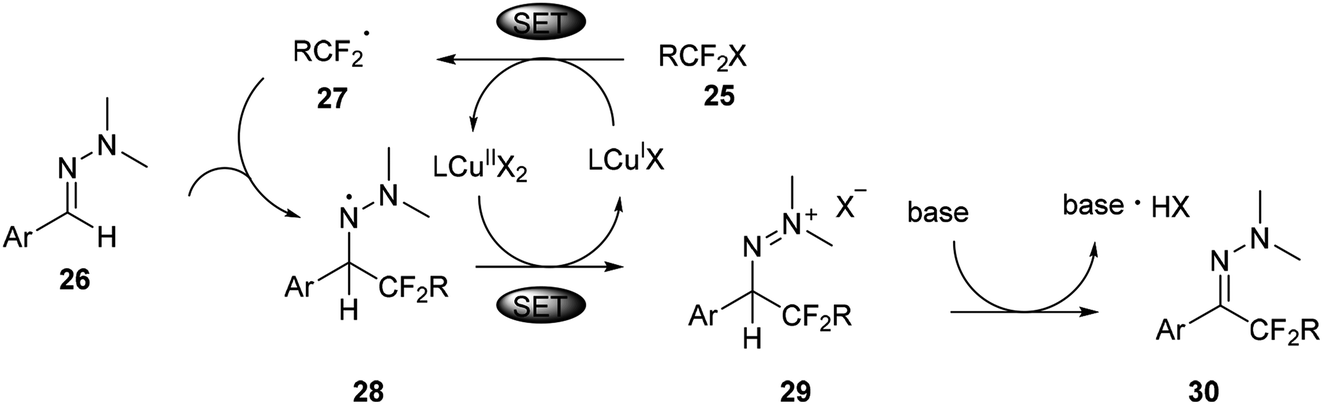 | ||
| Scheme 8 SET mechanism for copper catalysed difluoromethyl hydrazones in Scheme 7. | ||
Recently, the important gem-difluoromethylene azo compounds were also synthesised by the Hao's group. The protocol achieved gem-difluoromethylenation through radical addition of the in situ generated benzo-1,3-diazolic difluoromethylene to arenediazonium salts, with the mild silver salt as a catalyst. The method could provide a new synthetic strategy for the synthesis of gem-difluoromethylene azo compounds, but the use of benzo-1,3-diazolic difluoromethane sulfinates as starting materials would restrict the field of application.31
Visible-light photoredox-catalysed C–H activation to synthesise difluorohydrazone
In 2016, a visible-light photoredox-catalysed C–H activation to prepare difluorinated hydrazones was reported by Zhu and co-workers (Scheme 9).32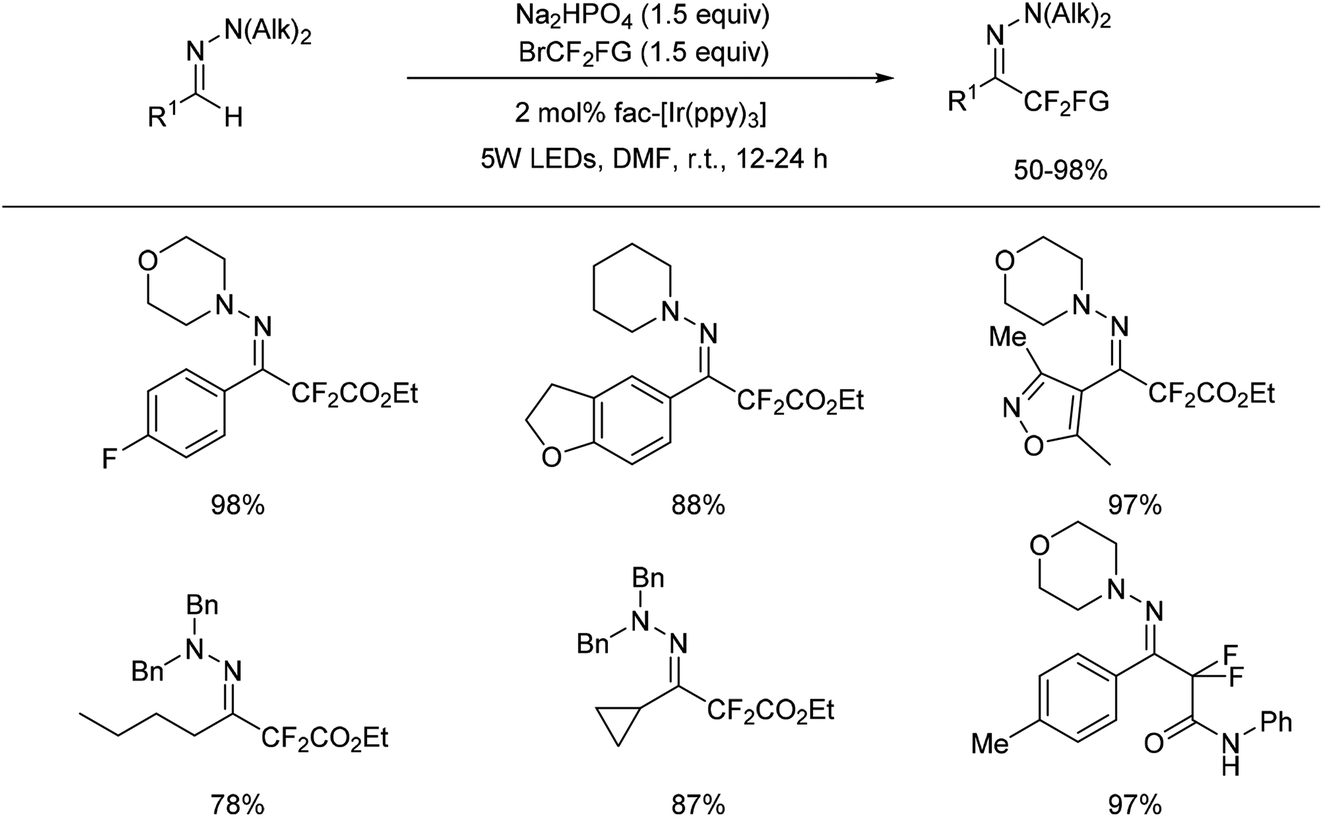 | ||
| Scheme 9 Visible light photoredox catalysed difluoromethylation hydrazones. | ||
The method was further highlighted by its feasibility of one-pot synthesis of a difluorinated hydrazone. Although the precise reaction mechanism was pending, a plausible aminyl radical/polar crossover mechanism was proposed. No product was detected when the radical inhibitor 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) was added to the reaction system, thus further implying a radical reaction pathway. Under visible-light irradiation, the photocatalyst fac-[Ir3+(ppy)3] could undergo a metal-ligand charge-transfer (MLCT) process to produce the strong reducing excited state Ir3+* (Scheme 10). A SET process from this species to 15 could generate the difluoroalkyl radical precursor 17 and Ir4+. Subsequent aminyl radical intermediate 32 could be generated through radical addition of 17 to the CN bond. The aminyl radical intermediate 32 might be stabilised by the adjacent nitrogen atom through a possible three-electron π-bonding interaction. Instead of hydrogen atom abstraction to generate the corresponding hydrazine 33, a key aminyl radical/polar crossover step could proceed between 32 and Ir4+, thus regenerating the photocatalyst Ir3+ and either the aminyl cation 34 or 35 (path a). Further tautomerization and deprotonation of aminyl cation would give the product 38. Alternatively, a light on/off experiment verified the necessity of continuous irradiation of visible light and suggested that the chain propagation was not the predominant mechanistic pathway. As the oxidation potential of radical intermediate could not be measured, DFT calculations were performed to exclude path b. And the results could account for the aminyl radical/polar mechanism (path a).
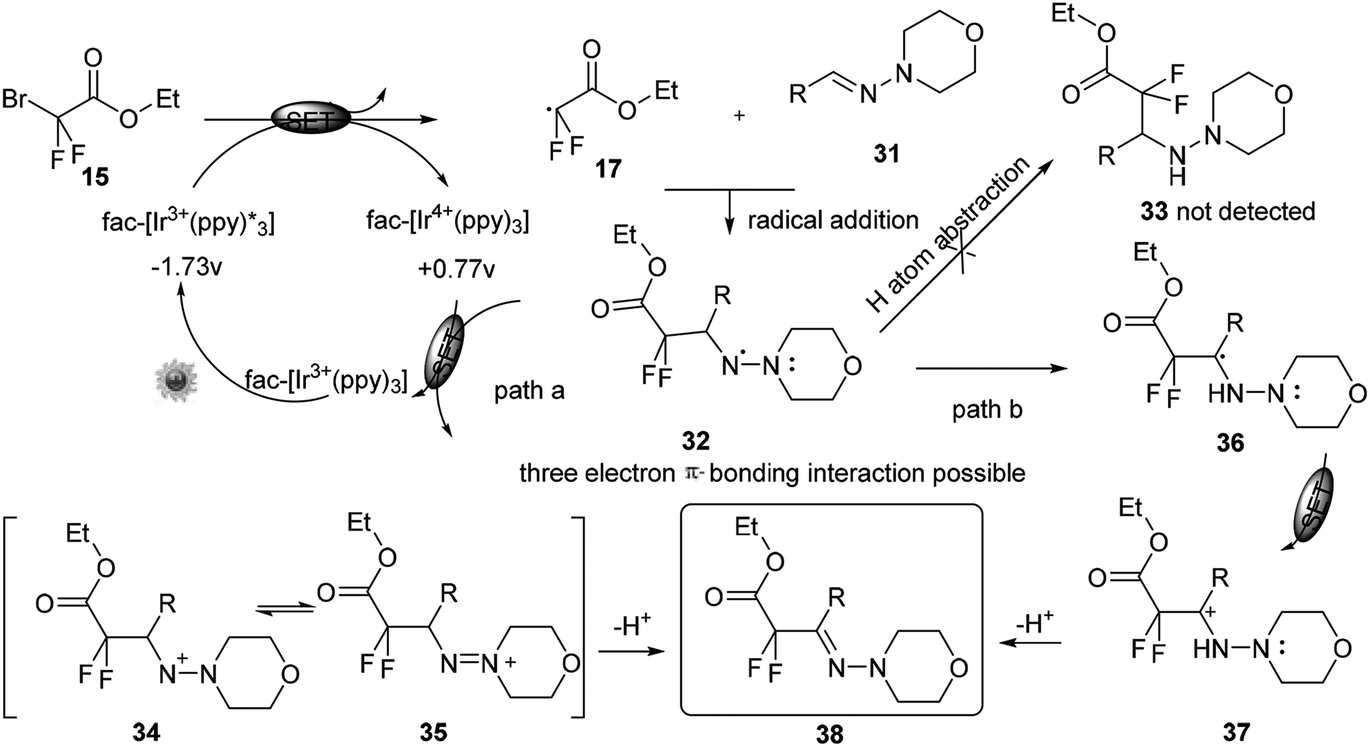 | ||
| Scheme 10 Mechanism for iridium catalysed C–H activation in the presence of visible light in Scheme 9. | ||
The present transformation is attractive as it enables direct C–H difluoroalkylation of aldehyde-derived hydrazones via visible light photoredox-catalysis. The application of this method would be, however, restricted by its limited substrate scope, for example, secondary amino groups, such as N-Boc and N-Bs hydrazones were not tolerated in the reaction conditions.
Shortly after, Hashmi and co-workers reported the first gold-catalysed intermolecular photoredox C(sp2)–H difluoroalkylation and perfluoroalkylation of aromatic aldehyde hydrazones with commercially available fluoroalkyl bromides (Scheme 11).33
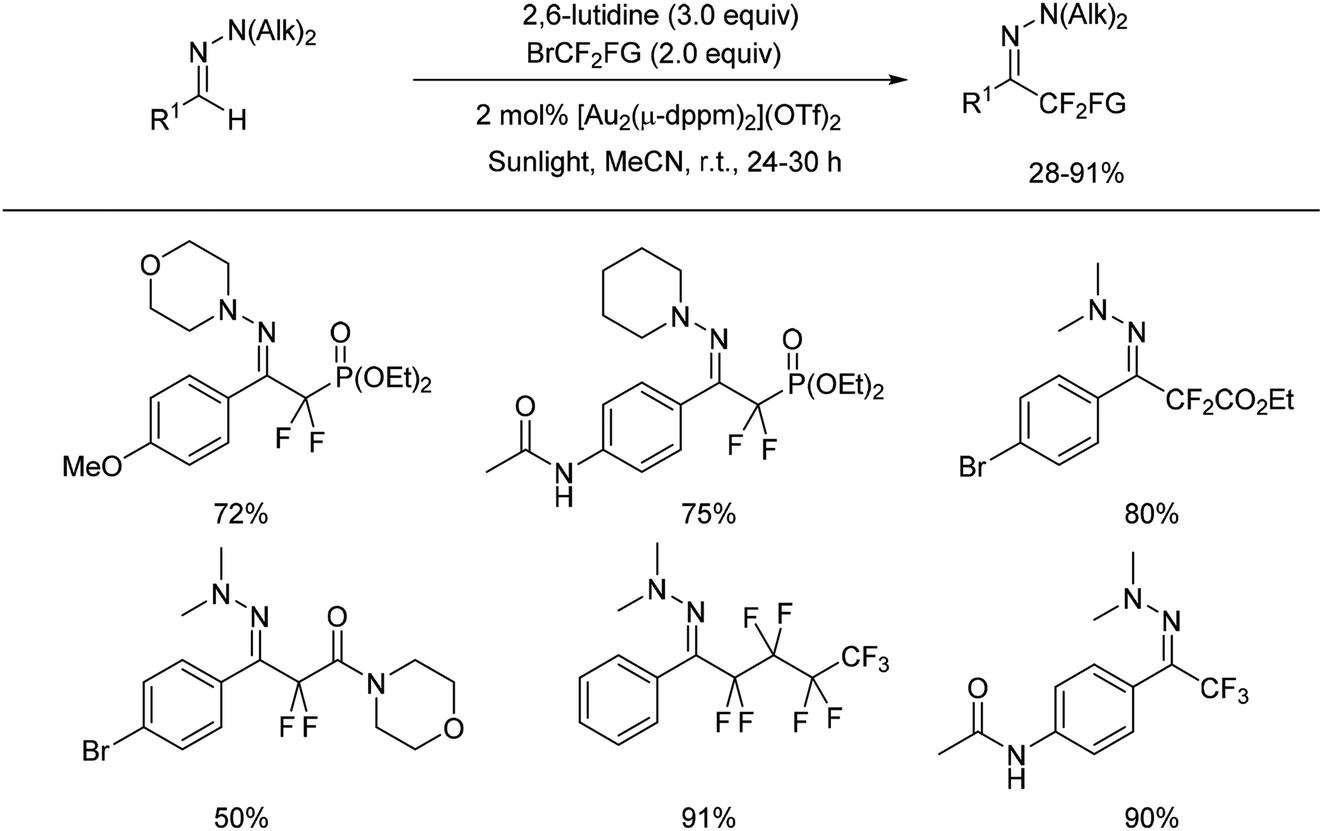 | ||
| Scheme 11 Gold catalysed photoredox reaction for difluoromethylated hydrazones formation. | ||
A possible mechanism is shown in Scheme 12. Firstly, a high-energy, long-lived photoexcited species, *[Au2(μ-dppm)2]2+ was proposed to be generated by irradiation of [Au2(μ-dppm)2](OTf)2. Next, a single electron transfer from the gold species to 39 could form difluoromethyl phosphonate radical 40 and gold intermediate. The electrophilic radical 40 could then attack hydrazone 31 to generate the three-electron π-bonding aminyl radical intermediate 41. Finally, oxidation of aminyl radical 41 by gold species followed by deprotonation could deliver the desired difluoromethylated product 44. An EPR spin-trapping experiment suggested the involvement of a difluoroalkyl radical intermediate. DFT calculations indicated that the products with the CN bond in E configuration were lower in energy than those with Z configuration, and the formation of the thermodynamic E-configured product was favoured.
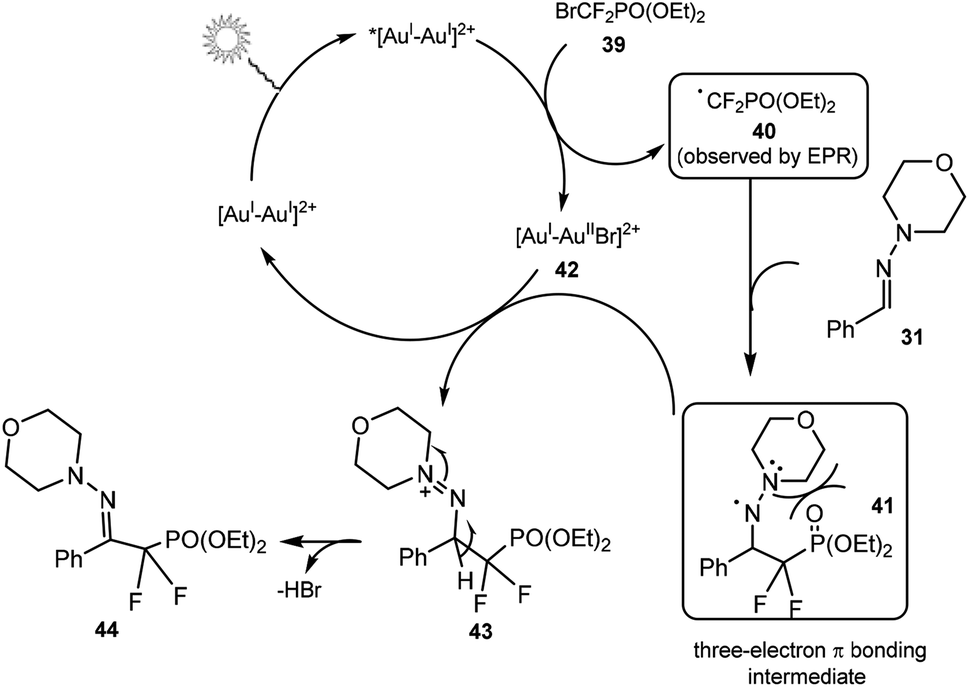 | ||
| Scheme 12 Proposed mechanism for the reaction in Scheme 11. | ||
This method is attractive for its mild reaction conditions, a broad substrate scope, and excellent functional group compatibility.
Synthesis of gem-difluorinated azo compounds
In 2015, Shen and co-workers developed a thermally stable NHC-ligated difluoromethylated silver complex [(SIPr)AgCF2H]. By reaction of the complex with a variety of activated electrophiles containing aryldiazonium salts, the corresponding difluoromethylated azo compounds were obtained in excellent yields.34Recently, we also reported the first example of a mild and tuneable cascade reaction of aryl diazonium salts and trialkylamine in the presence of Selectfluor to prepare gem-difluorinated azo compounds without metals (Scheme 13).24 A plausible mechanism was proposed as shown in Scheme 14. The early process was proposed to be the same as the formation of generating the monofluorinated intermediate 13 (Scheme 4). The formed intermediate 13 could react with Selectfluor to generate the gem-difluorinated azo intermediate 45, which could react with enamine 9 to form intermediate 46. After oxidation and elimination of 46, the gem-difluorinated iminium ion 48 would be formed. After chain extension, the formed 50 could undergo hydrolysis to produce the gem-difluorinated azo compound 51.
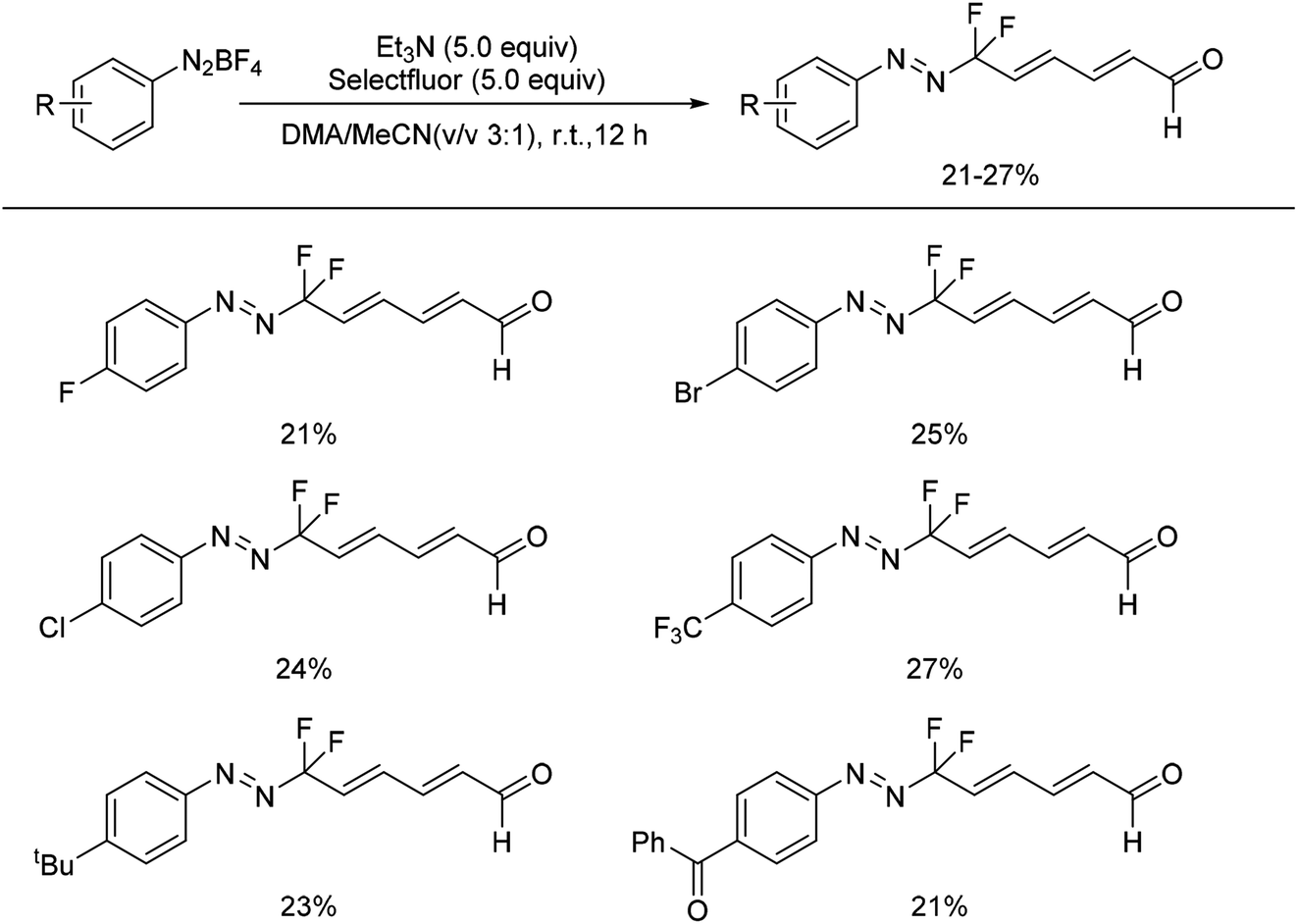 | ||
| Scheme 13 Examples of gem-difluorinated azo compounds from aryl diazonium salt. | ||
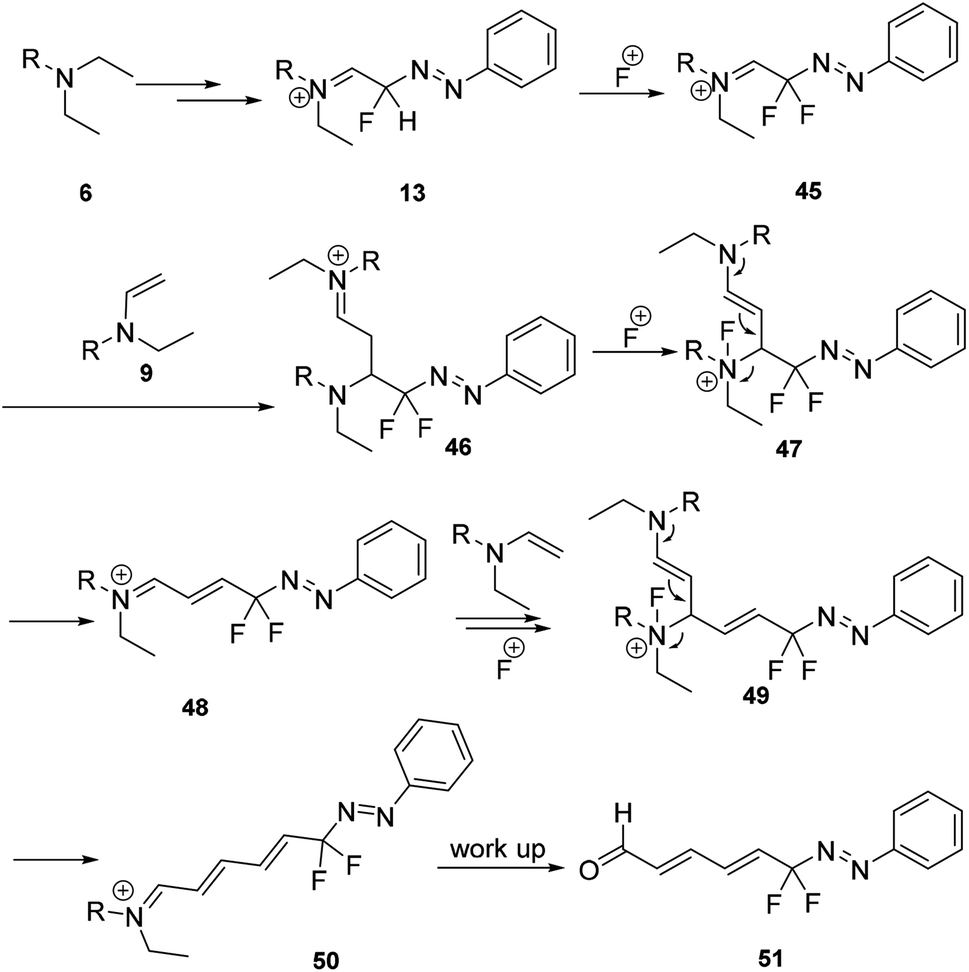 | ||
| Scheme 14 Mechanism of gem-difluorinated azo compound formation in Scheme 13. | ||
Except the desired products, the protonation byproduct of aryl diazonium salts and other unknown fluorinated byproducts were generated. Although low yields of gem-difluorinated azo compounds were found, the transformation itself attracts further investigation.
Synthesis of trifluoromethylated hydrazones
In 1992, Hamper and co-workers reported a convenient synthetic method to prepare 3-hydroxypyrazoles containing either perhaloalkyl or sterically bulky substituents in the 5-position of the pyrazole ring from β-substituted acetylenic esters. With this method, the trifluoromethylated hydrazone was synthesised (Scheme 15) from the corresponding trifluoromethylated alkynyl ester,35 and was directly collected from the water–methanol reaction mixture by filtration.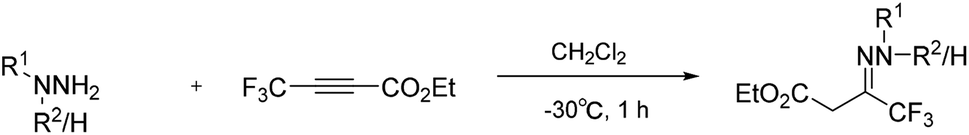 | ||
| Scheme 15 Trifluoromethylated hydrazones from substituted alkynyl ester. | ||
In 2003 and 2005, Boger and co-workers developed a method for trifluoromethylation of N,N-dimethylhydrazone. Trifluoromethyl ketone and N,N-dimethylhydrazine were used as precursors to obtain the substituted nitrogen-containing compounds (Scheme 16a).36–39 In 2009, Shi and Wei's group reported a method for preparing the corresponding bis(alkoxycarboxy)hydrazone from 1-(4-chlorophenyl)-2,2,2-trifluoroethanone. The reaction was suggested to proceed through an interesting Huisgen zwitterion intermediate, formed from the reaction of diisopropyl azodicarboxylate (DIAD) and P(OEt)3 (Scheme 16b).40 Although the yield of this methodology was moderate, the tactics of utilizing trifluorinated ketones as building blocks could provide another alternative for the synthesis of trifluoromethyl hydrazones.
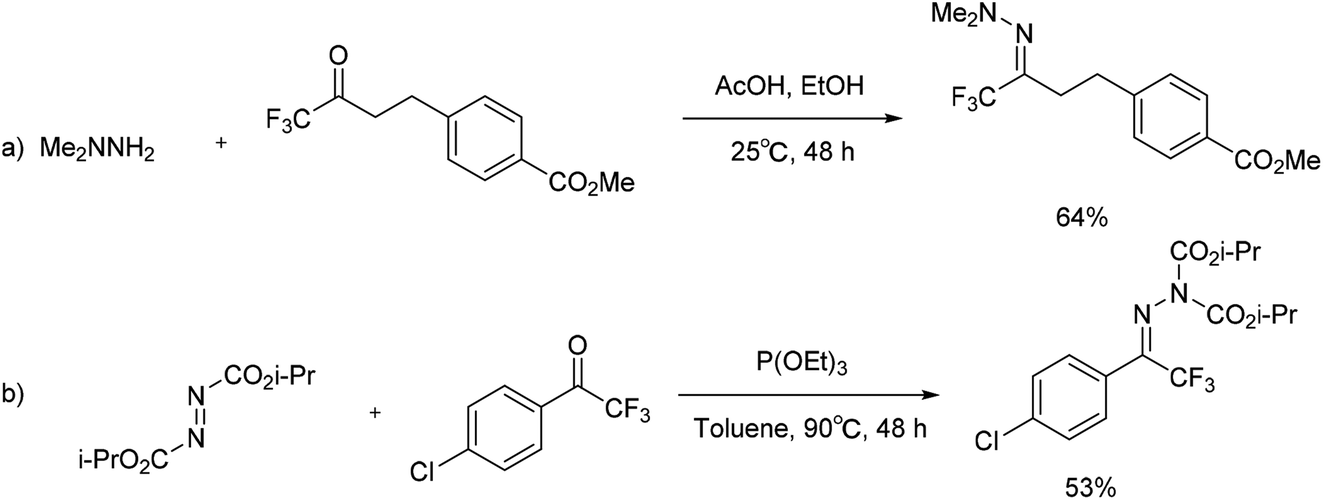 | ||
| Scheme 16 Formation of trifluoromethylation of hydrazones via substituted ketone. | ||
Other methods were also developed, for example, in 1989, Mison and co-workers reported the preparation of trifluoromethyl substituted secondary aziridines, by reacting Grignard reagents with oximes or a N,N,N-trimethylhydrazonium salt bearing a trifluoromethyl substituent.41 Kinds of trifluoromethyl hydrazones or trifluoromethyl pyrazoles as the condensation intermediators were synthesised between 1982 and 1991.42–47
In 2005, trifluoromethylpyrazoles were prepared successfully by Lahm's group by reacting a suitable 1,3-diketone with substituted hydrazine using AcOH as solvent.48 A similar work was developed by Fustero and coworkers in 2008 (Scheme 17).49 The use of fluorinated alcohols as solvents dramatically increased the regioselectivity of the pyrazole formation, and its modification could be applied to straightforward synthesis of fluorinated pyrazole.
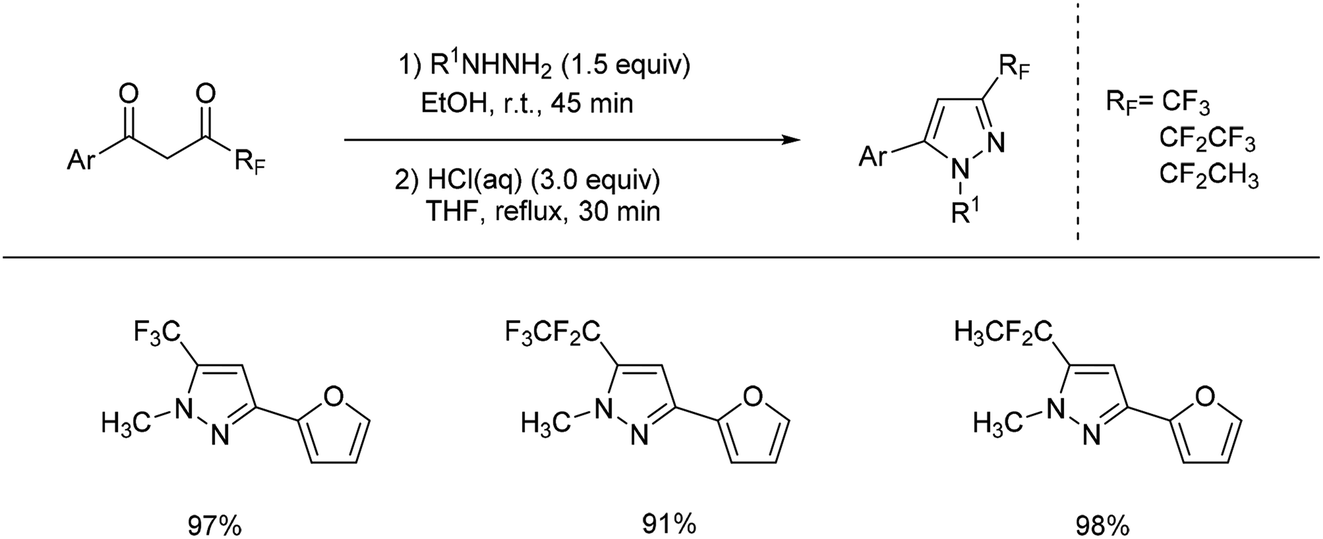 | ||
| Scheme 17 Formation of trifluoromethylation of hydrazones via 1,3-diketone compounds. | ||
Possible mechanism for the formation of fluorinated pyrazoles is showed in Scheme 18, which explains the excellent levels of regioselectivity when fluorinated alcohols were used as solvents. The process proceeded via as a step-by-step pathway. The highly electrophilic character of the carbonyl group adjacent to fluorinated functional groups was suggested to be enhanced by the high hydrogen bond donating ability of the fluorinated alcohol solvents 53, which could make the rate of the hydrazine NH2 group attack to be faster than that of the other carbonyl group, COAr.
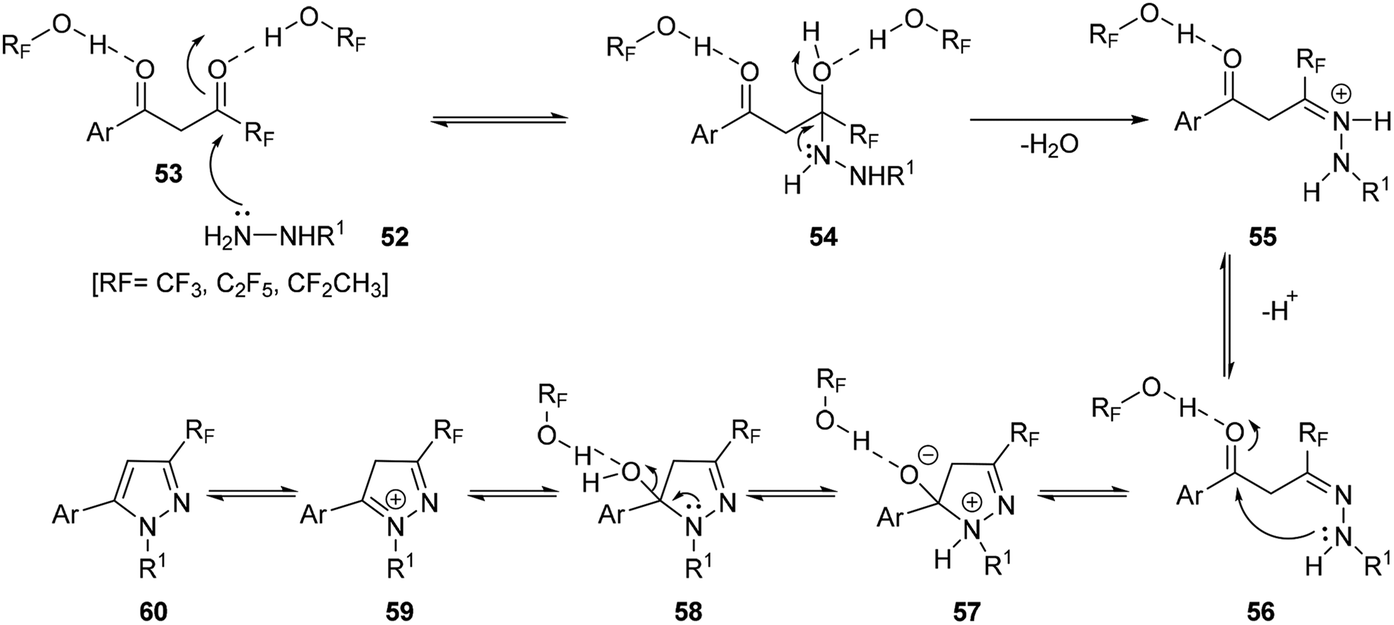 | ||
| Scheme 18 Mechanism for solvent hydrogen bonding enhanced region-selective N-methylpyrazoles formation. | ||
Subsequent loss of a water molecule from the intermediate 54 would generate the hydrazone intermediate 55, a process that could be facilitated by both the strong ionizing power of the fluorinated alcohols solvents and their ability to solvate water. Finally, the intramolecular attack of the substituted nitrogen of the hydrazone 56 to the activated COAr carbonyl, and the subsequent loss of water would produce the major isomer of the pyrazole derivative 60 (Scheme 18).
In 1995, Viehe and co-workers reported a methodology by which 3-trifluoroacetyllactams could cyclize with hydrazine or with its methyl or phenyl derivative to generate a trifluoromethylpyrazole.50
In 2015 and 2016, Leroux and co-workers also developed a general approach in the synthesis of fluoroalkyl pyrazoles by means of fluorinated iminium salts. Tuneable regioselectivity and broad substitution patterns was achieved by these methods.51,52
Between 2013 and 2016, Bouyssi and co-workers reported a mild procedure for trifluoromethylation of (hetero)aromatic aldehyde or aldehyde N,N-dialkylhydrazones using the Togni hypervalent iodine reagent with copper catalysis (Scheme 19).53–56
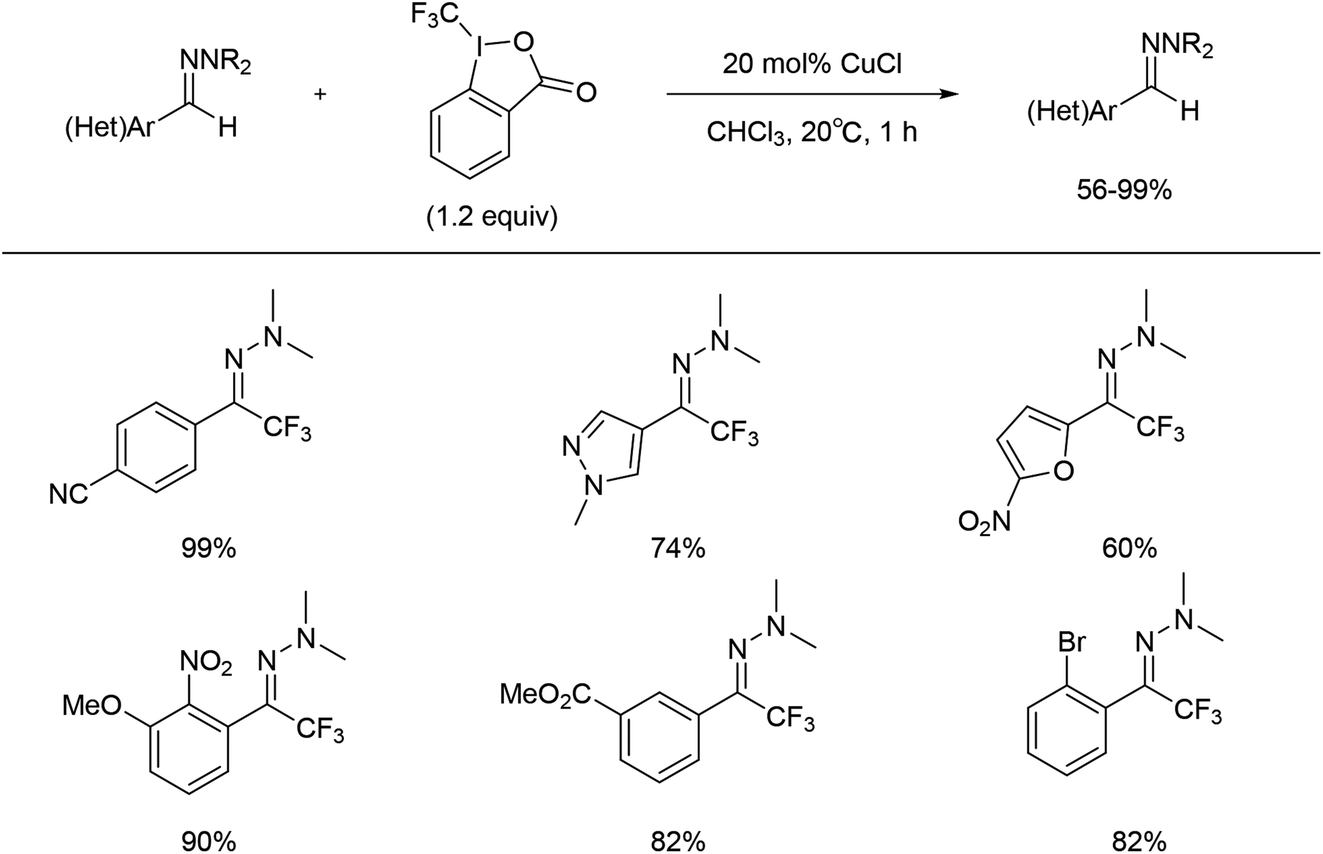 | ||
| Scheme 19 Copper catalysed trifluoromethylation of hydrazones with Togni's reagent as a fluorine source. | ||
The reaction pathway was proposed to begin with the activation of Tognís reagent 61 by CuI via single-electron transfer (SET) to form CuII species 62, which could act as a trifluoromethyl radical donor. Reaction with the hydrazine 64 would generate (2-iodobenzoyloxy)copper(II) chloride 63 and the trifluoromethylated aminyl radical intermediate 65, which could be stabilized by the lone pair of the adjacent nitrogen atom. Finally, oxidation of intermediate 65 with CuII could restore the hydrazone functional group to give the desired product 67 and recycle CuI (Scheme 20).
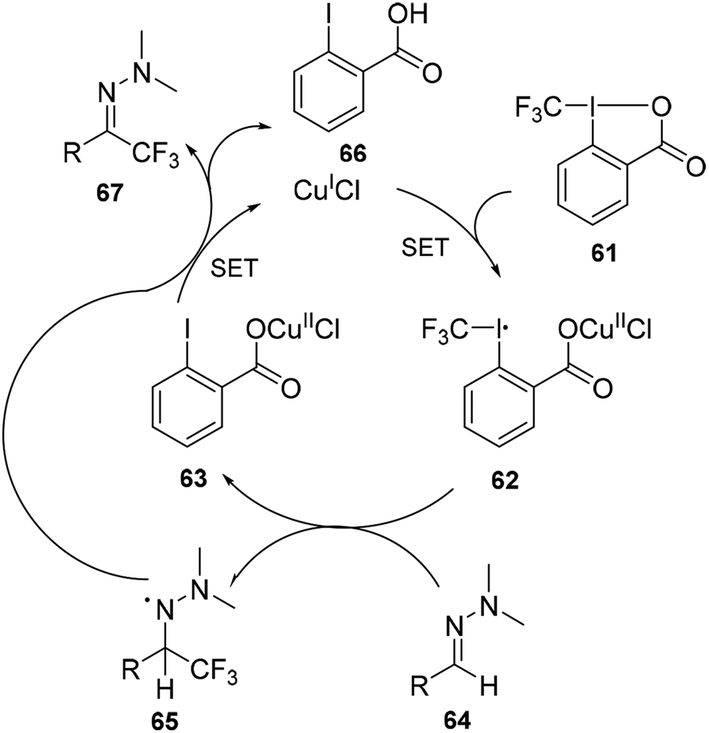 | ||
| Scheme 20 Mechanism for copper catalysed trifluoromethylation of hydrazones in Scheme 19. | ||
The copper-catalysed trifluoromethylation of hydrazone process was also effective for aliphatic aldehyde derivatives and heteroaryl derivatives, which demonstrated the wide applicability of this method. In addition, trifluoromethylated N-arylhydrazones have been proved to be suitable starting materials in the synthesis of trifluoromethyl ketones as well as 2-trifluoromethylindole derivatives.
Conclusion and future prospects
To summarize, significant advances have been made in the synthesis of fluorinated hydrazones during recent decades. Particularly, recent advances have allowed innovative approaches for C–H activation to prepare fluorinated hydrazones. Despite these excellent developments, some reactions still suffer from major drawbacks, such as harsh reaction conditions and limited substrate scope. The high reaction temperature required in some cases is a major concern. Moreover, gaining further insight into the reaction mechanism is essential for understanding and further developments. Developing cheaper metal catalysts like iron and cobalt into this chemistry would be more attractive. As valuable intermediates for organic-functional-group transformations, notably as precursors for substituted hydrazines or primary amines upon reductive cleavage of the N–N bond, and as chiral auxiliaries in asymmetric synthesis, the development of a general and practical method for the synthesis of fluorinated hydrazones is still highly desirable. The present challenges are expected to stimulate further development in the synthesis of fluorinated hydrazones.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the National Natural Science Foundation of China (No. 21707049), and the State Key Laboratory of Environmental Chemistry and Ecotoxicology, RCEES, CAS (KF2016-08).Notes and references
- S. Purser, R. P. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- V. V. Grushin, Acc. Chem. Res., 2010, 43, 160 CrossRef CAS PubMed.
- T. Furuya, S. A. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed.
- T. Liang, C. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed.
- J. Ma and S. Li, Org. Chem. Front., 2014, 1, 712 RSC.
- J. A. Cresswell, G. S. Davies, M. P. Robertsand and E. J. Thomson, Chem. Rev., 2015, 115, 566 CrossRef PubMed.
- G. M. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef PubMed.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634 CrossRef PubMed.
- X. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed.
- C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed.
- X. Yang, T. Wu, J. R. Phipps and D. F. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed.
- N. M. Essex, O. M. Connell and B. P. Brown, J. Int. Med. Res., 2012, 40, 2251 CrossRef PubMed.
- F. Giornal, S. Pazenok, L. Rodefeld, N. Lui, J. Vors and R. F. Leroux, J. Fluorine Chem., 2013, 152, 2 CrossRef CAS.
- J. Jiang and S. Zhu, J. Fluorine Chem., 2008, 129, 40 CrossRef.
- F. Li, C. Sun and N. Wang, J. Org. Chem., 2014, 79, 8031 CrossRef PubMed.
- O. V. Smirnov, I. M. Struchkova, E. D. Arkhipov, A. A. Korlyukov and D. A. Dilman, J. Org. Chem., 2014, 79, 11819 CrossRef PubMed.
- C. A. Day and C. M. Whiting, Org. Synth., 1988, 6, 10 Search PubMed.
- G. Stork and J. Benaim, Org. Synth., 1988, 6, 242 Search PubMed.
- R. F. Japp and F. Klingemann, Ber. Dtsch. Chem. Ges., 1887, 20, 2942 CrossRef.
- R. F. Japp and F. Klingemann, Ber. Dtsch. Chem. Ges., 1887, 20, 3284 CrossRef.
- R. F. Japp and F. Klingemann, Ber. Dtsch. Chem. Ges., 1887, 20, 3398 CrossRef.
- V. Y. Burgart, S. A. Fokin, G. O. Kuzueva, N. O. Chupakhin and I. V. Saloutin, J. Fluorine Chem., 1998, 92, 101 CrossRef.
- C. Reichardt and K. Halbritter, Chem. Ber., 1973, 106, 1661 CrossRef CAS.
- R. Guo, Z. Zhang, F. Shi and P. Tang, Org. Lett., 2016, 18, 1008 CrossRef CAS PubMed.
- S. Wagaw, H. B. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 6621 CrossRef CAS.
- S. Wagaw, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 10251 CrossRef CAS.
- O. R. Thiel, M. M. Achmatowicz, A. Reichelt and R. D. Larsen, Angew. Chem., Int. Ed., 2010, 49, 8395 CrossRef CAS PubMed.
- A. Prieto, R. Melot, D. Bouyssi and N. Monteiro, Angew. Chem., Int. Ed., 2016, 55, 1885 CrossRef CAS PubMed.
- A. Prieto, R. Melot, D. Bouyssi and N. Monteiro, ACS Catal., 2016, 6, 1093 CrossRef CAS.
- M. C. Belhomme, T. Poisson and X. Pannecoucke, Org. Lett., 2013, 15, 3428 CrossRef CAS PubMed.
- H. Jiang, Y. Chen, B. Chen, H. Xu, W. Wan, H. Deng, K. Ma, S. Wu and J. Hao, Org. Lett., 2017, 19, 2406 CrossRef CAS PubMed.
- P. Xu, G. Wang, Y. Zhu, W. Li, Y. Cheng, S. Li and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2939 CrossRef CAS PubMed.
- J. Xie, T. Zhang, F. Chen, N. Mehrkens, F. Rominger, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 2934 CrossRef CAS PubMed.
- Y. Gu, D. Chang, X. Leng, Y. Gu and Q. Shen, Organometallics, 2015, 34, 3065 CrossRef CAS.
- B. C. Hamper, M. L. Kurtzweil and J. P. Beck, J. Org. Chem., 1992, 57, 5680 CrossRef CAS.
- Y. Zhang, J. Desharnais, T. H. Marsilje, C. Li, M. P. Hedrick, L. T. Gooljarsingh, A. Tavassoli, S. J. Benkovic, A. J. Olson, D. L. Boger and I. A. Wilson, Biochemistry, 2003, 42, 6043 CrossRef CAS PubMed.
- J. Desharnais, I. Hwang, Y. Zhang, A. Tavassoli, J. Baboval, S. J. Benkovic, I. A. Wilson and D. L. Boger, Bioorg. Med. Chem., 2003, 11, 4511 CrossRef CAS PubMed.
- Y. Chong, I. Hwang, A. Tavassoli, M. E. Webb, Y. Zhang, I. A. Wilson, S. J. Benkovic and D. L. Boger, Bioorg. Med. Chem., 2005, 13, 3587 CrossRef CAS PubMed.
- H. Cheng, I. Hwang, Y. Chong, A. Tavassoli, M. E. Webb, Y. Zhang, I. A. Wilson, S. J. Benkovic and D. L. Boger, Bioorg. Med. Chem., 2005, 13, 3593 CrossRef CAS PubMed.
- X. Liu, Y. Wei and M. Shi, Org. Biomol. Chem., 2009, 7, 4708 CAS.
- K. Quinze, A. Laurent and P. Mison, J. Fluorine Chem., 1989, 44, 211 CrossRef CAS.
- H. M. Marsden, K. Yasufuku and J. M. Shreeve, Inorg. Chem., 1983, 22, 1202 CrossRef CAS.
- C. Aubert, J. P. Begue, M. Charpentier-Morize, G. Nee and B. Langlois, J. Fluorine Chem., 1989, 44, 377 CrossRef CAS.
- Y. Kamitori, M. Hojo, R. Masuda, Y. Kawamura and X. Fand, Heterocycles, 1990, 31, 2103 CrossRef CAS.
- K. Burger, O. Dengler and D. Huebl, J. Fluorine Chem., 1982, 19, 589 CrossRef CAS.
- C. Maliverney and H. G. Viehe, Tetrahedron Lett., 1990, 31, 6339 CrossRef CAS.
- P. E. Reed and J. A. Katzenellenbogen, J. Med. Chem., 1991, 34, 1162 CrossRef CAS PubMed.
- G. P. Lahm, T. P. Selby, J. H. Freudenberger, T. M. Stevenson, B. J. Myers, G. Seburyamo, B. K. Smith, L. Flexner, C. E. Clark and D. Cordova, Bioorg. Med. Chem. Lett., 2005, 15, 4898 CrossRef CAS PubMed.
- S. Fustero, R. Roman, J. F. Sanz-Cervera, A. Simon-Fuentes, A. C. Cunat, S. Villanova and M. Murguia, J. Org. Chem., 2008, 73, 3523 CrossRef CAS PubMed.
- J.-P. Bouillon, H. Frisque, M. Anne, Z. Janousek and H. G. Viehe, Heterocycles, 1995, 40, 661 CrossRef CAS.
- E. Schmitt, G. Landelle, J.-P. Vors, N. Lui, S. Pazenok and F. R. Leroux, Eur. J. Org. Chem., 2015, 6052 CrossRef CAS.
- E. Schmitt, A. Panossian, J.-P. Vors, C. Funke, N. Lui, S. Pazenok and F. R. Leroux, Chem.–Eur. J., 2016, 22, 11239 CrossRef CAS PubMed.
- E. Pair, N. Monteiro, D. Bouyssi and O. Baudoin, Angew. Chem., Int. Ed., 2013, 52, 5346 CrossRef CAS PubMed.
- A. Prieto, E. Jeamet, N. Monteiro, D. Bouyssi and O. Baudoin, Org. Lett., 2014, 16, 4770 CrossRef CAS PubMed.
- A. Prieto, M. Landart, O. Baudoin, N. Monteiro and D. Bouyssi, Adv. Synth. Catal., 2015, 357, 2939 CrossRef CAS.
- A. Prieto, D. Bouyssi and N. Monteiro, Asian J. Org. Chem., 2016, 5, 742 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |