
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA fluorescent calixarene-based dimeric capsule constructed via a MII–terpyridine interaction: cage structure, inclusion properties and drug release†
Jun-Fang
Wang
a,
Li-Yuan
Huang
b,
Jian-Hua
Bu
*c,
Shao-Yong
Li
d,
Su
Qin
b,
Yao-Wei
Xu
*b,
Jun-Min
Liu
 *b and
Cheng-Yong
Su
b
*b and
Cheng-Yong
Su
b
aFu Xing Hospital, Capital Medical University, Beijing, 100038, China
bSchool of Materials Science and Engineering, School of Chemistry, Sun Yat-sen University, Guangzhou, 510275, China. E-mail: liujunm@mail.sysu.edu.cn; xuyaowei3@mail.sysu.edu.cn
cXi'an Modern Chemistry Research Institute, Xi'an, 710065, China. E-mail: waltzbu@iccas.ac.cn
dSchool of Pharmacy, Tianjin Medical University Tianjin, 300070, China
First published on 20th June 2018
Abstract
Two analogues of capsule-like fluorescent cages have been constructed by dimerization of terpyridine-containing calixarene derivatives utilizing a MII–terpyridine (M = Zn and Cd) interaction. 1H NMR spectral studies show that the self-assembled molecular capsules Zn4L12 and Cd4L12 have a highly symmetrical D4h-structure. The encapsulation of the anticancer drug mercaptopurine in their cavities has been documented by NMR, ESI-TOF-MS, fluorescence switching, and molecular simulation, indicating that strong S–π and π–π interactions between drug and cage are of importance for the host–guest binding. The nanoscale cages exhibit excellent behaviors to control the release of mercaptopurine in phosphate buffered saline solution (pH = 7.4). These results further highlight the potential of self-assembled Zn4L12 cages for drug-carrier applications.
Self-assembled coordination cages with well-defined geometries and cavities have attracted much attention due to their potential applications in molecular recognition, catalytic reactions, biochemistry, and medicine.1 Concerning medicinal studies, metallocages are promising drug delivery carriers, which can interact with biomolecules, possess anticancer activity, and increase solubility in biological media.2 Recent reports have demonstrated that coordination cages can act as effective hosts for medicinal guests and have interesting biological properties.3 For example, Therrien et al. reported the first coordination cage, an arene–ruthenium-based metallocage, used as drug-delivery system, which showed anticancer effects against human A2780 ovarian cancer cells upon encapsulation of Pd and Pt acetylacetonato complexes.3a Lippard et al. developed a new strategy to use a hexanuclear supramolecular PtII cage as a drug delivery vehicle to deliver PtIV prodrugs to cancer cells.3b Briken and Isaacs et al. described that metal–organic polyhedron capped with cucurbit[8]uril could deliver doxorubicin to cancer cells and enhance the cytotoxicity, which was ascribed to a combination of increased cellular uptake of cage and doxorubicin release.3c Su and Jiang et al. investigated the binding behavior between drug molecule 5-fluorouracil and M4L4 type tetrahedral cages, and demonstrated that the porous cage nanoparticle could control release of 5-fluorouracil in simulated human body liquid of phosphate buffer solution.3d Despite this growing interest on drug encapsulation into metallocages as drug-delivery systems, the research on the drug adsorption/release processes is still in its infancy, and further studies are necessary to explore their uses in medicinal chemistry. In addition, fluorescent coordination cages, especially for the cages that turn-on their fluorescence in response to external stimuli, are very attractive for both therapeutic and imaging applications in the medicinal chemistry field, because they allow in vitro or in vivo imaging without specific handling.4 Therefore, the design of self-assembled cages that ensure the fluorescence response upon the release of an active molecule is more desirable.
Calixarenes are an important class of macrocyclic host molecules in supramolecular chemistry. Calixarene derivatives have been used for the recognition of various molecular species, such as sugars, amino acids, peptides, proteins, and nucleic acids,5 which are basic substrates in biological processes, and also used in drug delivery investigations.6 In terms of the calixarene cavity and cage cavity to encapsulate the guests, calixarene-based coordination cages may be employed as drug delivery systems through hydrophobic effects and/or ion-dipole or hydrogen bonding interactions.7 Moreover, compared with smaller sized coordination complexes studied previously (<1.5 nm), the larger sized calixarene-based supramolecular cages should exhibit slower renal clearance and longer circulation, which would be helpful for theranostic applications, because particle size influences the clearance rate of nanoparticles from the bloodstream.8 Only a few reports on the biological properties of calixarene-based nanoscale cages (>2.0 nm) have appeared.7,9 Recently, Liao and Hu et al. synthesized an extra-large octahedral coordination cage based on Co4-p-tert-butylsulfonyl calix[4]arene. The calixarene-based cage demonstrated good adsorption properties towards a small drug molecule, ibuprofen (Ibu), and the Ibu release experiment revealed that the cage exhibited a slow drug release behavior.9 Despite the remarkable size and well-known inclusion properties of calixarene-based cages, studies of their applications in drug delivery are rare.
Therefore, in this work, in order to obtain fluorescent calixarene-based nanoscale cages, we designed two self-assembled nanocapsules Zn4L12 and Cd4L12 based on upper rim terkis-phenyl-terpyridine substituted calix[4]arene derivatives which showed fluorescence properties due to the coordination bond of terpyridine (tpy) and Zn2+/Cd2+ ions. In general, the connectivity of tpy–M(II)–tpy (M = Zn, Cd, Fe, and Ru) is fixed at 180° and thus, limits the use of metal ions as cornered directing units. Therefore, a few previous studies of tpy–M(II)–tpy only concentrated on 0-D and 3-D supramolecular cages and prisms10 compared to the numerous reports of linear and 2-D structures based on tpy.11 On the other hand, to evaluate the capability of these cages as drug carriers, mercaptopurine was chosen as the drug guest. Mercaptopurine is a medication used for cancer and autoimmune diseases,12 the interactions of which with cage have rarely been thorough studied in the field of host–guest chemistry.
Herein, two highly symmetrical molecular capsules were constructed by dimerization of tpy-containing calixarene derivatives utilizing a Zn(II)/Cd(II)-tpy interaction for the first time. The D4h-structures and binding property of two Zn4L12 and Cd4L12 cages with mercaptopurine drug were investigated by NMR spectra, ESI-TOF-MS, AFM, UV-Vis and fluorescent spectra, and DFT calculations. The releasing drug experiments were carried out, revealing that nanoscale Zn4L12 and Cd4L12 cages can significantly delay release of mercaptopurine into the simulated body liquids.
In order to acquire capsules with nanoscale inner space, an upper rim terkis-terpyridine substituted calix[4]arene (L1) was used for constructing the capsules. Ligand 1 could be obtained according to our report.13
The two nanocapsules Zn4L12(OTf)8 and Cd4L12(OTf)8 could be obtained as yellow precipitation by adding a THF solution of L1 to a THF solution of zinc(II) or cadmium(II) trifluoromethanesulfonate (OTf−) with ligand/metal molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (Scheme 1). The counter-ions could be exchanged by adding a methanol of ammonium hexafluorophosphate (PF6−) to the acetonitrile solution of Cd4L12 to give Cd4L12(PF6)8.
:2 (Scheme 1). The counter-ions could be exchanged by adding a methanol of ammonium hexafluorophosphate (PF6−) to the acetonitrile solution of Cd4L12 to give Cd4L12(PF6)8.
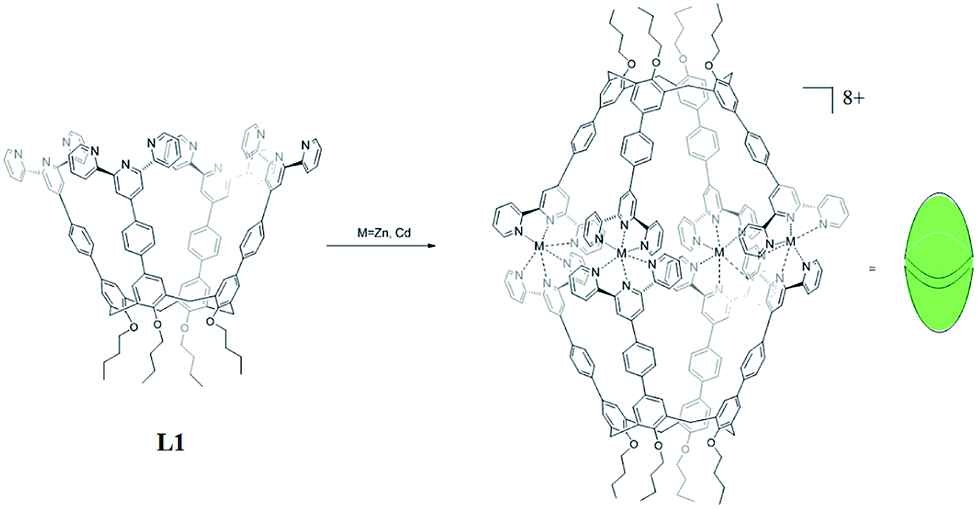 | ||
| Scheme 1 The synthesis of calix[4]arene based nanocapsules. | ||
All of the capsules were soluble in MeCN-d3, and then their NMR spectra were measured and showed in Fig. 1 and S1.† It could be seen that the signals in the 1H NMR spectra of Cd-capsules were well resolved, while the NMR spectra of Zn-capsules exhibited broad 1H signals. The 1H NMR of Cd4L12(PF6)8 (Fig. 1) showed the expected peaks of a tpy–metal complex. In the aromatic region, there were five sets of aromatic protons from tpy units, two sets from phenyl groups, and a single peak from calixarene, which were in agreement with the desired structure. The protons at 4,4′′, 5,5′′, and 6,6′′-position of tpy dramatically shifted upfield because the formation of complex led to the electron shielding effect. The 13C NMR of Zn4L12 and Cd4L12 showed only one series of clear and sharp peaks owing to the uniform and symmetrical architecture (Fig. S2†), implying no by-products and uncomplexed tpy moieties existed. At the same time, their DOSY spectra (Fig. 2 and S3†) confirmed only one species in the solution. The logD = −9.3 and −9.2 were observed and then diameter of 2.5 nm and 2.0 nm were determined in DOSY for Cd4L12 and Zn4L12, respectively, consistent with the following molecular modelling results. Cd4L12(PF6)8 and Zn4L12(OTf)8 were further characterized by ESI-MS to determine the proposed structures, which have a molecular weight of 5366.0 Da and 5202.2 Da, respectively. It was found that a series of peaks at m/z 2538.0, 1643.7, 1196.5, 928.2, 749.3 and 621.6 with charge states from 2+ to 7+ were detected for Cd4L12(PF6)8, which could be ascribed to the loss of a different number of counterion PF6− (Fig. 3). As shown in Fig. S4,† the isotope pattern of each peak of Cd4L12(PF6)8 was in good accordance with the corresponding simulated isotope distribution. Similarly, the peak series at m/z 2456.1, 1587.7, 1153.5, 893.0, 719.4 and 595.2 for [Zn4L12(OTf−)8−n]n+ (n = 2–7) verify formation of the same dimer cage (Fig. S5†).
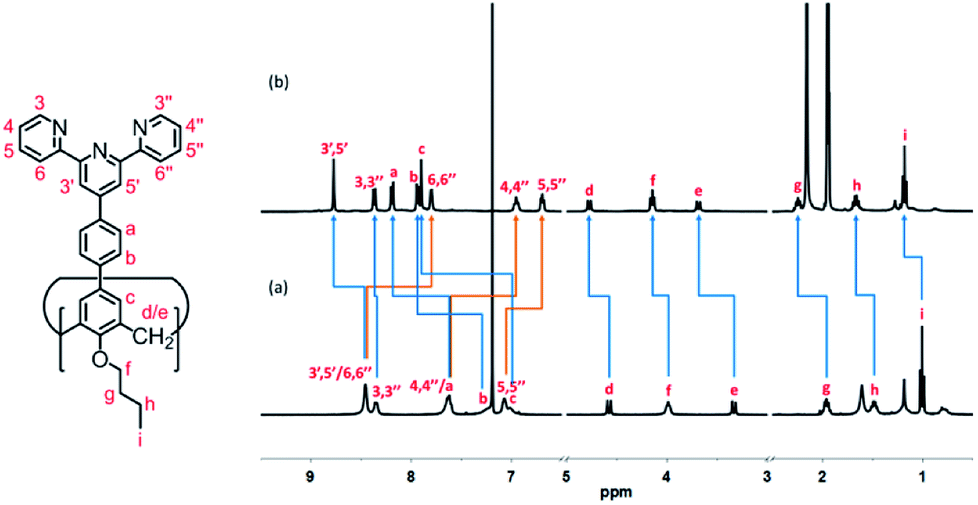 | ||
| Fig. 1 1H NMR spectra (400 MHz, 298 K) of (a) ligand L1 (CDCl3) and (b) Cd4(L1)2(PF6)8 MOC (CD3CN). | ||
 | ||
| Fig. 2 1H DOSY spectrum (400 MHz, CD3CN, 298 K) of Cd4(L1)2(PF6)8 MOC. | ||
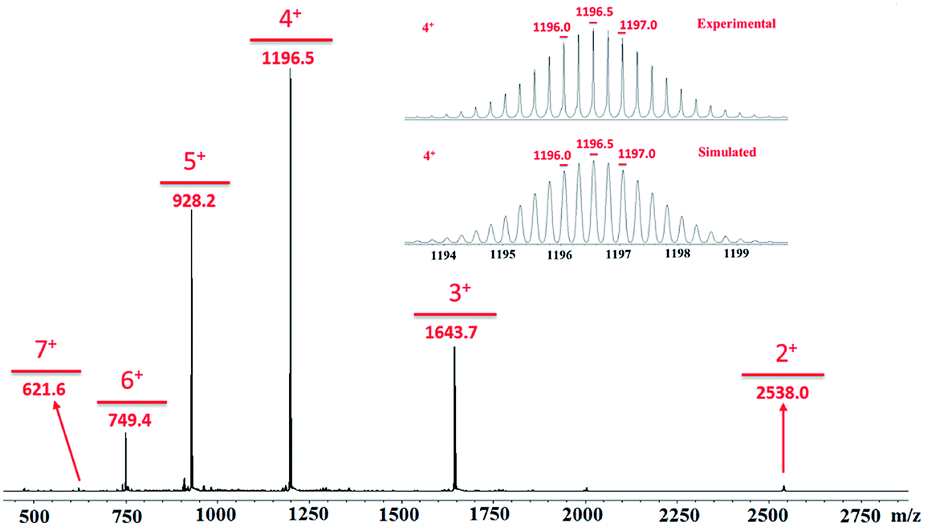 | ||
| Fig. 3 ESI-MS spectrum of Cd4(L1)2(PF6)8 MOC. | ||
Several attempts to obtain diffraction quality single crystals of Zn4L12 were unsuccessful. To incur further insight into the structural features, we obtained energy optimized structure of Zn4L12 using DFT (B3LYP, LANL2DZ) calculations. As illustrated in Fig. 4a and Table S1,† a cage in an approximate square bipyramid was constructed when the upper rims of two calix[4]arenes were close to each other and captured four Zn2+ ions. Six coordination bonds existed between each Zn2+ ion and each two terpyridine groups from two individual calix[4]arene arms. Four Zn2+ complex units were tightly interlocked into an approximate square through four π–π interactions between terpyridine groups of each adjacent Zn2+ complex unit. In fact, L1 was not favored for a dimer capsule and thus there was tension in the dimer capsules. Molecular modelling showed the structure distortion of the dimer was mostly neutralized by bending the phenyl group between calixarene and terpyridine. Therefore, the ligand bending was the major contribution to accommodate the strain. As additional evidence, the images from AFM showed the morphology of the dimer cage Zn4L12 as cone-shape dots on the mica surface (Fig. S6†). The measured height of these dots exhibited two different values: 3.9 ± 0.2 nm and 2.2 ± 0.3 nm, which were larger than cage height (3.1 nm) and length (2.0 nm) shown in molecular modelling, respectively, due to the unavoidable tip broadening effect.
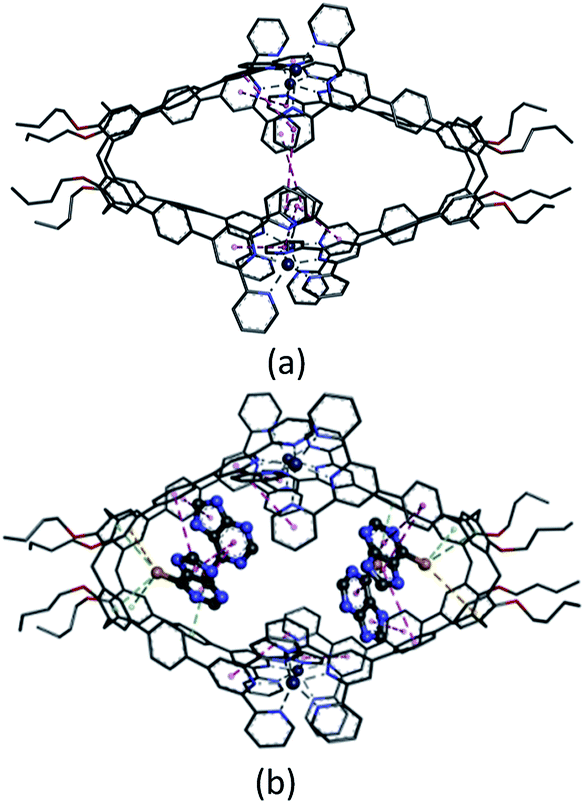 | ||
| Fig. 4 The optimized structures of Zn4L12 (a) and 4(mercaptopurine)@Zn4L12 (b). The interactions are colored in dotted lines: coordination bond in gray, S–π interaction in green, S–H–π interaction in yellow and π–π interaction in pink. All hydrogens are omitted for clarity. | ||
The host–guest properties of the dimer capsules and mercaptopurine were evaluated in solution. After the dimer capsules were dissolved in an aqueous acetonitrile solution (MeCN/H2O = 2/1), excessive mercaptopurine solids were added and stirred at room temperature for 2 h, and then the filtrate was separated for NMR and ESI-TOF-MS measurement. Considering the Cd-capsules had well-resolved signals and similar inclusion ability to Zn-capsules, Cd4L12 were substituted for Zn4L12 to study the binding interaction between the dimer capsules and mercaptopurine by 1H NMR (Fig. S7†) and 1H DOSY spectra (Fig. S8†). Mercaptopurine guests were observed to interact with cage Cd4L12 and were in slow exchange between cavity and bulk solution on the NMR time scale. Two proton signals of the mercaptopurine guests experienced an upfield shift when compared with their free 1H resonances measured in the solvent mixture of MeCN-d3 and D2O (2:1 v/v). This provided strong evidence for mercaptopurine binding within the cavities of the cages. Meanwhile, 1H NMR peaks of cage host corresponding to 3,3′′, 4,4′′, 5,5′′ and 3′,5′-H were observed to shift upfield, whereas 6,6′′-H shifted downfield. 1H DOSY measurements gave similar diffusion coefficients for 1H signals of host and guest, which further supported the formation of an inclusion complex. Integration of the guest peaks indicated the cages could accommodate about four mercaptopurine guests (Fig. S8†). These observations were consistent with what has been observed in other cases of hydrophobic guest binding in water.14 In addition, we tried to obtain further information on the stoichiometry of the host–guest interaction from ESI-MS experiments of the host–guest mixtures. As seen in Fig. S9 and S10,† the MS signals showed a series of binding species between Cd4L12/Zn4L12 and mercaptopurine with a general formula of [n(mercaptopurine)@Cd4L12/Zn4L12] (n ≤ 7), which exhibited higher stoichiometries than that observed for 1H NMR measurements (n ≈ 4). This suggested that the host–guest interaction of mercaptopurine and cage was strong. In addition to observing tightly bound mercaptopurine guests, the presumably weaker association of mercaptopurine with the exohedral binding sites on the exterior surface of the dimer cages could be detected because in the gas phase charged metallocages might produce ions related to extracage association of guests to the metal ions under the conditions of the mass spectral analysis. Similar mass spectra results were also observed in the reported literature.15 The control experiments of association of mercaptopurine with Zn(II)(phenyl-tpy)2(CF3SO3)2 and 25,26,27,28-tetrabutoxycalix[4] arene have been performed. As seen from Fig. S11,† no obvious shifts were observed in 1H NMR spectra of the mixture, compared with that of 25,26,27,28-tetrabutoxycalix[4]arene, mercaptopurine, and Zn(II)(phenyl-tpy)2(CF3SO3)2, respectively, supporting encapsulation of the guest molecules inside of the Zn4L12 cage.
From 1H NMR spectra (Fig. S8†), we speculated four mercaptopurine molecules could be encapsulated into the Zn4L12 cage. To further clarify stoichiometry of the mercaptopurine@Zn4L12 complex and explore their interaction modes, molecular simulation was executed in this drug-cage supramolecular system of mercaptopurine@Zn4L12 and the results of their geometry optimization were shown in Fig. 4b and Tables S2 and S3.† As illustrated in Fig. 4b, each pyramid unit encapsulated two mercaptopurine molecules. In one pyramid unit, one mercaptopurine orientated to cavity was captured by calix[4]arene cavity and two arms of the cage with three S–π interactions (3.93, 4.08 and 4.37 Å), one S–H–π interaction (4.37 Å) and one π–π interaction (4.55 Å). The other mercaptopurine was caught by the adjacent mercaptopurine and two arms of the cage with one S–π interactions (4.08 Å) and three π–π interactions (4.08, 4.47 and 4.76 Å). In another pyramid unit, there were similar interactions between the two encapsulated mercaptopurine molecules and the cage. There are two S–π interactions (3.95 and 4.25 Å), one S–H–π interaction (4.25 Å) and three π–π interactions (4.20, 4.55 and 4.58 Å) between the cage and the one mercaptopurine deepened into calix[4]arene cavity. And there are one S–π interactions (4.12 Å) and three π–π interactions (4.20, 4.27 and 4.52 Å) between the cage and the other mercaptopurine caught by the neighboring mercaptopurine. No coordination interaction appeared between mercaptopurine and Zn2+. Correspondingly, the symmetric geometrical structure of the cage was remarkably distorted in order to match the interactions with mercaptopurine. Moreover, the π–π interactions between adjacent terpyridine groups partially disappeared although the coordination interactions between terpyridine groups and Zn2+ still existed. Meanwhile, the stabilization energy −45.3 kcal mol−1 (see Table S3†) of 4(mercaptopurine)@Zn4L12 complex also confirmed the fact that four mercaptopurines can be stably captured by the cage.
It is well known that UV/Vis and fluorescent spectra are powerful methods for host–guest inclusion study. Therefore, the UV/Vis and fluorescent spectra of the Zn-capsules upon the addition of mercaptopurine were measured. In the absorption spectra, the titration of Zn4L12 with mercaptopurine resulted in the decrease of the absorption intensity at 256 nm and 366 nm, respectively, and increase of the intensity at 236 nm and 328 nm with a blue-shift, respectively, while the three isosbestic points at 247 nm, 285 nm, and 350 nm appeared, indicating complex formation (Fig. 5a). Interestingly, the Zn4L12 cage exhibited excellent fluorescent properties. In the fluorescent spectrum of Zn4L12, the maximum excitation and emission wavelengths were observed at 340 and 600 nm, respectively. The fluorescence titration of Zn4L12 with mercaptopurine showed a gradual decrease in the emission band at 600 nm with a slight blue-shift within 5 nm over mercaptopurine concentrations range of 6 × 10−4 to 7 × 10−2 mM, which confirmed the interaction between Zn4L12 and mercaptopurine further (Fig. 5b). Moreover, the interaction provided a fluorescence quenching pathway by the excitation energy transfer or charge transfer from Zn4L12 to the mercaptopurine guests. It is desirable that the Zn4L12 cages can make a fluorescence turn-on response upon the release of mercaptopurine.
 | ||
| Fig. 5 (a) UV-Vis and (b) fluorescent titrations of Zn4L12 (1.23 × 10−6 mol L−1) with mercaptopurine in MeCN and H2O (2:1 v/v). From a to u: 0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5 equiv.; from a to h: 0, 0.5, 1.0, 1.5, 2.0, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0, 15.0, 19.0, 23.0, 28.0, 38.0, 48.0, 58.0 equiv. | ||
Since Zn4L12 has lower toxicity in comparison to Cd4L12, Zn4L12 was chosen as candidate to test its drug delivery property. It is known that drug-carrying materials with the nanometer size can achieve effective drug delivery. Nanoscale mercaptopurine@Zn4L12 cages have been prepared by the method described above, and the loading amount was determined by 1H NMR. As seen from Fig. S12,† the loading amount of mercaptopurine encapsulated in Zn4L12 cages was estimated to be 13.0 ± 0.2 wt%, corresponding to [4(mercaptopurine)@Zn4L12].
To simulate drug sustained-release in human body liquid, the release of mercaptopurine from the mercaptopurine@Zn4L12 nanoparticles was performed in phosphate buffer solution (PBS, pH = 7.4) with dialysis bag and detected by UV/Vis spectrum. The release amount was calculated according to the calibration plots of standard curve of pure mercaptopurine in PBS. The mercaptopurine-loaded Zn4L12 samples used for the release experiments weighed 10.0 mg and 0.61 mg of Ibu was released from the sample. The release process was considered in two stages within 45 hours (Fig. 6). In the first stage (8 h), 0.35 mg of Ibu was released in the first stage and in the second stage 0.26 mg in the next 37 hours. The drug release became very slow after 45 hours. These release behaviors were similar to those for the reported coordination cages.9,16 For comparison, the pure mercaptopurine in solid state was dialyzed as a control-experiment which indicated that up to 95% of the total mercaptopurine was quickly released within 3 hours. These results suggested that the cage structures of Zn4L12 protracted the release of mercaptopurine. The slow release may be due to the slow diffusion rate of mercaptopurine from the windows of the cavities owing to the strong interaction of S–π and the π–π interactions between mercaptopurine and the aromatic skeleton of Zn4L12.
 | ||
| Fig. 6 The release of mercaptopurine from control (red circle) and mercaptopurine@Zn4L12 (black square). | ||
Conclusions
In conclusion, we have designed a fluorescent system, in which two capsule-like highly symmetrical cage (Zn4L12 and Cd4L12) based on calixarene were almost quantitatively constructed in a self-assembled manner by a terpyridine–MII–terpyridine (M = Zn and Cd) interaction. The nanoscale cages could encapsulate the anticancer drug mercaptopurine, as shown by NMR spectroscopy, AFM and ESI-TOF-MS. UV/Vis and fluorescent spectra confirmed the binding interaction and fluorescence switch behaviors between Zn4L12 and mercaptopurine further. Moreover, the theoretical simulation analysis verified the S–π and π–π interactions between the cage and mercaptopurine guests, in agreement with the spectroscopic results. Drug release experiment in PBS buffer solution demonstrated that the release process of drug molecules was able to last for 45 hours, effectively retarding burst release of mercaptopurine, which make these cages suitable candidates as drug carriers.Conflicts of interest
We have no competing interests.Ethics
An ethical assessment is not required for our research.Funding
Financial support came from the National Natural Science Foundation Project of China (grant no. 21572280), Science and Technology Plan Project of Guangzhou (grant no. 201707010114), and Fundamental Research Funds for the Central Universities (17lgzd01).Authors' contributions
J. M. L., J. F. W. and J. H. B. designed the study. J. F. W. and L. Y. H. prepared all samples for analysis. J. F. W., S. Y. L. and S. Q. collected and analysed the data. J. M. L., Y. W. X., and C. Y. S. interpreted the results and wrote the manuscript.Acknowledgements
We thank financial support from the National Natural Science Foundation of China and Guangzhou Science Technology and Innovation Commission.References
- (a) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS; (b) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; (c) S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS; (e) T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464–2474 CrossRef CAS.
- (a) A. Schmidt, V. Molano, M. Hollering, A. Pöthig, A. Casini and F. E. Kühn, Chem.–Eur. J., 2016, 22, 2253–2256 CrossRef CAS; (b) W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 625–631 RSC; (c) A. Mishra, S. Chang Lee, N. Kaushik, T. R. Cook, E. H. Choi, N. Kumar Kaushik, P. J. Stang and K.-W. Chi, Chem.–Eur. J., 2014, 20, 14410–14420 CrossRef CAS PubMed; (d) A. Mishra, Y. J. Jeong, J.-H. Jo, S. C. Kang, M. S. Lah and K.-W. Chi, ChemBioChem, 2014, 15, 695–700 CrossRef CAS PubMed; (e) J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC; (f) H. Ahmad, D. Ghosh and J. A. Thomas, Chem. Commun., 2014, 50, 3859–3861 RSC; (g) A. Pitto-Barry, N. P. E. Barry, O. Zava, R. Deschenaux, P. J. Dyson and B. Therrien, Chem.–Eur. J., 2011, 17, 1966–1971 CrossRef CAS; (h) F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS.
- (a) B. Therrien, G. Sìss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem. Int. Ed., 2008, 47, 3773–3776 ( Angew. Chem. , 2008 , 120 , 3833–3836 ) CrossRef CAS; (b) Y.-R. Zheng, K. Suntharalingam, T. C. Johnstone and S. J. Lippard, Chem. Sci., 2015, 6, 1189–1193 RSC; (c) S. K. Samanta, D. Moncelet, V. Briken and L. Isaacs, J. Am. Chem. Soc., 2016, 138, 14488–14496 CrossRef CAS; (d) W.-Q. Xu, Y.-Z. Fan, H.-P. Wang, J. Teng, Y.-H. Li, C.-X. Chen, D. Fenske, J.-J. Jiang and C.-Y. Su, Chem.–Eur. J., 2017, 23, 3542–3547 CrossRef CAS.
- (a) Y. Niko, Y. Arntz, Y. Mely, G. Konishi and A. S. Klymchenko, Chem.–Eur. J., 2014, 20, 16473–16477 CrossRef CAS; (b) A. Schmidt, M. Hollering, M. Drees, A. Casini and F. E. Kühn, Dalton Trans., 2016, 45, 8556–8565 RSC; (c) M. Wenzel, A. de Almeida, E. Bigaeva, P. Kavanagh, M. Picquet, P. Le Gendre, E. Bodio and A. Casini, Inorg. Chem., 2016, 55, 2544–2557 CrossRef CAS; (d) D. C. Crans, E. Nordlander, B. Bertrand, A. de Almeida, E. P. M. van der Burgt, M. Picquet, A. Citta, A. Folda, M. P. Rigobello, P. Le Gendre, E. Bodio and A. Casini, Eur. J. Inorg. Chem., 2014, 4532–4536 Search PubMed.
- (a) S. B. Nimse and T. Kim, Chem. Soc. Rev., 2013, 42, 366–386 RSC; (b) B. S. Creaven, D. F. Donlon and J. McGinley, Coord. Chem. Rev., 2009, 253, 893–962 CrossRef CAS; (c) X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- (a) J. Tian, P. K. Thallapally, S. J. Dalgarno, P. B. McGrail and J. L. Atwood, Angew. Chem. Int. Ed., 2009, 48, 5492–5495 ( Angew. Chem. , 2009 , 121 , 5600–5603 ) CrossRef CAS; (b) S. Alavi, T. K. Woo, A. Sirjoosingh, S. Lang, I. Moudrakovski and J. A. Ripmeester, Chem.–Eur. J., 2010, 16, 11689–11696 CrossRef CAS.
- L. Wang, L. L. Li, Y. S. Fan and H. Wang, Adv. Mater., 2013, 25, 3888–3898 CrossRef CAS.
- M. Elsabahy, G. S. Heo, S.-M. Lim, G. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS.
- S. Du, T.-Q. Yu, W. Liao and C. Hu, Dalton Trans., 2015, 44, 14394–14402 RSC.
- (a) C. Wang, X.-Q. Hao, M. Wang, C. Guo, B. Xu, E. N. Tan, Y.-Y. Zhang, Y. Yu, Z.-Y. Li, H.-B. Yang, M.-P. Song and X. Li, Chem. Sci., 2014, 5, 1221–1226 RSC; (b) T. Schröder, R. Brodbeck, M. C. Letzel, A. Mix, B. Schnatwinkel, M. Tonigold, D. Volkmer and J. Mattay, Tetrahedron Lett., 2008, 49, 5939–5942 CrossRef; (c) S. Cardona-Serra, E. Coronado, P. Gavina, J. Ponce and S. Tatay, Chem. Commun., 2011, 47, 8235–8237 RSC; (d) M. Schmittel and B. He, Chem. Commun., 2008, 4723–4725 RSC; (e) M. Schmittel, B. He and P. Mal, Org. Lett., 2008, 10, 2513–2516 CrossRef CAS.
- (a) A. Wild, A. Winter, F. Schlutter and U. S. Schubert, Chem. Soc. Rev., 2011, 40, 1459–1511 RSC; (b) U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892–2926 ( Angew. Chem. , 2002 , 114 , 3016–3050 ) CrossRef CAS; (c) H. Hofmeier and U. S. Schubert, Chem. Soc. Rev., 2004, 33, 373–399 RSC; (d) E. C. Constable, Chem. Soc. Rev., 2007, 36, 246–253 RSC; (e) E. C. Constable, Coord. Chem. Rev., 2008, 252, 842–855 CrossRef CAS; (f) S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555–1575 RSC; (g) M. C. Yeunga and V. W. Yam, Chem. Sci., 2013, 4, 2928–2935 RSC.
- S. Sahasranaman, D. Howard and S. Roy, Eur. J. Clin. Pharmacol., 2008, 64, 753–767 CrossRef CAS.
- J. Liu, M. Tonigold, B. Bredenkötter, T. Schröder, J. Mattay and D. Volkmer, Tetrahedron Lett., 2009, 50, 1303–1306 CrossRef CAS.
- (a) J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem. Int. Ed., 2013, 52, 7958–7962 ( Angew. Chem. , 2013 , 125 , 8116–8120 ) CrossRef CAS; (b) M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS; (c) L. Trembleau and J. Rebek, Science, 2003, 301, 1219–1220 CrossRef CAS; (d) J. M. Rivera, T. Martín and J. Rebek, Science, 1998, 279, 1021–1023 CrossRef CAS.
- T. Y. Kim, L. Digal, M. G. Gardiner, N. T. Lucas and J. D. Crowley, Chem.–Eur. J., 2017, 23, 15089–15097 CrossRef CAS PubMed.
- D. Zhao, S. W. Tan, D. Q. Yuan, W. G. Lu, Y. H. Rezenom, H. L. Jiang., L.-Q. Wang and H.-C. Zhou, Adv. Mater., 2011, 23, 90–93 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including synthesis, experimental procedure and supporting data. The datasets supporting this article have been uploaded as part of the supplementary material. See DOI: 10.1039/c8ra02146e |
| This journal is © The Royal Society of Chemistry 2018 |