
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew synthetic pathway leading to oxospirochlorins†
Justyna Śniechowska*a,
Piotr Palucha,
Tomasz Pawlak a,
Grzegorz D. Bujaczb,
Witold Danikiewiczc and
Marek J. Potrzebowski*a
a,
Grzegorz D. Bujaczb,
Witold Danikiewiczc and
Marek J. Potrzebowski*a
aCentre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363 Lodz, Poland. E-mail: jsniech@cbmm.lodz.pl; marekpot@cbmm.lodz.pl
bInstitute of Technical Biochemistry, Lodz University of Technology, Stefanowskiego 4/10, 90-924 Lodz, Poland
cInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
First published on 12th June 2018
Abstract
In this work we propose a completely new approach for the synthesis of spirochlorin derivatives based on the use of an imino-keto intermediate formed in situ from 2-amino-5,10,15,20-tetraphenylporphyrins and inverse electron demand Diels–Alder (iEDDA) cycloaddition with 3,6-di-2-pyridyl-1,2,4,5-tetrazine. The mechanism of reaction was analyzed employing theoretical methods by comparing the difference in energy of Frontier Molecular Orbitals (FMO) for appropriate reagents. Ground-state molecular electrostatic (ESP) potential maps were employed as additional tools allowing explanation of the reactivity of substrates. The new class of spirochlorin compounds was fully characterized by means of mass spectrometry, IR, liquid and solid state NMR and X-ray crystallography. Correlation between molecular structure and optical properties for the obtained title compounds is discussed.
Introduction
Porphyrinoids are a large group of macrocyclic heteroorganic compounds ubiquitous in nature. They play very important roles in multiple biological processes including cell respiration (hemoglobin), detoxification of xenobiotics, oxygen transport and the photosynthesis process.1 Chemical modifications of natural and synthetic porphyrinoids have been an area of intense interest for a number of scientific groups. Development of methods for structural transformations of the porphyrin macrocycle can provide a wide spectrum of new functional materials. Among the big family of porphyrinoid derivatives, chlorins (hydrochlorins) are a very important class of compounds due to their applicability in photodynamic therapy and diagnostics, catalysis, materials science, electronics and solar energy conversion.2 They have unique spectroscopic, optical and electronic properties and absorb light strongly in the near infrared part of the electromagnetic spectrum.3 From a chemical point of view an important issue is the fact that the physical features of porphyrinoids can be modified and tailored by changing their molecular structure. Hence, there is still great motivation for development of new methods of synthesis, which may open routes to production of attractive chlorin derivatives with unique properties.To the group of weakly recognized species with great application potential belong the spiro derivatives of chlorins (Fig. 1) which play key roles in nature. The naturally occurring chlorin with a spiro motif integral to pyrroline ring is heme d, which contains a spirolactone unit. During last decade significant efforts have been made toward the development of effective methods for obtaining of this class of compounds. According to the literature, most synthetic procedures leading to spirochlorins are based on reactions with use of gem-dialkylgroups.4 Kenner and co-workers found that acetamidoethylporphyrins can be converted into spirochlorins by phosphoryl chloride treatment in pyridine.5 Smith and colleagues described that spiroketochlorins can be obtained with high yields from porphyrin bearing a butyramide group.6 Kai and Suzuki prepared a spirochlorin by the reaction of tetracyanoetylene with bromovinylheptaetylporphyrin.7 Spirolactones can be obtained in reaction of vinyl porphyrins with hydrobromic acid and subsequent five-step procedure striving to intermolecular cyclization of propionate side chain.8
 | ||
| Fig. 1 Structures of some spiro derivatives of chlorins. | ||
In this work we propose a new approach for the synthesis of spirochlorin derivatives based on use of imino-keto intermediate as reagent in inverse electron demand Diels–Alder reaction (iEDDA). The starting point for development of our strategy is an observation reported over 30 years ago by Crossley and King9 who found that 2-amino-5,10,15,20-tetraphenylporphyrin 2a easily undergoes photo-oxidation forming compound 3a, which further on silica gel immediately undergoes hydrolysis to dioxochlorin 4a (Scheme 1).10 On the course of our studies we have noticed that compounds 3a–3f can be obtained under mild conditions, they are relatively stable and can be thought as active in situ precursors in chemical reactions. To the best of our knowledge to date this compound was not considered as a substrate in chemical synthesis. In our researches, we report the use of compounds 3a–3f in reaction with tetrazine to obtain new oxospirochlorins. The method reported herein is yield and efficient, can be employed for synthesis of novel functionalized spiro derivatives with desired spectroscopic properties. Thus, this reaction is a tool for preparing new dyes, scaffolds based on porphyrins and chlorins, molecular receptors and a biomimetic models of heme or another systems imitating the active sites of some enzymes.
 | ||
| Scheme 1 Synthesis of compounds 3a–3f and 4a–4f. | ||
Results and discussion
Synthesis of oxospirochlorins
The starting material, β-nitro porphyrin derivatives 1a–1f were obtained employing known procedure consisting several steps which include the synthesis of porphyrins from appropriate aldehyde and pyrrole;11 complexation reaction between porphyrin and copper acetate;12 nitration at β-position with copper(II) nitrate in mixture acetic acid/acetic anhydride.13 The key step in synthesis of β-amino porphyrins 2a–2f is demetallation reaction14 of 1a–1f and the reduction of nitro group with SnCl2 in hydrochloric acid.10,15 The formation of 2a and 3a were confirmed by mass spectrometry. The first hint that β-amino porphyrins 2a–2f are readily oxidized came from the routine ESI mass spectrum of the amine 2a (R = C6H5) recorded in acetonitrile (Fig. 2). Together with expected m/z 630.5 peak corresponding to the protonated amine 2a, an intense m/z 644.5 peak has been observed. An accurate mass measurement revealed the molecular formula C44H30N5O, which differs from the amine 2a by two lacking hydrogen atoms and one additional oxygen atom. Spontaneous oxidation of porphyrinoids during ESI MS measurements has been already observed in one of our groups.16 Additional experiments showed that they are oxidized most likely by the atmospheric oxygen and the porphyrinoid itself serves as the photosensitizer of this reaction. | ||
| Fig. 2 ESI mass spectrum of the acetonitrile solution of 2a. m/z 315.9 peak corresponds to the [M + 2H+]2+ ion of 2a. | ||
Further proof confirming formation of compounds 3a–3f is found by testing their chemical reactivity, in particular in the Diels–Alder (DA) cycloaddition. In this reaction the distribution of electron densities for both reagents is crucial. In a DA with normal electron demand, the dienophile typically bears an electron-withdrawing group (EWG), while the diene is electron-rich.17 Reversely, the iEDDA reaction is characterized by an electron-poor diene and an electron-rich dienophile.18 Compounds 3a–3f fulfill prerequisites expected for electron-rich dienophiles.
In our synthetic strategy the choice of diene is decisive. Very recently Mayer and Lang have reviewed the different aspects of tetrazines in iEDDA cycloadditions and theirs applications in synthesis of biological products.19 As reported 1,2,4,5-tetrazines containing strong EWG substituents are reactive species, commonly used in iEDDA.18–20 Due to their high reactivity these compounds are still of synthetic interest to organic chemists. Increase of the electron-withdrawing character of groups in 3,6 position in 1,2,4,5-tetrazine (e.g. pyridyl, CO2Me etc.) increased the reactivity of the diene. In this project we have used 3,6-di-2-pyridyl-1,2,4,5-tetrazine 5 (Scheme 2), because it has an ability to react as inverse electron demand diene. The compound 5 was synthesized from commercially available 2-pyridinecarbonitrile and hydrazine hydrate in a two-step procedure.21 Treatment of compounds 3a–3f generated in situ from 2a–2f solvent dramatically vary the reaction course. Formation of the oxospirochlorins 6a–6f was not observed when acetonitrile or dichloromethane were used as a solvents. We cannot exclude that in this case the high boiling point of toluene is critical parameter. Moreover, we tested the reactivity of compound 4a in the iEDDA reaction. Reaction of with equimolar amount of tetrazine 5 in refluxing toluene for 5 hours gave oxospirochlorins in 25–50% yields. Obtained products were isolated and purified by column chromatography. It has to be stressed that change of the 4a with tetrazine 5 carried out boiling toluene does not lead to the new product.
 | ||
| Scheme 2 Synthesis of oxospirochlorins 6a–6f. | ||
To test a generality of this process, we carried out reaction under the same conditions as mentioned above, using porphyrins containing different substituted aryl rings in meso position (Table 1). Steric and electronic character of groups in meso position influence the reactivity of the porphyrins as dienophile. The use of porphyrin, which is sterically hindered in reactive site, gave no product of reaction, even with longer heating time.
| Porphyrin | Tetrazine | Product | Yield [%] |
|---|---|---|---|
| 2a | 5 | 6a | 50 |
| 2b | 5 | 6b | 38 |
| 2c | 5 | 6c | 32 |
| 2d | 5 | 6d | 25 |
| 2e | 5 | 6e | — |
| 2f | 5 | 6f | — |
A plausible mechanism for the cycloaddition reaction is proposed in Scheme 3. The imino-keto intermediates 3 react with tetrazine 5 to give a transition state A and form the highly strained intermediate B. It has not been observed, because it instantly undergo a retro Diels–Alder step, which is responsible for the elimination of dinitrogen. The termination step of the reaction is hydrogen shift from compound C and compound D are formed.
 | ||
| Scheme 3 Plausible mechanism. | ||
Analysis of cycloaddition employing theoretical calculations
In the previous section we presented intuitive arguments which enable us to assume that reaction under investigation is classified as iEDDA despite that the substrates 3a–3f can be also thought as an electron deficient dienophile (double bond C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH conjugated with a CO group) and the 3,6-di-2-pyridyl-1,2,4,5-tetrazine 5 looks like an electron rich diene. In this section we provide proofs, based on theoretical approach which confirm our hypothesis. One of the main differences between classical DA and iEDDA reaction is defined trough analysis of frontier molecular orbitals (FMOs).22 In the iEDDA cycloaddition the azadiene has a highly stabilized HOMO orbital with energy below HOMO of dienophile (it is reverse situation comparing to nonpolarized DA reaction). It leads to decrease of energy gap between LUMO diene orbital and HOMO dienophile and significant electron delocalization from the dienophile to the azadiene with concomitant energy stabilization. This single FMO interaction is assumed to more than compensate for the increase in the forward FMO energy gap between the azadiene HOMO and the dienophile LUMO. Trough analysis of our computational data shown in Fig. 3 and Table 2 the mechanism of interaction between 3a–3d and 5 can be defined as iEDDA reaction. As seen HOMO dienophile-LUMO diene gap marked by dashed line arrows is significantly lower compared to opposite situation. Lack of formation of product in case of sample 3e can be explained on the ground of FMO. The situation with 3f is more complicated because the unfavorable steric effect of 2,6-difluorophenyl group most likely exceeds adequate character of HOMO–LUMO gap.
NH conjugated with a CO group) and the 3,6-di-2-pyridyl-1,2,4,5-tetrazine 5 looks like an electron rich diene. In this section we provide proofs, based on theoretical approach which confirm our hypothesis. One of the main differences between classical DA and iEDDA reaction is defined trough analysis of frontier molecular orbitals (FMOs).22 In the iEDDA cycloaddition the azadiene has a highly stabilized HOMO orbital with energy below HOMO of dienophile (it is reverse situation comparing to nonpolarized DA reaction). It leads to decrease of energy gap between LUMO diene orbital and HOMO dienophile and significant electron delocalization from the dienophile to the azadiene with concomitant energy stabilization. This single FMO interaction is assumed to more than compensate for the increase in the forward FMO energy gap between the azadiene HOMO and the dienophile LUMO. Trough analysis of our computational data shown in Fig. 3 and Table 2 the mechanism of interaction between 3a–3d and 5 can be defined as iEDDA reaction. As seen HOMO dienophile-LUMO diene gap marked by dashed line arrows is significantly lower compared to opposite situation. Lack of formation of product in case of sample 3e can be explained on the ground of FMO. The situation with 3f is more complicated because the unfavorable steric effect of 2,6-difluorophenyl group most likely exceeds adequate character of HOMO–LUMO gap.
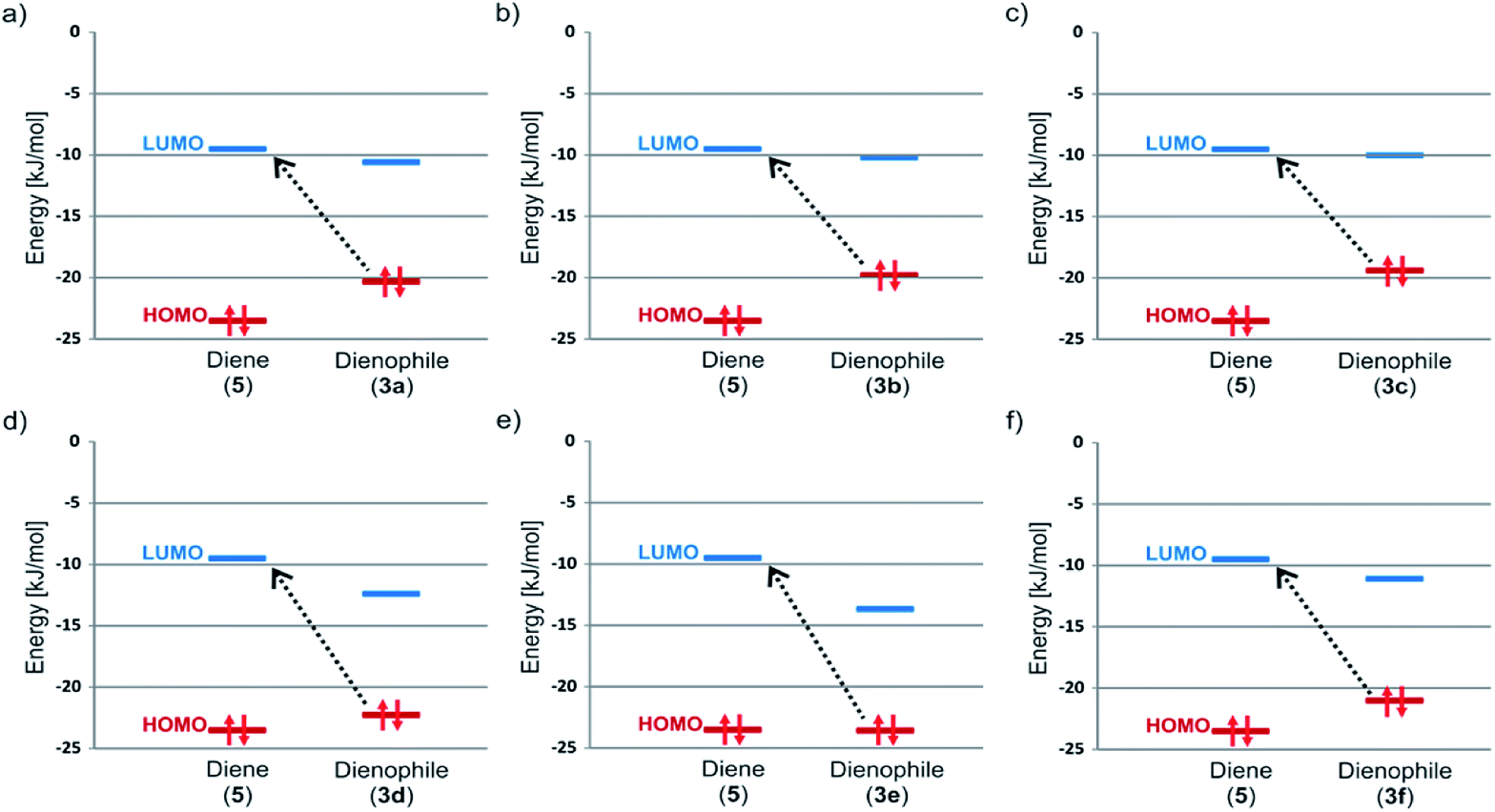 | ||
| Fig. 3 Comparison of FMO Energies for studied pair of diene (5) and dienophiles 3a (a), 3b (b), 3c (c), 3d (d), 3e (e) and 3f (f). | ||
| Compound | Energy [kJ mol−1] | |
|---|---|---|
| HOMO | LUMO | |
| 5 | −23.49 | −9.50 |
| 3a | −20.30 | −10.56 |
| 3b | −19.78 | −10.19 |
| 3c | −19.40 | −10.03 |
| 3d | −22.35 | −12.50 |
| 3e | −23.61 | −13.71 |
| 3f | −21.11 | −11.18 |
Another fast and convenient approach for analysis of the preference for cycloaddition is taking into account electrostatic potential (ESP) maps of studied models and evaluating the relative electron population of the atoms in imino-keto intermediates 3a–3f involved in bond formation during the reaction.23 Fig. 4 displays ESP maps for studied models which may be used to qualitative prediction the reactivity of the active sites of compounds 3a–3f. This strategy provides a useful device of relative wave function amplitude and electron density. Molecular ESPs are also advantageous because they are generally not highly computational method and basis set dependent. The electrostatic potentials at the surface are illustrated by different colors. While the yellow to red color parts point out the regions of negative electrostatic potential, the blue sites represent the regions of positive electrostatic potential and the parts with green color indicate the regions of close zero potential. In the ESP maps, one of the most negative region is located on the N–H group which can be considered as the most preferred site for electrophilic attack (reactive site of dienophile in the iEDDA cycloaddition).
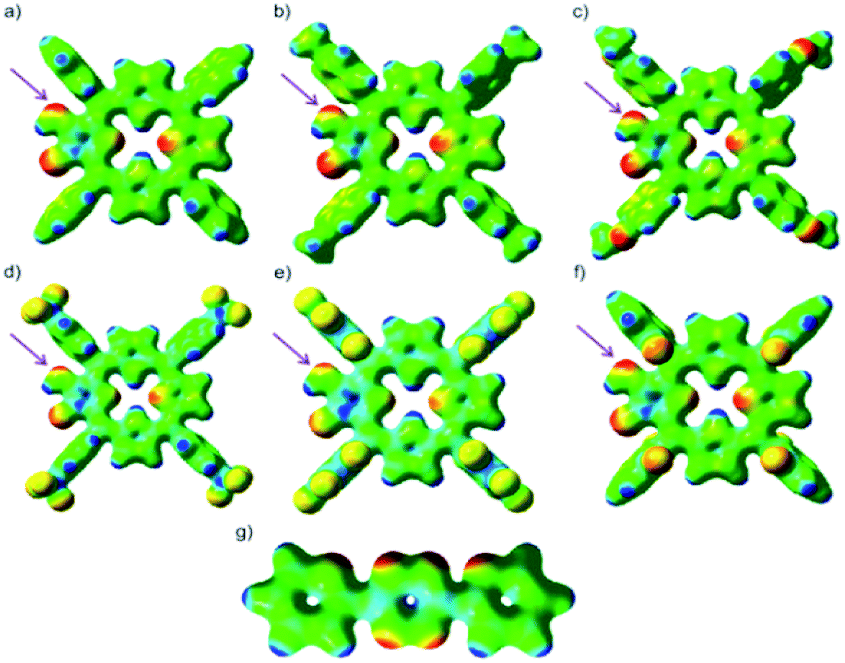 | ||
| Fig. 4 Comparison of ground-state molecular electrostatic potentials (ESPs) for studied models 3a (a), 3b (b), 3c (c), 3d (d), 3e (e), 3f (f) and 5 (g) plotted from −0.03 (red) to +0.3 (blue) Hartree. Violet arrows marked the reaction sites for imino-keto intermediates. | ||
Finally we wish to comment the last stage of synthetic pathway, leading from C to D (see Scheme 3) related with prototropic migration and formation of new thermodynamically stable tautomer. The question about reversibility of this process is justified. Our theoretical calculations of ground state energy for C and D shows significant difference between both forms. For D energy is lower ca. 74 kJ mol−1. Thus we can conclude that this process is irreversible.
Spectral and structural analysis of oxospirochlorins in the liquid and solid phases
The obtained title derivatives are new class of compounds hence their structure had to be proved in a doubtless manner. In the first approach, for structure analysis we employed techniques: NMR spectroscopy, UV-VIS and high-resolution mass spectrometry.The absorption spectra of synthesized oxospirochlorins 6a–6d were measured in dichloromethane at room temperature. Fig. 5 shows the spectra and the band maxima of absorption and log excitation coefficients (see Table 3). Similarly to others porphyrinoids, in the absorption spectrum it can be indicated Soret band and Q-bands. The position of the Soret bands and Q bands are nearly identical with each other. The slightly bathochromically shifted Soret bands for 6b and 6c, and hypsochromatically shifted for 6d have higher excitation coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ελmax = 5.58; 5.49 and 5.52, respectively) as compared to oxospirochlorin 6a (logελmax = 5.29). The spectra of synthesized compounds 6a–6d are different from spectrum of a typical tetraarylchlorin. For each sample the intensity of the red absorption band is much smaller than observed for chlorins. The difference in the absorption profile is associated with the existence of being of π-electron conjugation with the carbonyl group. This β-functionalization of chlorins renders the absorption spectrum neither chlorin-like. This electronic influence of the carbonyl group is also observed in the UV-VIS of dioxochlorins 4a.24 Despite of broadened Q-bands the compound 4a was classified as chlorin.24b,25
ελmax = 5.58; 5.49 and 5.52, respectively) as compared to oxospirochlorin 6a (logελmax = 5.29). The spectra of synthesized compounds 6a–6d are different from spectrum of a typical tetraarylchlorin. For each sample the intensity of the red absorption band is much smaller than observed for chlorins. The difference in the absorption profile is associated with the existence of being of π-electron conjugation with the carbonyl group. This β-functionalization of chlorins renders the absorption spectrum neither chlorin-like. This electronic influence of the carbonyl group is also observed in the UV-VIS of dioxochlorins 4a.24 Despite of broadened Q-bands the compound 4a was classified as chlorin.24b,25
 | ||
| Fig. 5 UV-VIS absorption spectra (CH2Cl2) at room temperature of obtained oxospirochlorins 6a–6d. λexcitation = λSoret. | ||
| Compound | λmax [nm] | logελmax in dichloromethane |
|---|---|---|
| 6a | 429 | 5.29 |
| 6b | 430 | 5.58 |
| 6c | 432 | 5.49 |
| 6d | 426 | 5.52 |
The obtained compounds were also characterized by mass spectrometry and NMR spectroscopy (see ESI† for original spectra). The ESI-MS spectra of 6a show the molecular ion peak at m/z 852.5 [M + H+]+ together with [M + H+ − H2O]+ and [M + H+ − CO]+ fragment peaks. The presence of the carbonyl group in 6a is confirmed by 1725 cm−1 absorption band in the IR spectrum. The structure of oxospirochlorin 6a was further confirmed by 1D and 2D NMR measurements. The 1H NMR spectrum contains two broad singlets near −2 ppm for the NH protons in typical for porphyrinoids region. In the 13C NMR spectrum (Fig. 6a) of compound 6a the most diagnostic and characteristic signals for the carbonyl carbon atom is observed at 202 ppm and the spiro carbon atom at 68 ppm. Moreover, the 1H–15N HSQC spectrum displays a singlet at 141.5 ppm corresponding to the imino NH group. These results confirm the structure of obtained compound 6a. The low symmetry of compound 6a removes the equivalency of the four meso-phenyl rings. In the 1H–13C HSQC spectrum not equivalent pyridyl rings are seen and recognized.
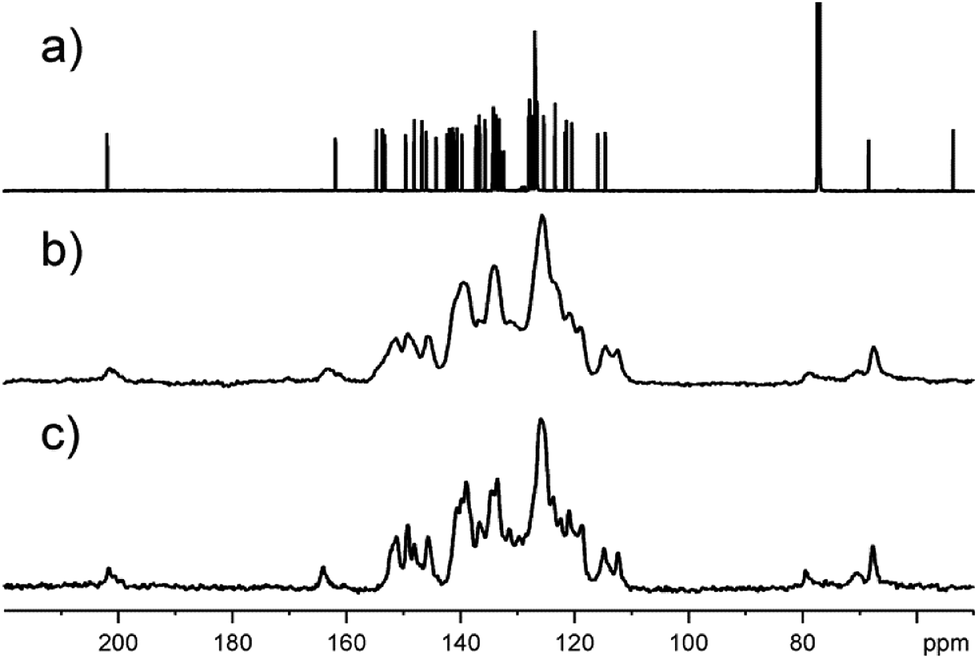 | ||
| Fig. 6 13C NMR spectra for sample 6a. (a) liquid sample dissolved in chloroform-d, (b) amorphous solid sample recorded employing 13C CP/TOSS MAS technique with spinning rate 8 kHz. (c) Precipitated solid sample recorded employing 13C CP/TOSS MAS technique with spinning rate 8 kHz. | ||
As in the case of almost all porphyrin derivatives, the obtained spirochlorins are solid. Testing the precipitated sample of 6a by means of 13C solid state NMR spectroscopy we were rather surprised by the quality of spectrum (Fig. 6c). Relatively sharp 13C signals suggest the significant contribution of crystalline phase in whole sample. Fig. 6 shows 13C NMR spectra with clearly seen characteristic resonances.
This promising observation, proving tendency of sample for crystallization, prompted us to focus on growing the single crystals with quality suitable for X-ray crystallographic studies. Solid state NMR was found to be a tool supporting optimization of crystallization process.26 It has to be stressed that to date deposited X-ray structures for spirochlorins do not exist in available crystallographic data bases.
Testing different approaches we have found that the best option for growing 6a crystals is recrystallization of compound by isothermal evaporation of dichloromethane at room temperature. The crystal structure and molecular packing of 6a is shown in Fig. 7. The structural information are attached as ESI.† The compound 6a crystallizes in triclinic form in centrosymmetric P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The independent unit contains one molecule of 6a and one molecule of crystallization solvent, dichloromethane. Two independent units related by center of symmetry fill unit cell. The molecules of 6a interact in crystal lattice mostly by hydrophobic contacts. The two solvent molecules are located in cage created by network of 6a. They are related by symmetry and interact with each other. The contacts with host molecule are by weak hydrogen bond between carbon hydrogen and ketone oxygen atoms and van der Waals contact of disordered C7 atom with disordered phenyl ring. In both positions carbon hydrogen atoms of dichloromethane with fractional occupancy are able to create contacts.
space group. The independent unit contains one molecule of 6a and one molecule of crystallization solvent, dichloromethane. Two independent units related by center of symmetry fill unit cell. The molecules of 6a interact in crystal lattice mostly by hydrophobic contacts. The two solvent molecules are located in cage created by network of 6a. They are related by symmetry and interact with each other. The contacts with host molecule are by weak hydrogen bond between carbon hydrogen and ketone oxygen atoms and van der Waals contact of disordered C7 atom with disordered phenyl ring. In both positions carbon hydrogen atoms of dichloromethane with fractional occupancy are able to create contacts.
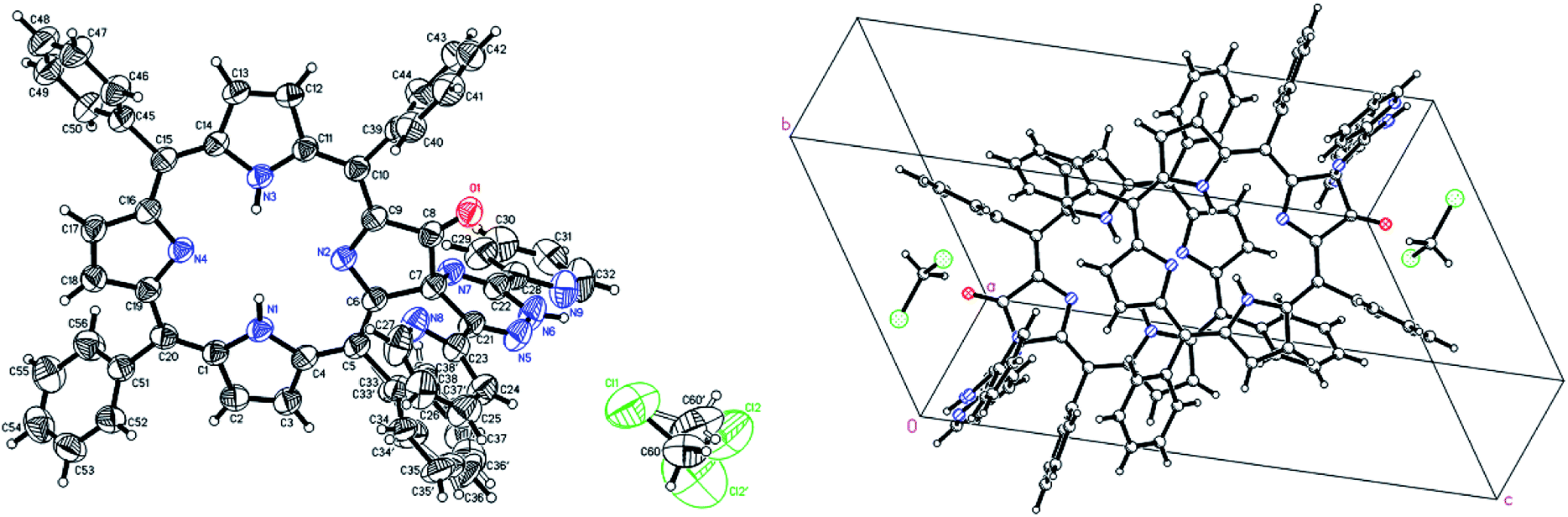 | ||
| Fig. 7 Crystal structure of obtained compound 6a (left) and unit cell (right). | ||
In the unit cell, molecule of 6a contains two main approximately planar systems perpendicular to each other. One of them, the chlorin ring, is connected with the other the 1,2,4-triazine ring, by a spiro C7 atom. The tetra phenyl chlorin moiety is slightly bound around the axis passing through the N2, N4 unprotonated nitrogen atoms. Two of four phenyl rings connected to the chlorin ring located in the vicinity of the modified pyrrole ring are perpendicular to it, while the other two, more distant from the modification, are angular. The disordered phenyl is parallel to the 1,2,4-triazine ring and the short distance between these two aromatic systems allows π–π interaction. One of the two pyridin-2-yl rings connected to the triazine ring is approximately coplanar with it and the other is slightly twisted. Only two nitrogens of the chlorin ring are protonated the positions of them were found from experimental data. The low no restricted isotropic thermal parameters and stabile position during refinement indicate that even at room temperature external proton exchange between nitrogen atoms doesn't occurs in a crystal.
Conclusions
In conclusion, novel and efficient synthesis of a series of oxospirochlorin derivatives has been achieved via the iEDDA reaction from β-substituted porphyrins and 1,2,4,5-tetrazine in reasonable yield. We revealed, that yield of reaction depends on the structure of substrates, position of the substituents in the meso ring and theirs electronic characters. The experimental data were explained by means of theoretical methods comparing the difference in energy of Frontiers Molecular Orbitals (FMO) for appropriate substrates. Ground-state molecular electrostatic (ESP) potential maps were employed as an additional tools allowing to describe the reactivity of substrates. Our experimental and computer results revealed that placing of atoms or groups in ortho position due to steric effect obstructs reactivity of system and in consequence the desired spiro product is not formed. The obtained compounds were fully characterized by spectroscopic methods. This reaction can be an excellent tool for preparing a novel chlorin derivatives. The method discovered in this work allows access to the libraries of compounds containing a tetrapyrrolic systems.Experimental
Materials and methods
The compounds were prepared using chemicals which were purchased from Sigma Aldrich, TCI and used without further purification. Reagent grade solvents were purchased from Polish Chemicals Reagents and were distilled prior to use. Silica gel columns for chromatography were prepared with silica (Kieselgel 60, 200–400 mesh). 5,10,15,20-tetraphenylporphyrin and 5,10,15,20-tetra(pentafluorophenyl)porphyrin were purchased from PorphyChem. 3,6-di-2-pyridyl-1,2,4,5-tetrazine 5 is known compound which was prepared according previous methods.21 β-Nitro porphyrins 1a and 1b were prepared following the reported procedures.11–15a,27NMR experiments were run on a 600 MHz Bruker Avance III spectrometer equipped with BBOF probhead operating at 600.13, 564.69, 150.92 MHz for 1H, 19F and 13C nuclei, respectively and 500 MHz Bruker AVANCE DRX. The following abbreviations were used to describe peak splitting patterns when appropriate: s = singlet, d = doublet, t = triplet, dd = doublet of doublets, ddd = doublet of doublet of doublets and m = multiplet. Coupling constants J are reported in Hz.
UV-VIS spectra were obtained on Specord S600 “Analytyk Jena”. Mass spectra of obtained compounds were registered by MALDI-TOF Mass Spectrometer – PerSeptive Biosystems and, additionally, on Synapt G2S HDMS (Waters) and 4000 Q TRAP (Sciex) mass spectrometers.
Theoretical calculations
All calculations in this work have been performed using Gaussian 16 (G16) program package.28 Starting geometries of molecules were generated de novo and minimized in vacuo using the hybrid Hartree–Fock/density functional theory (HF/DFT) method PBE1PBE (named also as PBE0)29 and the 6-311+G(d,p) basis set.30 The geometry was always optimized without any symmetry restrictions. Each stationary point was characterized by calculating the harmonic vibration frequencies in order to verify that they have no imaginary frequency. The distributions and energy levels of HOMO and LUMO orbitals as well as molecular electrostatic potential (ESP) map properties of the studied compounds are calculated using the same methodology.For optimized structures the energy gaps was also calculated using LC-ωHPBE functional (which is recommended version of the long-range-corrected ωPBE functional by G16),31 and aug-cc-pVTZ basis set.32 Such choice was made because it is well known that in DFT calculations, with common approximations used in the functionals is often not useful in practice. In other hand DFT is a compromise which allows for a treatment of quite large systems what would not be allowed using computationally more demanding methods.33 Recent papers shown that this weakness can be restored by using long-range corrected hybrid functionals where the HOMO and LUMO eigen values obtained from this new class of functionals have been demonstrated to be very accurate. It is found that tuned LC-ωPBE provides very small error that clearly improve upon standard exchange-correlation functionals.34 The results for LC-ωHPBE functional and aug-cc-pVTZ are presented in ESI† and shows the same tendencies as for PBE1PBE/6-311+G(d,p) level.
Single crystal X-ray measurements
Single crystal diffraction experiments for 6a were carried out on Oxford SuperNova single-crystal diffractometer with micro source CuKα radiation (λ = 1.5418 Å) and a Titan detector. The plate crystal of dimensions 0.18 × 0.09 × 0.02 was glued do a glass capillary by epoxy glue and measured at room temperature. The absorption correction was performed based on the crystal shape, orientation and absorption coefficient. Diffraction data collection, cell refinement, data reduction, and absorption correction were performed using the CrysAlis PRO program (Oxford Diffraction). The structure was solved by direct methods SHELXS and refined using full-matrix least-squares methods SHELX 2015 implemented in OLEX2 package.35 The hydrogen atoms ware set geometrically and refined as riding with the thermal parameter equal to 1.2 of the thermal vibration of the parental atom. The two hydrogen atoms connected with internal nitrogen atoms of the chlorin ring were found on the difference Fourier map and refined without any geometrical restrains with isotropic thermal parameters. Two disordered moieties were detected in the structure: the phenyl ring in the vicinity of 1,2,4-triazine ring and a solvent dichloromethane molecule. Both disordered fragments were modeled in two positions with the fractional occupation factors summing to one. The vibration of phenyl ring can be described as a swing of the pendulum in the ring plane. The dichloromethane molecule oscillates around the axis passing thought the Cl1 atom and the middle of the C60–Cl2 bond. The structure was validated by CheckCif (http://checkcif.iucr.org/) and deposited to the Cambridge Crystallographic Data Centre (CCDC) under accession number 1573589.General procedure for synthesis of β-amino porphyrins 2a–2f
β-Amino porphyrins 2a–2f were prepared in accordance to a previously described method,15a with slight modification. β-Nitro porphyrin 1a–1f (0.22 mmol) was dissolved in a concentrated HCl (15 mL) at room temperature. Afterwards, SnCl2·2H20 (1 mmol, 0.226 g) was added. The resultant dark green coloured mixture was heated at 65 °C for 2 h with stirring. The mixture was then cooled to room temperature and H2O was added. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with saturated NaHCO3(aq), before being dried over anhydrous K2CO3/MgSO4, filtered and concentrated to dryness in vacuo. The dark purple colored solid was used without purification. The formation of described compounds were confirmed by mass spectrometry. The attempts to isolation of compounds 2a–2f no give a pure products. Compound 2a: MS (MALDI TOF) m/z 630.8 [M + H]+; compound 2b MS (MALDI TOF) m/z 686.8 [M + H]+; compound 2c MS (MALDI TOF) m/z 750.7 [M + H]+; compound 2d MS (MALDI TOF) m/z 902.7 [M + H]+; compound 2e MS (MALDI TOF) m/z 990.8 [M + H]+; compound 2f MS (MALDI TOF) m/z 774.6 [M + H]+.General procedure for synthesis of oxospirochlorins
In round bottom flask a toluene (15 mL) solution of the β-amino porphyrin 2a–2f (0.18 mmol) and 3,6-di-2-pyridyl-1,2,4,5-tetrazine 5 (0.18 mmol, 0.043 g) was heated at reflux for 5 h with TLC monitoring (eluent: CHCl3). After being cooled to room temperature and evaporated, the reaction mixture was applied on the top of a silica gel column. Purification of the purple-brown coloured product using column chromatography and CHCl3 as eluent afforded the desired oxospirochlorin product as a dark purple colored solid.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been financially supported by the Polish National Center of Sciences (Grant No. 2013/11/N/ST5/02040). The computational resources were partially provided by the Polish Infrastructure for Supporting Computational Science in the European Research Space (PL-GRID).Notes and references
- (a) L. R. Milgrom, in The colours of life, Oxford University Press, Oxford, 1997 Search PubMed; (b) J. P. Hill, F. D'Souza and K. Ariga, in Supramolecular Chemistry: From Molecules to Nanomaterials ed. P. A. Gale, and J. W. Steed, John Wiley & Sons, Ltd, 2012 Search PubMed; (c) K. M. Kadish, K. M. Smith and R. Guilard in The Porphyrin Handbook, Academic Press, Burlington, MA, 2000 Search PubMed.
- (a) C. M. Che, V. K. Y. Lo, C. Y. Zhou and J. S. Huang, Chem. Soc. Rev., 2011, 40, 1950 RSC; (b) M. I. J. Stich, L. H. Fischer and O. S. Wolfbeis, Chem. Soc. Rev., 2010, 39, 3102 RSC; (c) C. M. Drain, K. C. Russell and J. M. Lehn, Chem. Commun., 1996, 337 RSC; (d) L. R. Milgrom, in The Colours of life, Oxford University, Oxford, U. K., 1997, ch. 7 Search PubMed; (e) K. Licha, Top. Curr. Chem., 2002, 222, 1 CrossRef; (f) T. Goslinski and J. Piskorz, J. Photochem. Photobiol., C, 2011, 12, 304 CrossRef.
- K. M. Kadish, K. M. Smith and R. Guilard, in Handbook of porphyrin science, with Applications to chemistry, Physics, Materials Science, Engineering, Biology and Medicine 2010, vol. 16–20 Search PubMed.
- (a) R. B. Woodward, Angew. Chem., Int. Ed., 1960, 72, 651 CrossRef; (b) R. W. Woodward, Pure Appl. Chem., 1961, 2, 383 CrossRef; (c) J. H. Fuhrhop, T. Lumantobing and J. Ullrich, Tetrahedron Lett., 1970, 3771 CrossRef; (d) H. H. Inhoffen, J. W. Buchler and P. Jager, Prog. Chem. Org. Nat. Prod., 1968, 26, 284 Search PubMed; (e) H. J. Callot, A. W. Johnson and A. Sweeney, J. Chem. Soc., Perkin Trans. 1, 1973, 1424 RSC; (f) M. Taniguchi and J. S. Lindsey, Chem. Rev., 2017, 117, 344 CrossRef PubMed.
- (a) G. L. Collier, A. H. Jackson and G. W. A. Kenner, Chem. Commun., 1966, 299 RSC; (b) G. L. Collier, A. H. Jackson and G. W. Kenner, J. Chem. Soc. C, 1967, 66 RSC.
- K. M. Smith, J. A. P. Baptista de Almeida and W. M. Lewis, J. Heterocycl. Chem., 1980, 17, 481 CrossRef.
- S. Kai and M. Suzuki, Tetrahedron Lett., 1996, 37, 5931 CrossRef.
- Y. Chang, P. S. Clezy and D. B. Morell, Aust. J. Chem., 1967, 20, 959 CrossRef.
- M. J. Crossley and L. G. King, J. Chem. Soc., Chem. Commun., 1984, 920 RSC.
- (a) J. E. Baldwin, M. J. Crossley and J. DeBernardis, Tetrahedron, 1982, 38, 685 CrossRef; (b) I. A. Abdulaeva, K. P. Birin, J. Michalak, A. Romieu, C. Stern, A. Bessmertnykh-Lemeune, R. Guilard, Y. G. Gorbunova and A. Y. Tsivadze, New J. Chem., 2016, 40, 5758 RSC.
- (a) A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476 CrossRef; (b) A. M. d’A. Rocha Gonçalves, J. M. T. B. Varejao and M. M. Pereira, J. Heterocycl. Chem., 1991, 28, 635 CrossRef.
- B. P. Bandgar and P. Gujarathi, J. Chem. Sci., 2008, 120, 259 CrossRef.
- R. Prasath, R. Butcher and P. Bhavana, Spectrochim. Acta, Part A, 2012, 87, 258 CrossRef PubMed.
- P. Wyrębek and S. Ostrowski, J. Porphyrins Phthalocyanines, 2007, 11, 822 CrossRef.
- (a) T. Khoury and M. J. Crossley, Chem. Commun., 2007, 4851 RSC; (b) J. P. C. Tome, A. M. V. M. Pereira, C. M. A. Alonso, M. G. P. M. S. Neves, A. C. Tome, A. M. S. Silva, J. A. S. Cavaleiro, M. V. Martinez-Diaz, T. Torres, A. G. M. Rahman, J. Ramey and D. M. Guldi, Eur. J. Org. Chem., 2006, 257 CrossRef.
- P. Świder, A. Nowak-Król, R. Voloshchuk, J. P. Lewtak, D. T. Gryko and W. Danikiewicz, J. Mass Spectrom., 2010, 45, 1443 CrossRef PubMed.
- (a) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef; (b) K. Alder and O. Diels, Justus Liebigs Ann. Chem., 1928, 460, 98 CrossRef; (c) D. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816 CrossRef; (d) S. Otto and J. B. F. N. Engberts, Pure Appl. Chem., 2000, 73, 1365 CrossRef.
- (a) M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518 CrossRef PubMed; (b) B. L. Oliveira, Z. Gou and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895 RSC; (c) Z. M. Png, H. Zeng, Q. Dr Ye and J. Dr Xu, Chem.–Asian J., 2017, 12, 2142 CrossRef PubMed; (d) E. Brachet and P. Belmont, Curr. Org. Chem., 2016, 20, 2136 CrossRef; (e) H. Wu and N. K. Devaraj, Top. Curr. Chem., 2015, 374, 1 Search PubMed; (f) R. A. Foster and M. C. Willis, Chem. Soc. Rev., 2013, 42, 63 RSC.
- S. Mayer and K. Lang, Synthesis, 2017, 49, 830 Search PubMed.
- (a) D. L. Boger, Chem. Rev., 1986, 86, 781 CrossRef; (b) N. Saracoglu, Tetrahedron, 2007, 63, 4199 CrossRef; (c) D. N. Kozhevnikov and A. M. Prokhorov, Prog. Heterocycl. Chem., 2011, 22, 427 Search PubMed; (d) A. M. Prokhorov and D. N. Kozhevnikov, Chem. Heterocycl. Compd., 2012, 48, 1153 CrossRef; (e) A. C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131 RSC.
- J. Sauer and D. K. Heldmann, Tetrahedron Lett., 1998, 39, 2549 CrossRef.
- K. Fukui, Angew. Chem., Int. Ed., 1982, 21, 801 CrossRef.
- A. Talbot, D. Devaian, S. J. Gustafson, I. Fernandez, F. M. Bickelhaupt and D. H. Ess, J. Org. Chem., 2015, 80, 548 CrossRef PubMed.
- (a) H. W. Daniel, S. C. Williams, H. A. Jenkins and C. Brückner, Tetrahedron Lett., 2003, 44, 4045 CrossRef; (b) C. Brückner, J. R. McCarthy, H. W. Daniell, Z. D. Pendon, R. P. Ilagan, T. M. Francis, L. Ren, R. R. Birge and H. A. Frank, Chem. Phys., 2003, 294, 285 CrossRef; (c) N. Hewage, M. Zeller and C. Brückner, Org. Biomol. Chem., 2017, 15, 396 RSC.
- (a) J. Akhigbe, M. Luciano, M. Zeller and C. Brückner, J. Org. Chem., 2015, 80, 499 CrossRef PubMed; (b) H. M. Rhoda, J. Akhigbe, J. Ogikubo, J. R. Sabin, C. J. Ziegler, C. Brückner and V. N. Nemykin, J. Phys. Chem. C, 2016, 120, 5805 CrossRef PubMed.
- (a) J. Śniechowska, P. Paluch, G. Bujacz, M. Gorecki, J. Frelek, D. T. Gryko and M. J. Potrzebowski, CrystEngComm, 2016, 18, 3561 RSC; (b) K. Trzeciak-Karlikowska, A. Bujacz, W. Ciesielski, G. D. Bujacz and M. J. Potrzebowski, J. Phys. Chem. B, 2011, 32, 9910 CrossRef PubMed; (c) K. Nowicka, A. Bujacz, P. Paluch, A. Sobczuk, A. Jeziorna, W. Ciesielski, G. D. Bujacz, J. Jurczak and M. J. Potrzebowski, Phys. Chem. Chem. Phys., 2011, 13, 6423 RSC.
- J. Śniechowska, P. Paluch and M. J. Potrzebowski, RSC Adv., 2017, 7, 24795 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef.
- (a) O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed; (b) O. A. Vydrov, J. Heyd, A. Krukau and G. E. Scuseria, J. Chem. Phys., 2006, 125, 074106 CrossRef PubMed; (c) T. M. Henderson, A. F. Izmaylov, G. Scalmani and G. E. Scuseria, J. Chem. Phys., 2009, 131, 044108 CrossRef PubMed.
- R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef.
- H. Sun and J. Autschbach, J. Chem. Theory Comput., 2014, 10, 1035 CrossRef PubMed.
- (a) T. Körzdörfer, J. S. Sears, C. Sutton and J.-L. Bredas, J. Chem. Phys., 2011, 135, 204107 CrossRef PubMed; (b) S. Refaely-Abramson, R. Baer and L. Kronik, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 075144 CrossRef; (c) J. Knight, X. Wang, L. Gallandi, X. Ren, P. Rinke, T. Körzdörfer, N. Marom, O. Dolgounitcheva and J. V. Ortiz, J. Chem. Theory Comput., 2016, 12, 615 CrossRef PubMed; (d) L. Gallandi, N. Marom, P. Rinke and T. Körzdörfer, J. Chem. Theory Comput., 2016, 12, 605 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for new compounds: NMR, ESI-MS, IR spectra, UV-VIS, crystal and structure details for 6a and supplementary theoretical studies. CCDC 1573589. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra02445f |
| This journal is © The Royal Society of Chemistry 2018 |