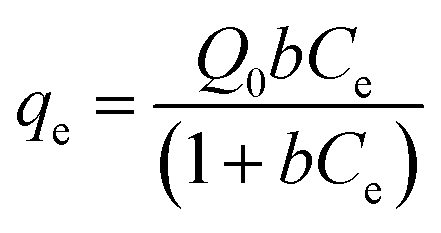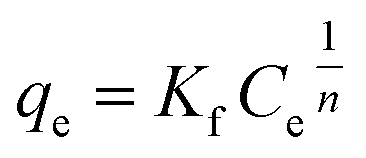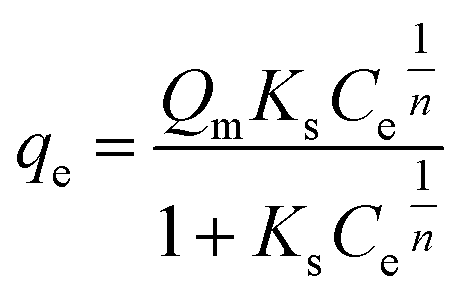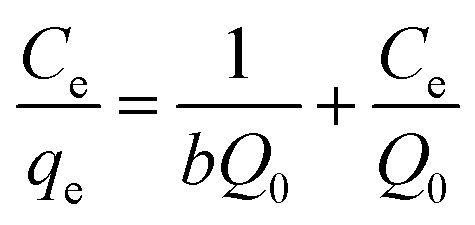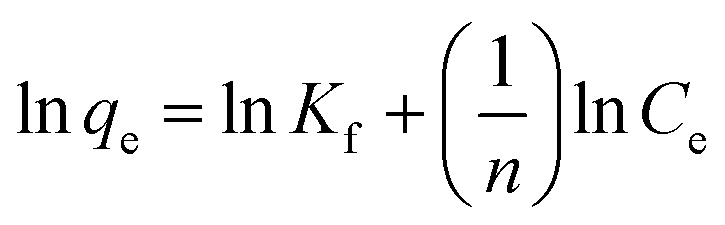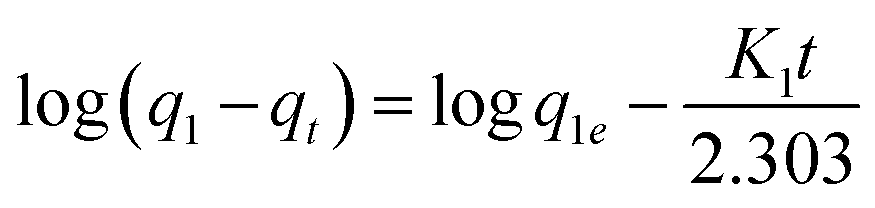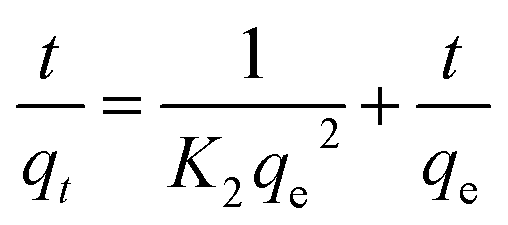DOI:
10.1039/C8RA02379D
(Paper)
RSC Adv., 2018,
8, 14280-14292
Computer-aided design of magnetic molecularly imprinted polymer nanoparticles for solid-phase extraction and determination of levetiracetam in human plasma†
Received
18th March 2018
, Accepted 12th April 2018
First published on 17th April 2018
Abstract
Analytical methods should be accurate and specific to measure plasma drug concentration. Nevertheless, current sample preparation techniques suffer from limitations, including matrix interference and intensive sample preparation. In this study, a novel technique was proposed for the synthesis of a molecularly imprinted polymer (MIP) on magnetic Fe3O4 nanoparticles (NPs) with uniform core–shell structure. The Fe3O4@MIPs NPs were then applied to separate and enrich an antiepileptic drug, levetiracetam, from human plasma. A computational approach was developed to screen the functional monomers and polymerization solvents to provide a suitable design for the synthesized MIP. Different analysis techniques and re-binding experiments were performed to characterize the Fe3O4@MIP NPs, as well as to identify optimal conditions for the extraction process. Adsorption isotherms were best fitted to the Langmuir model and adsorption kinetics were modeled with pseudo-second-order kinetics. The Fe3O4@MIP NPs showed reasonable adsorption capacity and improved imprinting efficiency. A validated colorimetric assay was introduced as a comparable method to a validated HPLC assay for the quantitation of levetiracetam in plasma in the range of 10–80 μg mL−1 after extraction. The results from the HPLC and colorimetric assays showed good precision (between 1.08% and 9.87%) and recoveries (between 94% and 106%) using the Fe3O4@MIP NPs. The limit of detection and limit of quantification were estimated to be 2.58 μg mL−1 and 7.81 μg mL−1, respectively for HPLC assay and 2.32 μg mL−1 and 7.02 μg mL−1, respectively for colorimetric assay. It is believed that synthesized Fe3O4@MIP NPs as a sample clean-up technique combined with the proposed assays can be used for determination of levetiracetam in plasma.
1. Introduction
Complex matrices as biological fluids constituting blood, plasma or urine contain a number of endogenous components, which if not removed may interfere and adversely affect the separation and identification of analyte(s) of interest.1,2 Such challenges made sample preparation one of the most important and crucial steps in the analytical process.3 However, one drawback of sample preparation steps is that they consume around two-thirds of the total analysis time and are considered the major source of error in the overall analytical process.2 Extraction techniques that are routinely used in sample preparation are solid-phase extraction (SPE) and liquid–liquid extraction (LLE).3 Currently, the most appropriate method for extraction of drugs in biological fluids is SPE due to its adaptability and simplicity while LLE suffers from high consumption of labor and solvents.2,4 Nevertheless, both extraction techniques lack specific adsorption ability.5
A molecularly imprinted polymer (MIP) is a synthetic polymer that reshapes its surface according to the molecular structure of the target molecule. The application of MIPs as sorbents in SPE is quite effective and appealing because they offer higher selectivity, reduce the influence of the matrix components and provide high sample enrichment factors.2,6,7 However, MIP as adsorbents suffers from some drawbacks as the heterogeneous distribution of binding sites, low binding capacity, and slow binding kinetic.8,9 Thus the development of molecular imprinting nano techniques with magnetic properties will provide a potential solution to overcome such drawbacks.9
Magnetic molecularly imprinted polymer nanoparticles (MMIP NPs) synthesis is a new technique achieved via encapsulating an inorganic magnetic particle with molecularly imprinted polymers. When the MMIP NPs bind the target analyte they can be easily separated from the matrix media by an external magnetic field without additional filtration or centrifugation.8,10 In addition, MMIP NPs are expected to improve the binding capacity and fasten binding kinetics over normal imprinting materials.8 Based on such assumptions many studies nowadays adopted the idea of incorporating magnetic nanoparticles with molecularly imprinted polymers for extraction of drugs from different media.5,11–18
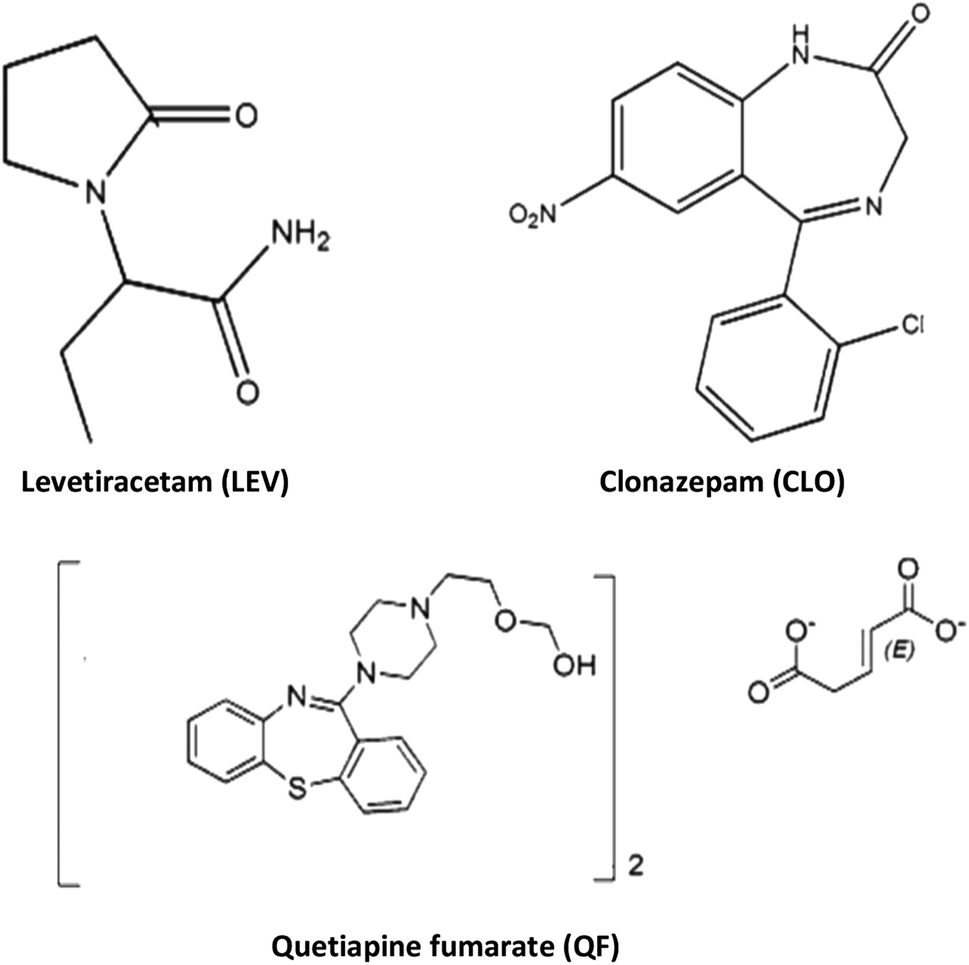
Levetiracetam (Fig. 1) (LEV) is an antiepileptic approved therapy for focal epilepsy and myoclonic and tonic-clonic seizures.19 LEV is also licensed for use as adjunctive therapy to other first line anti-epileptic drugs in adult patients with partial onset seizures and is often prescribed with antipsychotic drugs.20,21 LEV has a concentration ranging between 12 to 46 mg mL−1 in plasma.22,23 and its plasma protein binding is low (10%). LEV also undergoes insignificant hepatic metabolism to inactive metabolites and about 91% of the dose is excreted through renal route.24 Nevertheless, it is recommended to monitor the plasma concentrations of LEV to optimize its therapeutic effect, especially in patients with renal impairment, and in children, where the half-life of the drug is shortened and in the elderly where the half-life is extended.25,26 There are only a few papers published reporting therapeutic drug monitoring methods of LEV employing different chromatographic techniques including HPLC with UV-detection,27,28 and GC with NPD-detection.27,29 Chromatographic techniques are commonly sensitive and selective, yet, they require toxic solvents, expensive equipment, in addition to complex sample pretreatments. Thus, spectroscopic techniques are more convenient for routine laboratory analysis. Nevertheless, such spectroscopic techniques if not combined with sample pretreatment steps, may suffer from the influence of interfering biological samples components. Thus, sample preparation is considered a very crucial step.30
 |
| | Fig. 1 Chemical structure of levetiracetam and its co-administered drugs. | |
In this work, we present a novel method to prepare a molecularly imprinted polymer layer on magnetic Fe3O4 NPs to separate and enrich LEV from human plasma. To improve the properties of the synthesized MIP, a computer-aided study has been proposed. The morphology, chemical characteristics and adsorption properties of the Fe3O4@MIPs NPs were evaluated using different characterization techniques. LEV concentration in plasma was quantified via two novel validated assays; HPLC-UV and colorimetry.
2. Experimental
2.1. Materials
Levetiracetam, quetiapine fumarate, and clonazepam powders were kindly provided by Eva Pharmaceuticals, Chemi pharm, and Amoun pharmaceutical company respectively (Cairo, Egypt). Ferric chloride anhydrous (FeCl3), ferrous sulfate heptahydrate (FeSO4·7H2O), ammonia solution (25%, w/w) and tetraethoxysilane (TEOS) were purchased from Fisher Scientific (USA). Oleic acid was purchased from central drug house (India). Methacryloxypropyl trimethoxysilane (MPS) was obtained from Sigma Aldrich (China). Methacrylic acid (MAA), ethylene glycol dimethacrylate (EGDMA) were purchased from Aldrich (Germany) and azobisisobutyronitrile (AIBN) from Levochem (USA), human blank plasma was obtained from the Holding Company for Biological Products and Vaccines (VACSERA), Egypt. HPLC grade acetonitrile, acetone, and methanol were purchased from Fisher Scientific (UK). Potassium dihydrogen phosphate was purchased from Sigma Aldrich (Germany). All other chemicals and reagents used were of analytical grade or higher.
2.2. Instrumentation
The morphologies and dimensions of the synthesized nanoparticles were characterized using scanning electron microscopy (SEM) in a Zeiss instrument (Germany) and the transmission electron microscopy (TEM) on Tecani G20, FEI transmission electron microscope (USA). The structure of the synthesized nanoparticles was characterized by X-ray diffractometer (a Rigaku model Geigerflex apparatus). Fourier transform infrared (FT-IR) spectra (KBr pellets) were performed on a Perkin Elmer Spectrum GX spectrophotometer. Magnetic properties were analyzed using a vibrating sample magnetometer (Princeton EG and G Applied Research VSM, Model 155). The concentrations of levetiracetam in the solutions were determined by a Single beam UV-Vis model AE-S90-MD form A & E Lab (UK). High performance liquid chromatography (HPLC) was performed on an Agilent liquid chromatograph system model 1100 (Agilent Technologies, USA). The variable wavelength UV-Vis detector was operated at λmax for investigated drug determinations.
2.3. Computational approach
Computational chemistry tools were applied in order to estimate the affinities of different monomers to the template drug levetiracetam in different solvents. This strategy saves a lot of time and effort since one does not have to select monomers and solvents based on trial and error in the laboratory. In our computational study, the monomers tried were acrylamide (AAM), methacrylic acid (MAA), methyl methacrylate (MMA), acrylonitrile (AN) and divinylbenzene (DVB). The solvents tried were chloroform, methanol, acetonitrile, dimethyl sulfoxide (DMSO), and acetone. Firstly, a stochastic conformational search of the template–monomer complex was performed on Molecular Operating Environment (MOE)31 using the default settings. The MMFF94x force field was used with a distance-dependent dielectric model. In most of the cases, two possible 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes between template and monomer were obtained with a slight difference in energy, and only one reasonable 1:2 complex was obtained. These three complexes, involving 5 different monomers and in 5 different solvents were investigated using ab initio quantum mechanics. All the ab initio calculations were done using Gaussian 09 (ref. 32) on Pharmamatrix Cluster, University of Alberta. Geometry optimization of the template, monomer, and template–monomer complex was done employing the Hartree–Fock level of theory with the 6-31G(d) basis set. Frequency analysis was done to ensure the absence of imaginary frequencies at the optimized structures. The Polarizable Continuum Model (PCM)33 was employed to account for the different solvent effects. The interaction energy between the template and the monomer(s) was calculated using the following equation:
:1 complexes between template and monomer were obtained with a slight difference in energy, and only one reasonable 1:2 complex was obtained. These three complexes, involving 5 different monomers and in 5 different solvents were investigated using ab initio quantum mechanics. All the ab initio calculations were done using Gaussian 09 (ref. 32) on Pharmamatrix Cluster, University of Alberta. Geometry optimization of the template, monomer, and template–monomer complex was done employing the Hartree–Fock level of theory with the 6-31G(d) basis set. Frequency analysis was done to ensure the absence of imaginary frequencies at the optimized structures. The Polarizable Continuum Model (PCM)33 was employed to account for the different solvent effects. The interaction energy between the template and the monomer(s) was calculated using the following equation:
| ΔE = Etemplate–monomer complex − Etemplate − ΣEmonomers |
Images were created using VMD version 1.9.2.34 It is worth mentioning that counterpoise correction (CP) for the basis set superposition error (BSSE) was not employed. The utility of applying this correction is questionable especially if one only wants to rank the relative binding energies with no significance for absolute binding energies. It was shown that correction for BSSE only affects the absolute values of the binding energies without affecting the relative ranking of different monomers.35,36
2.4. Preparation of magnetic molecularly imprinted polymer nanoparticles (MMIP NPs) for LEV extraction
2.4.1. Synthesis of Fe3O4@oleic acid NPs. The synthesis of the magnetite (Fe3O4) NPs was done via a co-precipitation technique. 100 mL of 0.4 M FeCl3 aqueous solution were mixed with 100 mL of 0.2 M FeSO4 aqueous solution. Then, 100 mL of 3.0 M NaOH aqueous solution was added dropwise under continuous stirring. Finally, 2.0 mL of oleic acid was added and the reaction mixture was heated to 80 °C for 1 h. The black precipitate product was washed several times with ultrapure water and methanol to remove excess oleic acid from the solution and the precipitate was dried at 70 °C for 5 h.37
2.4.2. Synthesis of the Fe3O4@oleic acid@SiO2 NPs. 300 mg of Fe3O4@oleic acid NPs were dispersed in 40 mL ethanol and 4.0 mL of purified water via sonication for 15 min, followed by the addition of 2.0 mL ammonium hydroxide and 2.0 mL TEOS consecutively. The mixture was reacted for 18 h at room temperature under continuous stirring. The obtained product was collected by an external magnetic field, washed with methanol several times, and dried at 70 °C for 5 h.14
2.4.3. Synthesis of vinyl-modified Fe3O4 NPs. 250 mg of Fe3O4@SiO2 NPs was dispersed in 50 mL methanol by sonication for 10 min and 3.0 mL of MPS were added dropwise while stirring. The mixture was then reacted for 24 h at the room temperature under continuous stirring. The obtained product was collected by an external magnetic field and washed with methanol and dried in vacuum.13
2.4.4. Synthesis of the core–shell Fe3O4@MIP and Fe3O4@NIP NPs. The Fe3O4@MIP NPs were prepared using a non-covalent imprinting technique. The template molecule LEV (0.55 mmol) was mixed with the selected functional monomer MAA (2.2 mmol) in 30 mL chloroform as selected polymerization solvent, and the mixture was stirred for 12 h to form template–monomer complex. Vinyl modified Fe3O4 (200 mg), EGDMA (11 mmol) and AIBN (50 mg) were then added to the above solution. To remove dissolved oxygen, the solution was stirred under vacuum for 5 min. The mixture was then heated to reflux, 60 °C for 24 h. The Fe3O4@MIP NPs were rinsed with ethanol, acetonitrile and diethyl ether until the supernatant was clear. The template molecule LEV was extracted from the polymer with 90/10 (v/v) MeOH/acetic acid mixture in a Soxhlet extraction system during 72 h. Finally, the obtained product was dried in the air. Non-imprinted Fe3O4@NIP NPs were prepared following the same procedure in the absence of the template LEV.
2.5. Analysis techniques
2.5.1. Colorimetric method for binding experiments. Stock solution (0.1 mg mL−1) of LEV was prepared in methanol. Aliquots of the stock solution were transferred into 25 mL volumetric flasks. 0.5 mL of (1% (w/v)) ferric chloride; (freshly prepared by dissolving 500 mg of ferric chloride in 5 mL concentrated HCl and complete volume to 50 mL with distilled water), and 0.5 mL of (0.2% (w/v)) potassium ferricyanide; (prepared by dissolving 100 mg of Potassium ferricyanide in 50 mL distilled water) were added and final volume was made up to the mark with methanol. A Prussian blue colored product was formed immediately at room temperature. Absorbance of the samples was measured at 775 nm against the reagent blank prepared in the same manner. Calibration curve for LEV was obtained by plotting absorbance at the λmax = 775 nm against concentration. Various assay validation parameters were then calculated according to ICH guidelines.38–40
2.5.2. Spectrophotometric method for binding experiments. Standard solutions (0.1 mg mL−1) of quetiapine fumarate (QF) and clonazepam (CLO) were prepared in methanol separately. Solutions were scanned in the range of 200–400 nm and the wavelength of maximum absorption for each drug was determined. Accurate volumes of quetiapine fumarate and clonazepam stock solutions (0.3, 0.6, 1.2, 1.8, 2.4 and 3.0 mL) were transferred into 10 mL volumetric flasks separately and diluted to volume with methanol. Calibration curves for the investigated drugs were obtained by plotting absorbance at the λmax of each drug; 245 nm and 250 nm for QF and CLO respectively, against concentration. Various assay validation parameters were then calculated according to ICH guidelines.38–40
2.6. Binding experiments for optimization of molecular imprinting magnetic solid phase extraction (MI-MSPE) procedure
Preliminary studies were carried out to determine best solvent for extraction of the target drug LEV and the contact time required to reach equilibrium. 150 mg of Fe3O4@MIP NPs were added into 30 mL of LEV solution of initial concentration (100 mg L−1) at neutral pH and shaken at 25 °C. After defined time intervals, samples were removed and the absorbance of LEV left in the supernatant solutions after magnetic separation were determined using the validated colorimetric assay.
The influence of pH on the extraction of LEV was investigated using 100 mg L−1 of LEV solution over pH range of 3.0–11.0 adjusted using 0.1 M HCl and 0.1 M NaOH. To each of the pH-adjusted LEV solution, 25 mg of Fe3O4@MIP NPs was added and shaken for 30 min at 25 °C.
To study the effect of adsorbent concentration 25–200 mg of Fe3O4@MIP NPs was added to 5.0 mL of LEV solution (100 mg L−1) with a contact time of 30 min and pH adjusted at 7.0.
The quantity (Q) of the template bound to Fe3O4@MIPs or Fe3O4@NIPs was calculated according to the following equation:8
where
C0 and
Ct (mg L
−1) are the initial concentration and the residual concentration of LEV at time (
t), respectively;
V (L) is the initial volume of the solution, and
W (g) is the weight of the Fe
3O
4@MIPs or Fe
3O
4@NIPs.
Also, the amount of LEV extracted was expressed in percentage (R, %) and calculated using the equation R% = 100(C0 − Ct)/C0.41
2.7. Isothermal binding experiments
25 mg of Fe3O4@MIP NPs were added to 5.0 mL acetonitrile (ACN) solution of LEV of various concentrations from 25 to 125 mg L−1 and incubated for 30 min at 25 °C. The Fe3O4@MIP NPs were collected by an external magnetic field, and the concentration of LEV in the supernatants was measured colorimetrically at λmax 775 nm.
2.8. Kinetic adsorption experiments
25 mg of Fe3O4@MIP NPs were added to LEV solution (5.0 mL) of various concentrations (25, 50 and 100 mg L−1) and incubated at different time intervals (0–30 min) at 25 °C. The supernatant and imprinted polymers were separated by an external magnetic field, and the concentration of LEV in the supernatants was measured colorimetrically at λmax 775 nm.
2.9. Selectivity test
To evaluate the selectivity of levetiracetam magnetic imprinted polymer, several co-administered drugs such as quetiapine fumarate (QF) and clonazepam (CLO) were tested. The experiments were carried out by adding 25 mg of Fe3O4@MIP and Fe3O4@NIP NPs to 5.0 mL of investigated drugs standard solutions at a concentration of 100 mg L−1 separately and pH was adjusted at pH 7.0 using phosphate buffer solution. The solutions were shaken at 60 rpm for 30 min at 25 °C in a rotatory shaker. The supernatant of each drug and the magnetic imprinted polymers were separated by an external magnetic field and analyzed by UV-Vis spectroscopy.
2.10. Molecularly imprinted magnetic solid-phase extraction (MI-MSPE) of LEV from plasma samples
2.10.1 Calibration and control samples. A standard stock solution of LEV (1.0 mg mL−1) was prepared in ACN. Spiking solutions for calibration curve and quality controls (QC) were prepared by appropriate dilution in ACN. Spiking solutions (50 μL) were added to drug-free human plasma (950 μL) as a bulk, to obtain LEV spiking concentration levels of 10–80 μg mL−1. Each validation run consisted of blank sample (a plasma sample processed without drug), calibration curve consisting of six non-zero samples covering a range of 10 μg mL−1 to 80 μg mL−1 and quality control (QC) samples; LLOQ = 7.5 μg mL−1, QCL = 15 μg mL−1, QCM = 4 μg mL−1 and QCH = 70 μg mL−1. Validation experiments were done on three consecutive days and linearity was assessed by least square regression analysis. The acceptable calibration curve is expected to have a correlation coefficient (R2) of 0.999 or better. The acceptance criterion for each back-calculated standard concentration was 15% deviation from the nominal value except LLOQ, which was set at 20%.
2.10.2. Sample processing. 1 mL of plasma samples spiked with known variable amounts of LEV standard solution was vortexed with 2.0 mL of ACN for 5.0 min. The mixtures were then centrifuged for 15 min at 5000 rpm. The supernatants were totally pipetted and diluted to 5.0 mL with phosphate buffer where pH was adjusted at 7.0. Blank samples were prepared in the same way as above but without the LEV spiking step.50 mg of Fe3O4@MIP NPs was placed into a glass vial. Prior to each extraction, particles were conditioned with 1 mL ACN and 2 mL PBS (pH 7.0) and decanted under magnetic field. Then 5.0 mL of sample or standard LEV solution at pH 7.0 was added and the resulting mixture was shaken for 30 min at room temperature. Next, the solution was decanted by applying external magnetic field. The separated particles were washed by 2.0 mL ACN to remove any possible interference. LEV was extracted from Fe3O4@MIP NPs shell by eluting the adsorbent with 2.0 mL MeOH by sonication for 10 min. The desorbed solution was collected under an external magnetic field and was analyzed via two different techniques; HPLC assay and a colorimetric assay, to quantify the recovered analyte and compare the integrity of the two proposed techniques.
2.10.3. Method validation. Specificity of the proposed methods was determined using randomly selected six blank human plasma samples, which were collected under controlled conditions.Within-batch and between-batch accuracy and precision were evaluated by analysis of six replicates of the QC samples on the same day and on three consecutive days, respectively. Recovery of LEV was assessed by comparing the mean peak area or the mean absorbance of six extracted QC samples to those prepared in post-extraction blank plasma. In addition, the process efficiency was evaluated by comparing the mean peak areas or mean absorbance of extracted QC samples to those of LEV standards prepared in pure solvent.
2.10.4. Chromatographic assay. Separation of eluted spiked plasma samples was carried out using a reversed phase Agilent C18 column (250 × 4.6 mm, 5 μm). (Agilent, USA) with a mobile phase of methanol:PBS (pH 4.0 adjusted with phosphoric acid) in the ratio of 25:75 and a flow rate of 1.5 mL min−1 at 25 °C. Each component of the mobile phase was degassed before use in an ultrasonic bath for 10 min. The detection wavelength was set to 205 nm, injection volume was 20 μL and a total run time of 8 min. The retention time of LEV was 4.87 min under the described conditions.
2.10.5. Colorimetric assay. Eluted solutions of spiked plasma samples in methanol were transferred into 10 mL volumetric flasks. 0.5 mL of 1.0% (w/v) ferric chloride and 0.5 mL of 0.2% (w/v) potassium ferricyanide were added and final volume was made up to the mark with methanol. The colored product was formed immediately at room temperature. The absorbance of the samples was measured at 775 nm against the reagent blank prepared in the same manner.
3. Results and discussion
3.1. Theoretical study of the template–monomer interactions
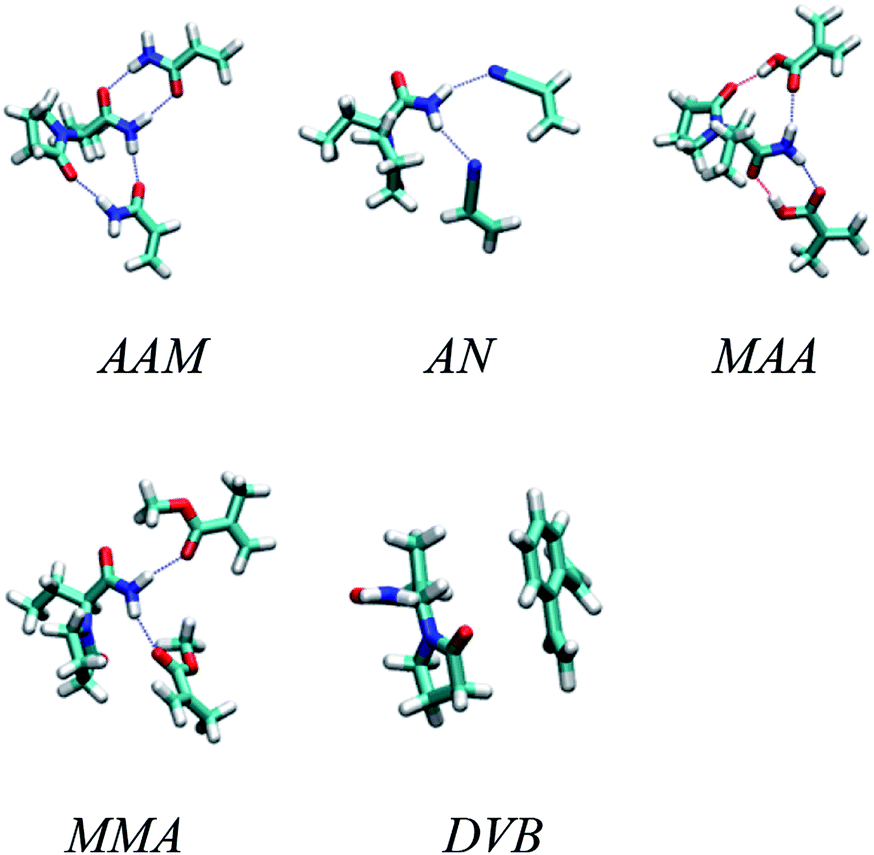
Generally, typical imprinting protocol is tedious and time consuming where the selection of optimum imprinting conditions, like functional monomers and polymerization solvent, depends on trial and error method. Therefore, a computer-aided study has been suggested as a rational and fast method to search for optimal imprinting conditions in order to improve the properties of the fabricated Fe3O4@MIP NPs, as mentioned in the methods section, we optimized several 1:1 and 1:2 complexes in different solutions. Fig. 2 shows the optimized geometries of the most stable complexes between LEV and the five monomers. The figure shows the 1:2 complex for all of them except divinylbenzene which could not form any stable 1:2 complex. As to the 1:1 complexes for the other four monomers they were simply formed by dropping either of the two monomers in the 1:2 complex, resulting in two possibilities for each monomer.
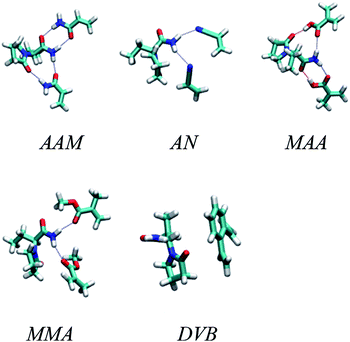 |
| | Fig. 2 Optimized geometries of the most stable complexes between LEV and acrylamide (AAM), methacrylic acid (MAA), methyl methacrylate (MMA), acrylonitrile (AN) and divinylbenzene (DVB) monomers. | |
The results for the 1:1 binding energy calculations are listed in Table S1.† For AAM and MAA, two 1:1 complexes were investigated and compared. For other monomers, the binding energies for the optimum 1:1 complexes were too low to encourage further investigation of other 1:1 complex geometries.
As the table shows, the highest binding energies are between LEV and MAA followed by AAM. The best solvent appears to be chloroform followed by acetone in both cases. This makes sense because chloroform offers a very hydrophobic environment which encourages the drug and the monomer to cluster together and form mutual hydrogen bonds with their hydrophilic hydrogen bonding groups. Other monomers gave very low binding energies and were thus considered inappropriate for molecular imprinting.
After that we investigated the 1:2 complexes formed between LEV and the monomers. Table S2† shows the results of these calculations where DVB was not considered due to its very poor binding energies in 1:1 calculations. As the table suggests, the best binding energy is obtained at the 1:2 ratio between LEV and MAA in chloroform.
3.2. Preparation of Fe3O4@MIP nanoparticles for LEV extraction
The fabrication technique of Fe3O4@MIP NPs constituted a multistep approach. Fe3O4@OA NPs were synthesized via a wet chemical co-precipitation technique. Oleic acid as surface modifier is very crucial for the successful creation of the SiO2 shell.12 The surface of Fe3O4@OA NPs was then coated with silica by TEOS. The SiO2 shell provided a hydrophilic and biocompatible surface, and prevented oxidation of Fe3O4.42 Moreover, the silanol groups allowed the existence of extra functional groups on the Fe3O4@SiO2 surface. Thus, double bonds were introduced onto Fe3O4@SiO2 using MPS to ensure firm growth of the imprinted layer.42 Finally Fe3O4@MIPs NPs were synthesized using the modified Fe3O4 NPs, MAA as monomer, EGDMA as crosslinker, levetiracetam as template and AIBN as initiator in chloroform as porogenic solvent. The resultant Fe3O4@MIP NPs provided access to the recognition sites of LEV and can be easily separated using an external magnetic field thus can be used as a clean-up technique in human plasma assay.
3.3. Characterization of Fe3O4@MIP nanoparticles
3.3.1. TEM. The size and shape of Fe3O4, Fe3O4@SiO2, Fe3O4@MPS and Fe3O4@MIP NPs were examined by TEM. It can be observed that the mean diameter of Fe3O4@OA NPs was about 9 nm (Fig. 3[a]). After modifying Fe3O4@OA with SiO2 (Fig. 3[b]), the diameter of the Fe3O4@SiO2 NPs increased to approximately 17 nm, corresponding to 8 nm thickness of the SiO2 layer on the surface of Fe3O4 NPs. Double bond modified Fe3O4@MPS NPs diameter is further increased to 24 nm (Fig. 3[c]) indicating that the coupling of MPS on the Fe3O4 surface was successful. After polymerization with LEV as the template, Fe3O4@MIP particle size increased to about 250–300 nm (Fig. 3[d]) and you can see the obvious core–shell junction structure which facilitates the mass transfer process between Fe3O4@MIP NPs surface and solution.
 |
| | Fig. 3 TEM images of: [a] Fe3O4@OA NPs, [b] Fe3O4@SiO2 NPs, [c] Fe3O4@MPS NPs and [d] Fe3O4@MIP NPs. | |
3.3.2. SEM. The nature of the Fe3O4@MIP and Fe3O4@NIP NPs was evaluated by SEM (Fig. 4). It was observed that the surface of the Fe3O4@MIP NPs is uneven with many holes; thus facilitating the adsorption of template molecule; LEV. On the other hand, the surface of Fe3O4@NIP NPs is smooth, thus lacking effective adsorption sites.
 |
| | Fig. 4 SEM images of: [a] Fe3O4@ MIP NPs and [b] Fe3O4@NIP NPs. | |
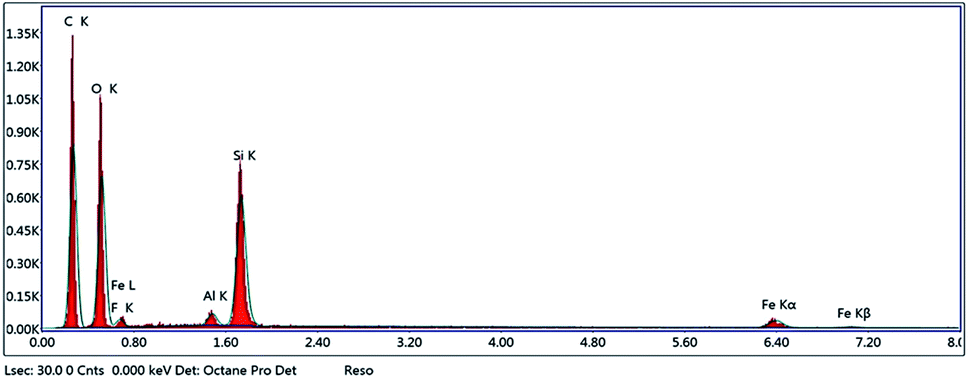
3.3.3. Energy-dispersive X-ray spectroscopy (EDX). EDX analysis was performed to confirm that polymerization took place. EDX spectra (Fig. 5) indicated the presence of Fe, Si, O and C, confirming that polymerization occurred on the surface of Fe3O4@MPS NPs.
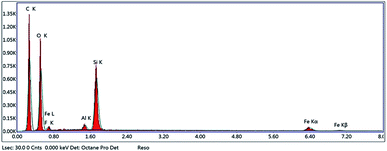 |
| | Fig. 5 EDX of Fe3O4@MIP NPs. | |
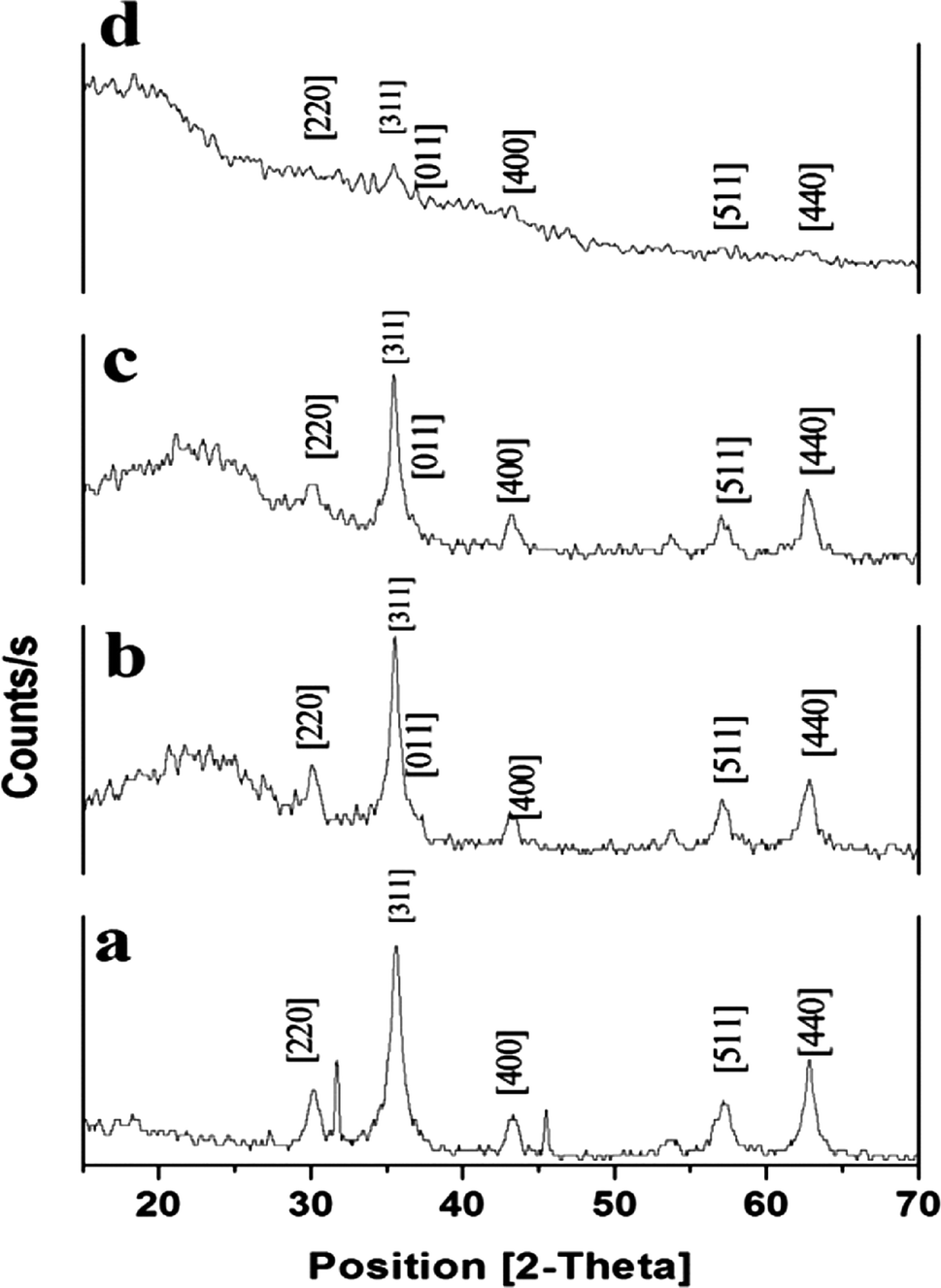
3.3.4. XRD. Structural and chemical properties of synthesized Fe3O4@MIP NPs were evaluated using X-ray diffraction (XRD). As shown in Fig. 6, XRD patterns of the synthesized Fe3O4, Fe3O4@SiO2, Fe3O4@MPS and Fe3O4@MIP NPs displayed several strong reflection peaks in the 2θ region of 20–70°. Six diffraction peaks can be indexed to (2 2 0), (3 1 1), (4 0 0), (4 2 2), (5 1 1) and (4 4 0), which match well with the database of magnetite in JCPDS (JCPDS card: 19-629) file. The XRD patterns also indicated that the synthesized polymeric particles are crystalline in shape.
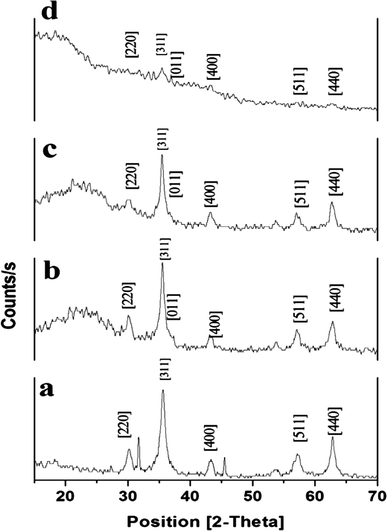 |
| | Fig. 6 XRD patterns of: [a] Fe3O4@OA NPs, [b] Fe3O4@SiO2 NPs, [c] Fe3O4@MPS NPs and [d] Fe3O4@MIP NPs. | |
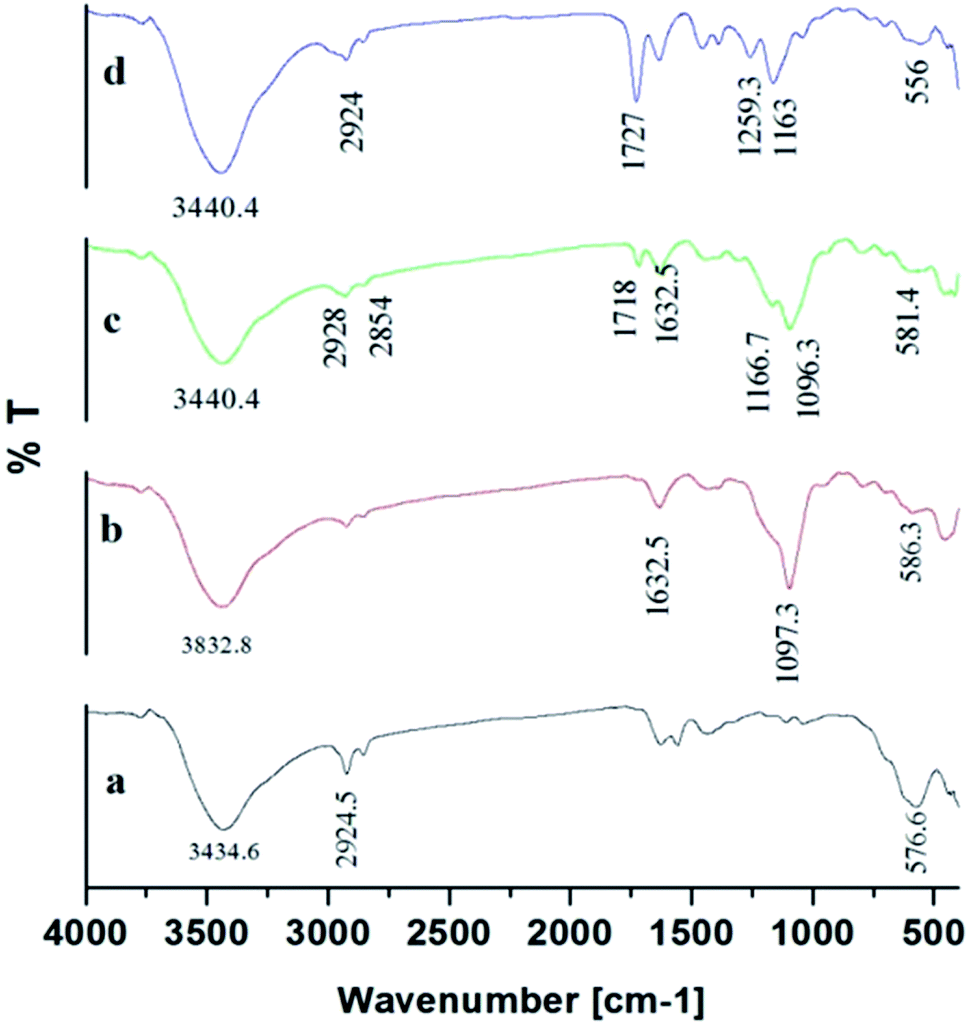
3.3.5. FTIR. The products of Fe3O4, Fe3O4@SiO2, Fe3O4@MPS, and Fe3O4@MIPs were investigated by FT-IR spectroscopy (Fig. 7). The characteristic absorption bands at 576.6 cm−1 were attributed to the Fe–O stretching vibration (Fig. 7[a]) and the C–H stretching vibrations of oleic acid were indicated by an absorption band at 2924.5 cm−1.8 After modification of SiO2 on Fe3O4@OA NPs surface, the Si–O–Si stretching vibration was observed at 1097.3 cm−1 and OH group at 1632.5 cm−1 and 3832.8 cm−1 (Fig. 7[b]) indicating the formation of silica coating on the surface of Fe3O4@OA NPs.13 As can be seen from Fig. 7[c], stretching vibrations of C–H at 2928 cm−1 and 2854 cm−1 together with the absorption band appearing at 1632.5 cm−1 indicated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond formation and that MPS had successfully modified Fe3O4@SiO2 surface.43 Also the peaks of C–O group at 1259.3 cm−1 and CO group at 1727.9 cm−1 and C–H group of methyl at 2924.5 cm−1 indicated that the methacrylic acid layer was formed on the surface of Fe3O4@MPS NPs (Fig. 7[d]) and thus confirmed the successful preparation of Fe3O4@MIPs NPs.13
C double bond formation and that MPS had successfully modified Fe3O4@SiO2 surface.43 Also the peaks of C–O group at 1259.3 cm−1 and CO group at 1727.9 cm−1 and C–H group of methyl at 2924.5 cm−1 indicated that the methacrylic acid layer was formed on the surface of Fe3O4@MPS NPs (Fig. 7[d]) and thus confirmed the successful preparation of Fe3O4@MIPs NPs.13
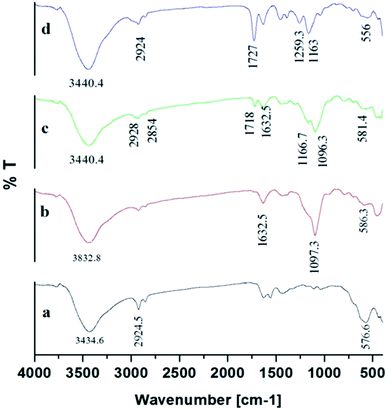 |
| | Fig. 7 FTIR spectra of: [a] Fe3O4@OA NPs, [b] Fe3O4@SiO2 NPs, [c] Fe3O4@MPS NPs and [d] Fe3O4@MIP NPs. | |
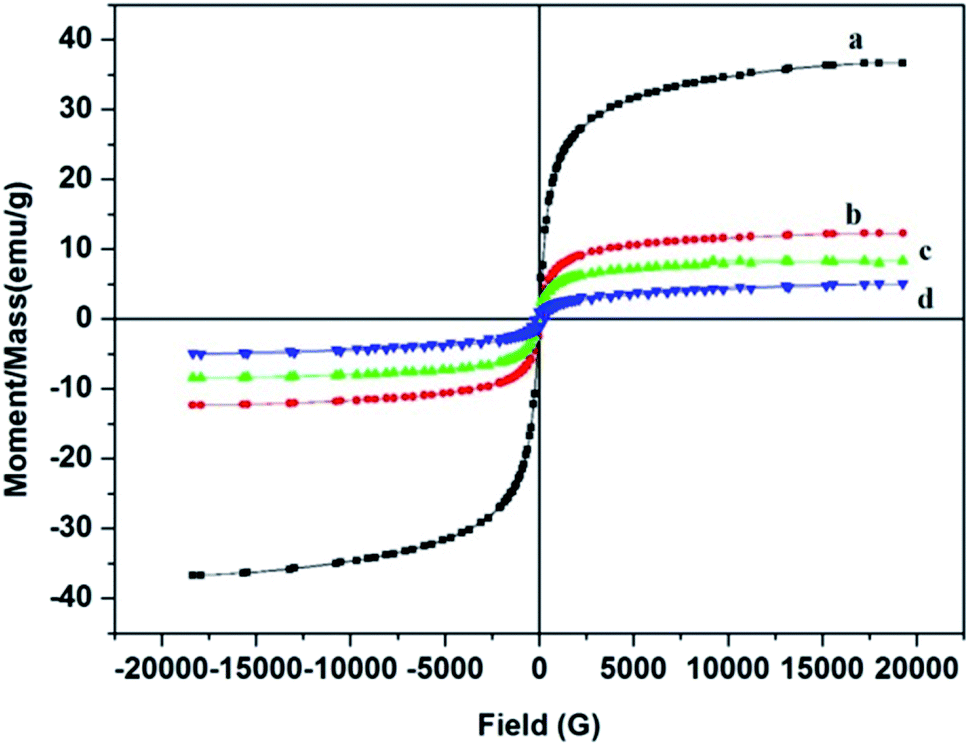
3.3.6. VSM. The magnetic properties of Fe3O4@MIP NPs were studied by VSM. The magnetic hysteresis loops of Fe3O4, Fe3O4@SiO2, Fe3O4@MPS and Fe3O4@MIP NPs at room temperature are shown in Fig. 8. Obviously, there is no hysteresis, both remanence and coercivity are almost zero, suggesting that the prepared NPs are superparamagnetic.9 The saturation magnetization values of the prepared Fe3O4, Fe3O4@SiO2, Fe3O4@MPS and Fe3O4@MIP NPs were 36.7, 12.3, 8.4 and 5.0 emu g−1, respectively. Thus, it can be concluded that the magnetization values decreased with increasing the number of non-magnetic coating layers on the surface of Fe3O4 NPs. Fe3O4@MIP NPs showed the least saturation magnetization yet, it can be separated using an external magnetic field.43,44
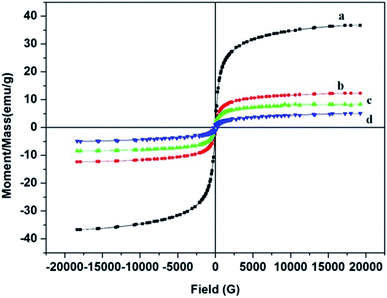 |
| | Fig. 8 Hysteresis loop of: [a] Fe3O4@OA NPs, [b] Fe3O4@SiO2 NPs, [c] Fe3O4@MPS NPs and [d] Fe3O4@MIP NPs. | |
3.4. Analysis techniques
3.4.1. Colorimetric method for binding experiments. The proposed colorimetric method for determination of LEV is based on the reduction of the Fe3+ in FeCl3 to Fe2+ by the drug in the presence of K3Fe(CN6). Consequently, the in situ formed Fe2+ reacts with K3Fe(CN6) under acidic conditions to form soluble Prussian blue (KF[Fe2(CN6)]) whose absorbance can be measured at 775 nm against the corresponding reagent blank.45 The method was validated according to ICH guidelines for validation of analytical procedures and the validation results are illustrated in Table S3.†
3.4.2. Spectrophotometric method for binding experiments. The wavelength of maximum absorption (λmax) was determined for the studied drugs (QF 245 nm and CLO 250 nm). The method was validated according to ICH guidelines for validation of analytical procedures regarding linearity, accuracy, precision (within and between days), limit of detection (LOD), and limit of quantification (LOQ).38–40 The validation results are summarized in Table S3.† Results indicated the suitability of the assay for accurate determination of the studied drugs in neat solvent for optimization purposes.
3.5. Optimization of the MI-MSPE procedure
To attain the best conditions for extraction of the target drug; LEV, various factors such as extraction solvent, extraction time, pH and amount of adsorbent were studied and optimized for LEV pre-concentration and clean-up.
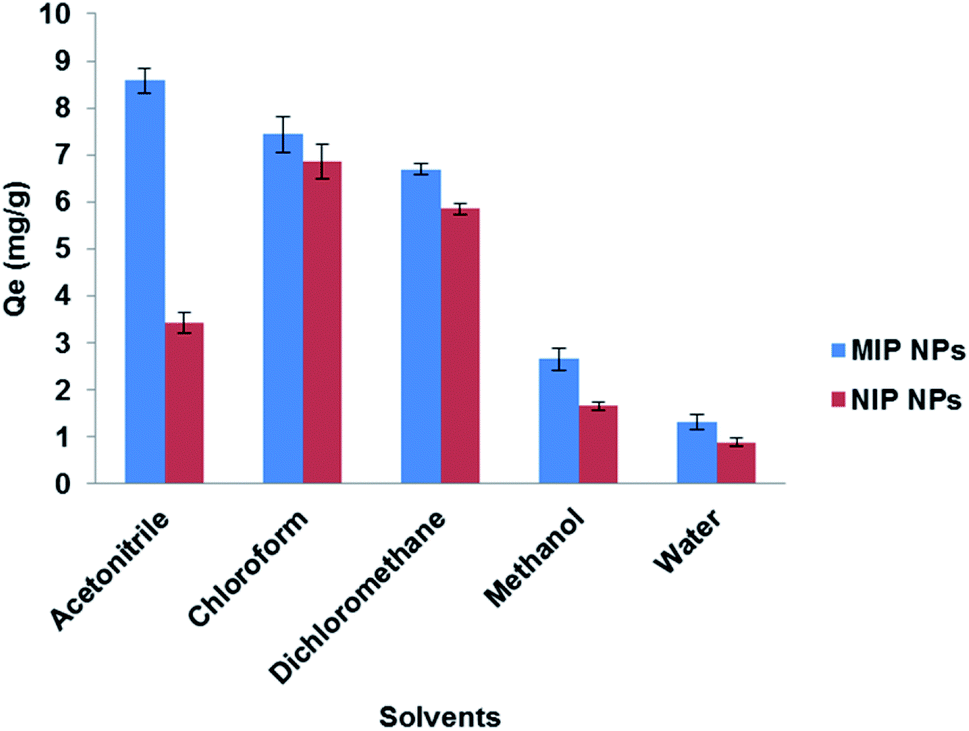
3.5.1. Extraction solvent. To find a suitable solvent for the rebinding of the template molecule LEV, several types of solvents were investigated (Fig. 9). Upon the use of chloroform (synthesis solvent) and dichloromethane, almost no difference between Fe3O4@MIP and Fe3O4@NIP NPs in extraction efficiency was found and this can be attributed to the formation of aggregates of the imprinted particles upon contact with these solvents leading to extraction by clogging effect rather than by adsorption. The results also showed that the addition of water or methanol disturbed the specific binding effect of the Fe3O4@MIP NPs, most probably because the strong polarity of water or methanol destroyed the hydrogen bond between the Fe3O4@MIP NPs and LEV.5 Nevertheless, ACN showed the best result in extraction and specificity and therefore was used in further studies.
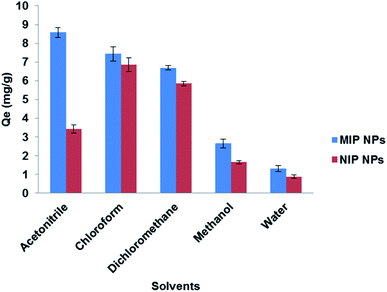 |
| | Fig. 9 Effect of different solvents on the extraction of LEV by Fe3O4@MIP NPs and Fe3O4@NIP NPs. | |
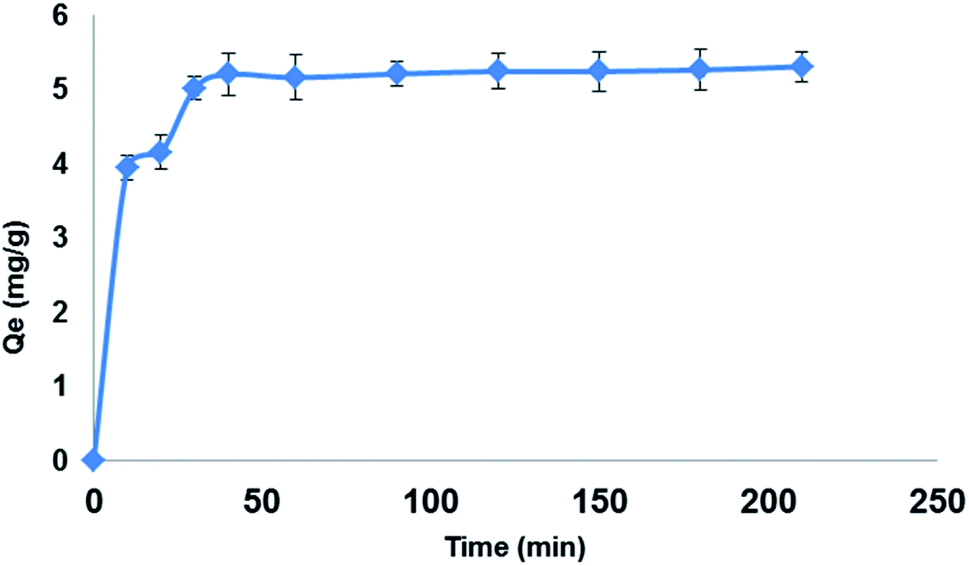
3.5.2. Extraction time. The effect of extraction time on the extraction efficiency of LEV (100 mg L−1) on 150 mg Fe3O4@MIP NPs in 30 mL ACN was studied in the range of 10–210 min. The optimum extraction was accomplished within 30 min, while longer contact times showed no further increase in the extraction efficiency (Fig. 10). The reason behind this is that at prolonged contact time, equilibrium was already attained on the surface of Fe3O4@MIP NPs resulting in no further extraction of LEV from the extraction solvent.37 In addition, 30 min is considered a short extraction time and this can be attributed to the good dispersion of Fe3O4@MIP NPs in the extraction solvent thus increasing the exposed surface area of the Fe3O4@MIP NPs for contact with target molecule; LEV.37 Based on the obtained results, an extraction of 30 min was adopted.
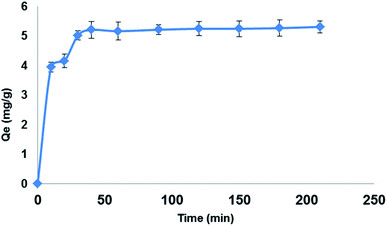 |
| | Fig. 10 Effect of extraction time on the adsorption capacity (qt) of LEV onto Fe3O4@MIP NPs; adsorbent: 150 mg, drug conc. 100 mg L−1, temperature: 25 °C, pH: natural pH of LEV, time: 210 min. | |
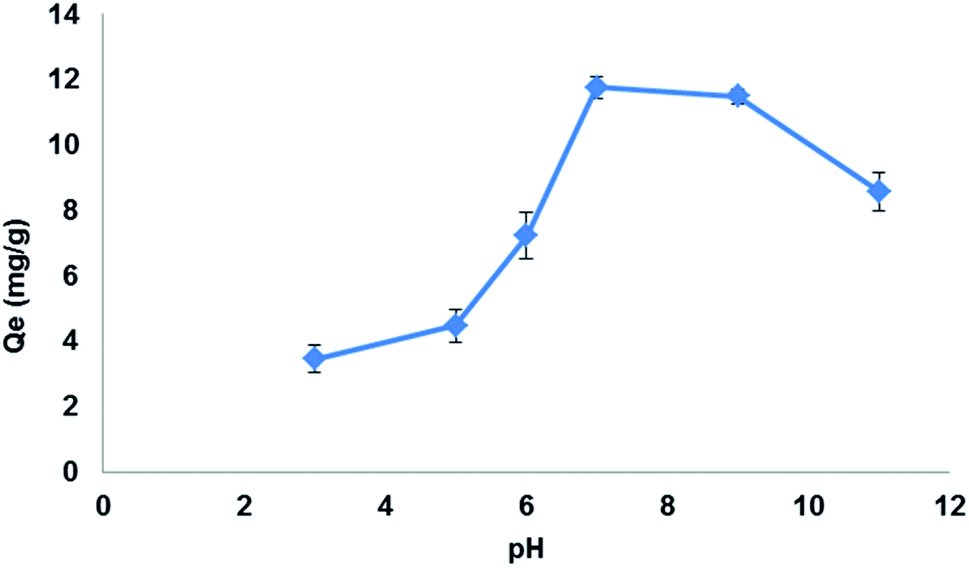
3.5.3. Effect of pH. pH of the sample solution played an important role in LEV adsorption by Fe3O4@MIP NPs. The carboxylic acid incorporated in the cavities of the Fe3O4@MIP NPs has very high hydrophilic affinity with respect to LEV.46 In addition, the amine group of LEV also possesses hydrogen bonding with the carboxylic acid group of the Fe3O4@MIP NPs. Thus, the extent of LEV extraction depends on its interaction with carboxylic groups of the Fe3O4@MIP NPs. To evaluate the effect of pH, a working solution with different pHs in the range of 3.0–11.0 was studied. As shown in Fig. 11, the highest adsorption capacity of LEV by Fe3O4@MIP NPs was at pH 7.0 and lesser adsorption was observed at lower and higher pH values. It can be estimated that at pH 7.0 the carboxyl groups of Fe3O4@MIP NPs were deprotonated while LEV functional groups become protonated thus allowing smooth interaction between LEV and the Fe3O4@MIP NPs.46 Therefore, the pH 7.0 was selected as optimized pH according to the findings of the present study.
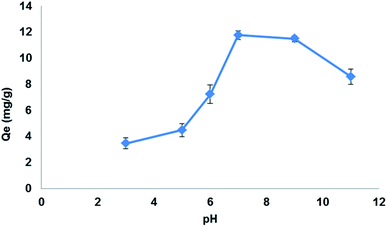 |
| | Fig. 11 Effect of pH on the adsorption capacity (qe) of LEV onto Fe3O4@MIP NPs. Adsorbent: 25 mg, drug conc. 100 mg L−1, temperature: 25 °C, time: 30 min. | |
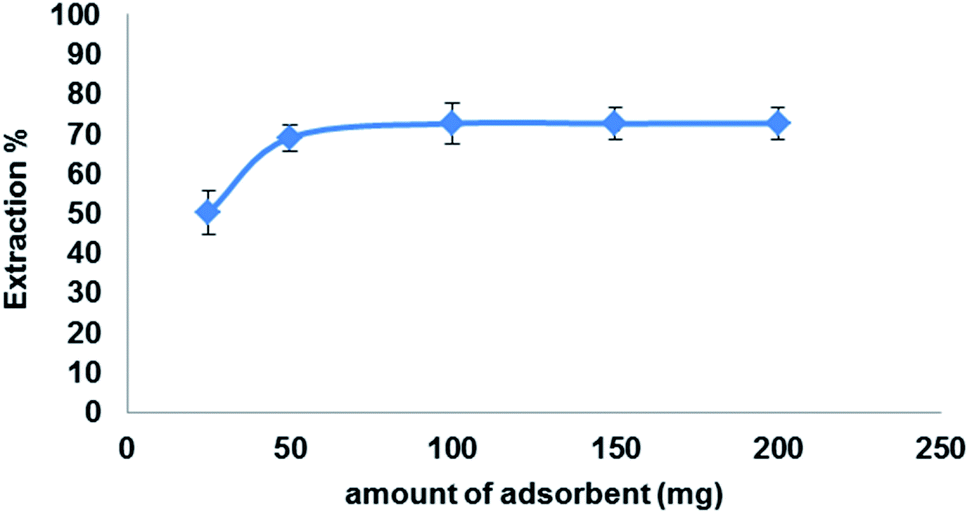
3.5.4. Effect of adsorbent amount. To investigate the optimum amount Fe3O4@MIP NPs on the extraction efficiency of LEV (100 mg L−1) in 5 mL ACN under the optimized conditions, various amounts of Fe3O4@MIP NPs were studied from 25 to 200 mg. The results shown in Fig. 12 indicate that the recovery of LEV has increased following an increase in the Fe3O4@MIP NPs amount up to 50 mg due to the increase in available adsorption sites. Further increase in the amount of Fe3O4@MIP NPs showed no significant increase in the extraction yields of LEV. Thus, a 50 mg amount was used as the optimum amount of Fe3O4@MIP NPs.
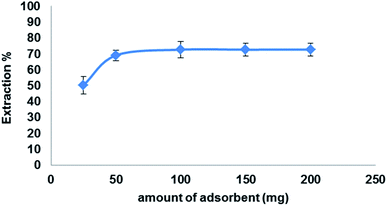 |
| | Fig. 12 Effect of adsorbent amount on the extraction efficiency of LEV onto Fe3O4@MIP NPs; drug conc. 100 mg L−1, temperature: 25 °C, pH: 7.0, time: 30 min. | |
3.6. Adsorption isotherms
Results of equilibrium studies were analyzed using Langmuir, Freundlich and Sips isotherms to find the best fitted model that can describe the adsorption process.
The original forms of the Langmuir,47 Freundlich48 and Sips49 isotherms are given by eqn (1), (2), and (3) respectively,
| |
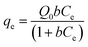 | (1) |
| |
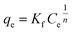 | (2) |
| |
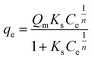 | (3) |
where
Ce is the equilibrium concentration of LEV in solution (mg mL
−1),
qe is the amount of LEV adsorbed per unit mass of adsorbent (mg g
−1), at equilibrium concentration,
Ce,
b is the Langmuir equilibrium constant (mL mg
−1).
Q0 signifies the maximum adsorption capacity (mg g
−1),
Kf and 1/
n are Freundlich constants where
n is related to the adsorption intensity, and
Kf (mg
1−1/n mL
1/n g
−1) indicates the adsorption capacity.
Qm is the maximum adsorption capacity calculated by Sips (mg g
−1),
Ks is the Sips constant (mL mg
−1) and
n is the Sips model exponent.
Corresponding linear forms of Langmuir and Freundlich isotherms can be expressed in eqn (4) and (5), respectively,47,50
| |
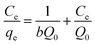 | (4) |
| |
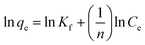 | (5) |
As shown in Fig. S4[a], [b] and [c],† the data were fitted by Langmuir, Freundlich, and Sips models respectively. It was observed that the equilibrium data were best matched by Langmuir isotherm with good correlation coefficient and χ2 values. The detailed values are reported in Table 1.
Table 1 Parameters of adsorption isotherms using Langmuir, Freundlich and Sips equations for Fe3O4@MIP NPs
| Langmuir |
Freundlich |
Sips |
| Q0 (mg g−1) |
B (mL mg−1) |
RL |
R2 |
Χ2 |
Kf (mL g−1) |
1/n |
R2 |
Χ2 |
Qm (mg g−1) |
Ks (mL mg−1) |
1/n |
R2 |
Χ2 |
| 26.04 |
14.20 |
0.36 |
0.9997 |
0.0008 |
85.05 |
0.69 |
0.9934 |
0.038 |
13.13 |
0.93 |
0.20 |
0.9986 |
0.0004 |
3.7. Adsorption kinetics
The kinetic data obtained were evaluated using pseudo-first-order and pseudo-second-order kinetics equations.
The kinetic data obtained were evaluated using pseudo-first-order and pseudo-second-order kinetics eqn (6) and (7) respectively.51,52
| |
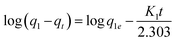 | (6) |
| |
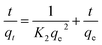 | (7) |
where
qe and
qt (mg g
−1) are the amounts of LEV adsorbed on Fe
3O
4@MIP NPs at equilibrium and at time
t (min), respectively.
K1 is the pseudo-first-order kinetics constant (min
−1) and
K2 is the pseudo-second-order kinetics constant (g mg
−1 min
−1).
All the linear regression correlation coefficients and rate constants of adsorption are presented in Table 2. The second-order model was more suitable to describe the kinetic data than the first-order model because of the favorable fit between experimental and calculated values of qe in the second-order model (all R2 values are above 0.999 at different initial concentrations). The plots of the experimental data of the sorption capacity against time for the pseudo-first-order and pseudo-second-order model were shown in Fig. S5[a] and [b],† respectively. It can be seen from Fig. S5† that the equilibrium adsorption amount increased with an increase of initial LEV concentration.
Table 2 Pseudo-first order, and pseudo-second-order kinetic models parameters for the adsorption of LEV by Fe3O4@MIP NPs
| |
Pseudo 1st order |
Pseudo 2nd order |
| qe (exp) (mg g−1) |
qe (cal) (mg g−1) |
K1 (min−1) |
R2 |
χ2 |
qe (cal) (mg g−1) |
K2 (g mg−1 min−1) |
R2 |
χ2 |
| 25 mg L−1 |
4.02 |
4.19 |
0.129 |
0.9943 |
0.010 |
5.09 |
0.025 |
0.9995 |
0.001 |
| 50 mg L−1 |
6.79 |
4.50 |
0.128 |
0.987 |
0.022 |
7.65 |
0.034 |
0.9991 |
0.014 |
| 100 mg L−1 |
11.76 |
7.81 |
0.147 |
0.9941 |
0.028 |
13.05 |
0.006 |
0.9991 |
0.030 |
3.8. Study of Fe3O4@MIP NPs selectivity
In order to evaluate the selectivity of the synthesized Fe3O4@MIP NPs, extraction of LEV and its co-administered drugs; QF and CLO, was evaluated. The initial concentration of drugs (5.0 mL, 100 mg L−1) was extracted by 50 mg of Fe3O4@MIP NPs and Fe3O4@NIP NPs at pH 7.0. The partition coefficient K was determined according to the following equation:13
where Cp is the amount of LEV bound by Fe3O4@MIP or Fe3O4@NIP NPs and Cs is the concentration of LEV remaining in solution.
Additionally, the imprinting factor (IF) and selectivity coefficient (SC) were determined to evaluate the selectivity properties of Fe3O4@MIP and Fe3O4@NIP NPs towards LEV and its co-administered drug (QF and CLO). The IF and SC were calculated by the following equations.53
| Imprinting factor (IF) = Ki/Kc |
| Selectivity coefficient (SC) = IFLEV/IFi |
where
Ki and
Kc represent the partition coefficients of the drug for the Fe
3O
4@MIP and Fe
3O
4@NIP NPs, IF
LEV and IF
i are the imprinting factor for LEV and co-administered drugs respectively.
The obtained results are illustrated in Table 3. The adsorption capacity of LEV for Fe3O4@MIP NPs was higher than those of the co-administered drugs, meaning that the template molecule had a higher affinity for Fe3O4@MIP NPs. Moreover, the higher IF obtained for LEV strongly confirmed a higher selectivity of Fe3O4@MIP NPs because the specific sites present in Fe3O4@MIP NPs are complementary in size, shape and spatial distribution to the template molecule LEV, thus LEV had the advantage to occupy the binding sites over the other drugs.13
Table 3 The adsorption capacity, partition coefficients, imprinting factors and selectivity coefficients of LEV and its co-administered drugs (QF and CLO) for the imprinted Fe3O4@MIP and control Fe3O4@NIP NPsa
| Analyte |
QMIP (mg g−1) |
QNIP (mg g−1) |
KMIP (mL g−1) |
KNIP (mL g−1) |
IF |
SC |
| In this experiment, 25 mg of Fe3O4@MIP or Fe3O4@NIP NPs were added to 5 mL of ACN solutions containing LEV, QF and CLO separately with a concentration of 100 mg L−1 for each drug. |
| LEV |
10.92 |
2.92 |
257.31 |
35.49 |
7.25 |
— |
| QF |
2.45 |
2.37 |
35.24 |
33.84 |
1.04 |
6.96 |
| CLO |
2.22 |
0.98 |
29.59 |
12.10 |
2.45 |
2.96 |
3.9. Molecularly imprinted magnetic solid-phase extraction (MI-MSPE) of LEV from plasma samples
3.9.1. Sample preparation. Matrix components, such as proteins can profoundly disrupt the molecular shape–selective interactions, thus decreasing extraction recoveries.54 Proteins can be easily removed from the biological fluids by simple precipitation procedures using methanol, ACN or some acids54 or by molecularly imprinted polymers.55 The aim of this study is to develop a selective procedure for the extraction of LEV from human plasma. Preliminary experiments were carried out in ACN to understand the retention mechanism on Fe3O4@MIP NPs in this medium. Extraction procedure concluded during the optimization step was then applied for human plasma experiments. ACN was employed as loading solvent, and the elution procedure was developed for obtaining maximum extraction of LEV using methanol as eluting solvent.
3.9.2. HPLC assay. Initially, HPLC-UV using C18 column and mobile phases of different pHs were tried. Optimum performance was obtained using Agilent RP C18 column and mobile phase of methanol:PBS (pH 4.0 adjusted with phosphoric acid), 25:75 with a flow rate of 1.5 mL min−1. Such separation resulted in a sharp and symmetrical peak (Fig. S6[c]†). Since levetiracetam lacks chromophores (Fig. 1), it can only be detected with sufficient sensitivity at very short wavelengths, in our case 205 nm. At this spectral range, UV-detection is very unspecific and selectivity has to be achieved either by chromatographic separation or sophisticated sample preparation. As it can be seen, in plasma free and spiked plasma samples chromatograms in Fig. S6[d] and [f]† respectively, no peaks from endogenous substances were observed, only the peaks of LEV and that of solvent appeared. This selectivity was achieved by the unique extraction mechanism of LEV using Fe3O4@MIP NPs. Confirming that any interference from endogenous compounds was suppressed by the sophisticated sample treatment (Fig. S6[f]†).
3.9.3. Colorimetric assay. Spectrophotometry may be used in both qualitative and quantitative assays. However, one of the major problems in applying spectrophotometry to complex matrices is that it is not a selective technique as most endogenous compounds mainly absorb in the UV part of the spectrum.56 On the other hand, visible spectrophotometry (colorimetry) is the first reliable quantitative method used for drug/metabolite analysis in blood.56 Such analysis can be performed through a chromogenic reaction directly in the sample with the condition of appropriate sample treatment including protein precipitation, solvent extraction or micro diffusion.56One of the colorimetric methods that have been applied for analysis of drugs in biological fluids is the potassium ferricyanide colorimetric assay.57,58 Such assay has been applied for the determination of LEV in its dosage form and was found to be a reliable method for LEV quantitative analysis.45 Fig. S7† illustrates the absorption spectra of free and spiked plasma samples at different concentrations of LEV after reaction with potassium ferricyanide. It can be observed that the blank plasma has no significant effect on potassium ferricyanide reaction where the absorbance at λmax is almost zero. Nevertheless, spiked plasma samples showed absorption spectra with varying intensities according to LEV concentration indicating that LEV is the one interacting with potassium ferricyanide and no interference from endogenous plasma components was found. Such results confirmed the findings of the HPLC chromatograms indicating that Fe3O4@MIP NPs allowed the extraction of LEV from human plasma samples with complete suppression of any other endogenous compound in the sample matrix thus omitted the problem of interference and supported the applicability of the proposed colorimetric assay for the determination of LEV in human plasma samples.
3.9.4. HPLC and colorimetric assays validation. A comparison was performed between the two proposed assays (HPLC and colorimetry) to evaluate the applicability of the colorimetric assay for the determination of LEV in human plasma samples after MI-MSPE using Fe3O4@MIP NPs.
I. Selectivity. Analysis of six batches of blank plasma and comparison to fortified plasma samples showed the absence of interference at LEV's retention time for HPLC assay and λmax of LEV's complex with potassium ferricyanide for the colorimetric assay. The chromatograms and the absorbance curves obtained for plasma samples (blank and spiked) are shown in Fig. S6 and S7† respectively. These results demonstrated the high selectivity of the method as it allowed cleaner extracts to be obtained with no interference from endogenous components of biological matrices.
II. Linearity and lower limit of quantitation. The average of six determinations at six concentration levels covering the range of 10–80 μg mL−1 for LEV was used to estimate the linearity. The mean values for the regression equations were (AUC = 1.2283conc + 20.753, r = 0.9993) and (Abs = 0.0091conc + 0.3031, r = 0.9994) for HPLC and colorimetric assay respectively. Blank samples were used to confirm the absence of interference and reproducibility of sample preparation. The LOD and the LOQ values of LEV were 2.58 g mL−1 and 7.81 g mL−1, respectively for HPLC assay and 2.32 g mL−1 and 7.02 g mL−1, respectively for colorimetric assay.
III. Accuracy and precision. Analysis of spiked plasma samples at LLOQ, QCL, QCM, and QCH revealed that the within-run accuracy was 94.61–106.16% and 94.71–105.35% with a precision of 2.72–8.53% and 2.57–6.77% for HPLC and colorimetric assay respectively (Table 4). The between-run accuracy was within 97.95–104.86% and 100.68–105.02% with RSD of 2.09–9.87% and 3.11–7.47% for HPLC and colorimetric assay respectively (Table 4). Obtained results indicated that both assays possess acceptable accuracy and precision.
Table 4 Accuracy and precision for the determination of LEV via HPLC and colorimetric assays
| |
HPLC assay |
Colorimetric assay |
| Within run |
Between run |
Within run |
Between run |
| Mean recovery% ± RSD% |
Mean recovery% ± RSD% |
| LLOQ |
94.61 ± 8.53 |
99.91 ± 9.87 |
105.35 ± 4.51 |
102.42 ± 3.19 |
| QCL |
98.85 ± 6.41 |
97.95 ± 7.26 |
101.90 ± 6.77 |
100.98 ± 7.47 |
| QCM |
99.71 ± 3.93 |
103.02 ± 6.52 |
107.21 ± 3.24 |
105.02 ± 4.15 |
| QCH |
106.16 ± 2.72 |
104.86 ± 2.09 |
94.71 ± 2.57 |
100.68 ± 3.11 |
IV. Recovery and process efficiency. MI-MSPE using Fe3O4@MIP NPs was found to be a suitable approach to achieve reliable recovery of LEV in plasma samples. The recoveries were measured at the QC levels in six replicates. Results demonstrated that the mean percentage recovery of LEV was in the range of 91.52–93.88% and 91.74–93.47% with RSD of 5.51–62% and 1.44–8.14%, as summarized in Tables 5 and 6 for HPLC and colorimetric assays respectively. The process efficiency was evaluated in the studied QC plasma samples and the mean peak areas or absorbances of LEV were compared to those obtained from analysis of neat standards of equivalent concentrations. The results of process efficiency for the two proposed assays are demonstrated in Tables 5 and 6 respectively. The mean percentage recovery of LEV was in the range of 89.17–91.88% and 89.17–90.59% and with RSD of 1.08–4.15% and 2.58–8.35%, for the HPLC and colorimetric assays respectively. In addition, the recovery of LEV after extraction via protein precipitation (90.22%) was almost similar to that after MI-MSPE (89.44%) which gives an indication that there was almost no loss of LEV after the 2-step extraction technique, yet a purer extract was obtained (Fig. S6[e] and [f]†).
Table 5 Recovery and process efficiency for the determination of LEV by HPLC assay
| |
Recovery |
Process efficiency |
| Mean RE% ± RSD% |
Mean PE% ± RSD% (after protein ppt.) |
Mean PE% ± RSD% (after MI-MSPE) |
| QCL |
92.02 ± 5.51 |
— |
91.88 ± 4.15 |
| QCM |
91.52 ± 6.52 |
90.22 ± 4.59 |
89.44 ± 2.26 |
| QCH |
93.88 ± 6.62 |
— |
89.17 ± 1.08 |
| |
n = 6 |
n = 6 |
n = 6 |
Table 6 Recovery and process efficiency for the determination of LEV by colorimetric assay
| |
Recovery |
Process efficiency |
| Mean RE% ± RSD% |
Mean PE% ± RSD% (after MI-MSPE) |
| QCL |
93.47 ± 1.44 |
89.31 ± 8.35 |
| QCM |
92.91 ± 3.29 |
90.59 ± 4.39 |
| QCH |
91.74 ± 8.14 |
89.17 ± 2.58 |
| |
n = 6 |
n = 6 |
4. Conclusions
In the present work, fully optimized and validated MI-MSPE/HPLC-UV and MIMSPE/colorimetric methods for extraction and determination of the anti-epileptic drug; levetiracetam in human plasma samples was presented. Fe3O4@MIP NPs were synthesized with a molecularly imprinted polymer layer on Fe3O4 NPs by surface imprinting and nano techniques. Results indicated the successfulness of using the computer-aided design for the rational selection of functional monomers and solvents to improve the properties of the fabricated MIP. Our Fe3O4@MIP NPs just needed 30 min to reach the adsorption equilibrium with a maximum adsorption capacity of 26.04 mg g−1. Moreover, the successful selective separation and enrichment of LEV in human plasma samples designate that the Fe3O4@MIP NPs has a unique extraction ability where the extract is almost free of any foreign bodies other than the desired template. Thus, the combination of the proposed sample preparation technique with simple analytical assays e.g.: spectrophotometry, can provide a fast, reliable, cost-effective approach for analysis of various types of chemical compounds in different kinds of matrices.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Dr Ahmed Refaat Gardouh from Pharmaceutics and Industrial pharmacy department, Faculty of Pharmacy, Suez Canal University, Egypt for his guidance in the synthesis of MIP NPs.
References
- C. Bylda, R. Thiele, U. Kobold and D. A. Volmer, Analyst, 2014, 139, 2265 RSC.
- S. Azodi-Deilami, M. Abdouss, E. Asadi, A. Hassani Najafabadi, S. Sadeghi, S. Farzaneh and S. Asadi, J. Appl. Polym. Sci., 2014, 131, 41209 Search PubMed.
- J. L. Benedé, A. Chisvert, D. L. Giokas and A. Salvador, Anal. Chim. Acta, 2016, 926, 63–71 CrossRef PubMed.
- P. Lucci, D. Derrien, F. Alix, C. Pérollier and S. Bayoudh, Anal. Chim. Acta, 2010, 672, 15–19 CrossRef CAS PubMed.
- X. Su, X. Li, J. Li, M. Liu, F. Lei, X. Tan, P. Li and W. Luo, Food Chem., 2015, 171, 292–297 CrossRef CAS PubMed.
- S. Azodi-Deilami, M. Abdouss and S. Hasani, Open Chem., 2010, 8, 861–869 CAS.
- E. Asadi, S. Azodi-Deilami, M. Abdouss, D. Kordestani, A. Rahimi and S. Asadi, Korean J. Chem. Eng., 2014, 31, 1028–1035 CrossRef CAS.
- S. Azodi-Deilami, M. Abdouss and D. Kordestani, Appl. Biochem. Biotechnol., 2014, 172, 3271–3286 CrossRef CAS PubMed.
- X. Wang, L. Wang, X. He, Y. Zhang and L. Chen, Talanta, 2009, 78, 327–332 CrossRef CAS PubMed.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- Y.-L. Zhang, J. Zhang, C.-M. Dai, X.-F. Zhou and S.-G. Liu, Carbohydr. Polym., 2013, 97, 809–816 CrossRef CAS PubMed.
- M. Díaz-Álvarez, E. Turiel and A. Martín-Esteban, J. Chromatogr. A, 2016, 1469, 1–7 CrossRef PubMed.
- X. Kong, R. Gao, X. He, L. Chen and Y. Zhang, J. Chromatogr. A, 2012, 1245, 8–16 CrossRef CAS PubMed.
- G. Sheykhaghaei, M. H. Sadr and S. Khanahmadzadeh, Bull. Mater. Sci., 2016, 39, 647–653 CrossRef CAS.
- R. Gao, X. Mu, Y. Hao, L. Zhang, J. Zhang and Y. Tang, J. Mater. Chem. B, 2014, 2, 1733–1741 RSC.
- R. Gao, Y. Hao, L. Zhang, X. Cui, D. Liu, M. Zhang, Y. Tang and Y. Zheng, Chem. Eng. J., 2016, 284, 139–148 CrossRef CAS.
- Y. Su, B. Qiu, C. Chang, X. Li, M. Zhang, B. Zhou and Y. Yang, RSC Adv., 2018, 8, 6192–6199 RSC.
- R. Gao, X. Cui, Y. Hao, L. Zhang, D. Liu and Y. Tang, Food Chem., 2016, 194, 1040–1047 CrossRef CAS PubMed.
- J. Martens-Lobenhoffer and S. M. Bode-Böger, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 819, 197–200 CrossRef CAS PubMed.
- F. M. C. Besag and D. Berry, Drug Saf., 2006, 29, 95–118 CrossRef CAS PubMed.
- J. J. Luszczki, M. M. Andres, P. Czuczwar, A. Cioczek-Czuczwar, J. Wojcik-Cwikla, N. Ratnaraj, P. N. Patsalos and S. J. Czuczwar, Eur.
Neuropsychopharmacol., 2005, 15, 609–616 CrossRef CAS PubMed.
- H. R. Adams, J. E. Riviere and M. G. Papich, Veterinary Pharmacology and Therapeutics., Wiley, 2013 Search PubMed.
- V. P. Shah, K. K. Midha, J. W. Findlay, H. M. Hill, J. D. Hulse, I. J. McGilveray, G. McKay, K. J. Miller, R. N. Patnaik, M. L. Powell, A. Tonelli, C. T. Viswanathan and A. Yacobi, Pharm. Res., 2000, 17, 1551–1557 CrossRef CAS.
- E. A. Wilson and M. J. Brodie, Baillieres Clin. Neurol., 1996, 5, 723–747 CAS.
- E. Perucca and M. Bialer, Clin. Pharmacokinet., 1996, 31, 29–46 CrossRef CAS PubMed.
- J. M. Pellock, T. A. Glauser, E. M. Bebin, N. B. Fountain, F. J. Ritter, R. M. Coupez and W. D. Shields, Epilepsia, 2001, 42, 1574–1579 CrossRef CAS PubMed.
- T. A. Vermeij and P. M. Edelbroek, J. Chromatogr. B: Biomed. Sci. Appl., 1994, 662, 134–139 CrossRef CAS PubMed.
- V. Pucci, F. Bugamelli, R. Mandrioli, A. Ferranti, E. Kenndler and M. A. Raggi, Biomed. Chromatogr., 2004, 18, 37–44 CrossRef CAS PubMed.
- T. A. Glauser, W. G. Mitchell, A. Weinstock, M. Bebin, D. Chen, R. Coupez, A. Stockis and Z. Lu, Epilepsia, 2007, 48, 1117–1122 CrossRef CAS PubMed.
- L. Wang and Z. Zhang, Talanta, 2008, 76, 768–771 CrossRef CAS PubMed.
- Molecular Operating Environment (MOE), 2012, Chemical Computing Group Inc., 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, et al., ( 2009), Gaussian 09 Revision D.01, Gaussian Inc., Wallingford Search PubMed.
- M. Cossi, V. Barone and R. Cammi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- W. Humphrey and A. Dalke, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- M. Tabandeh, S. Ghassamipour, H. Aqababa and M. Tabatabaei, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 898, 24–31 CrossRef CAS PubMed.
- N. T. Abdel Ghani, R. M. El Nashar and F. M. Abdel-Haleem, Electroanalysis, 2016, 28, 1530–1538 CrossRef CAS.
- J. L. Benedé, A. Chisvert, D. L. Giokas and A. Salvador, J. Chromatogr. A, 2014, 1362, 25–33 CrossRef PubMed.
- I. Guideline, Q2A Text on Validation of Analytical Procedures, Fed. Regist. (1994), http://www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/ich/qual/q2a-eng.php, accessed February 28, 2018.
- I. Guideline, Validation of analytical procedures: Text and Methodology, Q2. (2005), http://somatek.com/content/uploads/2014/06/sk140605h.pdf, February 28, 2018.
- International Conference on Harmonization (ICH), Q2B, Validation of Analytical Procedures: Definitions and Terminology, vol. 60, US FDA Federal Register, 1995 Search PubMed.
- O. A. Attallah, M. A. Al-Ghobashy, M. Nebsen and M. Y. Salem, RSC Adv., 2016, 6, 11461–11480 RSC.
- D. K. Yi, S. S. Lee, G. C. Papaefthymiou and J. Y. Ying, Chem. Mater., 2006, 18, 614–619 CrossRef CAS.
- L. Chen, Y. Liu, X. He and Y. Zhang, Chin. J. Chromatogr., 2015, 33, 481–487 CrossRef CAS.
- M. Safdarian, Z. Ramezani and A. A. Ghadiri, J. Chromatogr. A, 2016, 1455, 28–36 CrossRef CAS PubMed.
- M. Indupriya, R. S. Chandan, B. M. Gurupadayya and K. Sowjanya, Der Pharma Chemica, 2011, 3, 472–481 CAS.
- M. Abdouss, S. Azodi-Deilami, E. Asadi and Z. Shariatinia, J. Mater. Sci. Mater. Med., 2012, 23, 1543–1552 CrossRef CAS PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- H. Freundlich, Über die Adsorption in Lösungen, W. Engelmann, 1906 Search PubMed.
- R. Sips, J. Chem. Phys., 1948, 16, 490 CrossRef CAS.
- A. Z. M. Badruddoza, G. S. S. Hazel, K. Hidajat and M. S. Uddin, Colloids Surf., A, 2010, 367, 85–95 CrossRef CAS.
- S. Lagergren, About the theory of so-called adsorption of soluble substances, K. Sven. Vetenskapsakad. Handl., 1898, 24, 1–39 Search PubMed.
- Y. Ho and G. McKay, Process Biochem., 1999, 34, 451–465 CrossRef CAS.
- D.-M. Han, G.-Z. Fang and X.-P. Yan, J. Chromatogr. A, 2005, 1100, 131–136 CrossRef CAS PubMed.
- M. Lasáková, D. Thiébaut, P. Jandera and V. Pichon, J. Sep. Sci., 2009, 32, 1036–1042 CrossRef PubMed.
- M. A. A. El-Aal, M. A. Al-Ghobashy, F. A. A. Fathalla and Y. S. El-Saharty, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1046, 34–47 CrossRef PubMed.
- R. J. Flanagan and Wiley InterScience (Online service), Fundamentals of analytical toxicology, John Wiley & Sons, 2007 Search PubMed.
- C. Dong, Y. Zhang, L. Guo and Q. Li, J. Anal. Chem., 2010, 65, 707–712 CrossRef CAS.
- H. C. Yao, E. J. Xu, W. Y. Zeng, X. Y. Zeng, M. Zhang and J. Chen, J. Food Drug Anal., 2013, 21, 279–285 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02379d |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Medhat A. Al-Ghobashybc,
Ahmed Taha Ayouba,
Jack Adam Tuszynskid and
Marianne Nebsen*b
a,
Medhat A. Al-Ghobashybc,
Ahmed Taha Ayouba,
Jack Adam Tuszynskid and
Marianne Nebsen*b