
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeronamides G–L, polyene macrolactams from Streptomyces niveus†
Nan Dinga,
Li Han *a,
Yi Jiangb,
Guiding Lib,
Zehui Zhenga,
Bixuan Caoa,
Peipei Guana,
Yu Mua,
Bin Linc and
Xueshi Huang*a
*a,
Yi Jiangb,
Guiding Lib,
Zehui Zhenga,
Bixuan Caoa,
Peipei Guana,
Yu Mua,
Bin Linc and
Xueshi Huang*a
aInstitute of Microbial Pharmaceuticals, College of Life and Health Sciences, Northeastern University, Shenyang 110819, People's Republic of China. E-mail: hanli@mail.neu.edu.cn; huangxs@mail.neu.edu.cn
bYunnan Institute of Microbiology, Yunnan University, Kunming 650091, People's Republic of China
cSchool of Pharmaceutical Engineering, Shenyang Pharmaceutical University, Shenyang 110016, People's Republic of China
First published on 9th May 2018
Abstract
Six new polyene macrolactams, heronamides G–L (1–6), one new polyenoic acid derivative, niveamide B (10), together with four known compounds, BE-14106-6 (7), BE-14106 (8), GT32-B (9), and niveamide (11), were isolated from the fermentation broth of Streptomyces niveus. Their planar structures were elucidated by detailed analysis of spectroscopic data. The absolute configurations of compounds 1–6 were determined by calculated ECD spectra and analysis of the possible biosynthetic pathways. Compounds 1–6 and 8–11 did not exhibit any significant antimicrobial activities, cytotoxicities, or inhibitory effects on lipopolysaccharide-induced NO production in BV2 microglial cells.
Introduction
Polyene macrolactams refer to macrolactams possessing a 20- to 26-membered lactam ring with multiple double bonds in their scaffold.1 Heronamides are a class of 20-membered polyene macrolactam with methyl groups at C-6, C-14, and an unsaturated hydrocarbon tail at C-19. Recent studies have proven that a thermal or photochemical cycloaddition is involved in the formation of heronamide derivatives.1–3 Since the first heronamide, BE-14106, was reported in 1992,4 only ten natural members including BE-14106,4,5 GT32-B,5 ML-449,6 heronamides A–F,7,8 and 8-deoxyheronamide C9 had been isolated from actinomycetes and two derivatives of BE-14106 (BE-14106-6 and BE-14106-7) were semi-synthesized by O2 and UV irradiation.1 The heronamides exhibited a variety of biological activities, such as antiproliferative activity, weak antimicrobial activity, inhibitory activity against mixed lymphocyte reaction, and growth inhibition against fission yeast cells.1,5,9In the process of our continuous study on new bioactive metabolites from actinomycetes, the chemical constituents of Streptomyces niveus (YIM 32862) isolated from forest soil collected from Great Khingan Mountains were investigated. Six new polyene macrolactams, heronamides G–L (1–6), and one new polyenoic acid derivative, niveamide B (10), together with four known compounds, BE-14106-6 (7), BE-14106 (8), GT32-B (9), and niveamide (11), were isolated from the ethyl acetate extract of the fermentation broth of S. niveus. These new compounds were elucidated by extensive interpretation of spectroscopic data. The absolute configurations of compounds 1–6 were determined by calculated ECD spectra and analysis of the possible biosynthetic pathways. The antimicrobial, cytotoxic activities, and inhibitory effects on lipopolysaccharide-induced NO production of 1–6 and 8–11 were evaluated. Herein, we report the fermentation, isolation, structure elucidation, biological activities evaluation, and possible biosynthetic pathways of these compounds.
Results and discussion
Fermentation broth (140 L) of S. niveus was collected and centrifuged to yield a clarified supernatant, then the supernatant was extracted by EtOAc. Chemical constituents in EtOAc extract were isolated by sequential chromatographies over Sephadex LH-20, silica gel, ODS, and semipreparative HPLC to obtain pure compounds heronamides 1–9 and niveamides 10 and 11 (Fig. 1).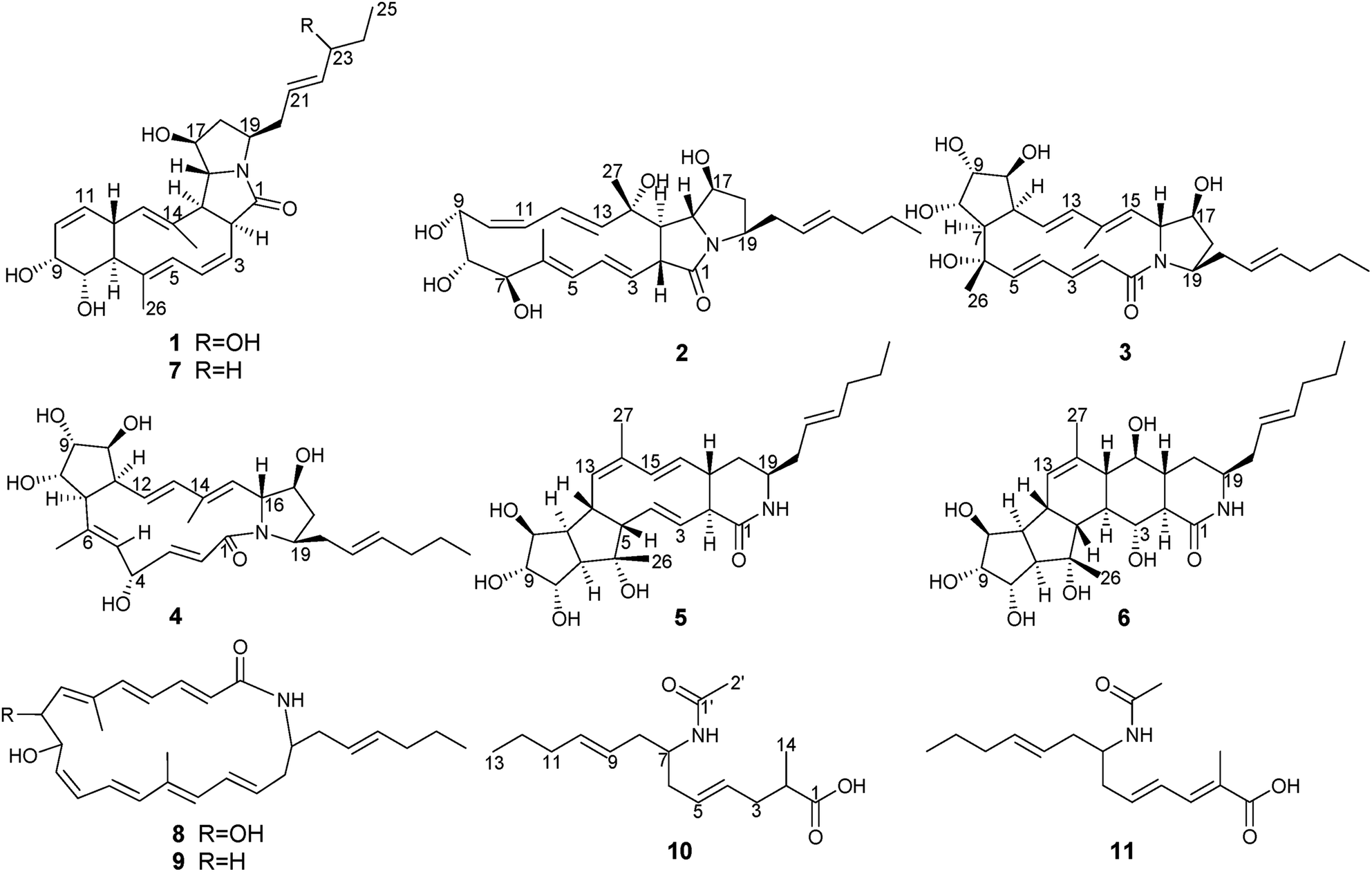 | ||
| Fig. 1 Structures of compounds 1–11. | ||
From the proposed biosynthesis of heronamides in the literature,3,6,7,10 the 9-hydroxy heronamide precursors were released from the PKS assembly; after that, transannular [6π + 4π] cycloaddition and SN2-mediated ring formation following olefinic epoxidation occurred to construct different heronamides. Compared with that of known heronamides,3,7 compounds 1–9 exhibited a similar biosynthetic relationship (Scheme 1).
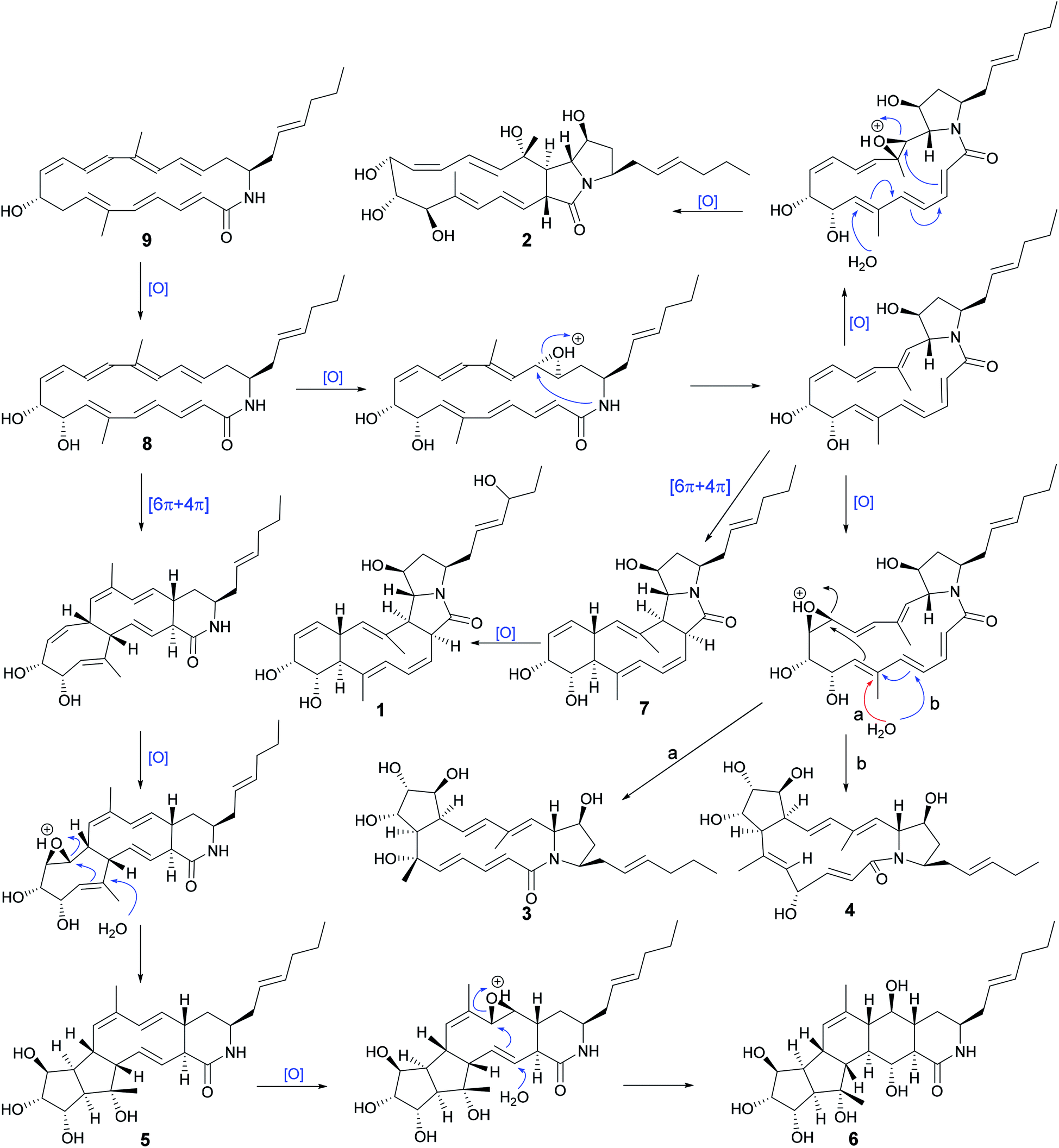 | ||
| Scheme 1 Possible biosynthetic pathways between compounds 1–9. | ||
Heronamide G (1) was obtained as an amorphous, colorless solid with a molecular formula of C27H37NO5 (ten degrees of unsaturation) based on the HRESIMS peak at m/z 456.2758 [M + H]+ and NMR data. Detailed analysis of 1H NMR, 13C NMR, and HSQC of 1 (Table 1) revealed it was almost identical to BE-14106-6 (7),7 which was also isolated from the same strain BE-14106-6/nivelactam was first isolated from S. niveus (YIM 32860) by Li L. et al. and named as nivelactam.11 BE-14106-6 was semi-synthesized from BE-14106 by O2 induced conversion.1 According to the same NMR data possessed by nivelactam and BE-14106-6, and the possible biosynthetic pathway in Scheme 1, the structure of BE-14106-6 was deemed reliable. The major difference between 1 and BE-14106-6 (7) is the presence of one more oxygenated methine signals (δC/H 71.6/3.80) and a hydroxy signal (δH 4.57) in 1 instead of a methylene signal in 7. The COSY correlations of H-22 (δH 5.47)/H-23 (δH 3.80)/H-24 (δH 1.42) and HMBC correlations from 23-OH (δH 4.57) to C-22 (δC 136.6), C-23 (δC 71.6), and C-24 (δC 29.6) showed that the hydroxyl was located at C-23 (Fig. 2A). The similar 13C NMR signals between 1 and 7 indicated they possessed the same relative configuration in the polyene macrolactam ring. Detailed analysis of 1H–1H coupling constants (Table 1) and NOE correlations (Fig. 2C) between H-2/H-5, H-13, H-15; H-7/H-13; H-15/H-13, H-17; H-4/H3-26; H-8/H-9, H-12, H3-26; H-12/H3-27, and H-16/H3-27 helped confirm the relative configuration of 1. The Z configuration of double bond Δ3,4 was assigned by the J3,4 values of 10.2 Hz. The E configurations of double bonds Δ5,6 and Δ13,14 were based on NOE correlations (Fig. 2C). The absolute configuration of 1 was determined by electronic circular dichroism (ECD) spectra with quantum chemical calculations using the time dependent density functional theory (TDDFT) method at the B3 LYP/6-311 ++ G(d) level. The experimental ECD spectrum of 1 matched the calculated spectrum of 2S,7S,8S,9R,12R,15S,16R,17S,19R-1 (Fig. 2B). Hence, the structure of 1 was established and named as heronamide G.
| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δC, type | δH, mult (J in Hz) | δC, type | δH, mult (J in Hz) | δC, type | δH, mult (J in Hz) | δC, type | δH, mult (J in Hz) | |
| a Overlapping signals. | ||||||||
| 1 | 173.7, C | 171.3, C | 167.1, C | 166.3, C | ||||
| 2 | 53.1, CH | 3.47, ddd (9.1, 6.8, 2.0) | 53.2, CH | 3.28, dd (12.8, 10.0) | 129.5, CH | 5.85, d (15.6) | 124.1, CH | 5.56, d (15.5) |
| 3 | 124.6, CH | 5.36, dd (10.2, 6.8) | 130.9, CH | 4.94, dd (14.9, 10.0) | 137.4, CH | 6.42, dd (15.6, 10.4) | 143.9, CH | 6.14, d (15.5, 8.3) |
| 4 | 131.2, CH | 6.60, ddd (10.2, 9.7, 2.0) | 130.3, CH | 6.17, dd (14.9, 11.0) | 127.6, CH | 5.92, dd (15.7, 10.4) | 68.3, CH | 4.61, m |
| 5 | 128.4, CH | 5.37, d (9.7) | 127.5, CH | 5.57, dd (11.0, 1.1) | 144.0, CH | 5.58, d (15.7) | 130.2, CH | 4.84, d (9.4) |
| 6 | 131.7, C | 133.7, C | 74.1, C | 136.8, C | ||||
| 7 | 51.8, CH | 1.95, t (10.5) | 78.9, CH | 3.38, brd (8.4) | 51.2, CH | 1.95, dd (12.3, 7.9) | 60.6, CH | 2.59, dd (12.1, 8.2) |
| 8 | 68.5, CH | 3.67, ddd (10.5, 7.8, 4.1) | 74.6, CH | 3.58, brd (8.4) | 74.7, CH | 4.29, m | 73.8, CH | 4.13, m |
| 9 | 65.2, CH | 3.97, ddd (4.9, 4.1, 3.7) | 65.0, CH | 3.89, brd (9.5) | 75.9, CH | 3.65, m | 78.1, CH | 3.68, m |
| 10 | 127.9, CH | 5.77, ma | 126.4, CH | 5.48, dd (11.0, 9.5) | 77.4, CH | 3.60, brd (3.6) | 75.2, CH | 3.77, m |
| 11 | 130.8, CH | 5.76, ma | 129.6, CH | 5.97, t (11.0) | 45.7, CH | 2.97, td (10.8, 4.6) | 48.0, CH | 3.05, ddd (12.1, 8.6, 5.2) |
| 12 | 41.5, CH | 2.72, t (11.0) | 124.9, CH | 5.76, dd (15.6, 11.0) | 125.5, CH | 5.64, dd (15.6, 10.8) | 125.4, CH | 5.68, dd (16.1, 8.8) |
| 13 | 130.6, CH | 4.95, d (11.0) | 141.1, CH | 5.42, d (15.6) | 138.0, CH | 5.89, d (15.6) | 136.3, CH | 5.63, d (16.1) |
| 14 | 135.9, C | 71.2, C | 136.3, C | 135.0, C | ||||
| 15 | 56.3, CH | 3.13, t (9.1) | 57.1, CH | 2.05, dd (12.8, 9.0) | 129.6, CH | 4.81, d (10.0) | 131.7, CH | 4.84, d (9.2) |
| 16 | 65.9, CH | 3.74, ma | 66.4, CH | 3.53, dd (9.0, 6.7) | 65.9, CH | 4.29, dd (9.8, 2.7) | 65.6, CH | 4.34, dd (9.2, 3.1) |
| 17 | 73.7, CH | 3.74, m | 73.6, CH | 3.79, m | 75.8, CH | 3.92,m | 76.2, CH | 3.89, m |
| 18 | 40.7, CH2 | 2.30, m | 39.3, CH2 | 2.31, m | 35.4, CH2 | 1.98, m | 35.4, CH2 | 1.99, m |
| 1.67, m | 1.67, m | 1.68, m | 1.67, ddd (12.8, 4.6, 4.6) | |||||
| 19 | 50.9, CH | 3.75, m | 51.6, CH | 3.72, m | 56.9, CH | 3.98, m | 57.0, CH | 4.02, m |
| 20 | 35.9, CH2 | 2.37, dt (13.7, 7.3) | 36.6, CH2 | 2.34, m | 36.8, CH2 | 2.51, m | 36.7, CH2 | 2.50, m |
| 2.25, m | 2.17, m | 2.34, m | 2.32, m | |||||
| 21 | 123.9, CH | 5.48, m | 125.1, CH | 5.36, m | 127.4, CH | 5.35, m | 127.3, CH | 5.34, m |
| 22 | 136.6, CH | 5.47, m | 132.3, CH | 5.45, m | 132.9, CH | 5.42, m | 132.9, CH | 5.41, m |
| 23 | 71.6, CH | 3.80, m | 33.5, CH2 | 1.96, q (7.0) | 34.7, CH2 | 1.96, q (7.3) | 34.7, CH2 | 1.95, q (7.3) |
| 24 | 29.6, CH2 | 1.42, m | 21.4, CH2 | 1.35, sxt (7.0) | 22.6, CH2 | 1.35, sxt (7.3) | 22.6, CH2 | 1.34, sxt (7.3) |
| 1.38, m | ||||||||
| 25 | 9.2, CH3 | 0.83, t (7.4) | 12.9, CH3 | 0.87, t (6.1) | 14.0, CH3 | 0.87, t (7.4) | 14.0, CH3 | 0.86, t (7.3) |
| 26 | 11.8, CH3 | 1.65, s | 10.8, CH3 | 1.66, s | 22.7, CH3 | 1.25, s | 15.1, CH3 | 1.44, s |
| 27 | 15.7, CH3 | 1.29, s | 19.2, CH3 | 1.29, s | 14.0, CH3 | 1.83, s | 12.9, CH3 | 1.71, s |
| 6-OH | 4.75, s | 4.88, d (4.3) | ||||||
| 7-OH | 4.74, brs | |||||||
| 8-OH | 4.26, d (7.8) | 4.73, brs | 4.33, brs | 4.25, brs | ||||
| 9-OH | 4.65, d (4.9) | 4.45, d (5.1) | 4.52, d (3.1) | 4.62, brs | ||||
| 10-OH | 4.79, d (3.7) | 4.99, brs | ||||||
| 14-OH | 4.67, brs | |||||||
| 17-OH | 5.11, d (4.7) | 5.67, brs | 5.28, d (3.8) | 5.33, d (6.4) | ||||
| 23-OH | 4.57, d (4.5) | |||||||
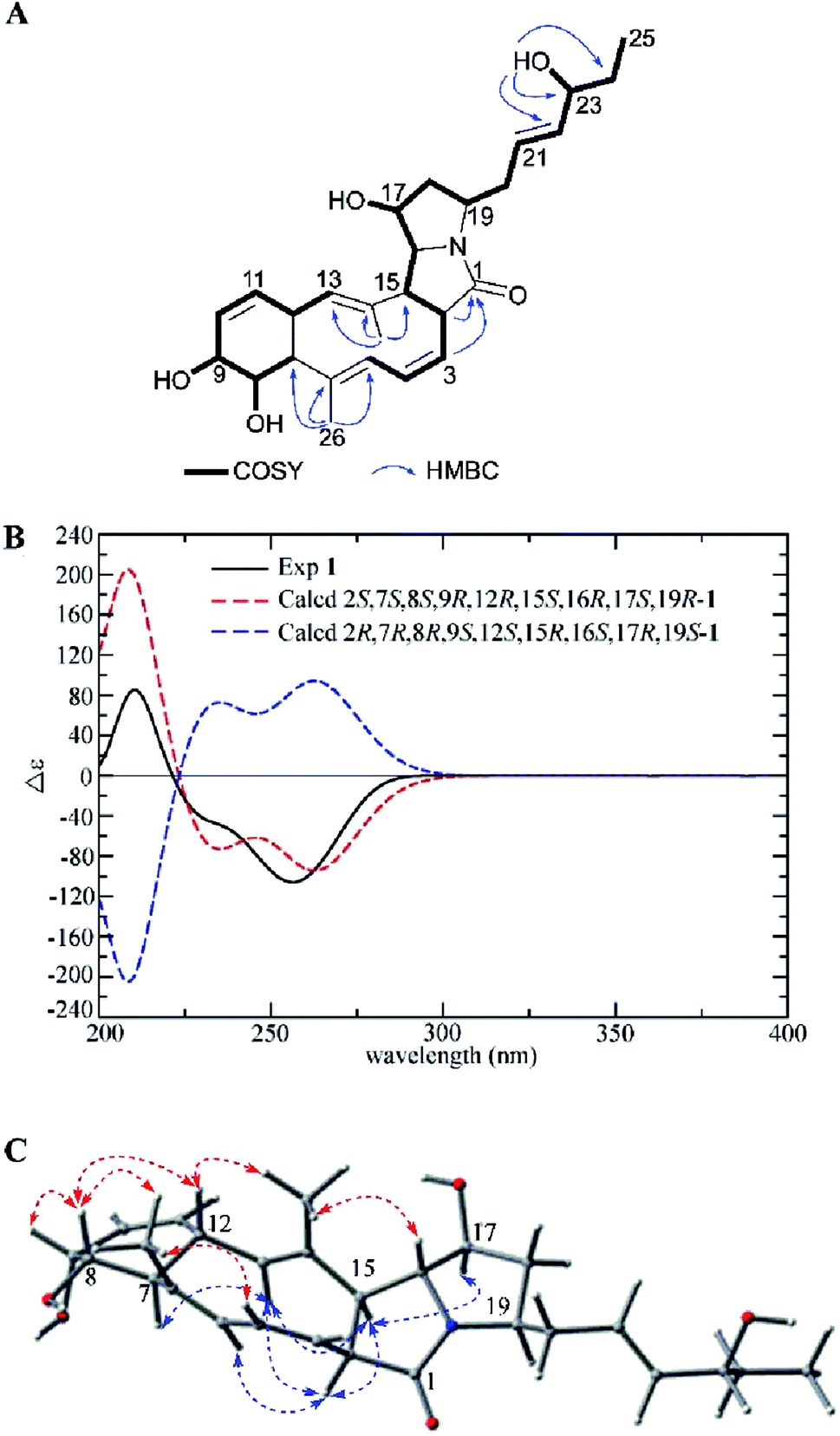 | ||
| Fig. 2 (A) COSY and key HMBC correlations of 1, (B) comparison of the experimental ECD spectra of 1 with the calculated ECD spectra, and (C) key NOEs observed in 1. | ||
Heronamide H (2) was purified as a white powder and assigned the molecular formula C27H39NO6 (nine degrees of unsaturation) from the HRESIMS peak at m/z 474.2861 [M + H]+. The 1H NMR, 13C NMR, and HSQC data of 2 (Table 1) showed the presence of two sp2 nonprotonated carbons (δC 171.3 and 133.7), nine sp2 methines (δC/H 141.1/5.42, 132.3/5.45, 130.9/4.94, 130.3/6.17, 129.6/5.97, 127.5/5.57, 126.4/5.48, 125.1/5.36, 124.9/5.76), one sp3 oxygenated nonprotonated carbon (δC 71.2), eight sp3 methines including four oxygenated (δC/H 78.9/3.38, 74.6/3.58, 73.6/3.79, and 65.0/3.89) and four other methines (δC/H 66.4/3.53, 57.1/2.05, 53.2/3.28, and 51.6/3.72), four methylenes (δC/H 39.3/2.31, 1.67; 36.6/2.34, 2.17; 33.5/1.96; and 21.4/1.35), and three methyls (δC/H 19.2/1.29, 12.9/0.87, and 10.8/1.66). In addition to an amide carbonyl and five double bonds accounting for six degrees of unsaturation, 2 was required to be a tricyclic skeleton according to its unsaturation. Careful interpretation of 1H–1H COSY of 2 revealed the presence of two substructures (subunits A and B) (Fig. 3A). HMBC correlations from H-2 (δH 3.28), H-3 (δH 4.94), H-19 (δH 3.72) to C-1 (δC 171.3) indicating the location of the amide carbonyl, from H3-26 (δH 1.66) to C-5 (δC 127.5), C-6 (δC 133.7), and C-7 (δC 78.9) and from H3-27 (δH 1.29) to C-13 (δC 141.1), C-14 (δC 71.2), and C-15 (δC 57.1) completing the connections between subunits A and B, helped establish the gross structure of 2 (Fig. 3A).
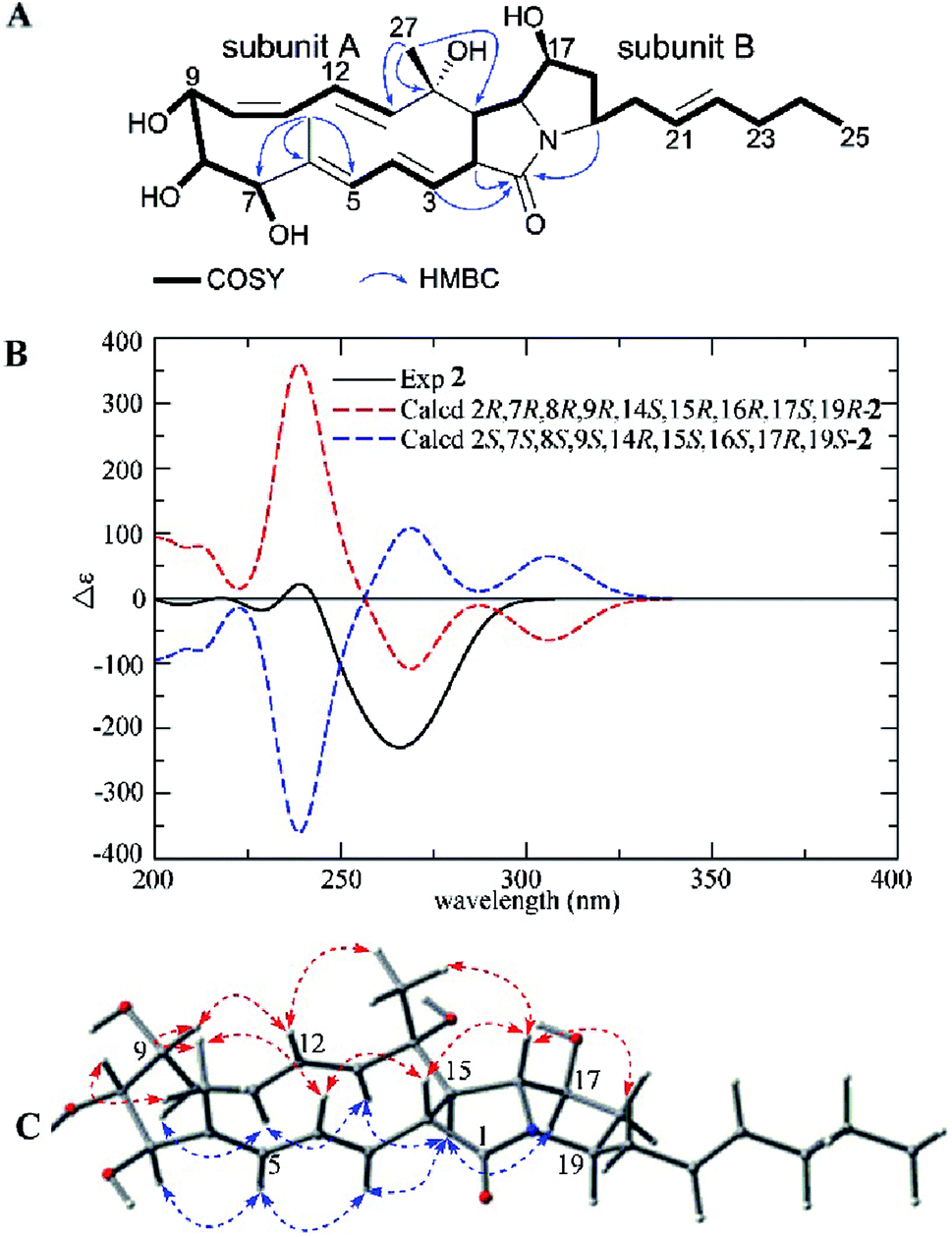 | ||
| Fig. 3 (A) COSY and key HMBC correlations of 2, (B) comparison of the experimental ECD spectra of 2 with the calculated ECD spectra, and (C) key NOEs observed in 2. | ||
A detailed NOESY analysis of 2 between H-2/H-4, H-16; H3-26/H-4, H-8, H-9; H-9/H-12, and H3-27/H-12, H-16 determined the H-2, H-4, 6-CH3, H-8, H-9, H-12, 14-CH3, H-16 were placed on the same face of the ring system, while the correlations between H-3/H-5, H-15; H-5/H-7; H-11/H-10, H-13; H-15/H-13, H-17, and H-16/H-20 revealed the H-3, H-5, H-7, H-10, H-11, H-13, H-15, H-17, H-19 were on the opposite side (Fig. 3C). Compared to 1, the difference of relative configurations of H-2 and H-15 was further supported by the JH-2/H-15 = 12.8 Hz in 2 instead of JH-2/H-15 = 9.1 Hz in 1. Analysis of the possible biosynthetic pathways of 1 and 2 indicated the two compounds were completely different in the formation of the five-membered lactam ring (Scheme 1), which further explained the different relative configuration of H-2/H-15 between 1 and 2. The 1H–1H coupling constants and NOE correlations (Fig. 3C) also helped establish the configuration of double bonds in 2: E Δ3,4 (J3,4 14.9 Hz), E Δ5,6 (NOE correlations between H-3/H-5, H3-26/H-4), Z Δ10,11 (J10,11 11.0 Hz), and E Δ12,13 (J12,13 15.6 Hz). The experimental ECD spectrum of 2 showed the same pattern as the calculated ECD spectrum 2R,7R,8R,9R,14S,15R,16R,17S,19R-2 (Fig. 3B). Hence, the absolute configuration of 2 was determined as shown.
Compound 3 was isolated as a white powder. The molecular formula of 3 was determined to be C27H39NO6 (nine degrees of unsaturation) based on the HRESIMS peak at m/z 474.2870 [M + H]+. The 1H NMR, 13C NMR, and HSQC spectra revealed the presence of one amide carbonyl (δC 167.1), one nonprotonated olefinic carbon (δC 136.3), and nine olefinic methines (δC/H 144.0/5.58, 138.0/5.89, 137.4/6.42, 132.9/5.42, 129.6/4.81, 129.5/5.85, 127.6/5.92, 127.4/5.35, and 125.5/5.64), explaining six degrees of unsaturation. Hence, 3 should be a tricyclic structure. Detailed analysis of the 1H–1H COSY correlations showed the presence of three substructures (C-2 to C-5, C-7 to C-13, and C-15 to C-25) (Fig. 4A). In addition, the HMBC correlations from H-2 (δH 5.85), H-3 (δH 6.42) to C-1 (δC 167.1), from H3-26 (δH 1.25) to C-5 (δC 144.0), C-6 (δC 74.1), C-7 (δC 51.2), from H-9 (δH 3.65) to C-7 (δC 51.2), C-10 (δC 77.4), C-11 (δC 45.7), and from H3-27 (δH 1.83) to C-13 (δC 138.0), C-14 (δC 136.3), C-15 (δC 129.6) connected three subunits in 3 (Fig. 4A).
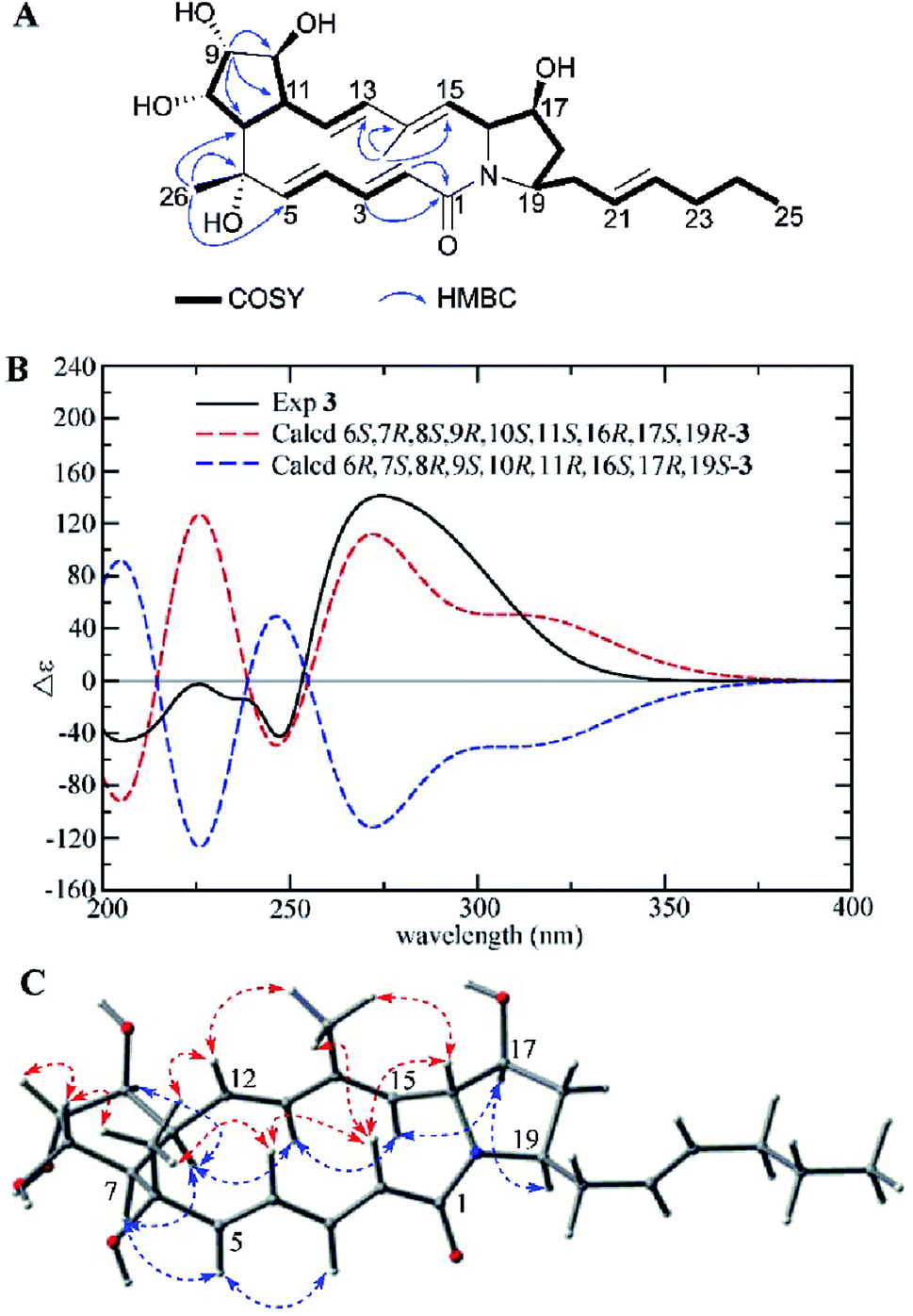 | ||
| Fig. 4 (A) COSY and key HMBC correlations of 3, (B) comparison of the experimental ECD spectra of 3 with the calculated ECD spectra, and (C) key NOEs observed in 3. | ||
The relative configuration of 3 was deduced by careful interpretation of the NOE correlations and coupling constants (Table 1). NOE cross peaks observed between H-2/H-4, H-16, H3-27; H3-26/H-4, H-8, H-12; H-8/H-9 and H3-27/H-12, H-16 demonstrated that the H-2, H-4, 6-CH3, H-8, H-9, H-12, 14-CH3, and H-16 were located on the same side, while the NOE cross peaks between H-5/H-3, H-7; H-11/H-7, H-10, H-13 and H-15/H-13, H-17; H-17/H-19 indicated that H-3, H-5, H-7, H-10, H-11, H-13, H-15, H-17, H-19 were on the other side (Fig. 4C). The large 3JHH values of double bonds Δ2,3 (J2,3 15.6 Hz), Δ4,5 (J4,5 15.7 Hz), and Δ12,13 (J12,13 15.6 Hz) together with the NOE correlations between H3-27/H-12, H-15/H-13 revealed the E configurations of the four double bonds in the macrolactam ring in 3. The experimental ECD spectrum of 3 was similar to the calculated ECD curve of 6S,7R,8S,9R,10S,11S,16R,17S,19R-3 (Fig. 4B). Therefore, the structure of 3 was established and named as heronamide I.
Heronamide J (4) was obtained as a white powder. The molecular formula of 4 was established as C27H39NO6 on the basis of HRESIMS at m/z 474.2869 [M + H]+, indicating nine degrees of unsaturation. The 1H and 13C NMR data of 4 were very similar to those of 3 except for the presence of an oxygenated methine (δC/H 68.3/4.61) and a nonprotonated olefinic carbon (δC 136.8) instead of an oxygenated quaternary carbon and an olefinic methine in 3. Interpretation of 1H–1H COSY correlations from H-2 to H-5 indicated the double bond at C-4/C-5 in 3 translocated to C-5/C-6 in 4 and the hydroxyl located at C-4 in 4. The HMBC correlations from H3-26 (δH 1.44) to C-5 (δC 130.2), C-6 (δC 136.8), and C-7 (δC 60.6) further confirmed the speculative structure of 4 (Fig. 5A). The relative configuration of 4 was determined by NOE correlations and coupling constants (Table 1). NOE interactions observed between H3-26/H-4, H-8; H-8/H-9, and H3-27/H-12, H-16 revealed H-4, 6-CH3, H-8, H-9, H-12, 14-CH3, and H-16 were located on the same side, and NOE correlations between H-5/H-7; H-11/H-7, H-10, and H-15/H-13, H-17 along with a comparison of 13C NMR with 3 presented H-5, H-7, H-10, H-11, H-13, H-15, H-17, and H-19 on the opposite (Fig. 5C). The E configurations of the double bonds in the macrolactam ring were assigned by the coupling constants Δ2,3 (J2,3 15.5 Hz), Δ12,13 (J12,13 16.1 Hz) and the NOE correlations (Fig. 5C). The experimental ECD was almost identical to the calculated ECD curve of 4R,7S,8S,9R,10S,11S,16R,17S,19R-4 (Fig. 5B). On the basis of the foregoing evidence, the structure of 4 was determined.
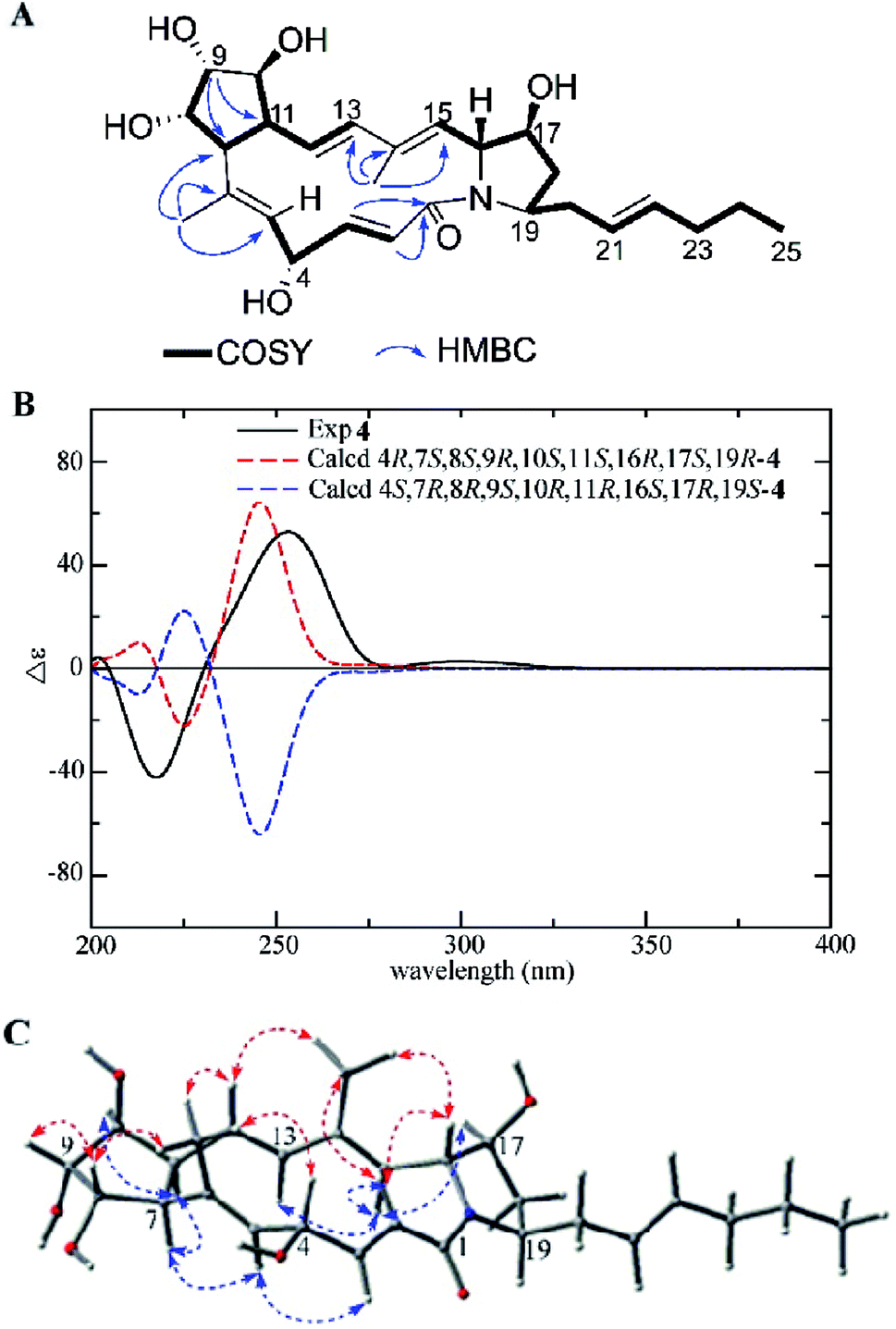 | ||
| Fig. 5 (A) COSY and key HMBC correlations of 4, (B) comparison of the experimental ECD spectra of 4 with the calculated ECD spectra, and (C) key NOEs observed in 4. | ||
Compound 5 was isolated as a white powder with a molecular formula of C27H39NO5 (nine degrees of unsaturation) as shown by the HRESIMS peak at m/z 458.2912 [M + H]+. The 13C NMR data (Table 2) indicated the presence of an amide carbonyl (δC 171.8) and eight sp2 olefinic carbons (δC 126.4 to 139.6), accounting for five degrees of unsaturation. Thus, there was a tetracyclic skeleton in 5. A close inspection of the 1H–1H COSY correlations disclosed three substructures A, B, and C (Fig. 6A). The multiple HMBC correlation signals from NH (δH 7.50) to C-1 (δC 171.8) and C-2 (δC 55.7), from H-16 (δH 4.93) to C-18 (δC 30.0) indicated the connection between subunit A, subunit B and the position of amide carbonyl; from H3-26 (δH 1.08) to C-5 (δC 62.0), C-6 (δC 79.5) and C-7 (δC 60.6), from H-12 (δH 2.92) to C-6 (δC 79.5), C-10 (δC 74.7) and C-11 (δC 51.3) revealed the connection of subunits B and C; while H-7 (δH 2.24) to C-9 (δC 79.5) suggested C-8 was linked to C-9; and H3-27 (δH 1.74) to C-13 (δC 134.3), C-14 (δC 130.8), C-15 (δC 139.6) showed C-14 was substituted by a methyl, and confirmed the gross structure of 5.
| No. | 5 | 6 | 10 | |||
|---|---|---|---|---|---|---|
| δC, type | δH, mult (J in Hz) | δC, type | δH, mult (J in Hz) | δC, type | δH, mult (J in Hz) | |
| a Overlapping signals. | ||||||
| 1 | 171.8, C | 175.1, C | 176.6, C | |||
| 2 | 55.7, CH | 2.17, t (9.1) | 47.8, CH | 1.83, ma | 38.7, CH | 2.31, m |
| 3 | 127.5, CH | 4.99, dd (15.9, 9.1) | 74.6, CH | 3.39, t (9.8) | 35.7, CH2 | 2.23, m |
| 2.02, m | ||||||
| 4 | 139.5, CH | 4.65, dd (15.9, 10.2) | 42.9, CH | 1.34, m | 128.7, CH | 5.36, m |
| 5 | 62.0, CH | 2.40, dd (10.2, 8.2) | 52.7, CH | 1.91, dd (8.5, 5.2) | 127.9, CH | 5.36, m |
| 6 | 79.5, C | 81.3, C | 36.2, CH2 | 1.98, m | ||
| 7 | 60.6, CH | 2.24, dd (9.7, 7.6) | 61.5, CH | 2.10, dd (9.5, 6.3) | 47.8, CH | 3.67, m |
| 8 | 74.6, CH | 3.64, ddd (7.6, 6.4, 4.3) | 73.9, CH | 3.58, q (5.9) | 36.2, CH2 | 2.06, m |
| 9 | 79.5, CH | 3.56, ddd (4.3, 3.4, 3.1) | 78.9, CH | 3.49, q (4.1) | 126.2, CH | 5.32, m |
| 10 | 74.7, CH | 3.78, ddd (6.8, 4.2, 3.1) | 75.3, CH | 3.82, dt (6.9, 4.3) | 131.4, CH | 5.40, m |
| 11 | 51.3, CH | 2.59, t (8.9) | 48.9, CH | 2.43, ddd (9.2, 7.2, 1.9) | 33.5, CH2 | 1.92, m |
| 12 | 42.8, CH | 2.92, t (7.4) | 37.4, CH | 2.50, ma | 21.5, CH2 | 1.32, m |
| 13 | 134.3, CH | 5.24, d (6.8) | 133.1, CH | 5.39, brs | 12.8, CH3 | 0.85, t (7.3) |
| 14 | 130.8, C | 136.7, C | 15.8, CH3 | 1.01, d (6.8) | ||
| 15 | 139.6, CH | 5.69, d (16.4) | 48.4, CH | 1.72, t (10.7) | ||
| 16 | 126.4, CH | 4.93, dd (16.4, 8.4) | 72.3, CH | 3.18, q (9.3) | ||
| 17 | 40.5, CH | 2.01, m | 38.3, CH | 1.52, m | ||
| 18 | 30.0, CH2 | 2.04, m | 27.7, CH2 | 2.17, m | ||
| 1.84, m | 1.44, m | |||||
| 19 | 50.0, CH | 3.37, m | 50.3, CH | 3.36, m | ||
| 20 | 39.7, CH2 | 2.23, m | 39.2, CH2 | 2.17, m | ||
| 2.14, m | 2.07, m | |||||
| 21 | 127.0, CH | 5.38, m | 126.6, CH | 5.35, m | ||
| 22 | 133.3, CH | 5.47, m | 133.7, CH | 5.48, m | ||
| 23 | 34.6, CH | 1.96, q (7.3) | 34.7, CH2 | 1.95, q (7.2) | ||
| 24 | 22.5, CH2 | 1.34, sxt (7.3) | 22.5, CH2 | 1.34, sxt (7.2) | ||
| 25 | 14.0, CH3 | 0.85, t (7.4) | 14.0, CH3 | 0.86, t (7.4) | ||
| 26 | 23.3, CH3 | 1.08, s | 24.0, CH3 | 1.15, s | ||
| 27 | 20.1, CH3 | 1.74, s | 20.3, CH3 | 1.82, s | ||
| 1′ | 167.8, C | |||||
| 2′ | 22.1, CH3 | 1.76, s | ||||
| –NH | 7.50, d (2.5) | 7.92, d (1.7) | 7.57, d (8.5) | |||
| –COOH | 12.11, brs | |||||
| 3-OH | 6.51, s | |||||
| 6-OH | 3.79, s | 2.70, s | ||||
| 8-OH | 4.18, d (6.4) | 4.12, d (5.9) | ||||
| 9-OH | 4.36, d (3.4) | 4.36, d (4.1) | ||||
| 10-OH | 4.70, d (4.2) | 4.69, d (4.3) | ||||
| 16-OH | 4.59, d (8.1) | |||||
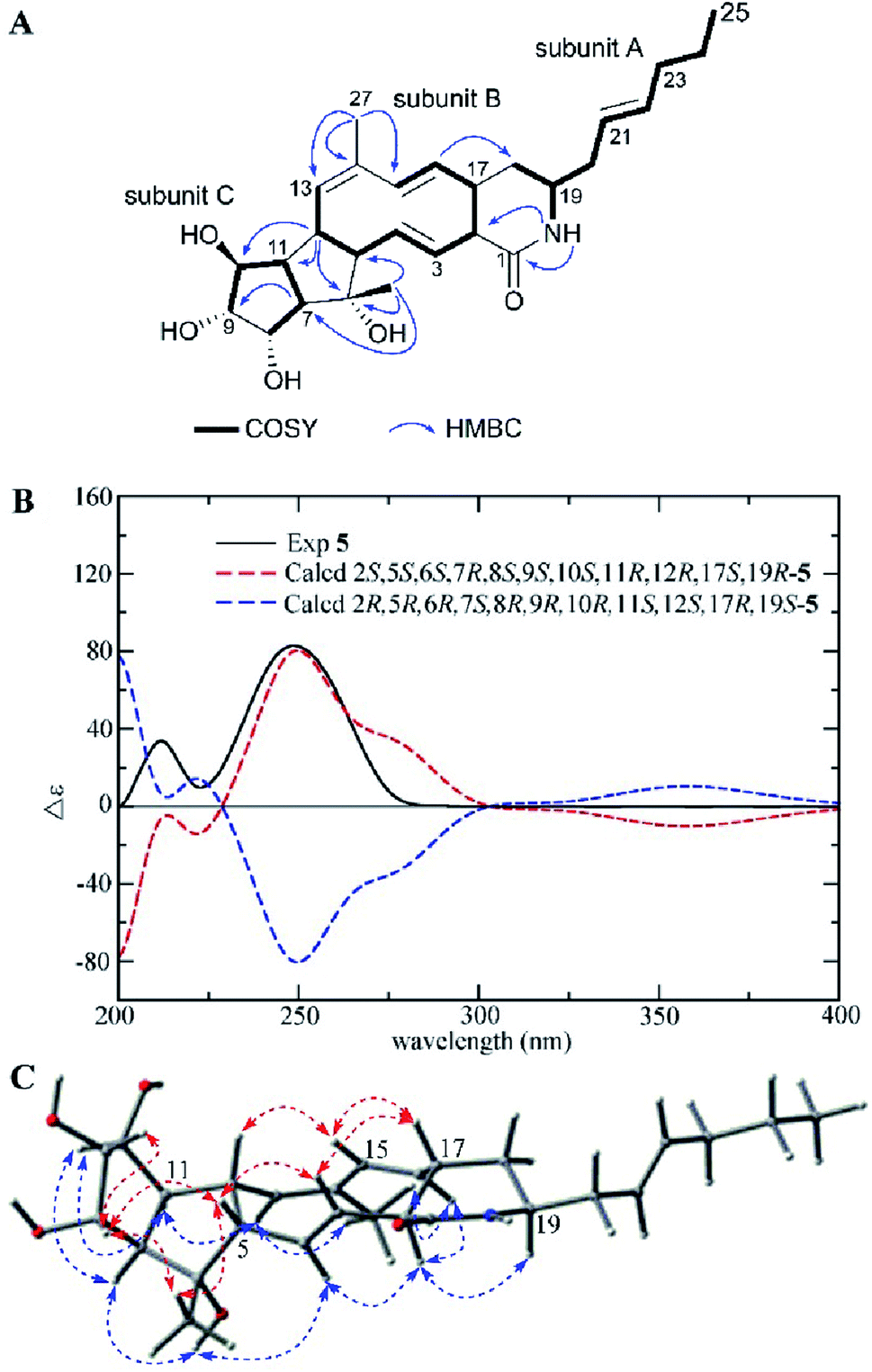 | ||
| Fig. 6 (A) COSY and key HMBC correlations of 5, (B) comparison of the experimental ECD spectra of 5 with the calculated ECD spectra, and (C) key NOEs observed in 5. | ||
The relative configuration of 5 was determined by detailed analysis of a NOESY experiment. NOE correlations between H-3/H-5, H-17; H-5/H3-26, H-8; H-8/H3-26, H-9 and H-15/H-12, H-17 positioned H-3, H-5, 6-CH3, H-8, H-9, H-12, H-15, and H-17 on the same side of the ring system, while the NOE cross peaks between H-2/H-4, H-16, H-19; H-4/6-OH; H-11/H-10, H-13 and H3-27/H-13, H-16 demonstrated H-2, H-4, 6-OH, H-7, H-10, H-11, H-13, 14-CH3, H-16, and H-19 were on the other face of the ring system (Fig. 6C).
The E configurations of double bonds Δ3,4, Δ15,16, and Z configuration of double bond Δ13,14 were established according to the J3,4 values (15.9 Hz), J3,4 values (16.4 Hz), and the NOE correlations between H3-27/H-13, respectively. The experimental ECD spectrum of 5 was conformed to the calculated ECD curve of 2S,5S,6S,7R,8S,9S,10S,11R,12R,17S,19R-5 (Fig. 6B). Thus, the structure including absolute configuration of 5 was determined and named heronamide K.
Heronamide L (6) was purified as a white powder owning a molecular formula of C27H41NO7 (eight degrees of unsaturation) as presented by the HRESIMS peak at m/z 492.2976 [M + H]+. The 1H NMR, 13C NMR, and HSQC spectra in DMSO-d6 (Table 2) revealed the presence of one amide carbonyl (δC 175.1), one nonprotonated olefinic carbon (δC 136.7), and three olefinic methines (δC/H 133.7/5.48, 133.1/5.38, and 126.6/5.35), satisfying three out of eight degrees of unsaturation. Therefore, 6 should have a pentacyclic skeleton to explain the remaining five degrees of unsaturation. Detailed analysis of 1H–1H COSY correlations provided substructures as shown in Fig. 7A. HMBC correlations from 3-OH to C-2, C-3, C-4, from H3-26 to C-5, C-6, C-7, from H3-27 to C-13, C-14, C-15, and from NH to C-1, C-2 further helped determine the planar structure of 6. The relative configuration of 6 was deduced by careful interpretation of the NOESY experiment (Fig. 7B). Despite the lack of a calculated ECD result, the absolute configuration of 6 could be determined by comparing the spectra data with 5 as well as analysis of the possible biosynthetic pathway (Scheme 1), which possessed high stereospecificity and deduced the same configurations of compounds 1–6. Thereby, the structure of 6 was established as shown.
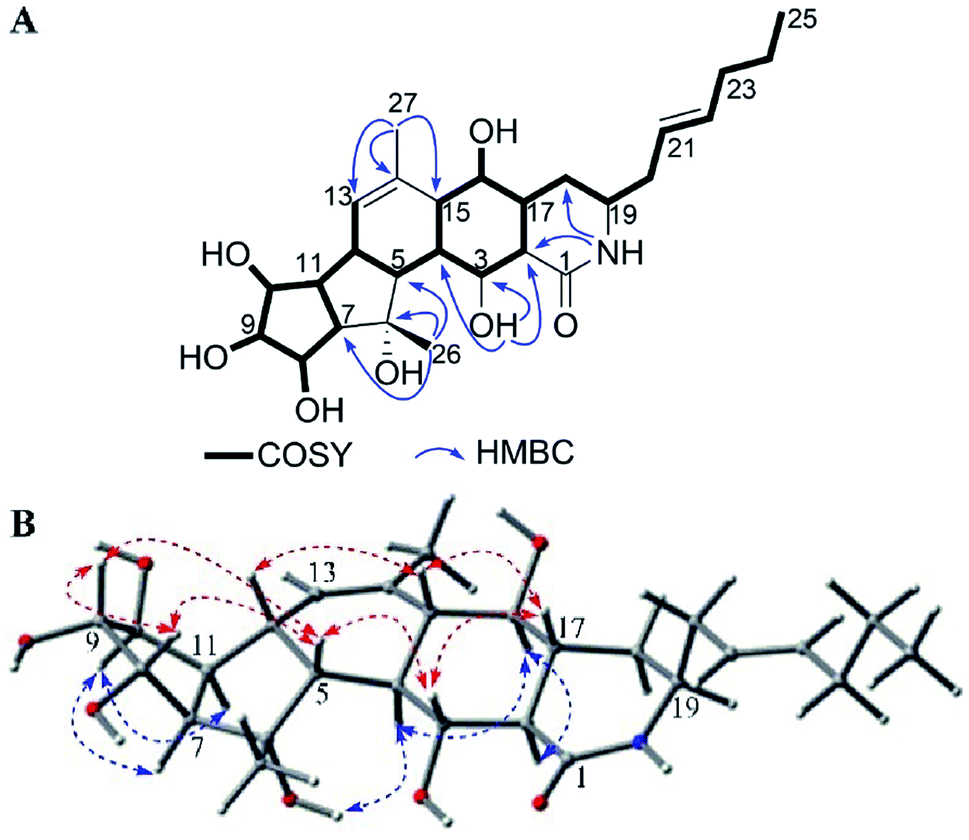 | ||
| Fig. 7 (A) COSY and key HMBC correlations of 6. (B) Key NOE correlations observed in 6. | ||
Niveamide B (10) possessed a molecular formula of C16H27NO3 deduced from HRESIMS peak at m/z 282.2075 [M + H]+. The 1H NMR, 13C NMR (Table 2) together with HSQC displayed one amide carbonyl carbon (δC 167.8), four olefinic methines (δC/H 131.4/5.40, 128.7/5.36, 127.9/5.36, and 126.2/5.32), two aliphatic methines (δC/H 47.8/3.67, 38.7/2.31), five methylenes (δC/H 36.2/2.06; 36.2/1.98; 35.7/2.23, 2.02; 33.5/1.92; and 21.5/1.32), three methyls (δC/H 22.1/1.76, 15.8/1.01, and 12.8/0.85). However, because of low sample quantity, the 13C NMR spectrum didn't present the carboxylic carbon signal at δC 176.6, which was supported by the HMBC correlations from H-2 (δH 2.31) and H3-14 (δH 1.01) to C-1 (δC 176.6). The 1H and13C NMR observed for 10 closely resembled the known compound niveamide (11)10 except for the presence of –CH2CH– and the absence of –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– moiety. 1H–1H COSY correlations between H3-14/H-2, H-2/H-3, and H-3/H-4 and HMBC correlations from H-2, H3-14 to C-1 demonstrated the structure of 10 and it was named as niveamide B.
C– moiety. 1H–1H COSY correlations between H3-14/H-2, H-2/H-3, and H-3/H-4 and HMBC correlations from H-2, H3-14 to C-1 demonstrated the structure of 10 and it was named as niveamide B.
The compound BE-14106 (8) and its deoxy analogue GT-32B (9) were reported to possess weak antibacterial, antifungal, and moderate cytotoxic activities.5,12 In 2016, BE-14106 was found to show potent growth inhibition against fission yeast cells with an MIC value of 0.50 μM compared to the clinically used amphotericin B (MIC = 0.27 μM).7 BE-14106-6 (7) was shown to exhibit moderate cytotoxicities.11 In this report, biological activities including antimicrobial activities against Bacillus subtilis, Staphylococcus aureus, Escherichia coli, Candida albicans, and Candida parapsilosis; cytotoxicities against BGC-823, H460, Ishikawa, SMMC-7721, and inhibitory effects on lipopolysaccharide-induced NO production in BV2 microglial cells of 1–6 and 8–11 were tested. However, except that compounds 8 and 9 showed weak cytotoxicities at 100 μM, no significant activity was detected for these compounds at 100 μg mL−1 for antimicrobial activities or 100 μM for cytotoxicities and NO production inhibitory activity.
Experimental section
General experimental procedures
Optical rotations were obtained using an Anton Paar MCP 200 polarimeter. Ultraviolet spectra were obtained with a Beckman Coulter DU 730 nucleic acid/protein analyzer. CD spectra were measured on a Biologic MOS-450 spectra polarimeter (Biologic Science, Claix, France). IR spectra were performed on a Bruker Tensor 27 FT-IR spectrophotometer (film). NMR spectra were recorded on a Bruker AV-600 spectrometer using tetramethylsilane as an internal standard, and the chemical shifts were recorded in δ values. HRESIMS were measured using a Bruker micro TOF-Q mass instrument (Bruker Daltonics, Billerica, MA, USA). ESIMS were performed on an Agilent 1290-6420 Triple Quadrupole LC-MS spectrometer. Silica gel (300–400 mesh, and 1200–1500 mesh, Qingdao Marine Chemical Ltd., Qingdao, China), Sephadex LH-20 (GE Healthcare Biosciences AB, Uppsala, Sweden), YMC*GEL ODS-A (S-50 μm, 12 nm) (YMC Co., Ltd., Kyoto, Japan), and MCI gel (CHP-20P, Mitsubishi Chemical Corp., Tokyo, Japan) were utilized for column chromatography. Semipreparative HPLC was performed using a ODS column (250 mm × 10 mm, 5 μm, YMC-ODS-A). Unless otherwise specified, all chemicals and solvents were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shenyang, China). Biological assays were analyzed using a microplate reader (BioTek Synergy H1, BioTek Instruments, Inc., Vermont, USA).Microbial material
The producing organism was isolated from a forest soil sample collected in Great Khingan Mountains, Northeastern of China in July 2003. The strain was assigned to be Streptomyces niveus based on morphological characteristics and 16S rRNA gene sequences. The Blast result showed that the sequence was most similar (99.78%) to the sequence of S. niveus (strain: NRRL 2466T, GenBank accession no. DQ442532). The strain (no. YIM 32862) was deposited in Yunnan Institute of Microbiology, Yunnan University, China.Fermentation, extraction, and isolation
The strain S. niveus was inoculated to a 100 mL seed medium consisting of 4 g L−1 yeast extract, 4 g L−1 glucose, 5 g L−1 malt extract, and 1.0 mL of trace element solution at a pH of 7.2 without adjustment. The flasks were cultivated for 2 days on a rotary shaker (180 rpm) at 28 °C, followed by inoculation to a fermentation medium (10 g L−1 soybean meal, 2 g L−1 peptone, 20 g L−1 glucose, 5 g L−1 soluble starch, 2 g L−1 yeast extract, 4 g L−1 NaCl, 0.5 g L −1 K2HPO4, 0.5 g L−1 MgSO4·7H2O, and 2 g L−1 CaCO3, pH = 7.8) with a 10% volume. The fermentation was incubated at 28 °C for 7 days on a rotary shaker at 180 rpm.The completed fermentation culture (140 L) was centrifuged (4000 rpm, 5 min) into supernatant and mycelium, and the supernatant was extracted by EtOAc four times and evaporated to yield crude extract 60 g. The dried extract was subjected to silica gel (300–400 mesh) column chromatography eluting with a gradient CH2Cl2–MeOH solvent system (from 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 10:1, and finally 4:1) to give seven fractions: Fr. A–G. Fractions A and B were filtrated to obtain compound 9 (100 mg) and compound 8 (35.0 mg), respectively. Fraction C was subjected to a Sephadex LH-20 column (MeOH) to produce nine sub-fractions, Fr. C1–C9. Fr. C3 was put on a silica gel (1200–1500 mesh) for column chromatography (CH2Cl2–MeOH, 20:1) and further purified by semipreparative HPLC (MeOH–H2O 65:35) to yield 10 (3.9 mg) and 11 (14.7 mg). Fraction F was subjected to Sephadex LH-20 chromatography (MeOH) to produce 2 sub-fractions, Fr. F1 and F2. Sub-fraction F2 was divided into six fractions, F2.1–F2.6 by silica gel (1200–1500 mesh) column chromatography (CH2Cl2–MeOH, 8:1). Sub-fraction F2.4 was further put on silica gel (1200–1500 mesh) eluting with CH2Cl2–acetone (1:1) followed by semipreparative HPLC (MeOH–H2O 55:45) to yield 1 (22.0 mg), 7 (20.0 mg). Sub-fraction F2.5 was subjected to silica gel (1200–1500 mesh) column chromatography (CH2Cl2–acetone, 1:1) and further purified by semipreparative HPLC (MeOH–H2O 65:35) to give 2 (5.6 mg). Fraction G was subjected to a MCI gel CHP-20P column and eluted with MeOH to produce 6 sub-fractions, Fr. G1–G6. Fr. G2–G5 was subjected to silica gel (1200–1500 mesh) column chromatography (CH2Cl2–MeOH, 6:1 or 8:1) separately and further separated by semipreparative HPLC (MeOH–H2O 65:35 or 60:40) to afford 6 (5.0 mg), 3 (4.5 mg), 4 (2.0 mg), and 5 (9.4 mg), respectively.
:1 to 10:1, and finally 4:1) to give seven fractions: Fr. A–G. Fractions A and B were filtrated to obtain compound 9 (100 mg) and compound 8 (35.0 mg), respectively. Fraction C was subjected to a Sephadex LH-20 column (MeOH) to produce nine sub-fractions, Fr. C1–C9. Fr. C3 was put on a silica gel (1200–1500 mesh) for column chromatography (CH2Cl2–MeOH, 20:1) and further purified by semipreparative HPLC (MeOH–H2O 65:35) to yield 10 (3.9 mg) and 11 (14.7 mg). Fraction F was subjected to Sephadex LH-20 chromatography (MeOH) to produce 2 sub-fractions, Fr. F1 and F2. Sub-fraction F2 was divided into six fractions, F2.1–F2.6 by silica gel (1200–1500 mesh) column chromatography (CH2Cl2–MeOH, 8:1). Sub-fraction F2.4 was further put on silica gel (1200–1500 mesh) eluting with CH2Cl2–acetone (1:1) followed by semipreparative HPLC (MeOH–H2O 55:45) to yield 1 (22.0 mg), 7 (20.0 mg). Sub-fraction F2.5 was subjected to silica gel (1200–1500 mesh) column chromatography (CH2Cl2–acetone, 1:1) and further purified by semipreparative HPLC (MeOH–H2O 65:35) to give 2 (5.6 mg). Fraction G was subjected to a MCI gel CHP-20P column and eluted with MeOH to produce 6 sub-fractions, Fr. G1–G6. Fr. G2–G5 was subjected to silica gel (1200–1500 mesh) column chromatography (CH2Cl2–MeOH, 6:1 or 8:1) separately and further separated by semipreparative HPLC (MeOH–H2O 65:35 or 60:40) to afford 6 (5.0 mg), 3 (4.5 mg), 4 (2.0 mg), and 5 (9.4 mg), respectively.
ε) 253 (4.31), 217 (4.73) nm; IR (film) νmax 3351, 3023, 2962, 2924, 1667, 1433, 1377, 1102, 1074, and 974 cm−1; CD (0.5 mg mL−1, MeOH) λmax (Δε) 210 (85.22), 256 (−105.99); 1H NMR and 13C NMR data, see Table 1; ESIMS m/z 456 [M + H]+, 478 [M + Na]+; HRESIMS m/z 456.2758 [M + H]+ (calcd for C27H38NO5, 456.2750).ε) 268 (4.50), 222 (5.19) nm; IR (film) νmax 3379, 2958, 2923, 1678, 1405, 1369, 1102, 1047, and 969 cm−1; CD (0.9 mg mL−1, MeOH) λmax (Δε) 229 (−18.39), 239 (22.10), and 266 (−229.20); 1H NMR and 13C NMR data, see Table 1; ESIMS m/z 474 [M + H]+, 496 [M + Na]+; HRESIMS m/z 474.2861 [M + H]+ (calcd for C27H40NO6, 474.2856).ε) 230 (5.20) nm; IR (film) νmax 3361, 2957, 2928, 1598, 1411, 1286, 1071, and 974 cm−1; CD (1.25 mg mL−1, MeOH) λmax (Δε) 247 (−42.88), 274 (141.07); 1H NMR and 13C NMR data, see Table 1; ESIMS m/z 474 [M + H]+, 496 [M + Na]+; HRESIMS m/z 474.2870 [M + H]+ (calcd for C27H40NO6, 474.2856).ε) 225 (4.68) nm; IR (film) νmax 3351, 2958, 2924, 1658, 1596, 1421, 1072, and 972 cm−1; CD (0.6 mg mL−1, MeOH) λmax (Δε) 218 (−42.13), 253 (52.73); 1H NMR and 13C NMR data, see Table 1; ESIMS m/z 474 [M + H]+, 496 [M + Na]+; HRESIMS m/z 474.2869 [M + H]+ (calcd for C27H40NO6, 474.2856).ε) 247 (4.47), 204 (5.00) nm; IR (film) νmax 3382, 2958, 2928, 1651, 1435, 1332, 1111, 1079, 975 cm−1; CD (0.5 mg mL−1, MeOH) λmax (Δε) 212 (33.91), 249 (82.86); 1H NMR and 13C NMR data, see Table 2; ESIMS m/z 458 [M + H]+, 480 [M + Na]+; HRESIMS m/z 458.2912 [M + H]+ (calcd for C27H40NO5, 458.2906).ECD calculations
The conformational search was performed by CONFLEX,13,14 based on molecular mechanics with MMFF94S force fields. Selected conformers whose energies were 3 kcal mol−1 higher than the conformer with the lowest energy were further optimized by the density functional theory method at the B3LYP/6-31G (d) level in Gaussian 09 program package,15 which was further checked by frequency calculation and resulted in no imaginary frequencies. The ECDs of the conformers of 1–5 were then calculated by the TDDFT method at B3LYP/6-311 ++ G (2d, p) level with the CPCM model in methanol solution. The final ECD curves were generated based on the rotatory strengths by SpecDis16 and calculated from the spectra of individual conformers according to their contribution to the Boltzmann weighting. The theoretical spectra have been corrected based on the UV correction.Antimicrobial assay
A micro broth dilution assay was used to evaluate the antimicrobial activity of 1–6 and 8–11 against Bacillus subtilis ATCC 6633, Staphylococcus aureus ATCC 25923, Escherichia coli ATCC 25922, Candida albicans ATCC MYA-2876, and Candida parapsilosis ATCC 22019 as previously reported.17 Ciprofloxacin and amphotericin B were used as positive controls against bacteria and fungi, respectively.Cytotoxicity assay
The human gastric carcinoma cell line (BGC-823), human large-cell lung carcinoma cell line (H460), human endometrial carcinoma cell line (Ishikawa), and human hepatocellular carcinoma cell line (SMMC-7721) were used to evaluate the cytotoxic effects of compounds 1–6 and 8–11 using the MTT method as previously reported.17 Adriamycin was assayed as a positive control.Inhibition of NO production assay
The inhibitory effects on lipopolysaccharide-induced NO production in BV2 microglial cells was measured using the Griess reaction to indicate NO production and examined according to a previously described protocol.18,19 The cell culture and data analysis of NO production inhibition followed a previously published method.20 Dexamethasone was used as the positive control.Conclusions
In summary, we isolated and characterized six new polyene macrolactams, heronamides G–L (1–6), one new polyenoic acid derivative, niveamide B (10), and four known compounds, BE-14106-6 (7), BE-14106 (8), GT32-B (9), and niveamide (11), from the fermentation broth of Streptomyces niveus. The absolute configurations of compounds 1–6 were determined by calculated ECD spectra and analysis of the possible biosynthetic pathways. However, compounds 1–6 and 8–11 did not exhibit any significant antimicrobial activities, cytotoxicities, or inhibitory effects on lipopolysaccharide-induced NO production in BV2 microglial cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by National Natural Science Foundation of China (No. 81573327, 31460005, and 81771167) and the Fundamental Research Funds for the Central Universities, China (No. N172004004 and N172008008).Notes and references
- K. Fujita, R. Sugiyama, S. Nishimura, N. Ishikawa, M. A. Arai, M. Ishibashi and H. Kakeya, J. Nat. Prod., 2016, 79, 1877–1880 CrossRef CAS PubMed.
- T. J. Booth, S. Alt, R. J. Capon and B. Wilkinson, Chem. Commun., 2016, 52, 6383–6386 RSC.
- P. Yu, A. Patel and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 13518–31523 CrossRef CAS PubMed.
- K. Katsuhisa, N. Shigeru, S. Hajime, K. Hisao and S. Hiroyuki, J. Antibiot., 1992, 45, 868–874 CrossRef.
- T. Isami, O. Yashushi, N. Yashushi, O. Keiko and M. Tamio, J. Antibiot., 1997, 50, 186–188 CrossRef.
- H. Jorgensen, K. F. Degnes, A. Dikiy, E. Fjaervik, G. Klinkenberg and S. B. Zotchev, Appl. Environ. Microbiol., 2010, 76, 283–293 CrossRef PubMed.
- R. Raju, A. M. Piggott, M. M. Conte and R. J. Capon, Org. Biomol. Chem., 2010, 8, 4682–4689 CAS.
- W. Zhang, S. Li, Y. Zhu, Y. Chen, Y. Chen, H. Zhang, G. Zhang, X. Tian, Y. Pan, S. Zhang, W. Zhang and C. Zhang, J. Nat. Prod., 2014, 77, 388–391 CrossRef CAS PubMed.
- R. Sugiyama, S. Nishimura, N. Matsumori, Y. Tsunematsu, A. Hattori and H. Kakeya, J. Am. Chem. Soc., 2014, 136, 5209–5212 CrossRef CAS PubMed.
- Y. Zhu, W. Zhang, Y. Chen, C. Yuan, H. Zhang, G. Zhang, L. Ma, Q. Zhang, X. Tian, S. Zhang and C. Zhang, ChemBioChem, 2015, 16, 2086–2093 CrossRef CAS PubMed.
- L. Li, Y. Cai, Y. Jiang, J. Liu, J. Ma, C. Yuan, Y. Mu, L. Han and X. Huang, Bioorg. Med. Chem. Lett., 2016, 26, 1599–1604 CrossRef CAS PubMed.
- H. Jørgensen, K. F. Degnes, H. Sletta, E. Fjaervik, A. Dikiy, L. Herfindal, P. Bruheim, G. Klinkenberg, H. Bredholt, G. Nyga, S. O. Døskeland, T. E. Ellingsen and S. B. Zotchev, Chem. Biol., 2009, 16, 1109–1121 CrossRef PubMed.
- H. Goto and E. Osawa, J. Am. Chem. Soc., 1989, 111, 8950–8951 CrossRef CAS.
- H. Goo and E. Osawa, J. Chem. Soc., Perkin Trans. 2, 1993, 187–198 Search PubMed.
- G. Mazeo, E. Santoro, A. Andolfi, A. Cimmino, P. Troselj, A. G. Petrovic, S. Superchi, A. Evidente and N. Berova, J. Nat. Prod., 2013, 76, 588–599 CrossRef PubMed.
- T. Bruhn, A. Schaumloeffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- Q. Ma, L. Han, X. Bi, X. Wang, Y. Mu, P. Guan, L. Li and X. Huang, Phytochemistry, 2016, 131, 150–157 CrossRef CAS PubMed.
- R. A. Hunter, W. L. Storm, P. N. Coneski and M. H. Schoenfisch, Anal. Chem., 2013, 85, 1957–1963 CrossRef CAS PubMed.
- Z. Li, Z. Li, J. Bai, D. Meng, N. Li, Y. Pei, F. Zhao and H. Hua, J. Nat. Prod., 2014, 25, 792–799 CrossRef PubMed.
- J. Xu, C. Yuan, G. Wang, J. Luo, H. Ma, L. Xu, Y. Mu, Y. Li, N. Seeram, X. Huang and L. Li, J. Agric. Food Chem., 2018, 66, 571–580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR, HRMS, and IR spectra for 1–6, and 10. See DOI: 10.1039/c8ra02167h |
| This journal is © The Royal Society of Chemistry 2018 |