
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorous Pt3Ni with enhanced activity and durability towards oxygen reduction reaction†
Shuying Mi,
Na Cheng,
Hao Jiang ,
Chunzhong Li* and
Haibo Jiang*
,
Chunzhong Li* and
Haibo Jiang*
Key Laboratory for Ultrafine Materials of Ministry of Education, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: jianghaibo@ecust.edu.cn; czli@ecust.edu.cn; Tel: +86 021 64250996 Tel: +86 021 6425094
First published on 24th April 2018
Abstract
The size of nanocrystals (NCs) is regarded as one of the vital factors determining their electrochemical performance. To achieve high electrochemical activity and durability at the same time still remains a big challenge. This work has demonstrated the successful synthesis of Pt3Ni nanocrystals of large size with porous characteristics (PNC-Pt3Ni). The mass and specific activity of the as-prepared catalyst are 6 and 6.6 times more than those of commercial Pt/C at 0.9 volts versus the reversible hydrogen electrode (RHE), respectively. More importantly, PNC-Pt3Ni prevails against a durability test (23.7% loss of mass activity after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential cycling) with little change to the porous morphology under harsh experimental conditions. Density functional theory calculations show a much lower activation energy for PNC-Pt3Ni during the process of dissociation of the oxygen molecule adsorbed on the surface of the catalyst, which may account for the improvement in the catalytic activity. The lower series resistance for PNC-Pt3Ni is also verified by electrochemical impedance spectroscopy (EIS) data, resulting from fewer grain boundaries for nanocrystals with large sizes. This exciting work contributes a new strategy for the optimization of electrochemical performance and durability.
000 potential cycling) with little change to the porous morphology under harsh experimental conditions. Density functional theory calculations show a much lower activation energy for PNC-Pt3Ni during the process of dissociation of the oxygen molecule adsorbed on the surface of the catalyst, which may account for the improvement in the catalytic activity. The lower series resistance for PNC-Pt3Ni is also verified by electrochemical impedance spectroscopy (EIS) data, resulting from fewer grain boundaries for nanocrystals with large sizes. This exciting work contributes a new strategy for the optimization of electrochemical performance and durability.
1. Introduction
Proton exchange membrane fuel cells (PEMFCs) are considered to be one of the most promising and clean devices with negligible emission.1–3 However, the extensive use and development of PEMFCs are mostly hindered by the high-cost and non-persistent Pt cathode catalyst for the oxygen reduction reaction (ORR).4–6 According to the 2017 target set by the U.S. Department of Energy (DOE) and the rapid emergence of energy, as well as environmental issues, continuing advancements are required to minimize precious metal loading.7,8 Therefore, during the last decade, Pt-based or Pt-free nanocrystals (NCs) have received a significant amount of attention mainly focusing on two crucial forms, namely enhancing the electrochemical performance and maintaining the durability.9–11In order to achieve the excellent electrochemical activity, synthesizing small size (mostly with a diameter of less than 10 nm) single-crystal catalysts with a high specific surface area is favoured as a valid method from the point of view of taking full advantage of the Pt atoms.12 For instance, Yamamoto et al. proposed that nanoscale Pt clusters consisting of small numbers of atoms exhibited different excellent physical properties with high catalytic activity from bulk metals.13 Moreover, Pt nanoparticles with a diameter of around 2.2 nm are regarded as the best catalysts for ORR.4 However, insufficient long-term operation durability still remains a severe problem.14 Typically, most works have exhibited aggregation and the Ostwald ripening process of commercial Pt/C (2–3 nm) during accelerated deterioration test (ADT). After 10000 cycles, the catalyst displays at least 40% loss of mass activity accompanied by a size growth of up to 10–30 nm.15,16
Generally, methods of modifying Pt with Au and increasing the average size of the catalysts are aimed at solving the above-mentioned problems.17,18 Research has verified that alloying and incorporating Pt with Au is expected to increase catalytic stability observably under highly acidic circumstances, because of OH-repulsive properties.19 Zhang et al. observed insignificant changes in the activity and surface area of Au-modified Pt under the oxidizing conditions of potential cycling between 0.6 and 1.1 volts over 30000 cycles.20 A decrease in the oxidation of Pt nanoparticles covered by Au was discovered compared with Pt nanoparticles without Au coverage, ascribing to the much higher Pt oxidation potential. Some researchers also have made an effort on developing large size catalysts. Zhang et al. have reported the synthesis of large Pd NCs supported on small Pt NCs that presented excellent durability against potential cycling.21 However, the ORR performance of either Pt–Au or large size catalysts barely reaches or is barely comparable to commercial Pt/C. Yu et al. put forward Pt concave nanocubes (edge lengths: 15–40 nm) with high-index facets which exhibited enhanced specific activity relative to the Pt nanocubes enclosed by low-index facets, yet it turned out that the mass activity was not as high as expected due to the low utilization of the Pt atoms.22
In light of the foregoing problems, a number of structures have been studied for balancing activities and durabilities,23–27 such as nanowires/tubes,28–31 carbon encapsulation,32 and hollow33,34 and open-framework structures.35,36 The results show that these structures possess excellent activities and durabilities. Li et al. have synthesized typical nanowires with a diameter of ∼5 nm and a length of ∼250 to 300 nm, which showed a 12% mass activity decline after 6000 cycles of ADT.37 Galeano et al. encapsulated Pt nanoparticles of approximately 3–4 nm within hollow graphitic spheres, conclusively proving that encapsulation remarkably suppresses detachment and agglomeration of Pt nanoparticles while still sustaining high activity.38
Combining the merits of high utilization of Pt atoms and exceptional durability, herein, we report a new class of porous Pt3Ni with a size of around 47 nm, which exhibits high activity as well as excellent durability towards ORR. Abundant branches and porous structure make it possible for the maximum utilization of Pt atoms,39 while the large size characteristic promises stability. The as-prepared PNC-Pt3Ni delivers high mass and specific activity compared to commercial Pt/C catalysts and prevails in a highly acidic ORR environment.
2. Experimental methods
2.1 Synthesis of PNC-Pt3Ni
The controlled synthesis of PNC-Pt3Ni was realized through a solvothermal method at 180 °C. Platinum(II) acetylacetonate ([Pt(acac)2], 10 mg) and nickel(II) acetylacetonate ([Ni(acac)2], 6.5 mg) were mixed with 3 ml oleylamine (OM) and 2 ml oleic acid (OA) as metal precursors in the presence of dodecyltrimethylammonium chloride (DTAC, 25.9 mg). The molar ratio of the two metal precursors was regulated at 1:1. The as-prepared mixture was sealed before being ultrasonicated for one hour. Afterward, the homogeneous mixture was heated at 180 °C for two hours (including the pre-heating time) in an oil bath before being cooled to room temperature. The product was collected by centrifugation and washed three times with a mixture of ethanol and cyclohexane for further characterization tests.
2.2 Characterization
Low and high resolution transmission electron microscopy (TEM) were conducted on a JEOL-2100 transmission electron microscope with an LaB6-cathode (200 kV). EDX mapping was carried out in a JEM-ARM200F by scanning transmission electron microscopy (STEM). Powder X-ray diffraction (XRD) was performed to analyse the crystallization of the catalysts. Inductively coupled plasma-atomic emission spectroscopy (ICP-AES, iCAP 6000 Radial, THERMO) was used to measure the chemical compositions of the catalysts. XPS spectra were obtained on a Thermo Scientific ESCALAB 250 XI X-ray photoelectron spectrometer.2.3 Electrocatalytic measurements
ORR measurements were performed with a rotating disk electrode (RDE) setup with a potentiostat (CHI 618D). A saturated calomel electrode (SCE) and platinum sheet were selected as the reference and counter electrodes, respectively. A modified glassy carbon RDE with a diameter of 5 mm was used as the working electrode. After dispersing PNC-Pt3Ni in 1 ml cyclohexane and Vulcan XC-72 carbon in 3 ml ethanol, these two homogeneous solutions were then mixed and ultrasonicated for 1 h. Later, the mixed solid was collected by centrifugation and redispersed in a mixture of acetic acid and ethanol. After being ultrasonicated for 1 h, the catalysts were washed with ethanol three times and transferred to quartz tubes for further annealing at 250 °C for 2 h under atmospheric conditions to remove organic surfactants. When the catalysts were cooled to room temperature, they were dispersed in an isopropanol and Nafion (5%) mixture (v/v = 50:1) to produce the homogeneous catalysts’ ink and they were ultrasonicated for 30 min. 16 μl catalysts’ ink was dispensed onto the RDE. Pt loading of each catalyst was controlled to be 20.4 μg cm−2 based on ICP-AES measurement. The thin films of the catalysts were formed at room temperature and remained on the glassy carbon surface for subsequent tests.
Cyclic voltammetry tests were performed in N2-saturated HClO4 (0.1 M) solution at a scan rate of 50 mV s−1 and between 0.06–1.1 V normalized to RHE. The polarization curves for the ORR were obtained in O2-saturated HClO4 (0.1 M) solution. The scan rate and rotation speed for the ORR measurements were 10 mV s−1 and 1600 rpm, respectively. The accelerated durability tests (ADT) were conducted at room temperature in 0.1 M HClO4 solution by applying cyclic potential sweeps between 0.6 V and 1.3 V versus RHE at a sweep rate of 100 mV s−1 for 10000 cycles. Mass and specific activities were calculated based on the kinetic current densities at 0.9 V (vs. RHE) and normalized to the electrochemically active surface area (ECSA) and the loading amount of Pt, respectively.
The EIS experiments were measured using an Autolab PGSTAT302N. The frequency range of EIS experiments was controlled from 100 kHz to 0.1 Hz (amplitude at 5 mV). The same amount of Pt loading was applied as in the ORR measurements.
3. Results and discussion
PNC-Pt3Ni was synthesised via a facile solvothermal approach. The metal precursors were added at the same time and were reduced at a relatively high reduction rate. It could be observed that the colour of the homogeneous solution turned from light yellowish-green to dark brown after being kept at 180 °C for 5 min, indicating a relatively rapid co-reduction process of the metal precursors. As shown in Fig. 1, the representative morphology of the as-prepared PNC-Pt3Ni was characterized by HRTEM and STEM. As can be seen from Fig. 1A, generally, the PNC structure dominated among the NCs with a mean size of around 47 nm. According to the HRTEM results, the obtained NCs were composed of 3–5 nm small units as well as the nanoporous structure which may create a number of active sites for electrocatalytic performance. The atomic ratio of the overall catalyst was measured to be 3:1 (Pt/Ni) by ICP-AES. STEM-electron energy-loss spectroscopy (EELS) mapping clearly confirmed the even distribution of each element throughout the single particle, where red represented Pt and green represented Ni. It should be noted that the Pt content was rich in the fringe of the NCs (may be stated as another possible reason for enhanced electrocatalytic performance) on the basis of the overlapped element image, and this was also verified by XPS later. Furthermore, the selected area electron diffraction (SEAD) pattern from one complete NC (Fig. 1B) showed a typical periodic lattice pattern and was consistent with the continuous lattice shown by HRTEM. Fig. 1D–G exhibited highly ordered parallel fringe patterns, representing four equally distributed sites recorded from a single NC (Fig. 2B, from site 1 to 4, respectively). The identical Fourier-transform patterns were also associated with the same crystal.
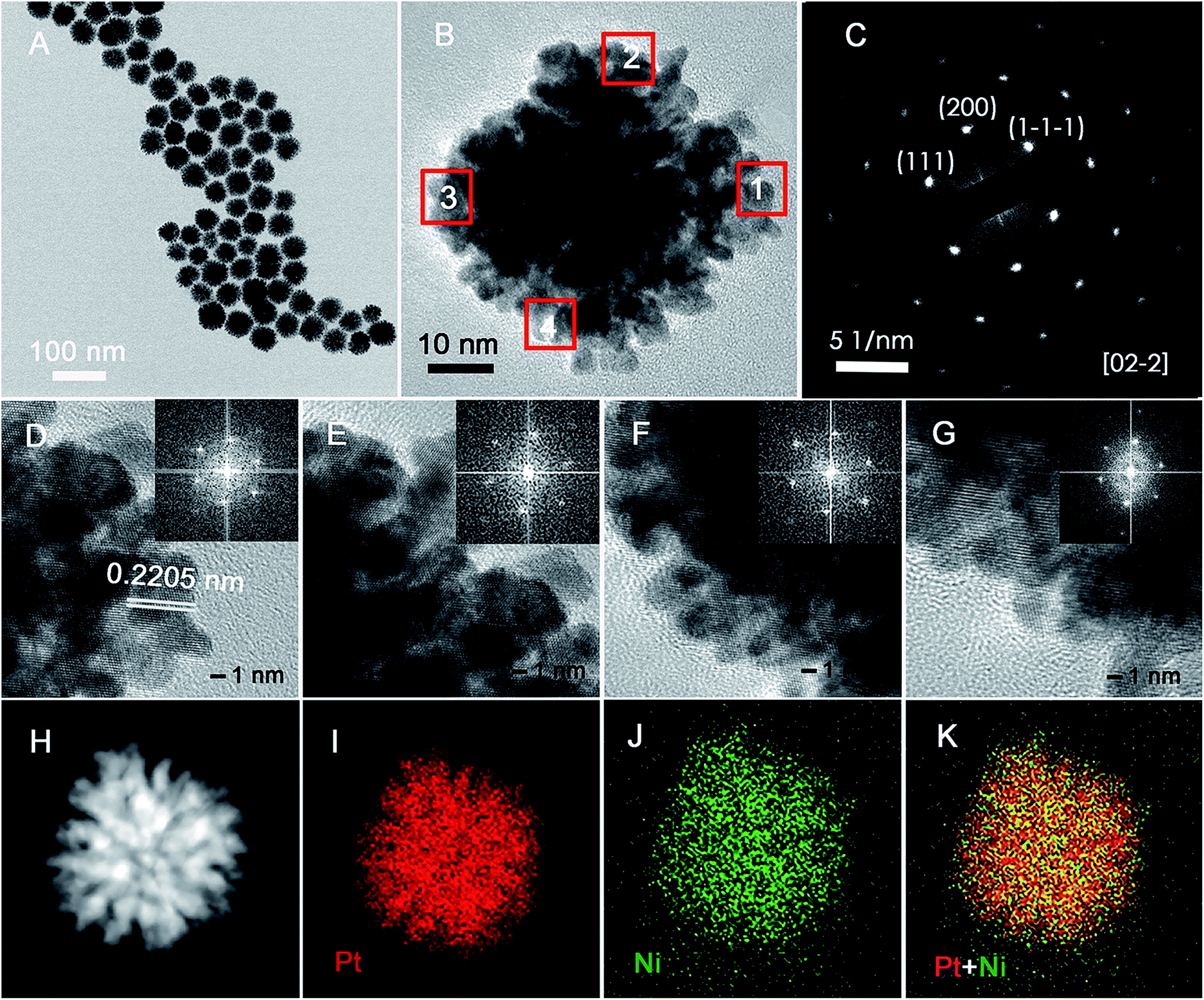 | ||
| Fig. 1 Structural analysis of PNC-Pt3Ni. (A) TEM image of uniformly distributed NCs, (B) HRTEM image, (C) SEAD image of a single crystal and (D to G) HRTEM images recorded from sites 1, 2, 3 and 4 marked in (B), respectively. The identical FT patterns shown in the insets reveal that a complete single crystal has the same lattice orientation. (H to K) STEM and elemental mapping images for Pt, Ni and composite Pt versus Ni, respectively. | ||
 | ||
| Fig. 2 TEM images of products captured at different time stages. (A) 3 min, (B) 5 min, (C) 20 min, (D) 30 min, (E) 1 h, and (F) 2 h. | ||
X-ray diffraction (XRD) patterns of PNC-Pt3Ni before and after annealing at 250 °C were further obtained to determine the crystal structure of PNC-Pt3Ni (Fig. S1A†). Three typical diffraction peaks, corresponding to (111), (200) and (220) planes, were observed between pure Pt (PDF#04-0802Pt) and Ni (PDF#04-0850Ni) standard peaks, revealing the alloy phase and face-centred cubic (fcc) structure of PNC-Pt3Ni and the high crystallinity of the Pt–Ni phase. No structural difference was detected for PNC-Pt3Ni before and after annealing at 250 °C, which is also verified by the TEM images in Fig. S2.† The molar ratio of Pt/Ni was obtained by the ICP-AES test after annealing, which was 2.93, generating the same result as the non-annealed sample. The surface components after annealing at 250 °C were analysed by XPS (Fig. S1B†) and two peaks in the Pt spectrum which appeared at 71.25 eV and 74.6 eV corresponded to Pt 4f5/2 and Pt 4f7/2, which could be deconvoluted into four peaks, 71.25 eV, 74.6 eV, 72.05 eV and 75.3 eV, respectively, suggesting the presence of metallic Pt and oxidized Pt.40 The surface Pt/Ni atom ratio disclosed by XPS produced higher results than when it was measured by ICP-AES, and this was 4.12:1 versus 3.15:1 (ICP-AES versus Pt:Ni, respectively), obtaining the same consequence indicated by elemental mapping previously. When combining all the test results above, the conclusion could be made that Pt3Ni with a porous structure was successfully synthesized.
To gain an understanding of the morphological evolution of PNC-Pt3Ni, the product at different time stages was captured and characterized by TEM (Fig. 2). The schematic diagram of this process is illustrated in Fig. 3. The colour of the homogeneous solution started to change after three minutes under 180 °C and the product was composed of small irregular particles with a size of 8–10 nm. Two minutes later, regular shapes of products overtook the irregular particles to form orderly piled cubes. These piled cubes provided the opportunity for self-assembly in the later growth process. When the reaction time reached 20 minutes, flower-shaped products dominated the overall products, the sharp edges and corners of the piled cubes disappeared followed by the epitaxial and mellow petal shapes, yet the pores formed between the orderly piled cubes remained to the end. Ultimately, after 2 h, porous single-crystals were conserved with the diameter of about 47 nm without further growing. In this case, during the process of the transformation from piled cubes to flower-shaped products, the primary control parameter was the relatively long reaction time. After the Pt precursors were consumed and reduced to Pt(0) monomers, the minimum reduction temperature (180 °C) for the Pt precursors ensured the successive formation of monomers in the presence of oleylamine.41 Further growth of the products led to the consumption of monomers, resulting in a gradient of monomer concentration.42 The piled cubes were inclined to stick out further as a flower shape to maintain the high monomer concentration, forced by minimum surface energy principles as well. With more monomers reaching the corners and edges, the corners and edges grew even faster, resulting in the phenomenon of the cubes transforming into small branches. Fig. S3† shows the tendency of the content and molar ratio of Pt/Ni. Generally, both broken lines of Pt/Ni showed overall growth accompanied by fluctuation and no sign of significant content loss of Ni was witnessed, which helped to exclude the possibility of an O2/Cl− etching growth mechanism occurring here.
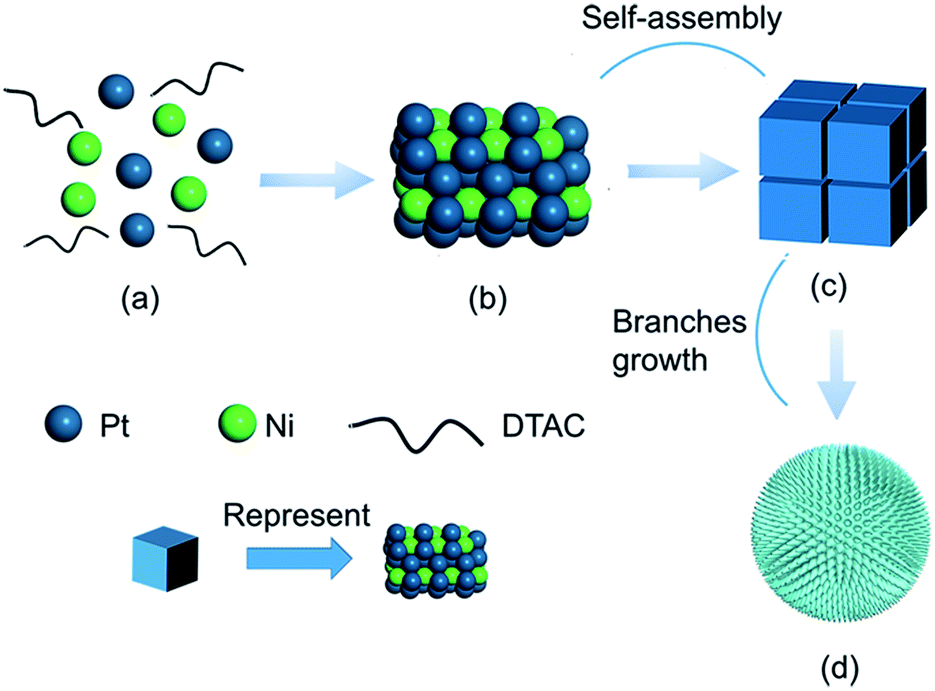 | ||
| Fig. 3 Schematic illustration of the synthesis of PNC-Pt3Ni. (a) Homogeneous mixture of metal precursors and DTAC. (b) Individual units. (c) Self-assembled cubes. (d) The final structure of PNC-Pt3Ni. | ||
To gain further insight into the growth mechanism, different conditions were applied to understand the reaction process comprehensively. DTAC, serving as a capping agent, was considered to be critical for the synthesis of PNC-Pt3Ni.43,44 Fig. S4† shows the morphology change with different doses of DTAC from 0 to 51.8 mg while other conditions were preserved. The morphology of the final products turned into a snowflake shape without any DTAC added into the solvent, while the Pt/Ni atomic ratio of the final component was unchanged based on ICP-AES measurement. Furthermore, as we raised the DTAC quantity to 5 mg, the average size of the synthesized NCs extended to 50–60 nm with a compact surface structure. In this case, the porous characteristic was substituted by an irregular cubic shape as well as a small amount of the configuration being projected as triangles. When additional DTAC was added, the solid product exhibited a spherical structure accompanying the porous characteristic. Nevertheless, the essential feature of the catalyst was unaltered as a result of the further adding of DTAC, which means that if the amount of DTAC exceeds absolute maximum, the morphology is retained. Well-dispersed PNC-Pt3Ni was successfully obtained due to the sufficient morphology direction during the nucleation and growth processes. The control force or the mechanism of this growth process may result from substantial interactions between Pt and Ni atoms and DTAC, but the profound intrinsic principles still remain to be solved.
Moreover, the composition of the final catalysts can be tailored by the molar ratio of the precursors whilst maintaining the similar contours in the presence of both precursors. With the increase of Ni precursors, the shape changed from compact spheres with a jagged surface to porous NCs (Fig. S5†). As more Ni precursors were employed, the catalyst became increasingly porous. However, it failed to ensure the uniformity of particle size and dispersion. It should be noted that as the molar ratio of metal precursors was adjusted for the component dominated by Ni, the average diameter of the final NCs declined from 72.5 nm to around 29 nm. Surprisingly, in the absence of Pt precursors, no catalysts were collected under the same conditions, indicating the possible facilitating role of Pt particles in the early stage.
In an effort to obtain an in depth understanding of the behavior of OA and OM, attempts were made to tune the size and shape of the NCs, shown in Fig. S6.† Clusters with a diameter as small as 2–3 nm were collected by employing 5 ml OM only. Slight aggregation and poor dispersion were found in this case. As the ratio of OM/OA declined to 2:3, the final color of the homogeneous mixture developed into an atramental black colour, and lead to the formation of nanorods. Only when both solvents satisfied the equilibrium relationship, did it result in the porous structure, otherwise a mild rise or reduction in the solvent ratio, for instance OM/OA = 2.5:2.5 instead of the optimum condition of OM/OA = 3:2, would give rise to the irregularities of morphology and agglomeration.
In addition, cyclic voltammetry (CV) and linear sweep voltammetry (LSV) measurements were carried out to evaluate the electrocatalytic performance of PNC-Pt3Ni towards the ORR. PNC-Pt3Ni and Pt/C were uniformly dispersed on supported carbon via ultrasonication of the catalysts and a carbon solution (Fig. S7†). The electrochemical surface area (ECSA) was calculated by the peak areas of adsorption and desorption of HUPD between 0–0.4 V after eliminating the double-layer charging correction (Table S1†). Although the particle size of PNC-Pt3Ni/C was approximately 22 times more than that of Pt/C, it exhibited comparable ECSA (75.3 m2 g−1) to that of commercial Pt/C (81.6 m2 g−1), which may be due to its porous and branch structure.45 CV curves are presented in the insets of Fig. 4A and B. It is notable that the redox potential of hydroxyl in the high potential region of PNC-Pt3Ni/C was remarkably more positive than that of Pt/C during the backward sweep, suggesting the weaker adsorption of oxygenate species on the surface of PNC-Pt3Ni/C.40 Thus, more active sites on the surface of PNC-Pt3Ni/C for O2 adsorption were available for the ORR process, making it favorable for enhanced electrocatalytic activities. Fig. 4A compared typical ORR polarization curves of PNC-Pt3Ni/C and Pt/C. The measurements were performed at room temperature unless otherwise indicated. The diffusion-limiting current region was shown to be below 0.8 V. The PNC-Pt3Ni/C catalysts displayed an ORR onset potential of around 1.0 V. It is intriguing that the half-wave potential of PNC-Pt3Ni/C (0.926 VRHE) was 60 mV more positive than the commercial catalyst (0.866 VRHE). To compare the ORR performance quantitatively, the mass activity and specific activity were calculated at 0.9 VRHE by applying the Koutecky–Levich equation (Table S1†). PNC-Pt3Ni/C displayed excellent mass and specific activity (0.594 A mgPt−1, 0.789 mA cm−2, respectively), which were 6 and 6.6 times higher than the corresponding performances of Pt/C (0.098 A mgPt−1, 0.120 mA cm−2, respectively). This pronounced enhancement may have resulted from multiple reasons, such as maximized utilization of Pt atoms due to the large number of branches and the porous structure, leading to fewer grain boundaries under the condition of the same thickness of catalyst being deposited on the RDE, which was also verified in the subsequent experiment.
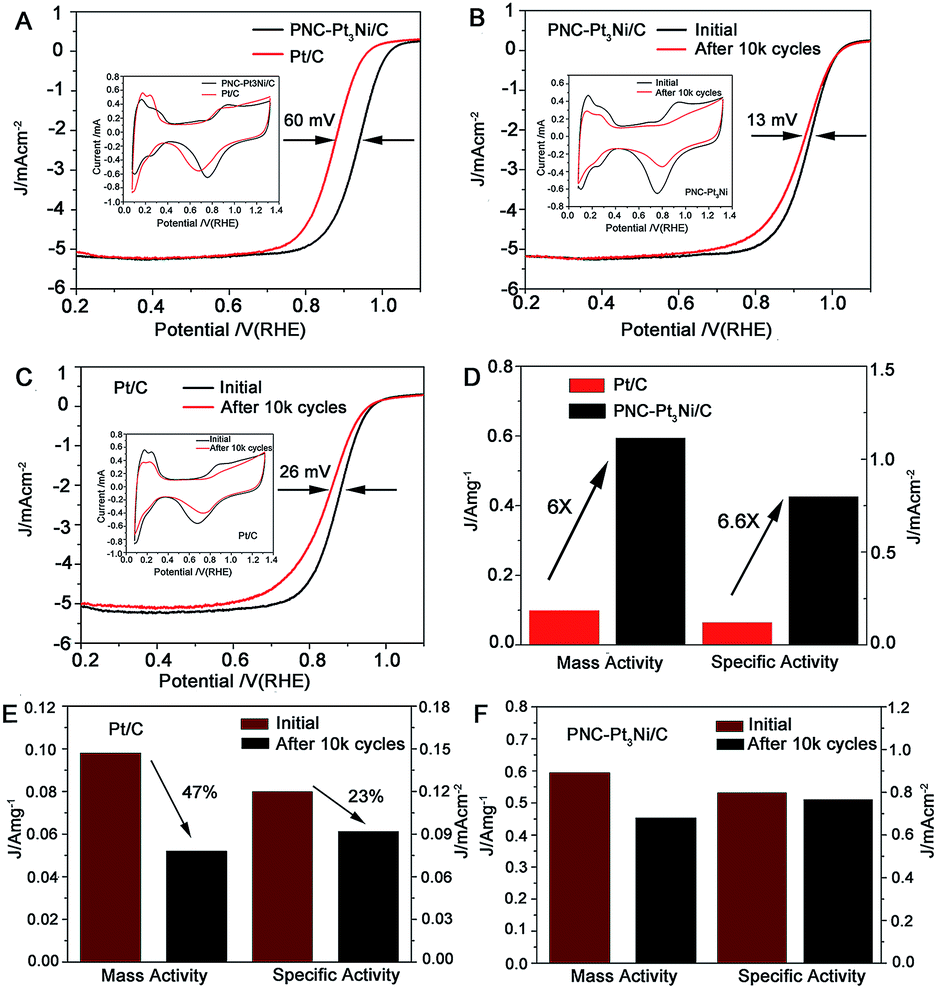 | ||
| Fig. 4 Electrocatalytic performance of PNC-Pt3Ni/C and Pt/C towards the ORR. (A) ORR polarization curves, the inset is the CV curves of these two catalysts. (B) ORR polarization curves of the PNC-Pt3Ni/C catalyst before and after 10000 potential cycles between 0.66 and 1.3 V versus RHE. (C) ORR polarization curves of the commercial Pt/C catalyst before and after 10000 potential cycles between 0.66 and 1.3 V versus RHE. (D) The difference between mass activity and specific activity of PNC-Pt3Ni/C and Pt/C. (E) The decrement of mass and specific activity of Pt/C before and after 10000 potential cycles. (F) The decrement of mass and specific activity of PNC-Pt3Ni/C before and after 10000 potential cycles. | ||
An investigation into the electrochemical stability of the as-prepared catalysts in comparison to Pt/C has been conducted by carrying out ADTs. The CV and LSV curves were recorded before and after ADT (shown in Fig. 4B and C). This showed the diminutive influence on the diffusion-limited current and only a 13 mV decrement of half-wave potential was detected for PNC-Pt3Ni/C. A double half-wave potential decrement was observed for commercial Pt/C, as well as an upper shift in the diffusion-limited current. The mass activities of PNC-Pt3Ni/C and Pt/C decreased to 0.453 A mgPt−1 and 0.052 A mgPt−1, respectively, which were 76.26% and 53.06% of their original values. Accordingly, the specific activity of PNC-Pt3Ni/C showed a slight reduction. It maintained 97.08% of its initial values, while commercial Pt/C lost 23.33% of its specific activity. On account of the above-calculated results, it can be concluded that the as-prepared catalysts exhibited improved durability towards ORR compared to commercial Pt/C. The outstanding stability was probably ascribed to the large size, which prevented the catalysts from aggregation. As shown in Fig. S8A,† after 10000 cycles, commercial Pt/C underwent serious agglomeration, and wider average sizes were seen from the TEM images afterwards, compared to the original morphology, disclosing a strong Ostwald ripening process during the durability test. As for PNC-Pt3Ni/C, although slight growth in the small units was seen (Fig. S8B†), the porous structure remained and prevailed throughout the strict conditions, which was also in accordance with no loss in the specific activity. Furthermore, ICP-AES data were collected to study the content of Pt/Ni after the ADT test. It showed an increased molar ratio (Pt/Ni: 4.34:1) compared to the initial status (Pt/Ni: 3:1), which was probably due to the leaching of the transition metal Ni.
In order to further demonstrate the benefit from the porous structure towards ORR activity, the as-prepared PNC-Pt3Ni was annealed at 400 °C for 3 h under a H2/Ar atmosphere, and this subsequently acted as the benchmark for ORR activity (recorded as Pt3Ni-400). The mass occupation of Pt and Ni in the final catalysts was controlled at 20%, which was consistent with the as-prepared PNC-Pt3Ni/C. Calcination caused serious damage to the branches, which could no longer be seen in the TEM (Fig. S8C†). The porous structure was replaced by the spherical framework, whilst maintaining the similar size distribution and element ratio according to the ICP-AES results. In this case, the electrocatalytic performance of Pt3Ni-400 was also tested under identical conditions, as illustrated in Fig. S8D.† The half-wave potential from the ORR polarization curve shifted negatively in comparison with the initial catalysts. The peak areas of adsorption and desorption of HUPD between 0–0.4 V after eliminating the double-layer charging correction decreased significantly. Nevertheless, the reduction current peak in the high potential region remained the same as that of the primary catalysts, suggesting a similar adsorption force of the oxygenate species on the surface. Unfortunately, a lack of active sites for ORR gave rise to inferior electrochemical activities. This provided definite proof that the porous structure with high exposure of the active sites was an ideal way to maximize the utilization of Pt atoms and boost the electrochemical activities for large size NCs. Moreover, the EIS results of PNC-Pt3Ni/C and Pt3Ni-400/C were compared under the same experiment conditions (Fig. S9†), which showed almost no difference in the porous and non-porous morphologies. This supported the conclusion that the porous structure helped to enhance the electrochemical performance.
Furthermore, the electrochemical impedance spectroscopy (EIS) data is shown in Fig. 5. On the basis of the Nyquist plots, the series resistance (stated as R1 here) can be determined.46,47 The R1 values of PNC-Pt3Ni/C and Pt/C were 18.56 and 19.83 Ω, respectively. It was considered that both catalysts shared identical general resistance under the same experimental conditions. Therefore, the series resistance represented the internal resistance and the grain boundaries of the catalysts. The as-prepared PNC-Pt3Ni/C possessed a lower R1 value due to fewer grain boundaries existing in the electrode, thus it obtained a higher current in the reaction. Both catalysts displayed limited R1 values, which may derive from the outstanding conductivity of the metal-based electrode materials.48
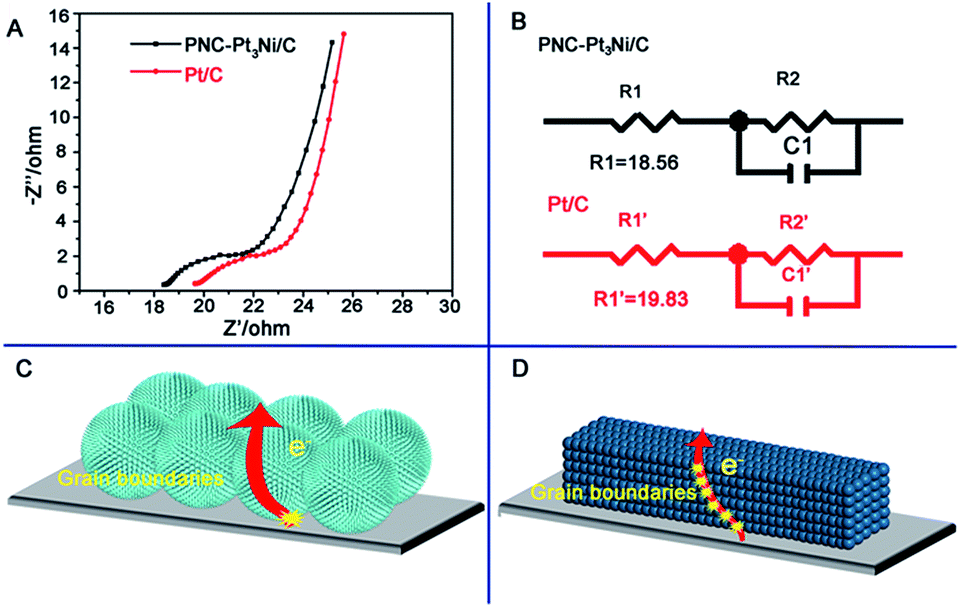 | ||
| Fig. 5 (A) The associated electrochemical impedance spectroscopy (EIS) of PNC-Pt3Ni/C and Pt/C recorded at −0.05 V within a frequency range of 100 kHz to 0.1 Hz (amplitude at 5 mV). (B) The equivalent circuit model for PNC-Pt3Ni/C and Pt/C. (C and D) Schematic illustration of PNC-Pt3Ni/C and Pt/C. Adequate grain boundaries exist in the electrode of the Pt/C single crystal compared to that of PNC-Pt3Ni within the same membrane thickness, which increase the internal resistance of the catalysts in the process of ORR and result in the drop in electrochemical performance. | ||
To understand the excellent electrochemical performance of the as-prepared PNC-Pt3Ni, density theory calculations were also carried out. It is generally considered that ORR is a multi-electron transfer reaction, including the adsorption and dissociation of oxygen, as well as the transfer of electrons and protons.49,50 The dissociation of the oxygen molecule adsorbed on the surface of the catalyst is regarded as the rate-determining step of the ORR reaction.51,52 As a consequence, the activation energy of oxygen molecular decomposition has a vital effect on the performance of ORR.
The application of the DFT model and calculations are explained in detail in the ESI.† According to the principle of thermodynamic stability, the surface with the lowest surface energy among the three possible step surfaces was chosen here. The low-coverage binding energies (BEs) of O2* (0.3–0.5 eV) and O* (3.47–3.73 eV)53 have been determined by thermal desorption spectroscopy (TDS)54 and electron energy-loss spectroscopy (EELS).55 According to these experiments, the dissociation barrier for O2* → 2O* on Pt (111) is assessed to be equivalent to the BE of O2*. In this work, the BEs of O2*(0.5 eV) and O* (3.5 eV) on the Pt (111) facets were obtained in order to manifest the acceptable accuracy of our calculations. Amplified diagrams from different projections of the reactant, transition state and product on Pt (111) and PNC-Pt3Ni (111) are illustrated in Fig. S10.†
Fig. 6 shows the activation energy for the dissociation of O2 on the surface of Pt (111) and PNC-Pt3Ni (111). Both of the patterns showed characteristics of the exothermic reaction. The O2* dissociation on PNC-Pt3Ni released relatively more energy (19.84 kcal mol−1) than the one on Pt (111) (16.85 kcal mol−1). On PNC-Pt3Ni (111), the activation energy was only one-sixth compared to the energy displayed on Pt (111), suggesting that it was more accessible for oxygen to dissociate on the surface of the as-prepared PNC-Pt3Ni, which was probably the fundamental principle that helped boost the electrochemical performance of the as-prepared PNC-Pt3Ni.
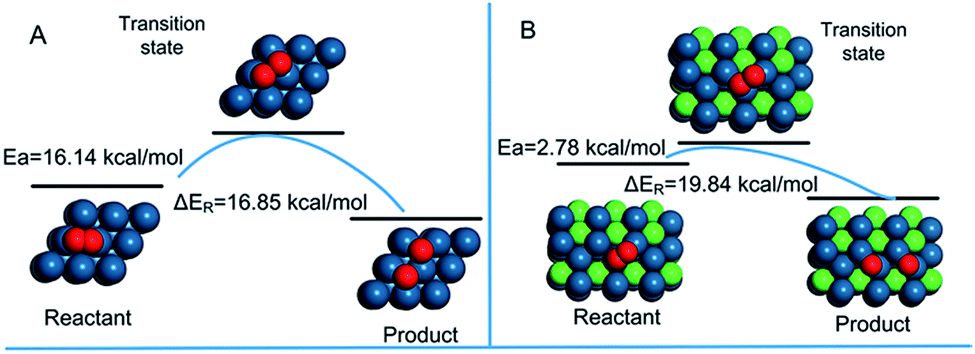 | ||
| Fig. 6 Reaction activation energy diagrams for the dissociation of O2 on (A) Pt (111) and (B) PNC-Pt3Ni (111). | ||
4. Conclusions
In summary, we have demonstrated the synthesis of PNC-Pt3Ni with enhanced activity and durability towards ORR under highly acidic circumstances. The particular morphology not only makes the utmost usage of Pt atoms but also ensures significant stability for long-term consideration. The half-wave potential of the as-prepared PNC-Pt3Ni is 60 mV more positive than that of Pt/C, and this is also verified by EIS results that show that less series resistance is detected for PNC-Pt3Ni/C when it shares the same amount of catalyst as Pt/C on the electrode. Thus, the large size characteristic helps to decrease the grain boundaries compared to small sizes. DFT calculations have shown that the activation energy of oxygen dissociation on the surface of PNC-Pt3Ni (111) is much lower than that on Pt/C (111), generating the same conclusion from LSV measurement results. In addition, after 10000 potential cycles, the mass activity of PNC-Pt3Ni/C maintains 76.3% of its original value, while Pt/C loses almost half of its initial activity. This work has demonstrated an effective way of simultaneously tackling electrochemical activity and durability issues by taking advantage of the large size property. It should be stated that this work shall be continued later for further exploration regarding the improvement of mass activity during the stability test, which may be due to the dissolution and redeposition of transition metal in the compound. It is also expected that the PNC-Pt3Ni/C catalysts will have great potential to be applied as enduring catalysts towards ORR.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21236003, 21322607, 21406072, 21522602, 91534202, and 91534122), the Basic Research Program of Shanghai (14JC1490700 and 15JC1401300), the Program for New Century Excellent Talents in University (NCET-13-0796), the Shanghai Shuguang Scholars Program (13SG31), the International Science and Technology Cooperation Program of China (2015DFA51220), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, the Research Project of Chinese Ministry of Education (113026A), the Shanghai Rising-Star (15QA1401200), and the Fundamental Research Funds for the Central Universities.References
- M. Winter and R. J. Brodd, ChemInform, 2004, 104, 4245 CAS.
- T. Fu, J. Fang, C. Wang and J. Zhao, J. Mater. Chem. A, 2016, 4, 8803–8811 CAS.
- R. Illathvalappil, V. M. Dhavale, S. N. Bhange and S. Kurungot, Nanoscale, 2017, 9, 9009–9017 RSC.
- M. Shao, Q. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- P. Mani, A. Ratndeep Srivastava and P. Strasser, J. Phys. Chem. C, 2008, 112, 2770–2778 CAS.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund and H. Ogasawara, Nat. Chem., 2010, 2, 454 CrossRef CAS PubMed.
- U.S. Department of Energy, Technical Plan: Fuel Cells, 2016, http://www.energy.gov/sites/prod/files/2016/06/f32/fcto_myrdd_fuel_cells_0.pdf.
- H. Zhang, M. Jin, J. Wang, W. Li, P. H. Camargo, M. J. Kim, D. Yang, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2011, 133, 6078–6089 CrossRef CAS PubMed.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- Y. Kang, J. B. Pyo, X. Ye, R. E. Diaz, T. R. Gordon, E. A. Stach and C. B. Murray, ACS Nano, 2013, 7, 645 CrossRef CAS PubMed.
- M. Takahashi, T. Imaoka, Y. Hongo and K. Yamamoto, Angew. Chem., Int. Ed. Engl., 2013, 52, 7419–7421 CrossRef CAS PubMed.
- D. Li, C. Wang, D. S. Strmcnik, D. V. Tripkovic, X. Sun, Y. Kang, M. Chi, J. D. Snyder, D. van der Vliet, Y. Tsai, V. R. Stamenkovic, S. Sun and N. M. Markovic, Energy Environ. Sci., 2014, 7, 4061–4069 CAS.
- S. Fu, C. Zhu, Q. Shi, D. Du and Y. Lin, Catal. Sci. Technol., 2016, 6, 5052–5059 CAS.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J. Y. Ma and D. Su, Science, 2016, 354, 1410 CrossRef CAS PubMed.
- X. X. Du, Y. He, X. X. Wang and J. N. Wang, Energy Environ. Sci., 2016, 9, 2623–2632 CAS.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732 CrossRef CAS PubMed.
- M. Schrinner, M. Ballauff, Y. Talmon, Y. Kauffmann, J. Thun, M. Mller and J. Breu, Science, 2009, 323, 617 CrossRef CAS PubMed.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 38, 220 CrossRef PubMed.
- G. Zhang, W. Lu, L. Cao, X. Qin, F. Ding, S. Tang, Z.-G. Shao and B. Yi, J. Power Sources, 2016, 326, 23–34 CrossRef CAS.
- T. Yu, D. Y. Kim, H. Zhang and Y. Xia, Angew. Chem., Int. Ed. Engl., 2011, 50, 2773–2777 CrossRef CAS PubMed.
- Y. Nie, S. Chen, W. Ding, X. Xie, Y. Zhang and Z. Wei, Chem. Commun., 2014, 50, 15431–15434 RSC.
- C. Galeano, J. C. Meier, V. Peinecke, H. Bongard, I. Katsounaros, A. A. Topalov, A. Lu, K. J. Mayrhofer and F. Schuth, J. Am. Chem. Soc., 2012, 134, 20457–20465 CrossRef CAS PubMed.
- L. Wang, S. Guo, J. Zhai and S. Dong, J. Phys. Chem. C, 2008, 112, 11372–11377 Search PubMed.
- W. Zhou, J. Wu and H. Yang, Nano Lett., 2013, 13, 2870–2874 CrossRef CAS PubMed.
- M. Zhou, H. Wang, M. Vara, Z. D. Hood, M. Luo, T.-H. Yang, S. Bao, M. Chi, P. Xiao, Y. Zhang and Y. Xia, J. Am. Chem. Soc., 2016, 138, 12263–12270 CrossRef CAS PubMed.
- H. Huang, K. Li, Z. Chen, L. Luo, Y. Gu, D. Zhang, C. Ma, R. Si, J. Yang, Z. Peng and J. Zeng, J. Am. Chem. Soc., 2017, 139, 8152–8159 CrossRef CAS PubMed.
- J. Zhu, M. Xiao, X. Zhao, C. Liu, J. Ge and W. Xing, Nano Energy, 2015, 13, 318–326 CrossRef CAS.
- G. Wang, L. Lei, J. Jiang, Y. Zhou, Q. Huang, Z. Zou, S. P. Jiang and H. Yang, Electrochim. Acta, 2017, 252, 541–548 CrossRef CAS.
- H. Huang, A. Ruditskiy, S. I. Choi, L. Zhang, J. Liu, Z. Ye and Y. Xia, ACS Appl. Mater. Interfaces, 2017, 9, 31203–31212 CAS.
- Q. Shi, C. Zhu, M. H. Engelhard, D. Du and Y. Lin, RSC Adv., 2017, 7, 6303–6308 RSC.
- N. Sui, K. Wang, X. Shan, Q. Bai, L. Wang, H. Xiao, M. Liu, V. L. Colvin and W. W. Yu, Dalton Trans., 2017, 46, 15541–15548 RSC.
- R. Xing, T. Zhou, Y. Zhou, R. Ma, Q. Liu, J. Luo and J. Wang, Nano-Micro Lett., 2018, 10, 3 CrossRef.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339 CrossRef CAS PubMed.
- X. Wang, A. Ruditskiy and Y. Xia, Natl. Sci. Rev., 2016, 3, 520–533 Search PubMed.
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C. Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov and Z. Lin, Science, 2016, 354, 1414 CrossRef CAS.
- C. Galeano, J. C. Meier, M. Soorholtz, H. Bongard, C. Baldizzone, K. J. J. Mayrhofer and F. Schüth, ACS Catal., 2014, 4, 3856–3868 CrossRef CAS.
- H. Xu, B. Yan, S. Li, J. Wang, C. Wang, J. Guo and Y. Du, Chem. Eng. J., 2017, 10, 175 Search PubMed.
- K. Y. Cho, Y. S. Yeom, H. Y. Seo, A. S. Lee, X. Huy Do, J. P. Hong, H.-K. Jeong, K.-Y. Baek and H. G. Yoon, Electrochim. Acta, 2017, 257, 412–422 CrossRef CAS.
- L. Guo, L.-B. Huang, W.-J. Jiang, Z.-D. Wei, L.-J. Wan and J.-S. Hu, J. Mater. Chem. A, 2017, 5, 9014–9021 CAS.
- Y. Jang, K. H. Choi, D. Y. Chung, J. E. Lee, N. Jung and Y. E. Sung, ChemSusChem, 2017, 10, 3063–3068 CrossRef CAS PubMed.
- J. Ding, L. Bu, S. Guo, Z. Zhao, E. Zhu, Y. Huang and X. Huang, Nano Lett., 2016, 16, 2762–2767 CrossRef CAS PubMed.
- Y. Mizukoshi, E. Takagi, H. Okuno, R. Oshima, Y. Maeda and Y. Nagata, Ultrason. Sonochem., 2001, 8, 1–6 CrossRef CAS PubMed.
- L. Han, P. Wang, H. Liu, Q. Tan and J. Yang, J. Mater. Chem. A, 2016, 4, 18354–18365 CAS.
- C. W. Wang, S. Yang, W. Q. Fang, P. Liu, H. Zhao and H. G. Yang, Nano Lett., 2016, 16, 427–433 CrossRef CAS PubMed.
- Y. Shen, Y. Zhou, D. Wang, X. Wu, J. Li and J. Xi, Adv. Energy Mater., 2018, 8, 1701759 CrossRef.
- L.-Y. Jiang, X.-Y. Huang, A.-J. Wang, X.-S. Li, J. Yuan and J.-J. Feng, J. Mater. Chem. A, 2017, 5, 10554–10560 CAS.
- L. Ou and S. Chen, J. Phys. Chem. C, 2013, 117, 1342–1349 CAS.
- X. Zhang, Z. Lu, Z. Fu, Y. Tang, D. Ma and Z. Yang, J. Power Sources, 2015, 276, 222–229 CrossRef CAS.
- A. B. Anderson and T. V. Albu, J. Electrochem. Soc., 2000, 147, 4229–4238 CrossRef CAS.
- A. B. Anderson, J. Roques, S. Mukerjee, V. S. Murthi, N. M. Markovic and V. Stamenkovic, J. Phys. Chem. B, 2005, 109, 1198–1203 CrossRef CAS PubMed.
- G. B. Fisher and J. L. Gland, Surf. Sci., 1980, 94, 446–455 CrossRef CAS.
- J. L. Gland, B. A. Sexton and G. B. Fisher, Surf. Sci., 1980, 95, 587–602 CrossRef CAS.
- S. Lehwald, H. Ibach and H. Steininger, Surf. Sci., 1982, 117, 342–351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: DFT models and calculations, XRD and XPS patterns of PNC-Pt3Ni, morphology change with different doses of DTAC, structural analysis recorded under different precursor ratios, structure of Pt–Ni alloy with different ratios of solvent, TEM images of PNC-Pt3Ni/C and Pt/C, TEM images after 10k potential cycles, and the illustration of O2* adsorption. See DOI: 10.1039/c8ra02219d |
| This journal is © The Royal Society of Chemistry 2018 |