DOI:
10.1039/C8RA02227E
(Paper)
RSC Adv., 2018,
8, 18016-18022
Chloramphenicol-borate/boronate complex for controlling infections by chloramphenicol-resistant bacteria†
Received
14th March 2018
, Accepted 6th May 2018
First published on 16th May 2018
Abstract
Increasing bacterial resistance to antibiotics is a pressing problem worldwide, with many health organisations prioritizing this issue. Whilst there is a desperate need for new effective antimicrobials, it is also important to understand the mechanisms and epidemiology of the resistant pathogens currently present in the community. Chloramphenicol is one such well known antibiotic which had lost its efficacy due to bacterial resistance. In this paper, we report the design, synthesis, and bio-studies of novel chloramphenicol-borate/boronate derivatives which showed the ability to control the infections caused by chloramphenicol-resistant bacteria. Activity profiling against P. aeruginosa strain EXR1 with catB gene indicated the inability of acetyl transferase to acetylate the chloramphenicol-borate/boronate complex, unlike chloramphenicol. Results obtained from the antimicrobial assays were further rationalized by molecular docking studies. The latter revealed that the probable reason for the enhanced antibacterial activity may be attributed to the change in the binding site of chloramphenicol-borate/boronate with chloramphenicol acetyl transferase (CAT) with respect to chloramphenicol itself. Hemolytic and genotoxic studies established the reduced toxicity of these synthetic derivatives with respect to chloramphenicol.
1. Introduction
Chloramphenicol-resistant common Gram-negative bacilli (GNB) like Enterobacteriaceae (such as Escherichia coli, Klebsiella spp., Enterobacter spp.), Pseudomonas aeruginosa, and Acinetobacter spp. have evolved to be menacing pathogens, raising concern in recent times. These are the leading cause of infections related to the nosocomial blood stream, urinary tract, intra-abdominal and respiratory tract.1 The major reason behind the rise in bacterial infection has been the development of antibiotic resistance in bacteria which is quite prevalent amongst the Gram-negative bacilli.2 They are difficult to control, and have become a major health problem worldwide. Infections caused by multi-antibiotic resistant bacteria are the cause of greater mortality and longer hospitalization.3,4 Recent studies have shown that most GNB bacteria have become resistant to several antibiotics. GNBs have developed various survival strategies to combat against antibiotics due to their indiscriminate and improper use. Some bacteria have developed efflux systems to pump out the invading antibiotics from their cells. Others produce enzymes which can “chew up” the antibiotics by hydrolysis or by altering the target site thus rendering them ineffective.5
This impending antibiotic crisis motivated us towards the development of new analogues of conventional antibiotics by appropriate modification. Such strategies leading to newer analogues of old antibiotics are often quite effective against antibiotic resistant pathogens. Chloramphenicol is a semi-synthetic, structurally simple (relative to other types), broad-spectrum antibiotic produced by Streptomyces spp. However, emergence of chloramphenicol-resistant GNBs has become a major problem.6 GNBs become resistant to chloramphenicol in different ways: the major pathway is enzymatic inactivation via acetyl transferases or in few cases via phosphotransferases.7–9 Other mechanisms involve alteration of target site of ribosome by mutation or modification,10 and the development of efflux pump systems thereby reducing the intracellular drug concentration.11,12 Almost all multi-drug resistant GNBs are able to produce chloramphenicol acetyl transferase (CAT) enzyme and get classified on the basis of their specificity.13 Chloramphenicol inhibits protein synthesis by binding reversibly to the peptidyl transferase of 50S ribosomal subunit which prevents the peptide chain elongation by blocking the transpeptidation process.14 Chloramphenicol acetyl transferase detoxifies chloramphenicol by covalently attaching an acetyl group from acetyl-CoA.6 Therefore, the acetylated chloramphenicol is unable to bind to ribosomal subunit. The C-terminal region of chloramphenicol acetyl transferase plays a central role in its catalytic mechanism of acetylation of chloramphenicol.
To negate this process, chloramphenicol should be structurally modified so that it does not undergo the detoxifying pathway without compromising its antibiotic action by binding to the ribosome. It may be pointed out that Mubarak et al.15 in 1984, first reported the retention of some antibacterial activity in chloramphenicol-phenylboronates only against chloramphenicol sensitive strains. In this work, we report the synthetic modification of chloramphenicol 1 into its corresponding borate 2 derivative. This along with the corresponding phenyl boronate derivative 3 exhibited remarkable activity against chloramphenicol-resistant GNB clinical isolates. Activity was also confirmed against chloramphenicol acetyl transferase producing strain which revealed inability of the transferase to acetylate the chloramphenicol borate/boronate complex unlike what happens in case of chloramphenicol. Molecular docking studies and UV-Vis spectroscopy based DNA-binding studies were done which validated that the synthesized derivatives are potential semi-synthetic antibiotic agents against resistant GNB pathogens.
2. Experimental
Synthesis of the compounds
All reactions were conducted with oven-dried glassware under an atmosphere of nitrogen. All common reagents were commercial grade reagents and used without further purification. TLC was performed on aluminum-backed plates coated with Silica gel 60 with F254 indicator. Locally available UV-lamp chamber and I2-blower were used as the TLC spot indicator. The 1H NMR spectra were recorded at 600 MHz and 13C NMR spectra were measured at 150 MHz using CD3CN. Proton and carbon spectra were referenced internally to solvent signals, using values of δ = 1.94 for proton and δ = 118.3 and 1.3 for carbons in CD3CN. The following abbreviations are used to describe peak patterns where appropriate: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet. All coupling constants (J) are given in Hz.
Method for the preparation of chloramphenicol-borate derivative (2)
To a solution of boric acid (A) (20 mg, 0.32 mmol) in acetonitrile (10 mL), chloramphenicol 1 (209 mg, 2 eq., 0.64 mmol) was added slowly with vigorous stirring at room temperature under nitrogen. The reaction was allowed to stir for 18 h after which the reaction mixture was filtered and the filtrate was evaporated to dryness. The reaction mixture was purified by precipitation from acetonitrile–dichloromethane mixture to obtain the target compound 2 (194 mg, 91%).
Spectral data. 1H NMR (600 MHz, CD3CN) δ 8.19–8.17 (m, 1H), 7.62–7.59 (m, 1H), 7.36 (d, J = 9.5 Hz, 1H), 5.89 (s, 1H), 5.56 (d, J = 2.7 Hz, 1H), 5.49 (bs, 1H), 4.56 (ddd, J = 9.4, 4.5, 2.7 Hz, 1H), 4.39 (dd, J = 11.7, 2.7 Hz, 1H), 3.95 (dd, J = 11.7, 1.8 Hz, 1H); 13C NMR (150 MHz, CD3CN) δ 164.6, 148.4, 147.4, 127.8, 124.1, 74.7, 67.1, 66.7, 50.2.
Method for the preparation of chloramphenicol-phenyl boronate ester (3)
To a solution of phenylboronic acid (B) (30 mg, 0.246 mmol) in dichloromethane (12 mL), chloramphenicol 1 (80 mg, 1 eq., 0.246 mmol) was added slowly with vigorous stirring at room temperature under nitrogen. The reaction was allowed to stir for 18 h after which the mixture was filtered. The filtrate was evaporated to dryness. The reaction mixture was purified by precipitation from acetonitrile–dichloromethane mixture to obtain the final product 3 (93 mg, 92%).
Spectral data. 1H NMR (600 MHz, CD3CN) δ 8.23–8.21 (m, 2H), 7.91 (dd, J = 8.0, 1.5 Hz, 1H), 7.74–7.72 (m, 2H), 7.54 (t, J = 7.5 Hz, 1H), 7.52–7.49 (m, 1H), 7.45–7.43 (m, 2H), 5.84 (s, 1H), 5.73 (d, J = 2.8 Hz, 1H), 4.76 (ddd, J = 9.4, 4.5, 2.7 Hz, 1H), 4.58 (dd, J = 11.9, 2.7 Hz, 1H), 4.12 (dd, J = 11.8, 1.8 Hz, 1H); 13C NMR (150 MHz, CD3CN) δ 164.7, 148.5, 147.3, 134.8, 132.0, 129.0, 128.7, 128.0, 124.1, 74.0, 67.1, 66.4, 50.3.
Antimicrobial susceptibility testing
After having access to the borate and boronate derivatives (2 and 3), we went forward with our antibacterial assay. The GNB strains, Escherichia coli, Pseudomonas aeruginosa, Klebsiella spp., and Acinetobacter sp. were isolated, cultured and tested in Molecular Microbiology Laboratory under hospital settings (Priyamvada Birla Aravind Eye Hospital, Kolkata, WB). Minimum inhibitory concentration (MIC) values of all derivatives were determined according to guidelines of Clinical and Laboratory Standards Institute (CLSI) using Muller Hinton Broth (MHB) medium; Seventeenth Informational Supplement Document M100-S17, CLSI, Wayne, PA: CLSI, 2010.16 The culture conditions and bacterial growth were monitored as described earlier by Roymahapatra et al.17
Amplification and sequencing of chloramphenicol acetyl transferase gene in chloramphenicol-resistant bacteria
Several clinical isolates of P. aeruginosa were grown in LB medium at 37 °C for 24 h. Whole cell DNA was extracted following the method as described earlier Sarkar et al.18 To detect the presence of chloramphenicol acetyl transferase (catB) gene in isolates, PCR amplification was performed using degenerate primers like HS223 (5′-CGGAATTCCGAGCTAYTAYTCYGGCTAYTA-3′) and HS224 (5′-GGGATCCCGCCGGGCATGAHCATBGCCTC-3′) as described earlier by White et al.19 Only the DNA of catB+ strain, the clinical isolate, P. aeruginosa strain EXR1 were used for the amplification of ‘Ps’ open reading frame (orf) using the primers HS240 (5′-GGGGATCCTATGGGCAACTATTTCGAG-3′) and HS241 (5′-GGGGATCCATCAGGCCGAGGCCTGGCG-3′).
The PCR product was visualized by electrophoresis on 1.0% agarose gel electrophoresis analysis, the band was eluted using quick gel extraction kit following the manufacturer protocol QIAGEN, USA and cloned into pCR2.1-TOPO vector. Recombinant plasmid was sequenced with M13 forward and reverse primer using BigDye® Terminator v3.1 Cycle Sequencing Kit according to manufacturer's instructions. Reaction products were analysed in 3500 gene analyzer (Life technologies). The sequence of catB gene of strain P. aeruginosa EXR1 is given in ESI (Fig. S5–S7).†
In silico analysis
The PDB structure of 2XAT (acetyl transferase of Pseudomonas aeruginosa) was downloaded from RCSB protein data bank. The acetyl transferase enzyme was co-crystallized with desulfo-coenzyme A and chloramphenicol.20 The amino acid chain was kept, the water molecules and co-crystallized ligands were removed, subsequently the missing atom types were repaired using prepare protein module under receptor–ligand interaction in Discovery Studio 3.5. Prepared protein and the ligands were saved in .pdb file and used for docking studies.21 Docking calculations were done in PatchDock server22 and the pictures were prepared using Discovery Studio 3.5. The most favourable poses were selected according to the minimum patch dock score of the receptor-small molecule complex and that was used for the analysis of receptor–ligand interactions.
DNA-binding assay
DNA-drug interaction is one of the important methods to identify the genotoxicity level that amend the pharmacodynamics of drug in blood. Accordingly, a simple study23 to determine the binding interaction of the chloramphenicol (1) and the synthesized borate derivative (2) with DNA was carried out. A Jasco V 730 spectrophotometer was used for absorption spectral studies. Solutions of Calf Thymus DNA (CT DNA) were prepared in Tris–HCl buffer (1 mM; pH = 7.2). The ratio of UV absorbances at 260 and 280 nm (A260/A280) was found to be 1.83. The DNA concentrations were determined by using an extinction coefficient of 6600 M−1 cm−1 at 260 nm and were expressed in terms of base molarity. UV absorption titrations were carried out by keeping the concentration of CT DNA (dissolved in Tris–HCl buffer) fixed and by adding a known concentration of chloramphenicol (1) and chloramphenicol-borate (2) (solution in Tris–HCl buffer) into both the cuvettes in increasing amounts until saturation was observed. Absorbance values were recorded after each successive addition of chloramphenicol (1) and chloramphenicol-borate (2) solution and equilibration.
Hemolytic assay
Hemocompatibility study was performed using standard protocol as described earlier24 by Mandal et al. Hemolytic activity was determined by measuring the absorption at 570 nm (Biorad Microplate reader 5804R). Control samples of 0% lysis (in PBS 1X buffer) and 100% lysis (in 1% TritonX-100) were used in the experiment. All assays were repeated thrice and data presented with their mean value. Hemolytic effect of each treatment was expressed as percent of cell lysis relative to the +ve control cells (% control) using the following formula: [(Abs570 of samples)/(Abs570 of (+)ve control cells)] × 100, where absorbance is abbreviated to Abs.
3. Results and discussion
Synthesis of target compounds (2 and 3)
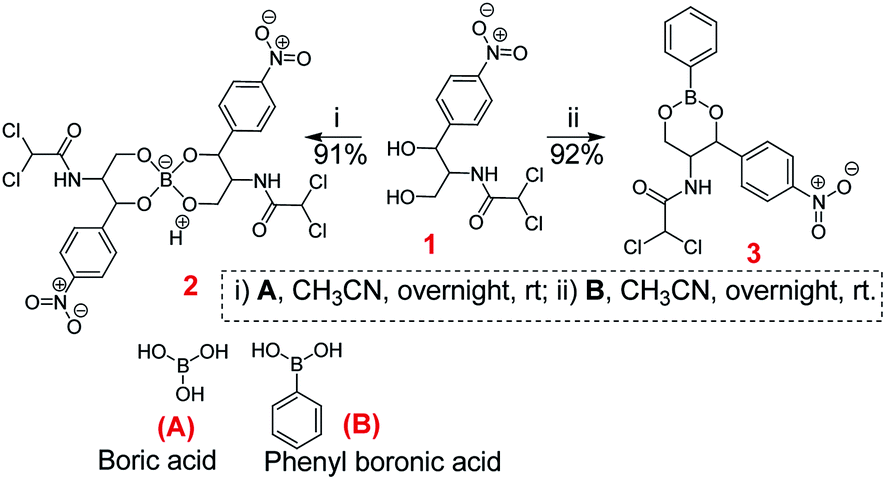
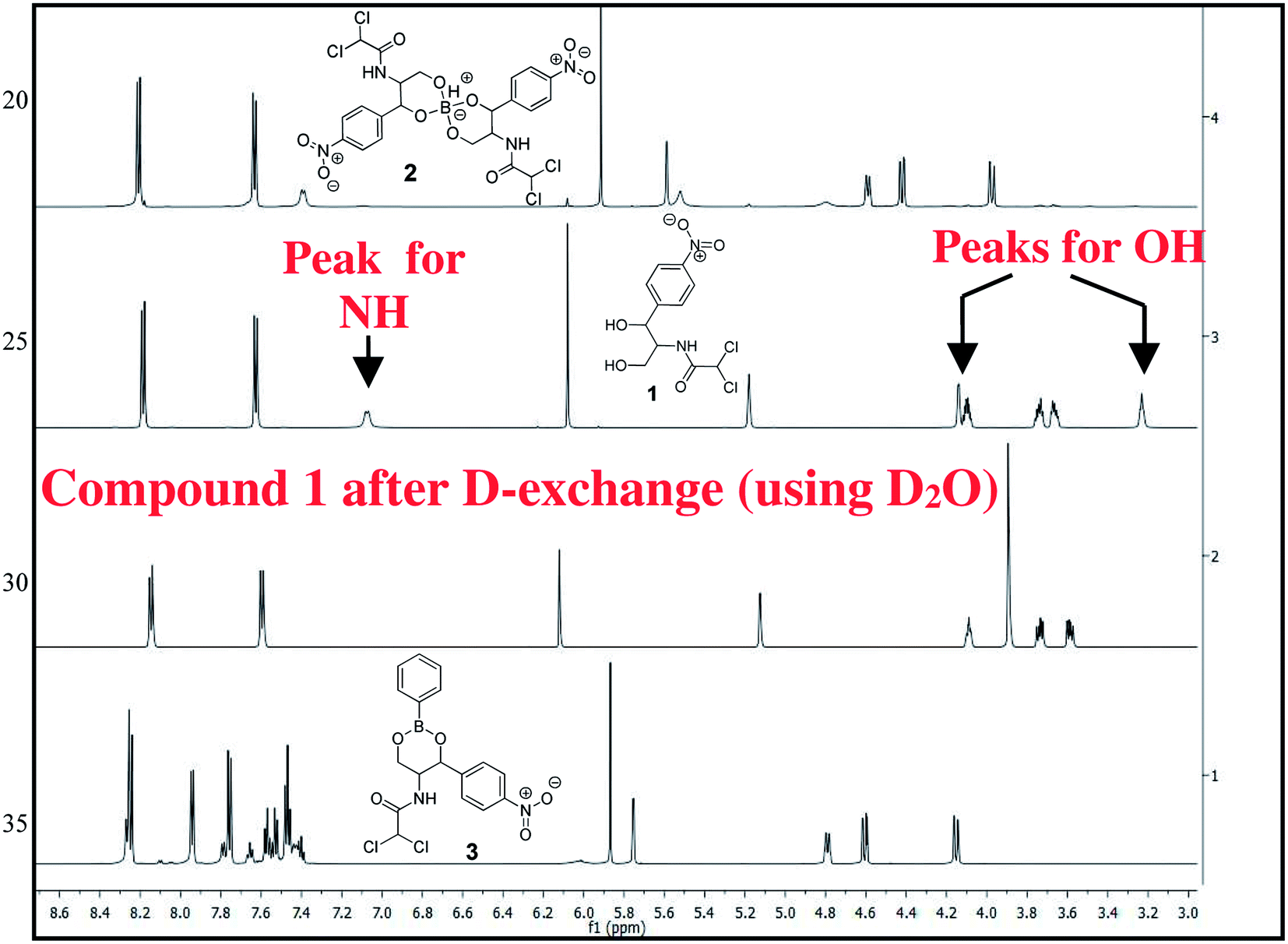
The chloramphenicol-borate and phenyl-boronate (2 and 3) were synthesized via the condensation reaction between 1,3-diol moiety in chloramphenicol 1 and boric acid (A) or phenyl boronic acid (B) respectively (Scheme 1). The reaction were carried out in acetonitrile by stirring at room temperature for 18 h, followed by subsequent precipitation using dichloromethane to afford the target compounds 2 and 3 respectively. These were fully characterized using 1H and 13C NMR (Fig. 1) (full spectra are provided in ESI, Fig. S1–S4†).
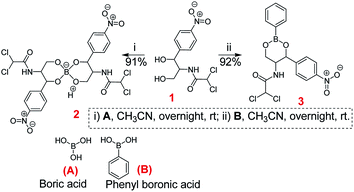 |
| | Scheme 1 Synthesis of chloramphenicol borate 2 and boronate 3. | |
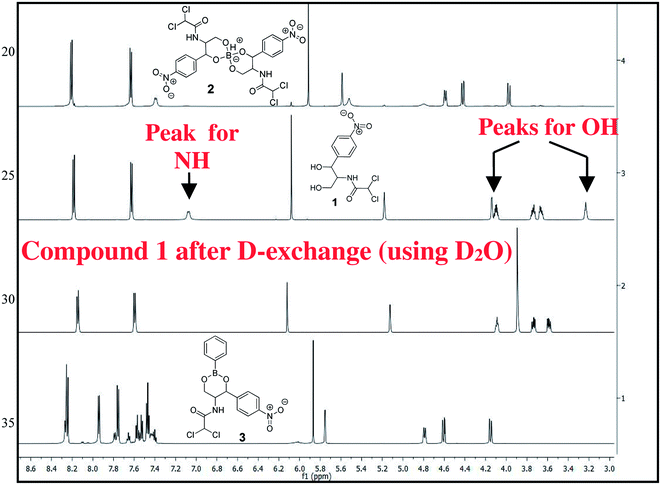 |
| | Fig. 1 Stacked 1H NMR spectra of compounds 1–3 in CD3CN, recorded in 600 MHz. | |
Antimicrobial study
Antimicrobial activity of the chloramphenicol (1) and borate/boronate (2 and 3) complexes were tested against several chloramphenicol resistant GNB clinical isolates following CLSI guidelines. The obtained MIC values revealed the potential of the new derivatives to control chloramphenicol-resistant pathogens. The results are shown in Table 1.
Table 1 Minimal inhibitory concentration (MIC) of chloramphenicol and chloramphenicol derivatives for chloramphenicol-resistant clinical isolates and mutant forms
| Bacteria (GNBs) |
MIC (μg mL−1) |
| Chloramphenicol (1) |
Chloramphenicol-borate (2) |
Chloramphenicol-phenylboronate (3) |
| P. aeruginosa EXR1 |
>512 |
4 |
8 |
| Acinetobacter sp. |
>512 |
4 |
8 |
| E. coli |
>512 |
2 |
4 |
| Enterobacter sp. |
64 |
2 |
4 |
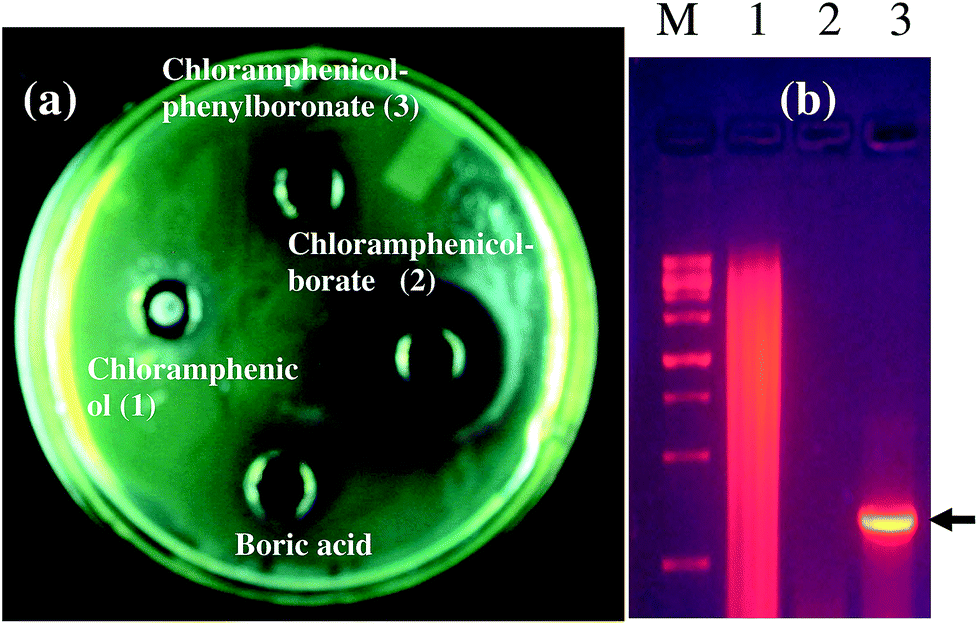
As a representative example, we have shown the agar diffusion assay plate against different chloramphenicol-resistant pathogens like A. baumannii, E. coli, and E. cloacae all of which showed extensive antibiotic-resistance profile towards chloramphenicol (Fig. 2). Between the two complexes, chloramphenicol-borate derivative 2 showed the highest antibacterial activity (Fig. 3a).
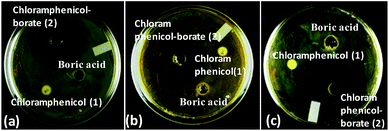 |
| | Fig. 2 Images are showing the activity of chloramphenicol-borate derivative against different chloramphenicol resistant pathogens. Images are represented as A. baumannii (a); E. coli (b) and E. cloacae (c). In all lanes the sample concentration was 2 μg mL−1 of 15 μL. | |
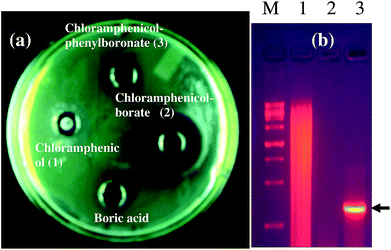 |
| | Fig. 3 (a) Screening of synthesized compounds against chloramphenicol resistant Gram-negative bacilli, Pseudomonas aeruginosa. 30 μg of each compound was used except for chloramphenicol from High Media, India for which 100 μg was used. (b) PCR amplification of chloramphenicol acetyl transferase gene from chloramphenicol resistant clinical isolate, P. aeruginosa EXR1. Lane M corresponding to marker; lane 1: genomic DNA of P. aeruginosa SS3; lane 2: PCR amplification of chloramphenicol susceptible strain; lane 3: PCR amplification of chloramphenicol resistant strain. | |
In our study, the presence of catB genetic system in the selected P. aeruginosa strain EXR1 was confirmed Fig. S5–S7 (in the ESI),† as the major cause for the development of chloramphenicol resistance. A positive amplification (Fig. 3b) product was observed after amplification with catB degenerate primers in Pseudomonas isolates and confirmed with their sequence, having high level of chloramphenicol (MIC > 500 μg mL−1) resistance.
In silico analysis
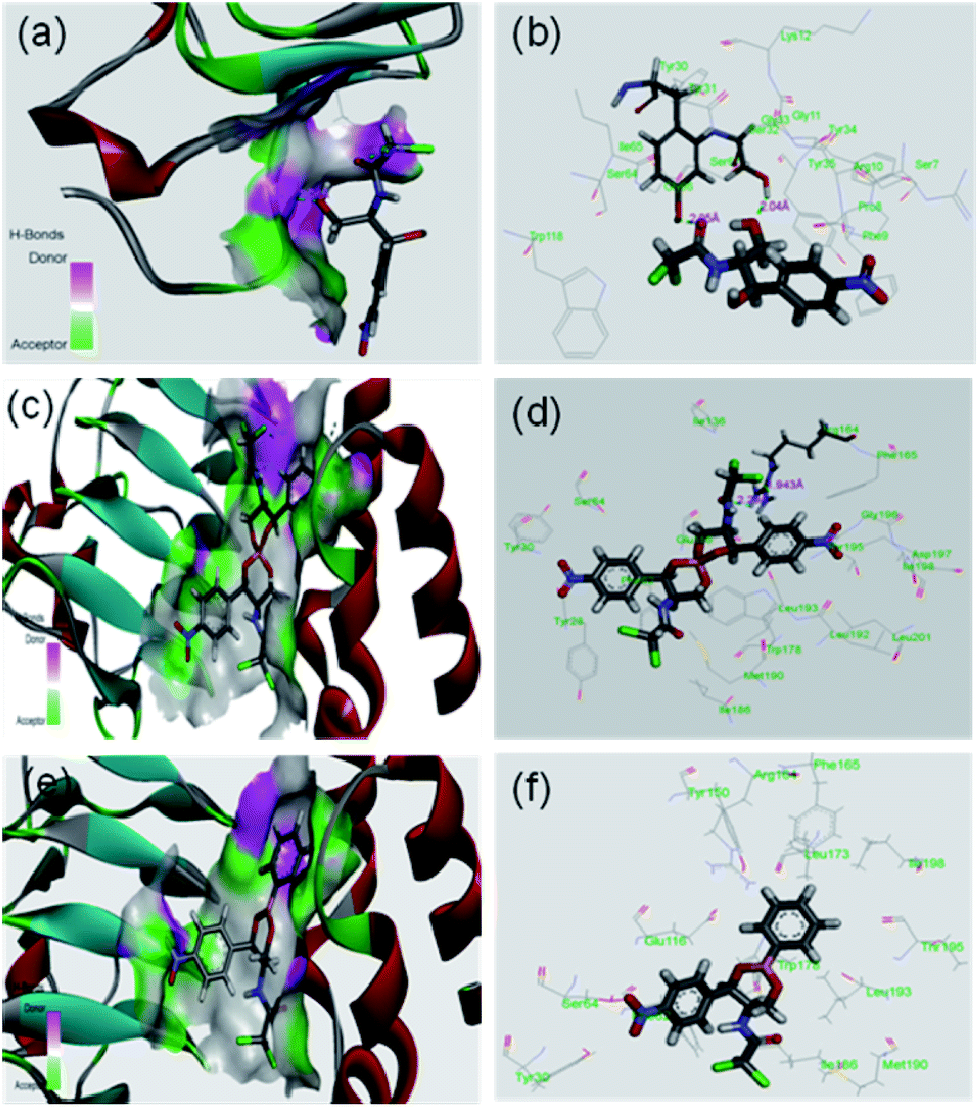
It was expected that after conversion of chloramphenicol to its corresponding borate/boronate derivative (2 and 3 respectively), there will be no scope for further acetylation as the –OH groups are tied up with boron. However, we were curious to know the binding motif of chloramphenicol vis-à-vis the derivatives 2 and 3. Thus, we have taken up the in silico approach to determine the binding efficiency of chloramphenicol 1 and its borate derivative 2 and chloramphenicol-phenyl boronate derivative 3 with chloramphenicol acetyl transferase. The xenobiotic acetyl transferase from Pseudomonas aeruginosa has been used for docking. The crystallographic structure with good resolution (3.2 Å) of ligand bound protein has been downloaded from RCSB protein data bank (http://www.rcsb.org). Chloramphenicol acetyl transferase (as receptor)–chloramphenicol (as ligand) interaction suggests that (according to crystal structure) it gets bound by two hydrogen bonds with Ser32 (2.04 Å) and Tyr30 (2.05 Å) whereas both the borate and phenyl boronate derivatives bind at Arg164 (Fig. 4). One of the two carbonyl oxygens and one of the two chlorine atoms form hydrogen bond with Arg164 in the borate complex 2. On the other hand, phenyl boronate complex 3 shows mainly van der Waals interactions in the most stable complex with acetyl transferase. The key parameters of docking study are shown in Table 2. The binding sites of acetyl transferase to chloramphenicol are Ser32 and Tyr30 whereas the binding sites with chloramphenicol-borate are different with Arg164 making two H-bonds alone. No hydrogen bonding interactions was observed for phenyl boronate ester. Therefore, the enzyme acetyl transferase is unable to acetylate both borate and boronate complex.
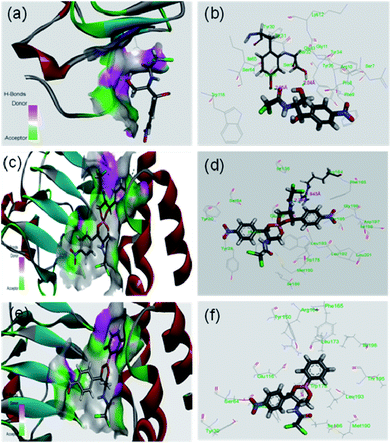 |
| | Fig. 4 In silico analysis of docking between compounds with protein (PDB id 2XAT). Surface area of the receptor where compounds got bound to protein is shown: (a) chloramphenicol 1, (c) chloramphenicol-borate 2 (e) and chloramphenicol-phenyl boronate 3. The various interactions inside the binding pocket of the protein are also shown: (b) chloramphenicol 1, (d) chloramphenicol-borate 2 and (f) chloramphenicol-phenyl boronate 3. | |
Table 2 Results of in silico study of binding
| Compound |
Hydrogen bonds |
| No. of H-bonds |
Amino acids interacting with the substrate |
Bond distance |
Bond angle (°) |
| Chloramphenicol (1) |
2 |
Ser32[OH⋯O of primary alcohol] |
2.04 (Å) |
140 |
| Tyr30[OH⋯O (carbonyl) of chloramphenicol] |
2.05 (Å) |
139 |
| Chloramphenicol-borate ester (2) |
2 |
Arg164[NH⋯Cl of chloramphenicol moiety] |
1.94 (Å) |
111 |
| Arg164[NH⋯O (carbonyl) of chloramphenicol moiety] |
2.29 (Å) |
90 |
| Chloramphenicol-phenyl boronate ester (3) |
— |
— |
— |
— |
Toxicity analysis
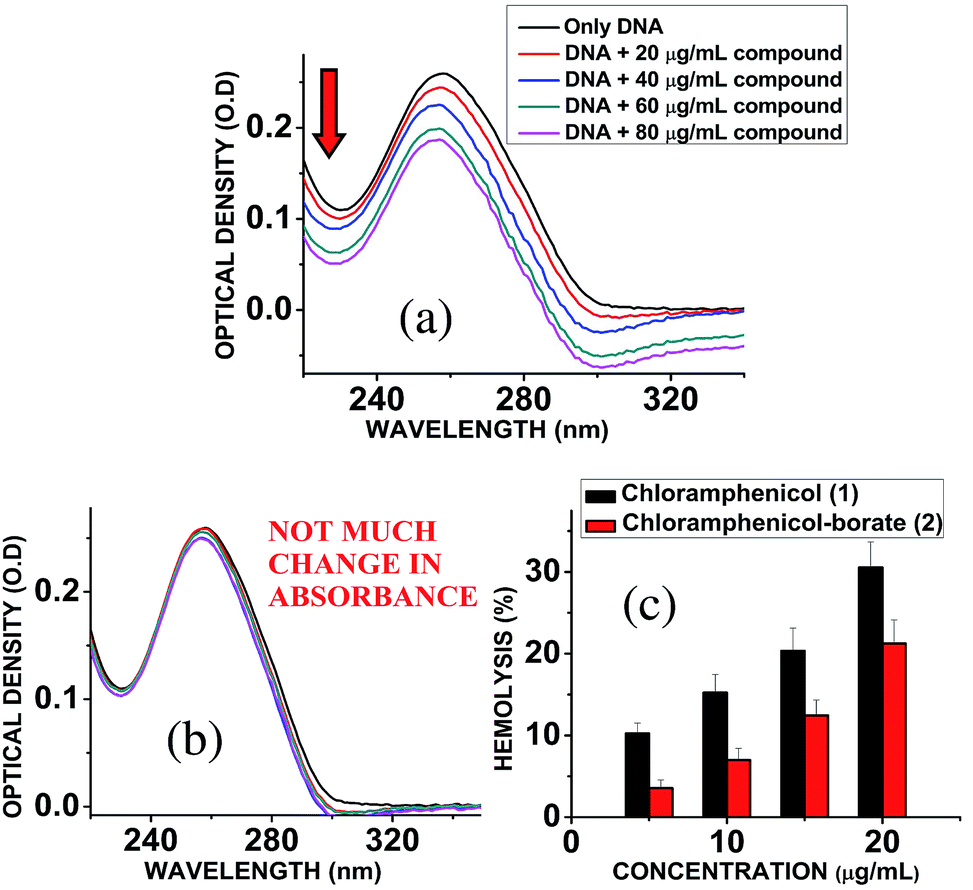
Chloramphenicol induces toxicity in mammalian system as aplastic anaemia, bone marrow disorder25 and apoptosis in hematopoietic stem cells.26 Different types of mechanism for their toxicity have been proposed earlier.27,28 Thus it is a matter of paramount importance and offers a crucial challenge to reduce the toxicity level during the designing and synthesis of its derivatives. Hemolytic assay was performed according to literature reported methods29 to check the compatibility level in comparison to native chloramphenicol. The results show that increasing the concentration of chloramphenicol and chloramphenicol-borate derivate, the hemolysis percentage was also increased. In all tested concentrations, the hemolysis percentage for the borate 2 was significantly lower than chloramphenicol only (Fig. 5c). We have also determined the genotoxic effect by studying the binding ability of chloramphenicol 1 and chloramphenicol-borate derivative 2 to DNA using UV-Vis absorption studies.30,31 The characteristic hypochromic shift was observed with chloramphenicol 1 only (Fig. 5a), whereas no such effect was observed upon addition of borate 2 (under same concentrations) to DNA (Fig. 5b). These results indicated that compound 2 has less binding affinity to DNA and less genotoxicity in comparison to only chloramphenicol 1.
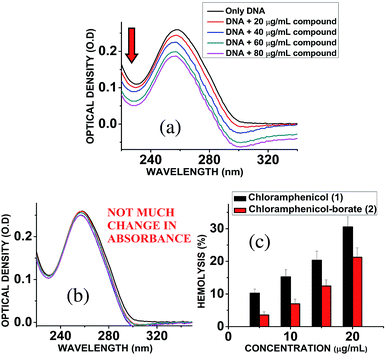 |
| | Fig. 5 (a) Interaction of chloramphenicol 1 with calf-thymus DNA, showing hypochromic shift in the absorbance of DNA. (b) Interaction of chloramphenicol-borate 2 with calf-thymus DNA, showing almost unchanged absorbance of DNA. (c) Effects of chloramphenicol and chloramphenicol-borate derivative on hemolytic activity. Effects of chloramphenicol on hemolysis represented with black color and its derivative with red color. | |
4. Conclusions
Chloramphenicol is a bacteriostatic antibiotic that binds to the 50S ribosomal subunit and inhibits the peptidyl transferase step in protein synthesis. Indiscriminate use of antimicrobials led to the bacterial resistance which is mostly due to inactivation of the antibiotic by chloramphenicol acetyltransferase (CAT) enzymes that acetylate the antibiotic. Understanding the mechanism of bacterial resistance at the molecular level led us to the successful design and synthesis of borate (2) and phenyl boronate (3) derivatives of chloramphenicol, both of which showed lower MIC values against chloramphenicol-resistant strains than the parent drug chloramphenicol. Among the two derivatives, the borate derivative 2 was found to be more active. The increase in activity observed against resistant GNBs was attributed to alteration in binding mode of this complex to the acetyl transferase thereby preventing acetylation. The hemolytic and genotoxic assays further established their lesser toxicity than the parent drug (chloramphenicol). It is understandable that sensible use of antibiotics and the search for effective alternative measures are of high importance in order to minimize the effect due to existing and emerging antimicrobial resistant microbes. It can be expected that the results obtained from this study will foster the research in this domain, stimulating the re-design and subsequent synthetic modification of a wide range of anti-bacterial compounds with a renewed therapeutic interest.32
Conflicts of interest
There is no conflict of interest.
Acknowledgements
P. B. is grateful to IIT Kharagpur for project fellowship. DST is acknowledged for an SERB grant (SC/S1/OC-94/2013) and for JC Bose Fellowship to A. B. which supported this research.
Notes and references
- J. Kallen and A. Srinivasan, Infect. Control Hosp. Epidemiol., 2010, 31, S51 CrossRef PubMed.
-
(a) National Nosocomial Infections Surveillance (NNIS) System, Am. J. Infect. Control, 1999, 27, 520 CrossRef;
(b) S. B. Levy and B. Marshall, Nat. Med., 2004, 10, S122 CrossRef PubMed;
(c) M. Lukačišinová and T. Bollenbach, Curr. Opin. Biotechnol., 2017, 46, 90 CrossRef PubMed.
- V. Aloush, S. Navon-Venezia, Y. Seigman-Igra, S. Cabili and Y. Carmeli, Antimicrob. Agents Chemother., 2006, 50, 43 CrossRef PubMed.
-
(a) T. H. Wong, B. H. Tan, M. L. Ling and C. Song, Burns, 2002, 28, 349 CrossRef PubMed;
(b) V. Waters, E. Larson, F. Wu, P. S. Gabriel, J. Haas, J. Cimiotti, P. Della-Latta and L. Saiman, Clin. Infect. Dis., 2004, 48, 1682 CrossRef PubMed.
- S. M. Mandal, A. Roy, A. K. Ghosh, T. K. Hazra, A. Basak and O. L. Franco, Front. Pharmacol., 2014, 13, 105 Search PubMed.
-
(a) W. V. Shaw, L. C. Packman, B. D. Burleigh, A. Dell, H. R. Morris and B. S. Hartley, Nature, 1979, 282, 870 CrossRef PubMed;
(b) A. Vasudevan, A. Mukhopadhyay, J. Li, E. G. Y. Yuen and P. A. Tambyah, BMC Infect. Dis., 2014, 14, 615 CrossRef PubMed.
- A. Aakra, H. Vebo, U. Indahl, L. Snipen, O. Gjerstad, M. Lunde and N. S. Nes, Int. J. Microbiol., 2010, 483048 Search PubMed.
- S. Schwarz, C. Kehrenberg, B. Doublet and A. Cloeckaert, FEMS Microbiol. Rev., 2004, 28, 519 CrossRef PubMed.
-
(a) B. Khameneh, R. Diab, K. Ghazvini and B. S. F. Bazzaz, Microb. Pathog., 2016, 95, 32 CrossRef PubMed;
(b) M. Fernández, S. Conde, J. de la Torre, C. Molina-Santiago, J.-L. Ramos and E. Duque, Antimicrob. Agents Chemother., 2012, 56, 1001 CrossRef PubMed.
- C. I. Montero, M. R. Johnson, C. J. Chou, S. B. Conners, S. G. Geouge, S. Tachdjian, J. D. Nichols and R. M. Kelly, Appl. Environ. Microbiol., 2007, 73, 5058 CrossRef PubMed.
- O. T. Magnusson, J. M. RoseFigura, H. Toyama, R. Schwarzenbacher and J. P. Klinman, Biochemistry, 2007, 46, 7174 CrossRef PubMed.
- C. Daniels and J. L. Ramos, Clin. Microbiol. Infect., 2007, 15, 32 CrossRef PubMed.
- S. Schwarz, C. Kehrenberg, B. Doublet and A. Cloeckaert, FEMS Microbiol. Rev., 2004, 28, 519 CrossRef PubMed.
-
(a) J. D. C. Yao and J. R. Moellering, Antibacterial Agents, in Manual of Clinical Microbiology, ed. J. Versalovic, ASM Press, Washington, 10th edn, 2011, vol. 1, p. 1043 Search PubMed;
(b) S. Sood, J. Clin. Diagn. Res., 2016, 10, DC01 Search PubMed.
- S. I. M. Mubarak, J. B. Stanford and J. K. Sugden, Drug Dev. Ind. Pharm., 1984, 10, 1617 CrossRef.
- Clinical and Laboratory Standard Institute (CLSI), Performance Standards for Antimicrobial Susceptibility Testing; 20th Informational Supplement, CLSI document M100-S20, Clinical and Laboratory Standard Institute, Wayne, PA, 2010 Search PubMed.
- G. Roymahapatra, S. M. Mandal, W. F. Porto, T. Samanta, S. Giri, J. Dinda, O. L. Franco and P. K. Chattaraj, Curr. Med. Chem., 2012, 19, 4184 CrossRef PubMed.
- S. K. Sarkar, A. Bhattacharyya and S. M. Mandal, Indian J. Med. Microbiol., 2015, 33, 139 CrossRef PubMed.
- P. A. White, H. W. Stokes, K. L. Bunny and R. M. Hall, FEMS Microbiol. Lett., 1999, 175, 27 CrossRef PubMed.
- T. W. Beaman, M. Sugantin and S. L. Roderick, Biochemistry, 1998, 37, 6689 CrossRef PubMed.
- S. Mondal, S. M. Mandal, T. K. Mondal and C. Sinha, Spectrochim. Acta, Part A, 2015, 50, 268 CrossRef PubMed.
- D. Schneidman-Duhovny, Y. Inbar, R. Nussinov and H. J. Wolfson, Nucleic Acids Res., 2005, 33, W363–W367 CrossRef PubMed.
-
(a) A. Paul and S. Bhattacharya, Curr. Sci., 2012, 102, 212 Search PubMed;
(b) M. Sirajuddin, S. Ali and A. Badshah, J. Photochem. Photobiol., B, 2013, 124, 1 CrossRef PubMed.
- S. M. Mandal, L. Migliolo, S. Das, M. Mandal, O. L. Franco and T. K. Hazra, J. Cell. Biochem., 2012, 113, 184 CrossRef PubMed.
- C. S. Ambekar, B. Cheung, J. Lee, L. C. Chan, R. Liang and C. R. Kumana, Eur. J. Clin. Pharmacol., 2000, 56, 405 CrossRef PubMed.
- C. T. Kong, D. E. Holt, S. K. Ma, A. K. Lie and L. C. Chan, Hum. Exp. Toxicol., 2000, 19, 503 Search PubMed.
- R. Holt, Lancet, 1967, 10(7502), 1259 CrossRef.
- A. A. Yunis, Am. J. Med., 1989, 87, 44N Search PubMed.
- S. M. Mandal, L. Migliolo, O. L. Franco and A. K. Ghosh, Peptides, 2011, 32, 1741 CrossRef PubMed.
- S. M. Mandal, A. Chakraborty, M. Hossain, D. Mahata, W. F. Porto, R. Chakraborty, C. K. Mukhopadhyay, O. L. Franco, T. K. Hazra and A. Basak, Mol. BioSyst., 2015, 11, 2551 RSC.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469 CrossRef PubMed.
- N. Cassir, J. M. Rolain and P. Brouqui, Front Microbiol., 2014, 5, 551 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02227e |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a