
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntioxidant properties of flavonoid derivatives and their hepatoprotective effects on CCl4 induced acute liver injury in mice†
Cen Xiang‡
a,
Yuou Teng‡ a,
Chaoran Yaoa,
Xuehui Lia,
Menglin Caoa,
Xuzhe Lia,
Guojun Pana,
Kui Lua,
Hervé Galons*b and
Peng Yu*a
a,
Chaoran Yaoa,
Xuehui Lia,
Menglin Caoa,
Xuzhe Lia,
Guojun Pana,
Kui Lua,
Hervé Galons*b and
Peng Yu*a
aChina International Science and Technology Cooperation Base of Food Nutrition/Safety and Medicinal Chemistry, State Key Laboratory of Food Nutrition and Safety, Tianjin University of Science and Technology, Tianjin 300457, China. E-mail: yupeng@tust.edu.cn
bUCTBS, INSERM U1022, Université Paris Descartes, 4 Avenue de l'Observatoire 75006, France
First published on 24th April 2018
Abstract
Excessive accumulation of free radicals in the body can cause liver damage, aging, cancer, stroke, and myocardial infarction. Anastatin B, a skeletal flavonoid, was reported to have antioxidant and hepatoprotective effects. Anastatin B derivatives, compound 1 and 2, were synthesized by our group previously. In this study, their antioxidant activity and hepatoprotective mechanism were studied using chemical evaluation methods, a cellular model of hydrogen peroxide (H2O2)-induced oxidative damage, and a mouse model of carbon tetrachloride (CCl4)-induced liver injury. Results from the chemical evaluation suggested that both compounds had good antioxidant power and radical scavenging ability in vitro. MTT assay showed that both compounds had cytoprotective activity in H2O2-treated PC12 cells. Moreover, their hepatoprotective activities evaluated using a mouse model of CCl4-induced liver injury that compared with the model group, pretreatment with compound 1 and 2 significantly decreased alanine transaminase (ALT), aspartate transaminase (AST), lactate dehydrogenase (LDH), and malondialdehyde (MDA) levels; reduced the liver tissue damage; and increased glutathione content. However, compound 2 was a more effective hepatoprotectant than compound 1 was. Finally, the amount of TNF-α and cytochrome P450 2E1 (CYP2E1) were significantly downregulated in compound 1 and 2 pretreatment groups. Collectively, our findings demonstrate that both compounds have potential antioxidant activity and hepatoprotective effect in vitro and in vivo. Further chemo-biological study and investigation of the compounds' enzymatic targets are ongoing.
Introduction
Accumulation of ROS in our body could disturb the metabolic redox balance causing detrimental effects that finally lead to liver injury (liver fibrosis and cirrhosis), neurodegenerative damage (Alzheimer's disease, Parkinson's disease, and Huntington's disease), aging, cancer, and diabetes.1–8 In the last several decades, antioxidants have generated considerable interest as potential therapeutic drugs against a wide variety of chronic diseases.9–14 Excessive accumulation of free radicals in the liver can lead to liver damage, fibrosis, and cirrhosis, and can eventually cause liver cancer. Consequently, antioxidants have been proposed as promising compounds for the prevention and treatment of hepatic damage.15Free radicals contain unpaired electrons, and they are generated by the normal metabolic pathways in the body. They mainly act in the body by targeting unsaturated fatty acids in biomembranes, and cause membrane lipid peroxidation (a hallmark of hepatotoxicity), decrease membrane fluidity, suppress enzyme and receptor activity, and damage membrane proteins, which finally triggers cell inactivation and death. Therefore, free radical scavenging is one of the major antioxidation mechanisms inhibiting the chain reaction of lipid peroxidation.16 Antioxidant capacity can be reflected by the ability to inhibit or remove free radicals, such as total reducing antioxidant power, and scavenging of ABTS˙+, DPPH˙, and other radicals.17–19
PC12 cells model of H2O2-induced oxidative-damage is useful for evaluating the properties of antioxidants in vitro.20 H2O2, a major ROS with high cellular membrane permeability, causes lipid peroxidation and DNA damage in cells. It is often used as a toxicant to mimic in vitro oxidative stress-induced injury. PC12 cell line was derived from a rat pheochromocytoma of the adrenal medulla, and is a model system for neurological and neurochemical studies.21
CCl4-induced hepatic injury is an experimental model widely used for hepatoprotective drug screening.22,23 In damaged liver cells, cell membrane integrity is damaged and ALT and AST leak into the blood stream leading to an increase in their plasma levels. CCl4 targets the unsaturated fatty acids in biomembranes forming lipid peroxides such as MDA, releasing LDH, and decreasing cellular GSH levels. Therefore, AST, ALT, MDA, LDH, and GSH levels can indirectly reflect the extent of liver damage caused by CCl4. The main mechanism of CCl4-induced liver injury involves CCl4 metabolites and free radicals. CCl4 is catalyzed by CYP2E1 to produce unstable free radicals of trichloro-methyl (˙CCl3), proxyl trichloromethyl (˙OOCCl3), and reactive oxygen species (ROS).24
Flavonoids have many bioactivities, such as antioxidant, antitumorigenic, antidiabetic and anti-inflammatory activities.25–28 Flavonoids can prevent injury caused by free radicals and display potent antioxidant activity in vitro by the mechanisms including direct scavenging of reactive oxygen species, activation of antioxidant enzymes, inhibition of oxidases, et al.24,29–31 The antioxidant capacities of many flavonoids are much stronger than those of vitamins C and E.31 Flavonoids are thought to have health-promoting properties due to their high antioxidant capacity in vivo.32,33
Anastatin B, a skeletal flavonoid compound that was isolated from the desert plant Anastatica hierochuntica, has shown hepatoprotective activity in injured primary rat hepatocytes.34 Compound 1 and 2, two anastatin B analogs, were first synthesized in our laboratory (Fig. 3A).35 This study was designed to investigate the protective effects of compound 1 and 2 by using chemical evaluation assays in vitro, a PC12 cell model of H2O2-induced oxidative damage, and a mouse model of CCl4-induced liver injury.
Materials and methods
Cell lines and culture conditions
PC12 cell line was obtained from the Shanghai Institutes of Biological Sciences (Shanghai, China). Cells were grown at 37 °C in RPMI-1640 supplemented with 10% fetal bovine serum, 2.05 mM glutamine, and 1% penicillin/streptomycin in a humidified atmosphere containing 5% CO2. The medium was replaced once every third day.MTT assay for H2O2-treated PC12 cells
H2O2-induced oxidative-damage PC12 cell model was established and the cell viability was evaluated by MTT assay as a measure of the antioxidant activity of test compounds31 gallic acid (GA), a well-known potent antioxidant, was used as a positive control in MTT assay. For each treatment, the mean cell viability was calculated from three independent experiments.Flow cytometric analysis of apoptosis
Apoptotic cells were assayed by using the Annexin-V-FITC Apoptosis Detection Kit (BD Biosciences, USA) according to the manufacturer's instruction. H2O2-induced oxidative-damage PC12 cell model was established and it was treated with DMSO, GA, compound 1 or compound 2 (10 μM) for 0.5 h. Cells were then harvested, washed twice with ice-cold PBS and resuspended in 1× binding buffer to reach a final concentration of 1 × 106 cells per mL. Cells were stained by adding 5 μL of Annexin-V-FITC and 5 μL of PI (50 μg mL−1), sit for 15 min at room temperature in the dark and analyzed by flow cytometry.Antioxidant activity by FRAP assay
FRAP assay measures the antioxidant capacity to reduce ferric 2,4,6-tripyridyl-s-triazine (TPTZ) complex to its ferrous form. Antioxidant activity by FRAP assay of compound 1 and 2 were performed as described previously. The vitamin C (Vc) standard was diluted to different concentrations (100–600 mg mL−1) for the experiment. The ferric-reducing antioxidant power in the reaction medium was calculated from the calibration curve derived from a serial dilution of the Vc standard. Equivalent amounts of Vc for the test compounds were calculated from three independent experiments.Antioxidant activity by ABTS˙+ assay
The ABTS radical cation assay is one of the most commonly used methods for screening and evaluating antioxidants currently. Radical scavenging activities of test compounds were measured using a modified ABTS˙+ assay. EC50 values for the test compound were calculated from three independent experiments by nonlinear regression using Graph Pad Prism 5.0.Antioxidant activity by DPPH˙ assay
For the DPPH radical scavenging activity assay, different concentrations of test compounds (50 μL, 0.02–20 mM) were incubated with 50 μL DPPH˙ (78.8 mM) at 37 °C for 30 min and absorption measured at 540 nm. EC50 values were calculated as described above.Animals and experimental design
Kunming mice (male, 4 weeks) were obtained from PLA Military Academy of Medical Sciences Laboratory Animal Center (Beijing, China). All experiment involving animal use were performed with the approval or the institutional animal care and use committee at Tianjin University or Science and Technology. The mice were housed at 25 ± 2 °C under a 12 h light/12 h dark cycle with access to food and water ad libitum. After acclimation for 1 week, the animals were randomly divided into nine groups (n = 8): normal (distilled water without CCl4 injection), model (distilled water and CCl4 injection), compound 1 treatment groups (10, 30, and 100 mg kg−1 of compound 1 and CCl4 injection), compound 2 treatment groups (10, 30, and 100 mg kg−1 of compound 2 and CCl4 injection), and positive control group (200 mg kg−1 of biphenyldicarboxylate pills (BP) and CCl4 injection). Compounds 1 and 2, and BP were dissolved in distilled water. Distilled water, compounds 1 and 2, or BP was orally administered for 10 consecutive days before CCl4 injection. One hour after the final drug treatment, severe, acute liver injury was induced by i.p. injection with CCl4 (0.25% (v/v) peanut oil mixture; 0.3 mL). The normal group was intraperitoneally injected with peanut oil. All mice were starved for 20 h afterwards, and were then sacrificed for collection of whole blood and liver samples. Liver tissue was removed, immediately weighed, and then fixed or stored in 10% neutral buffered formalin, or frozen for histopathological analysis and determination of biochemical parameters.All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “Tianjin University of Science and Technology” and approved by the Animal Ethics Committee of “Tianjin University of Science and Technology”.
Histological analysis
For evaluation of hepatotoxicity, the tissue fixed in 10% formalin was embedded in paraffin and cut into 5 μm-thick sections for histomorphological examination. After drying, hepatic tissue section slides were stained with hematoxylin–eosin (H&E). The stained sections were visualized using a microscope.Biochemical analysis
Serum was analyzed for ALT, AST, LDH, MDA, and TNF-α levels according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Glutathione (GSH) levels were determined in hepatic homogenates using a commercial kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The results were corrected for their protein content.Western blot
Briefly, liver tissues were homogenized in cold saline. Equal amount of protein were separated by 10% SDS-PAGE and electro-transferred onto a PVDF membrane. After blocking with 5% (w/v) nonfat milk, the membranes were incubated with CYP2E1 antibodies (Tianjin Sungene Biotech Co. Ltd., China; diluted 1000 fold) overnight at 4 °C, and then incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h. The immunoblots were visualized using an Odyssey infrared imaging system. An anti-tubulin antibody (Sigma-Aldrich; diluted 2000 fold) was used as a control for equal loading. The densities of protein bands were determined using ImageJ software.Statistical analysis
All data were expressed as mean ± SD. Results were analyzed by one-way analysis of variance (ANOVA), and significant differences were determined by post-hoc Tukey test using SPSS 21.0 software. #P < 0.05 compared to control group; ##P < 0.01 compared to control group; *P < 0.05 compared to model group; and **P < 0.01 compared to model group. P value < 0.05 was considered as significant.Results
Antioxidant ability of compounds 1 and 2 in vitro
Antioxidant ability was examined using the ABTS˙+ and DPPH˙ scavenging and total reduction capacity assays (Table 1). Vc was taken as the standard antioxidant for the chemical evaluation method. In the ABTS˙+ assay, EC50 values of Vc, compounds 1 and 2 were 2.40, 2.40, and 2.74 mM, respectively. Vc, compounds 1 and 2 at 0.13, 0.13, and 0.16 mM, respectively, were able to inhibit 50% of DPPH radicals in the assay. Similar patterns of antioxidant activity were observed in the FRAP assay.| Test compound | Scavenging activity (EC50, mM) | Equivalent amount of Vc (mg mmol−1) | |
|---|---|---|---|
| ABTS˙+ | DPPH˙ | FRAP | |
| Compound 1 | 2.4 ± 0.16 | 0.13 ± 0.02 | 221.56 ± 25.64 |
| Compound 2 | 2.74 ± 0.13 | 0.16 ± 0.03 | 193.66 ± 17.96 |
| Vc | 2.4 ± 0.03 | 0.04 ± 0.01 | 192.19 ± 12.33 |
Compounds 1 and 2 protect PC12 cells against H2O2-induced injury
To investigate whether cytoprotection could be ascribed to the antioxidant effect, we assessed the effect of compounds 1 and 2 on H2O2-mediated PC12 cell injury. As shown in Fig. 1B, compounds 1 and 2 at 0.1–10 μM exhibited no or low cytotoxicity. However, the viability of PC12 cells when exposed to H2O2 (100 μM, 2 h) significantly decreased to 31.67% as compared to that reported for the control group (100%) (Fig. 1C). When PC12 cells were pretreated with compounds 1 in the presence of 100 μM H2O2, a reversal in the decrease of cell viability caused by H2O2 was observed and the cell viability increased to 90.4% and 80.2% at doses of 3 and 10 μM, respectively. Compound 2 showed a stronger cytoprotective effect in PC12 cells with H2O2-induced injury than compound 1 did and significantly increased cell viability from 0.3 to 10 μM. The positive control GA exhibited no cytotoxicity in PC12 cells. The viability of the gallic acid-pretreated PC12 cells was 63.59% at 10 μM. The results in Fig. 1 suggest that both test compounds, especially compound 2 at low concentration, have better antioxidant activity than gallic acid did.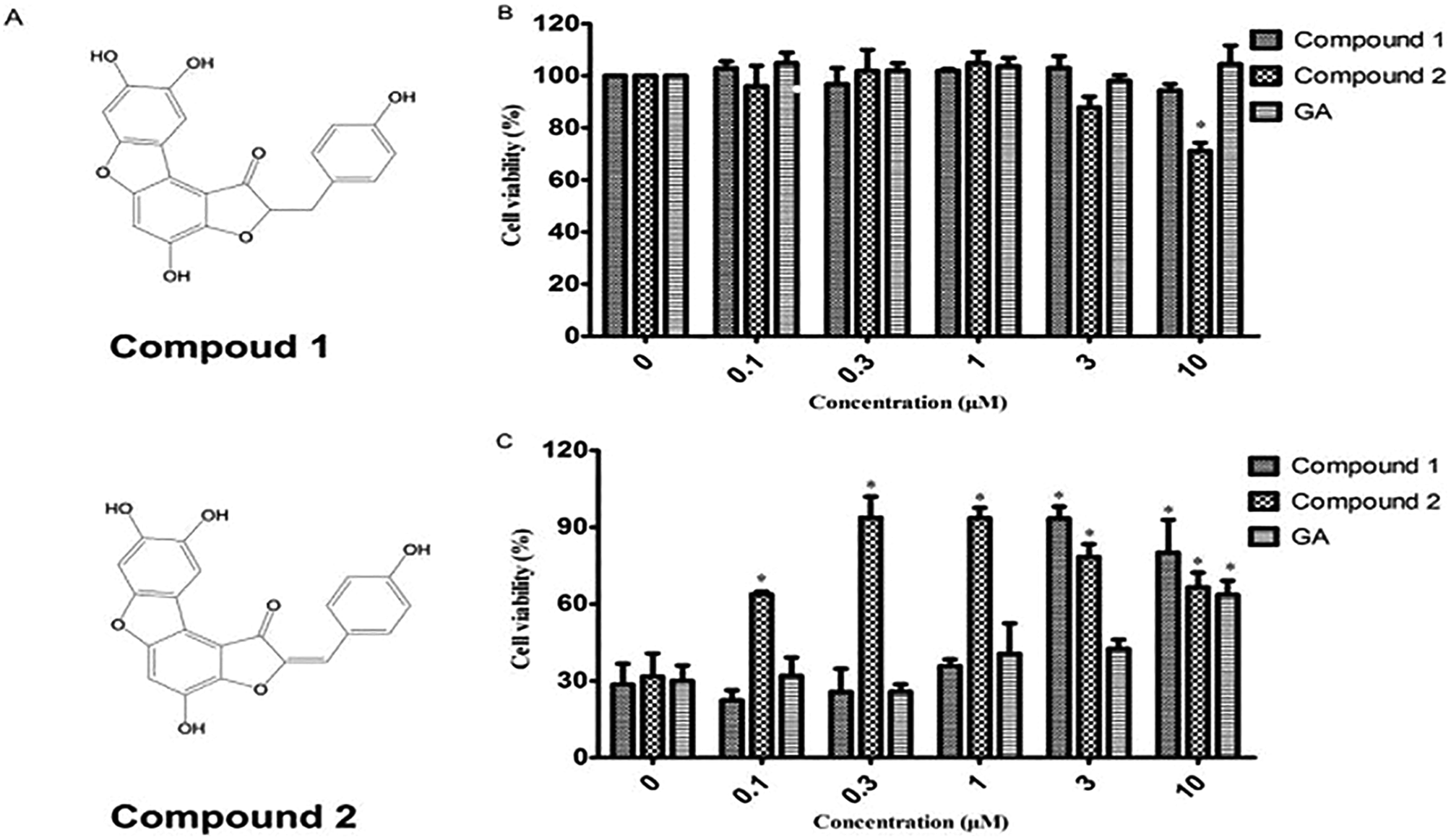 | ||
| Fig. 1 The in vitro antioxidant activities of compound 1 and 2 against H2O2-damaged PC12 cells. Cell viability of compound 1 and 2 (A)-treated PC12 cells was evaluated by MTT assay with or without H2O2-damaged (B and C). IC50 was calculated to reflect the cytotoxicity of compound 1 and 2. The DMSO-treated controls were assigned a cell viability value of 100%. Gallic acid was used as a positive control. *P < 0.05 compared to DMSO-treated control cells. | ||
Compounds 1 and 2 protect PC12 cells against H2O2-induced apoptosis
It was obvious that treatment with H2O2 caused a typical apoptotic morphology change in PC12 cells, such as cell shrinkage and/or blebbing. As shown in Fig. 2A, the apoptotic cells were clearly observed in PC12 cells after treatment with H2O2 for 2 h. Nevertheless, the PC12 cells pretreatment with GA, compound 1 or 2, the normal cells was increased. Flow cytometric analysis also showed that GA, compound 1 or 2 can protect PC12 cells against H2O2-induced injury apoptosis, when the cells were double labelled with annexin-V-FITC and PI (Fig. 2B). After pretreatment with GA, compound 1 or 2, the apoptotic rates (annexin V+) in H2O2-treated PC12 cells were declined to 39.4%, 1.2% and 26.0% of the total cells (10 μM, 0.5 h) whereas 65.4% of the total cells were observed as apoptotic cells in the control. Fig. 2 suggest that both test compounds, especially compound 1 at low concentration, have better antiapoptotic activity than gallic acid did.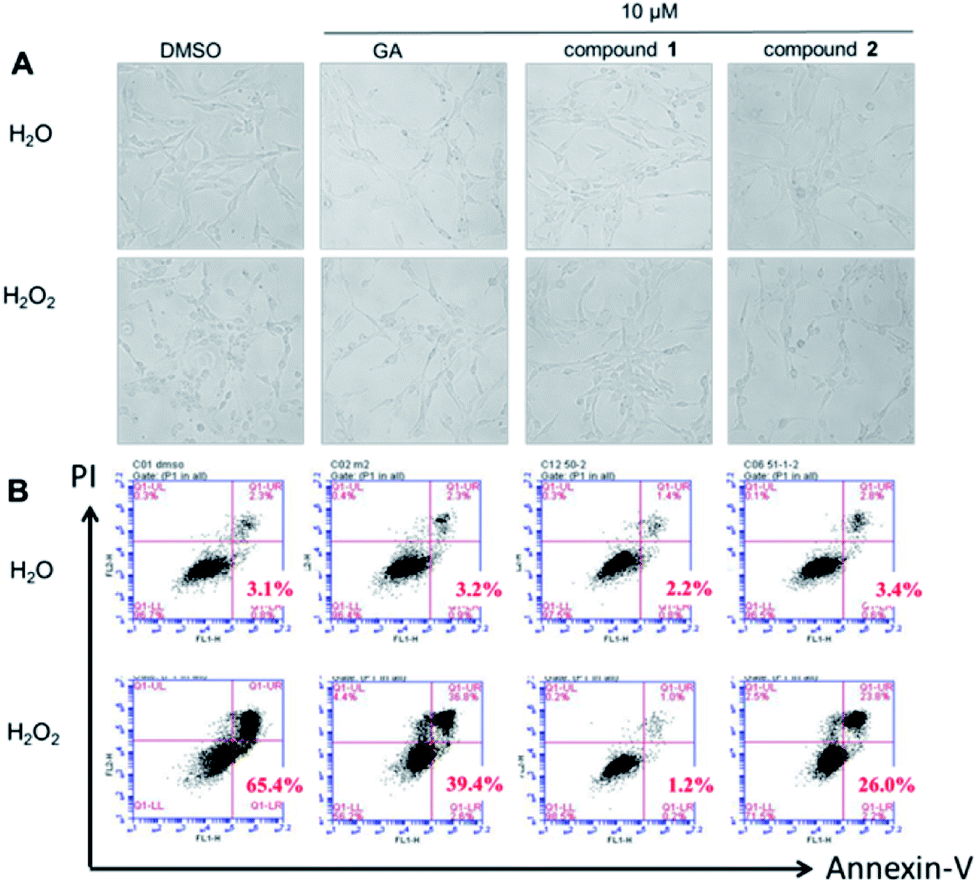 | ||
| Fig. 2 The in vitro antiapoptotic activities of compound 1 and 2 against H2O2-damaged PC12 cells. (A) Phase contrast images of H2O2-damaged PC12 cells after pretreatment with GA, compound 1 or 2 (10 μM) for 0.5 h (B) apoptosis was evaluated by annexin-V and PI staining of H2O2-damaged PC12 cells cultured in the GA, compound 1 or 2 of 10 μM for 0.5 h. | ||
Compounds 1 and 2 ameliorated CCl4-induced liver injury
Next, to determine the protective effects of compounds 1 and 2 on CCl4-induced hepatotoxicity, mice were pretreated with different doses of compounds 1 and 2 by gavage once daily for ten days. H&E staining showed that CCl4 administration caused massive necrosis in the liver. Pretreatment with compounds 1 and 2 (10, 30, and 100 mg kg−1) ameliorated CCl4-induced hepatic necrosis (Fig. 3A). Both AST and ALT levels, markers of liver injury, dramatically increased after CCl4 treatment. Consistent with the histological data, pretreatment with compounds 1 and 2 significantly reduced serum AST and ALT levels in CCl4-treated mice in a dose-dependent manner (Fig. 3B). These results suggest that pretreatment with compounds 1 and 2 effectively protected the liver against CCl4 toxicity.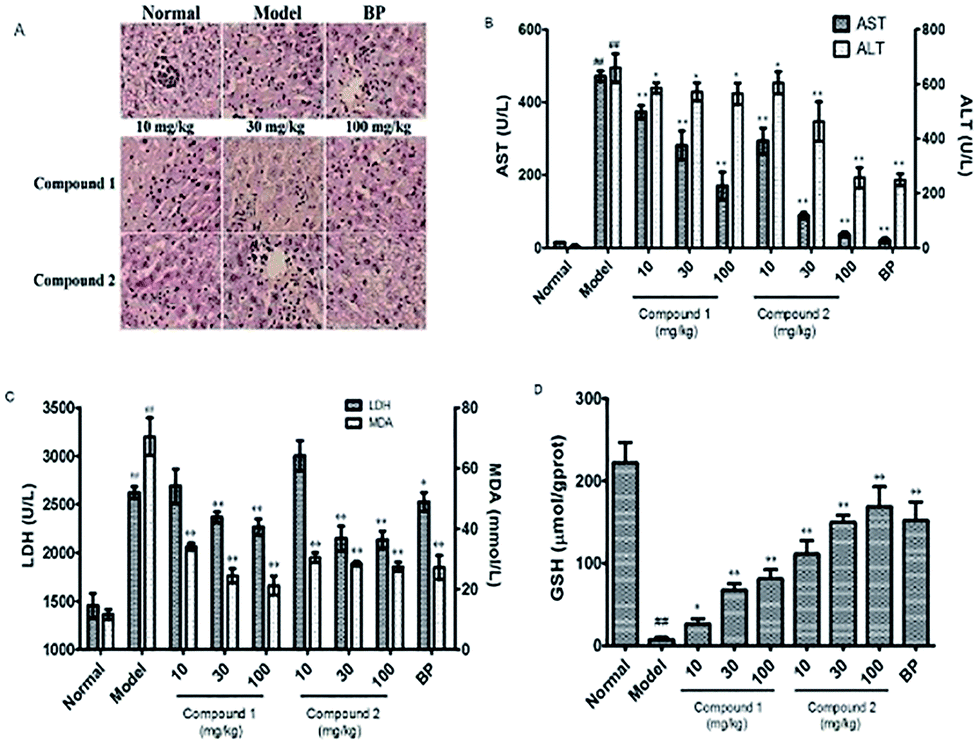 | ||
| Fig. 3 The in vivo hepatoprotectives and antioxidant effects of compound 1 and 2 against CCl4-induced liver injury in mice. (A) Preventive effect of compound 1 and 2 against CCl4-induced liver histopathological changes in mice (original magnification of 200×). (B) Effect of compound 1 and 2 on serum enzyme activities of ALT and AST. (C) Effect of compound 1 and 2 on the levels of LDH and hepatic MDA. (D) Effect of compound 1 and 2 on serum enzyme activity of GSH. All data are presented as means ± SD, n = 8. #P < 0.05 compared to control group; ##P < 0.01 compared to control group; *P < 0.05 compared to model group; **P < 0.01 compared to model group. P value < 0.05 was considered as significant. | ||
Oxidative stress and inflammation could be induced by CCl4 administration and play an initial, important role in the development of CCl4 hepatotoxicity. Twenty hours after CCl4 administration, liver samples were removed to assess oxidative stress. The liver oxidative stress parameters LDH and MDA significantly increased in the CCl4 group compared to those in the normal group. However, the reduced LDH and MDA levels in mice pretreated with compounds 1 and 2 (Fig. 3C) suggest that these compounds could alleviate CCl4-mediated oxidative stress. To further investigate the mechanisms of antioxidation affected by pretreatment with compounds 1 and 2, we evaluated the body's antioxidant defense system such as liver levels of the antioxidant GSH. In CCl4-treated mice, there was a significant decrease in GSH level (7.39 μmol g−1 prot) compared to that observed for the control group (222 μmol g−1 prot). However, pretreatment with compounds 1 and 2 suppressed this decrease, especially compound 2 at a high concentration. GSH level in compound 2 high-dosage group (100 mg kg−1) was 168.29 μmol g−1 prot compared to 151.69 μmol g−1 prot observed in the BP-treated positive control group (Fig. 3D).
To investigate whether pretreatment with compounds 1 and 2 could alleviate CCl4-mediated inflammation, we measured the serum TNF-α level in these mice. As noted in Fig. 4A, compounds 1 and 2 alleviated the CCl4-induced increased serum TNF-α level. Next, to investigate whether modification of the CCl4 metabolism pathway is involved in the observed hepatoprotective effect of compounds 1 and 2, we checked the protein expression levels of hepatic CYP2E1 by western blotting. It is known that CCl4 toxicity results from its reductive dehalogenation by cytochrome P450 into highly reactive free radicals such as trichloromethyl. As shown in Fig. 4B, hepatic CYP2E1 protein expression was significantly upregulated in CCl4-treated model group. However, pretreatment with compounds 1 and 2 effectively suppressed this increase.
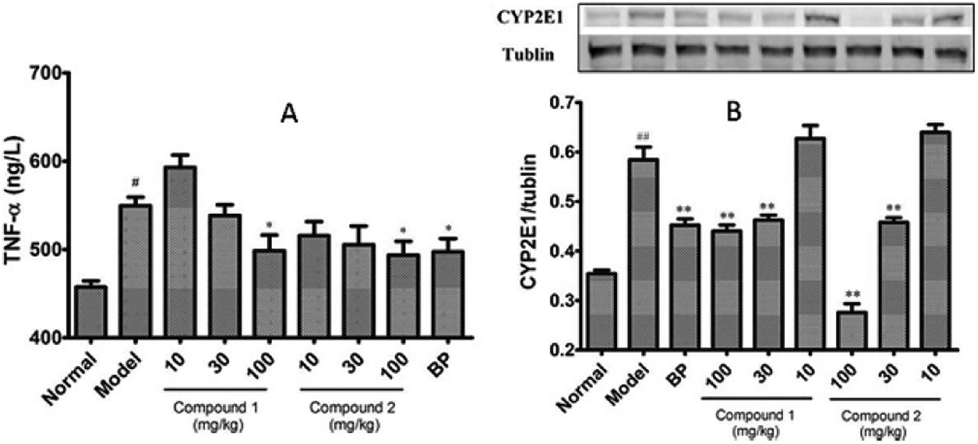 | ||
| Fig. 4 Effect of compound 1 and 2 on TNF-α (A) and CYP2E1 activity (B) in the liver microsomes of mice treated with CCl4. All data are presented as means ± SD, n = 8. #P < 0.05 compared to control group; ##P < 0.01 compared to control group; *P < 0.05 compared to model group; **P < 0.01 compared to model group. P value < 0.05 was considered as significant. | ||
Discussion
The number of studies has suggested protective effects of flavonoids against many infectious and degenerative diseases such as cardiovascular diseases, cancers, liver disease, diabetes and other age-related diseases.36,37A new skeletal flavonoid anastatin B, which was isolated from the methanolic extract of an Egyptian medicinal herb, was found to be hepatoprotective in primary cultured mouse hepatocytes. Compound 1 and compound 2, two flavonoid anastatin B derivatives, were synthesized by our group previously.37 In the present study, the antioxidant capacity and hepatoprotective activity of compound 1 and 2 were evaluated using chemical evaluation assays, an H2O2-induced PC12 cell model, and a CCl4-induced liver toxicity model.
The in vitro antioxidant activity of compounds 1 and 2 in the DPPH˙ and ABTS˙+ scavenging activity assays as well as the FRAP assay were comparable to those of the positive control Vc, suggesting both compounds have favorable antioxidant properties and the ability to scavenge free radicals. Furthermore, both compounds 1 and 2 could reverse the decrease of cell viability caused by H2O2 and remarkably increase the cell viability. In addition, compounds 1 and 2 (especially compound 2) showed hepatoprotective effects against CCl4-induced liver injury in mice. The present study shows that compounds 1 and 2 possess potential antioxidant activity and hepatoprotective effect both in vitro and in vivo.
Antioxidant capacity can be reflected by free radical scavenging activity, which is one of the major antioxidation mechanisms inhibiting the chain reaction of lipid peroxidation. In our study compound 1 and 2 showed potent antioxidant properties to scavenge free radicals (Table 1). The results indicated that compound 1 and 2 might proposed as promising compounds for the prevention and treatment of oxidative stress caused chronic diseases. On the basis of this analysis and previous work, we suppose that both two compounds might prevent the oxidative stress-induced injury in cell level, even in vivo. To verify our supposition, we next study the antioxidant capacity of compound 1 and 2 using H2O2-induced injury in PC12 cell model. As we expected, both test compounds, especially compound 2, have better antioxidant activity than GA did on H2O2-mediated PC12 cell injury. To further study whether compound 1 and 2 could ameliorate oxidative stress-induced injury in vivo, we a CCl4-induced liver injury mice model was used to against CCl4-induced liver injury in further chemo-biological studies of these compounds and their enzymatic targets are ongoing in our laboratory.
Conclusions
In the present study, the antioxidant activities and hepatoprotective mechanisms of compounds 1 and 2 were assessed. Both compounds 1 and 2 had good antioxidant and radical scavenging abilities in vitro. Both compounds showed cytoprotective activity in H2O2-treated PC12 cells. Both of them were potent hepatoprotectants in a mouse model of CCl4-induced hepatotoxicity. Compound 2 was a more effective hepatoprotectant than compound 1.Conflicts of interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work. There is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled.Acknowledgements
The authors sincerely thank the financial support from the National Natural Science Foundation of China (31301142), the International Science & Technology Cooperation Program of China (2013DFA31160).References
- S. Federico, Aging Dis., 2016, 7, 130–135 CrossRef PubMed.
- F. He and L. Zuo, Int. J. Mol. Sci., 2015, 16, 27770 CrossRef CAS PubMed.
- M. J. Jackson, Mol. Aspects Med., 2016, 50, 33 CrossRef CAS PubMed.
- D. Limongi and S. Baldelli, Oxid. Med. Cell. Longevity, 2016, 2016, 6547248 Search PubMed.
- G. Noctor, A. Mhamdi and C. H. Foyer, Plant, Cell Environ., 2016, 39, 1140 CrossRef CAS PubMed.
- I. G. Onyango, D. Jameel and S. M. Khan, Aging Dis., 2016, 7, 201–214 CrossRef PubMed.
- K. Sugimoto and Y. Takei, Hepatol. Res., 2017, 47, 70 CrossRef PubMed.
- T. Yahata and K. Hamaoka, Rheumatology, 2017, 56, 6 CrossRef PubMed.
- Z. Allameh and J. Salamzadeh, J. Res. Pharm. Pract., 2016, 5, 79–85 CrossRef CAS PubMed.
- A. Bielli, M. G. Scioli, D. Mazzaglia, E. Doldo and A. Orlandi, Life Sci., 2015, 143, 209–216 CrossRef CAS PubMed.
- K. Hagymási, A. Egresi and G. Lengyel, Orv. Hetil., 2015, 156, 1884 CrossRef PubMed.
- D. Singh, W. C. Cho and G. Upadhyay, Frontiers in Physiology, 2016, 6, 363 CrossRef PubMed.
- E. A. Fraunberger, G. Scola, V. L. M. Laliberté, A. Duong and A. C. Andreazza, Oxid. Med. Cell. Longevity, 2015, 2016, 4729192 Search PubMed.
- K. A. Wojtunik-Kulesza, A. Oniszczuk, T. Oniszczuk and M. Waksmundzka-Hajnos, Biomed. Pharmacother., 2016, 78, 39 CrossRef CAS PubMed.
- L. Sha, H. Y. Tan, W. Ning, Z. J. Zhang, L. Lao, C. W. Wong and Y. Feng, Int. J. Mol. Sci., 2015, 16, 26087–26124 CrossRef PubMed.
- W. Jiang, M. Gao, S. Sun, A. Bi, Y. Xin, X. Han, L. Wang, Z. Yin and L. Luo, Biochem. Biophys. Res. Commun., 2012, 422, 344 CrossRef CAS PubMed.
- I. F. F. Benzie and J. J. Strain, Anal. Biochem., 1996, 239, 70–76 CrossRef CAS PubMed.
- A. Cano, J. Hernández-Ruíz, F. García-Cánovas, M. Acosta and M. B. Arnao, Phytochem. Anal., 2015, 9, 196–202 CrossRef.
- B. Li and D. A. Pratt, Free Radical Biol. Med., 2015, 82, 187–202 CrossRef CAS PubMed.
- M. Gassen and M. B. Youdim, J. Neural Transm., Suppl., 1999, 56, 193–210 CAS.
- J. Z. Wu, C. C. Cheng, L. L. Shen, Z. K. Wang, S. B. Wu, W. L. Li, S. H. Chen, R. P. Zhou and P. H. Qiu, Int. J. Mol. Sci., 2014, 15, 18525 CrossRef CAS PubMed.
- C. S. Lieber, Adv. Pharmacol., 1996, 38, 601–628 Search PubMed.
- L. W. D. Weber, M. Boll and A. Stampfl, Crit. Rev. Toxicol., 2003, 33, 105–136 CrossRef CAS PubMed.
- J. Liu, J. F. Lu, X. Y. Wen, J. Kan and C. H. Jin, Int. J. Biol. Macromol., 2015, 72, 1479 CrossRef CAS PubMed.
- M. Akhlaghi, Phytother. Res., 2016, 30, 1559–1571 CrossRef CAS PubMed.
- J. Dong, Q. Zhang, Q. Cui, G. Huang, X. Pan and S. Li, ChemMedChem, 2016, 11, 2102–2118 CrossRef CAS PubMed.
- V. Teresa, R. N. Alba, A. Francesca, U. M. Pilar, R. C. M. Elena and G. Julio, Nutrients, 2016, 8, 211 CrossRef PubMed.
- X. Xiao, X. Wang, X. Gui, L. Chen and B. Huang, Chem. Biodiversity, 2016, 13, 1427 CAS.
- V. P. Menon and A. R. Sudheer, Adv. Exp. Med. Biol., 2007, 595, 105–125 CrossRef PubMed.
- D. Procházková, I. Boušová and N. Wilhelmová, Fitoterapia, 2011, 82, 513–523 CrossRef PubMed.
- R. L. Prior and G. H. Cao, HortScience, 2000, 35, 588–592 CAS.
- N. C. Cook and S. Samman, J. Nutr. Biochem., 1999, 7, 66–76 CrossRef.
- C. A. Riceevans, N. J. Miller, P. G. Bolwell, P. M. Bramley and J. B. Pridham, Free Radical Res., 1996, 22, 375–383 CrossRef.
- M. Yoshikawa, F. Xu, T. Morikawa, K. Ninomiya and H. Matsuda, ChemInform, 2003, 13, 1045–1049 CAS.
- G. Pan, X. Li, L. Zhao, M. Wu, C. Su, X. Li, Y. Zhang, P. Yu, Y. Teng and K. Lu, Eur. J. Med. Chem., 2017, 138, 577–589 CrossRef CAS PubMed.
- A. K. Pandey, Natl. Acad. Sci. Lett., 2007, 30, 383–386 Search PubMed.
- S. Kumar, A. Gupta and A. K. Pandey, ISRN Pharmacol., 2013, 2013, 691372 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02523a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |