DOI:
10.1039/C8RA03879A
(Paper)
RSC Adv., 2018,
8, 22515-22529
Synthesis, spectroscopic, thermal, antimicrobial and electrochemical characterization of some novel Ru(III), Pt(IV) and Ir(III) complexes of pipemidic acid
Received
7th May 2018
, Accepted 9th June 2018
First published on 20th June 2018
Abstract
Three new solid complexes of pipemidic acid (Pip–H) with Ru3+, Pt4+ and Ir3+ were synthesized and characterized. Pipemidic acid acts as a uni-dentate chelator through the nitrogen atom of the –NH piperazyl ring. The spectroscopic data revealed that the general formulas of Pip–H complexes are [M(L)n(Cl)x]·yH2O ((1) M = Ru3+, L: Pip–H, n = 3, x = 3, y = 6; (2) M = Pt4+, L: Pip–NH4, n = 2, x = 4, y = 0 and (3) M = Ir3+, L: Pip–H, n = 3, x = 3, y = 6). The number of water molecules with their locations inside or outside the coordination sphere were assigned via thermal analyses (TG, DTG). The DTG curves refer to 2–3 thermal decomposition steps where the first decomposition step at a lower temperature corresponds to the loss of uncoordinated water molecules followed by the decomposition of Pip–H molecules at higher temperatures. Thermodynamic parameters (E*, ΔS*, ΔH* and ΔG*) were calculated from the TG curves using Coats–Redfern and Horowitz–Metzeger non-isothermal models. X-ray powder diffraction (XRD), scanning electron microscopy (SEM) and transmission electron microscopy (TEM) techniques were carefully used to assign properly the particle sizes of the prepared Pip–H complexes. The biological enhancement of Pip–H complexes rather than free chelate were assessed in vitro against four kinds of bacteria G(+) (Staphylococcus epidermidis and Staphylococcus aureus) and G(−) (Klebsiella and Escherichia coli) as well as against the human breast cancer (MCF-7) tumor cell line.
1 Introduction
Pipemidic acid (Pip–H; Fig. 1) has the IUPAC formula 8-ethyl-5,8-dihydro-5-oxo-2-(1-piperazinyl)-pyrido[2,3-d]pyrimidine-6-carboxylic acid.1 Pip–H has medical efficacy in the treatment of urinary tract infections because of its antibacterial agency against Gram-negative and some Gram-positive bacteria.2 Concerning the Pip–H moiety, the –COOH group existing at the C6-position gives this drug an acidic nature, while the piperazine ring in the C2-position includes an –NH group that is basic in character. In aqueous media solution, 2-piperazinyl quinolone exists in three different species: acidic for pH values below pKa1 = 5.4, neutral for pH values close to the isoelectric point (pH = 6.8) and alkaline for pH values higher than pKa2 = 8.2.3 The Pip–H antibiotic drug is important as an antibacterial agent against Pseudomonas aeruginosa.4 It robustly damages DNA in the absence of an exogenous metabolizing system and it can act as a multi-dentate chelator, binding with metal ions.5
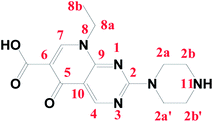 |
| | Fig. 1 Structure and numbering of the pipemidic acid (Pip–H) antibiotic drug. | |
Quinolone-metal chelates have the potential ability to interact with various metal ions and record biological activity towards different microorganisms.6,7 Quinolones can be act as uni-dentate, bi-dentate and bridging chelates. In general, quinolones are coordinated in bi-dentate fashion, and this takes place through one oxygen atom upon deprotonation of the –COOH group and carbonyl oxygen atom (Fig. 2a). Rarely, quinolones can act as bi-dentate chelators via two carboxyl oxygen atoms (Fig. 2b) or through both piperazinic nitrogen atoms (Fig. 2c). Quinolones can also form complexes via uni-dentate ligand binding to the metal ion through the terminal piperazinyl nitrogen (Fig. 2d).8
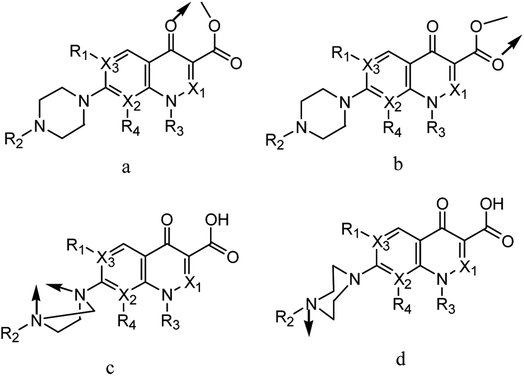 |
| | Fig. 2 (a–d) Chelation modes of quinolone drugs towards metal ions. | |
The medicinal usage of platinum(II,IV) compounds was limited by the development of tumor resistance to the drugs9 and by their side effects.10 These limitations caused further research to be oriented toward finding new, more active, less toxic, anti-tumor agents based on other chelates. Ruthenium(III) complexes had shown ligand exchange kinetics similar to those of the platinum anti-tumor agents currently used in the clinic and exhibited a reduced toxicity.11 The interactions of different metal ions with pipemidic acid as an antimicrobial agent have been discussed12 and can be referenced by chemical formulas with different molar ratios between Pip–H and some metal ions as follows (Table 1):
Table 1 Different Pip–H complexes with different metal ions, chemical formulas and molar ratios
| |
Molar ratio |
Formula |
Reference |
| Pip–H complexes |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
[Fe(PPA)(HO)2(H2O)] |
17–19 |
| 1:2 |
[VO(PPA)2(H2O)], [Mn(PPA)2(H2O)2], [Co(PPA)2(H2O)2], [Ni(PPA)2(H2O)2], [Zn(PPA)2(H2O)2], [MoO2(PPA)2], [Cd(PPA)2(H2O)2], [UO2(PPA)2], [Cu(PPA)2(H2O)] |
| 1:3 |
[Fe(PPA)3] |
Metal ions are considered essential to the human body, being an integral part of organic moieties in performing actions of physiological interest and having vital properties in the body.13 The synthesis and assignment of new metal complexes with quinolones are of wide importance for a better understanding of drug-metal interactions.14 Complexes of quinolone drugs and metal ions are synthesized with different chemical structures and different types of chelation,15,16 so, this article refers to the synthesis and spectroscopic characterizations of a new complexation mode between ruthenium(III), platinum(IV) and iridium(III) metal ions with the Pip–H drug as a 1st generation of quinolone antibacterial drugs. The new complexes were synthesized in CH3OH solvent with ratios of 1:3 and 1:2. These complexes have been interpreted using micro-analytical, FT-IR, conductance, magnetic, UV-vis, 1H-NMR and TG-DTG analyses. The morphological surface and particle size of these three new complexes were investigated using XRD, SEM and TEM.
2 Experimental
2.1 Chemical materials
Pipemidic acid (Pip–H) was purchased from the Aldrich-Sigma Chemical Company. RuCl3, H2PtCl6·6H2O and IrCl3·xH2O and solvents like CH3OH and dimethylsulfoxide (DMSO) were purchased from the Fluka Chemical Company and used without further purification. Tetrabutylammonium perchlorate (TBAP) (Sigma-Aldrich) was used as supporting electrolyte at various scan rates (50–2000 mV s−1).
2.2 Synthesis of pipemidic acid complexes
A hot methanolic solution (3 mmol, 20 mL) of pipemidic acid (91 mg) was added to an aqueous solution (20 mL) of RuCl3 (1 mmol, 20.8 mg) and IrCl3·xH2O (1 mmol, 29.9 mg). The reaction mixtures were refluxed for 1–2 h at ∼70–80 °C. The solutions were filtered off and left to slowly evaporate. After a day a black-dark green microcrystalline product was deposited, collected by filtration, washed with methanol and dried under vacuum. The same procedure was repeated for the platinum(IV) complex with a molar ratio 1:2 of H2PtCl6·6H2O (1 mmol, 51.8 mg) and Pip–H (2 mmol, 60.7 mg). However, then an aliquot of aqueous ammonia was added to Pip–H/H2PtCl6·6H2O and an ammonia-based complex was consequently formed.
[Ru(Pip–H)3(Cl)3]·6H2O complex (1). Yield: 96.15 mg, 86%. Anal calc. for complex (1) (C42H63Cl3N15O15Ru) (MW = 1225.47 g mol−1): calcd: C, 41.16; H, 5.18; N, 17.14; Ru, 8.25. Found: C, 41.04; H, 5.09; N, 17.12; Ru, 8.19%. The complex is black in color, soluble in (DMSO and DMF) and is a non-electrolyte (0.022 mS).
[Pt(Pip–NH4)2(Cl)4] complex (2). Yield: 90 mg, 80%. Anal calc. for complex (2) (C28H40Cl4N12O6Pt) (MW = 977.58 g mol−1): calcd: C, 34.40; H, 4.12; N, 17.19; Pt, 19.96. Found: C, 34.22; H, 4.06; N, 17.06; Pt, 19.90%. The complex is dark green, soluble in (DMSO and DMF) and is a non-electrolyte (0.042 mS).
[Ir(Pip–H)3(Cl)3]·6H2O complex (3). Yield: 100.35 mg, 83%. Anal calc. for complex (3) (C42H63Cl3IrN15O15) (MW = 1316.62 g mol−1): calcd: C, 38.31; H, 4.82; N, 15.96; Ir, 14.60. Found: C, 38.29; H, 4.57; N, 15.92; Ir, 14.55%. The complex is dark green in color, soluble in (DMSO and DMF) and is a non-electrolyte (0.011 mS).
2.3 Instrumentation and analytical measurements
The micro-analytical analyses of % C, % H and % N were made using a PerkinElmer CHN 2400 instrument (USA). Metal contents (%) were determined by conventional gravimetric analysis methods as the stable metal oxides MO2 formed. A Jenway 4010 conductivity meter was used for recording the molar conductivities of the complexes (1.0 × 10−3 mol cm−3) in DMSO. A UV2-Unicam UV/vis spectrophotometer fitted with a quartz cell of 1.0 cm path length was used for recording the electronic spectra of the prepared metal complexes. Infrared spectra with KBr discs were recorded on a Bruker FT-IR Spectrophotometer (4000–400 cm−1). Cyclic voltammetric measurements were carried out using an Autolab potentiostat PGSTAT 302 (Eco Chemie, Utrecht, The Netherlands) driven by General Purpose Electrochemical Systems data processing software (GPES, software version 4.9, Eco Chemie). A three-compartment electrochemical cell was used with Pt wires as the working and counter electrodes and Ag/AgCl as the reference electrode. The electrochemical experiments were performed in DMS–TBAP (tetrabutylammonium perchlorate). A Shimadzu thermogravimetric analyzer under N2 at 800 °C was used for recording thermal measurements (TG/DTG-50H) using a single loose top-loading platinum sample pan under nitrogen atmosphere at 30 mL min−1 flow rate and a 10 °C min−1 heating rate in the temperature range 25–800 °C. 1H-NMR spectra were recorded in DMSO solution on a Bruker 600 MHz spectrometer using TMS as the internal standard. A Jeol Jem-1200 EX II scanning electron microscope (SEM) at an acceleration voltage of 25 kV was used for recording the SEM images of the complexes. An X Pert Philips X-ray diffractometer was used for recording X-ray diffraction (XRD) patterns of the samples using CuKα1 radiation, with a graphite monochromator at 0.02° min−1 scanning rate. Transmission electron microscopy (TEM) images were performed on a JEOL 100 s microscope. The mass susceptibility (Xg) of the complexes was measured at room temperature using a Gouy magnetic balance. The effective magnetic moment (μeff) value was measured as reported earlier.17
2.4 Molecular modeling studies
The calculations of quantum chemical and molecular modeling studies for the Pip–H drug chelate are useful to support the suggested structures.18 The geometrical optimization and conformational analysis were performed using semi-empirical PM3 level19 as implemented in the software of the HyperChem 7.5 program20 (Fig. 3 and Table 2). A number of quantum chemical parameters have been estimated:18,19 the highest occupied molecular orbital energy (EHOMO), the lowest unoccupied molecular orbital energy (ELUMO), the difference between the HOMO and LUMO energy levels (ΔE), Mulliken electronegativity (χ), chemical potential (Pi), global hardness (η), global softness (S), global electrophilicity (ω), absolute softness (σ) and electronic charge (ΔNmax). The high value of the energy gap (ΔE) led to hard molecules as well as low reactivity, but, the low energy gap data led to soft molecules, with more reactivity and highly flexible transfer of electrons from the donor (ligand) to the acceptors (metal ions). Lower values of ELUMO gave high acceptability for electrons,19 and high values of EHOMO allowed electrons to be easily liberated. High values of EHOMO confirmed that the Pip–H ligand has powerful donation behavior. The difference between the energy gaps for the ligands reflects the presence of a complexation status. The global electrophilicity (ω) of Pip–H increases to that attributed to the highest capacity of accepted electrons. The calculation of both global hardness (η) and absolute softness (σ) parameters is useful for recognizing their molecular stability and reactivity. The negative data for both ELUMO and EHOMO were assigned to the stability of the synthetic complexes.
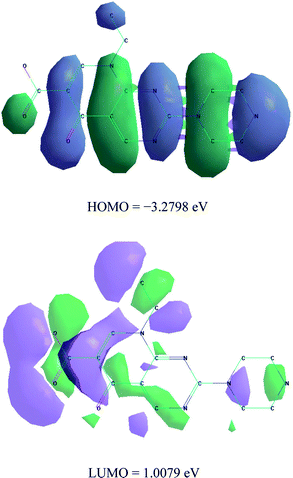 |
| | Fig. 3 HOMO and LUMO structure of Pip–H free drug. | |
Table 2 The quantum chemical parameters of Pip–H drug chelate
| Parameters |
Pip–H |
| Total energy (a.u) |
164.3 |
| Binding energy (a.u) |
282.4 |
| Heat formation (a.u) |
287.4 |
| Electronic energy (a.u) |
−948.1 |
| Dipole moment (debye) |
568.30 |
| EHOMO (eV) |
−3.279 |
| ELUMO (eV) |
1.0078 |
| ΔE (eV) |
4.2876 |
| χ (eV) |
1.1359 |
| η (eV) |
2.1438 |
| σ (eV) |
0.4665 |
| Pi (eV) |
−1.136 |
| S (eV) |
0.2332 |
| ω (eV) |
0.3009 |
| ΔNmax (eV) |
0.5299 |
2.5 Biological experiments
In accordance with Gupta et al.,21 the hole well method was applied. The investigated isolates of the bacteria were seeded in tubes with a nutrient broth (NB). The seeded NB (1 cm3) was homogenized in the tubes with 9 cm3 of melted (45 °C) nutrient agar (NA). The homogeneous suspensions were poured into Petri dishes. Holes (diameter 4 mm) were made in the cooled medium. After cooling in these holes, 2 × 10−3 dm3 of the investigated compounds were applied using a micropipette. After incubation for 24 h in a thermostat at 25–27 °C, the inhibition (sterile) zone diameters (including the disc) were measured and expressed in mm. An inhibition zone diameter over 7 mm indicates that the tested compound is active against the bacteria under investigation. The antibacterial activities of the investigated complexes were tested against bacteria G(+) (Staphylococcus epidermidis and Staphylococcus aureus) and G(−) (Klebsiella and Escherichia coli).
Mammalian cell lines: MCF-7 cells (human breast cancer cell line) were obtained from VACSERA Tissue Culture Unit. Chemicals used: dimethyl sulfoxide (DMSO), crystal violet and trypan blue dye were purchased from Sigma (St. Louis, Mo., USA). Fetal bovine serum, DMEM, RPMI-1640, HEPES buffer solution, L-glutamine, gentamycin and 0.25% trypsin–EDTA were purchased from Lonza. Crystal violet stain (1%): composed of 0.5% (w/v) crystal violet and 50% methanol made up to volume with ddH2O and filtered through a Whatman no. 1 filter paper.
The cells were propagated in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 1% L-glutamine, HEPES buffer and 50 μg ml−1 gentamycin. All cells were maintained at 37 °C in a humidified atmosphere with 5% CO2 and were subcultured twice a week. Cytotoxicity evaluation was undertaken using viability assay: for the cytotoxicity assay, the cells were seeded in a 96-well plate at a cell concentration of 1 × 104 cells per well in 100 μL of growth medium. Fresh medium containing different concentrations of the test sample was added after 24 h of seeding. Serial two-fold dilutions of the tested chemical compound were added to confluent cell monolayers, dispensed into 96-well, flat-bottomed microtiter plates (Falcon, NJ, USA) using a multichannel pipette. The microtiter plates were incubated at 37 °C in a humidified incubator with 5% CO2 for a period of 48 h. Three wells were used for each concentration of the test sample. Control cells were incubated without test sample and with or without DMSO. The small percentage of DMSO present in the wells (maximum 0.1%) was found not to affect the experiment. After incubation of the cells at 37 °C, various concentrations of sample were added, the incubation was continued for 24 h, and the yield of viable cells was determined by a colorimetric method. In brief, after the end of the incubation period, media were aspirated and the crystal violet solution (1%) was added to each well for at least 30 minutes. The stain was removed and the plates were rinsed using tap water until all the excess stain had been removed. Glacial acetic acid (30%) was then added to all the wells and mixed thoroughly, and then the absorbance of the plates was measured after gentle shaking on a microplate reader (TECAN, Inc.), using a test wavelength of 490 nm. All the results were corrected for background absorbance detected in wells without added stain. Treated samples were compared with the cell control in the absence of the tested compounds. All experiments were carried out in triplicate. The cell cytotoxic effect of each tested compound was calculated. The optical density was measured with a microplate reader (SunRise, TECAN, Inc, USA) to determine the number of viable cells and the percentage viability was calculated as [(ODt/ODc)] × 100% where ODt is the mean optical density of wells treated with the tested sample and ODc is the mean optical density of untreated cells. The relation between surviving cells and drug concentration is plotted to get the survival curve of each tumor cell line after treatment with the specified compound. The 50% inhibitory concentration (IC50), the concentration required to cause toxic effects in 50% of intact cells, was estimated from graphic plots of the dose response curve for each concentration using Graphpad Prism software (San Diego, CA. USA).22,23
3 Results and discussion
3.1 Micro-analytical and conductance measurements
This work aimed to synthesise, and make chemical and electrochemical characterizations of Ru(III), Pt(IV) and Ir(III) complexes with pipemidic acid as a first generation of quinolone drugs. The main target of this study was to identify the chelation mode under the influence of mixed solvent CH3OH/H2O (50/10 v/v). The percentages of essential elements (C, H, and N) are in agreement with the general formulas of three new Pip–H complexes as [M(L)n(Cl)x]·yH2O ((1) M = Ru3+, L: Pip–H, n = 3, x = 3, y = 6; (2) M = Pt4+, L: Pip–NH4, n = 2, x = 4, y = 0 and (3) M = Ir3+, L: Pip–H, n = 3, x = 3, y = 6) (Fig. 4). The Pip–H complexes are stable in air and have high melting points above 250 °C. The conductance data of complexes 1–3 is within the range for non-electrolytic nature (0.022–0.110 mS) with a low limit comparable with electrolyte materials as a ref. 24. From the conductance values it is deduced that the chlorine atoms are exhibited inside the chelation sphere.
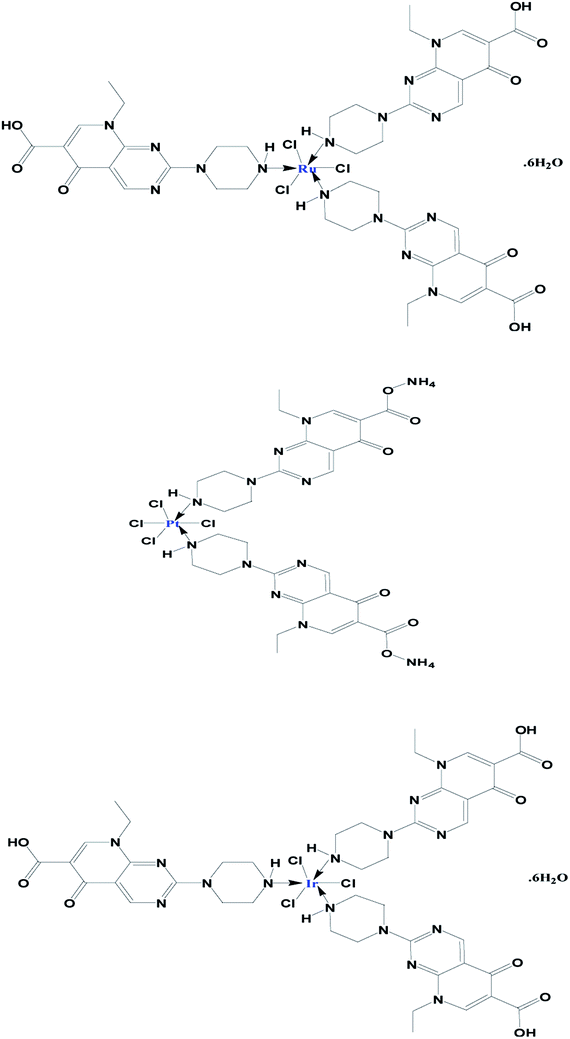 |
| | Fig. 4 Proposed chemical structures of the prepared Ru(III), Pt(IV) and Ir(III) Pip–H complexes 1–3. | |
3.2 Infrared spectra
The FT-IR spectra of Ru(III), Pt(IV) and Ir(III) Pip–H complexes 1–3 were compared with free Pip–H in order to discuss the coordination modes in three different Ru3+, Pt4+ and Ir3+ metal ions (Table 3). The FT-IR spectrum of free Pip–H drug does not have a characteristic stretching vibration band ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)COOH at wavenumber ∼1700 cm−1, because the carboxylic group is deprotonated and present in the zwitterionic state.25,26 The other distinguishable bands, which exist at 2971 and 1640 cm−1, can be assigned to the stretching vibrations of ν(N–H)piperazyl, and ν(CO)pyridine,25 respectively. An N–H bond is less polar than an O–H bond because nitrogen is less electronegative than oxygen. From this, the N–H stretch is less intense, and the peak narrower because the hydrogen bonding is not as strong. After chelation towards Ru3+, Pt4+ and Ir3+ metal ions (complexes 1–3), the ν(N–H)piperazyl band has disappeared or shifted to a lower wavenumber (∼2930 cm−1), the characteristic band at 1613–1624 cm−1 due to ν(CO)COOH has still not shifted and the stretching vibration band of ν(CO)pyridone has shifted to a higher wavenumber 1723–1731 cm−1 compared with free Pip–H (1640 cm−1). These assignments mean that the Pip–H drug acts as a uni-dentate chelator through the nitrogen atom of the piperazyl ring. The new absorption bands present within the range of 2720–2472 cm−1 are assigned to the stretching vibrations of crystallization water molecules under the effects of hydrogen bonds. Since the ν(O–H)COOH vibration changes little, it can be inferred that there is no coordination between the metal ions and the oxygen atoms. The O–H stretch of a carboxylic acid is also broadened by hydrogen bonding and it is associated with two characteristic infrared stretching absorptions which change markedly with hydrogen bonding. The assignments of the other stretching vibration motions ν(M–N) in Ru(III), Pt(IV) and Ir(III) Pip–H complexes 1–3 are observed at 500–400 cm−1.26
O)COOH at wavenumber ∼1700 cm−1, because the carboxylic group is deprotonated and present in the zwitterionic state.25,26 The other distinguishable bands, which exist at 2971 and 1640 cm−1, can be assigned to the stretching vibrations of ν(N–H)piperazyl, and ν(CO)pyridine,25 respectively. An N–H bond is less polar than an O–H bond because nitrogen is less electronegative than oxygen. From this, the N–H stretch is less intense, and the peak narrower because the hydrogen bonding is not as strong. After chelation towards Ru3+, Pt4+ and Ir3+ metal ions (complexes 1–3), the ν(N–H)piperazyl band has disappeared or shifted to a lower wavenumber (∼2930 cm−1), the characteristic band at 1613–1624 cm−1 due to ν(CO)COOH has still not shifted and the stretching vibration band of ν(CO)pyridone has shifted to a higher wavenumber 1723–1731 cm−1 compared with free Pip–H (1640 cm−1). These assignments mean that the Pip–H drug acts as a uni-dentate chelator through the nitrogen atom of the piperazyl ring. The new absorption bands present within the range of 2720–2472 cm−1 are assigned to the stretching vibrations of crystallization water molecules under the effects of hydrogen bonds. Since the ν(O–H)COOH vibration changes little, it can be inferred that there is no coordination between the metal ions and the oxygen atoms. The O–H stretch of a carboxylic acid is also broadened by hydrogen bonding and it is associated with two characteristic infrared stretching absorptions which change markedly with hydrogen bonding. The assignments of the other stretching vibration motions ν(M–N) in Ru(III), Pt(IV) and Ir(III) Pip–H complexes 1–3 are observed at 500–400 cm−1.26
Table 3 FT-IR assignments of Ru3+, Pt4+ and Ir3+–Pip–H complexes 1–3
| Compounds |
Assignments (cm−1) |
| ν(O–H)COOH |
ν(CO)COOH |
ν(CO)pyridone |
δ(C–H)aromatic |
ν(N–H)piperazyl |
| Pip–H |
3459 |
1617 |
1640 |
890 |
2971 |
| Complex 1 |
3430 |
1617 |
1728 |
912 |
2931 |
| Complex 2 |
3424 |
1613 |
1731 |
906 |
2939 |
| Complex 3 |
3426 |
1624 |
1723 |
918 |
— |
3.3 Magnetic measurements and electronic spectra
The electronic UV-vis. spectra of Pip–H free drugs and their Ru3+, Pt4+ and Ir3+ complexes 1–3 at a concentration of 10−3 M in DMF solvent in the 800–200 nm range were scanned. These spectra of three new Pip–H complexes have some distinguishable bands within two regions at 280–350 nm and 350–390 nm. The first range may be assigned to the π → π* electronic transition for the aromatic hydrocarbon rings and the second range includes some electronic absorption bands which may be attributed to the n → π* transition for the ketonic, –NH imine and carboxylic groups, respectively.27 In a solid sample, the electronic spectral values of the ruthenium(III) Pip–H complex are listed in Table 4. The ground state of Ru(III) is 2T2g and the first excited doublet levels in order of increasing energy are 2A2g and 2T1g, which are known to arise from the t2g 4e1g configuration.28 The Ru(III) complex shows three electronic transitions at 13568 cm−1, 19048 cm−1 and 22124 cm−1, which may be assigned to 2T2g → 4T1g (ν1), 2T2g → 4T2g (ν2), and 2T2g → 2A2g (ν3) in increasing order of energy. The interelectronic repulsion parameters (B, and C) and ligand field parameters (10Dq) parameters were calculated using the following equations:29
| 2T2g → 4T1g (ν1) = 10Dq − 5B − 4C, |
| 2T2g → 4T2g (ν2) = 10Dq + 3B − 4C, |
Table 4 Electronic spectral bands (cm−1) and ligand field parameters of the Pip–H complexes
| Complex |
λmax (cm−1) |
Assignments |
ν2/ν1 |
10Dq (cm−1) |
B (cm−1) |
C (cm−1) |
β |
| 1 |
13568 |
2T2g → 4T1g (ν1) |
1.40 |
25661 |
685 |
2167 |
1.08 |
| 19048 |
2T2g → 4T2g (ν2) |
| 22124 |
2T2g → 2A2g, 2T1g (ν3) |
| 2 |
30675 |
1A1g → 3T1g |
— |
— |
— |
— |
— |
| 28249 |
1A1g → 3T2g |
| 26738 |
1A1g → 1T1g |
| 25707 |
1A1g → 1T2g |
| 3 |
30675 |
1A1g → 1T1g (ν1) |
1.12 |
31567 |
223 |
892 |
0.34 |
| 34246 |
1A1g → 1T2g (ν2) |
The values of the ligand field parameters are comparable to those of different ruthenium(III) complexes reported in the literature.30 The Racah interelectronic repulsion parameter (B) observed for this complex is less than that for a Ru(III) ion (B′ = 630 cm−1).30 The 10Dq value is high and the nephelauxetic parameter 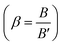 is ≤1.0. The decrease in β value is also associated with a reduction in the effective positive charge of the metal ion and with an increasing tendency to reduction to the lower oxidation state.31 These results suggest the presence of strong covalent bonding between the Pip–H donor and Ru3+ ions.30 The magnetic susceptibility of [Ru(Pip–H)3(Cl)3]·6H2O complex (1) was measured using a Gouy magnetic balance at room temperature. The ruthenium(III) complex being d5 (low spin), S = 1/2 has a paramagnetic character. The magnetic moment of the ruthenium(III) Pip–H complex is 1.90 B.M., which confirms the exhibition of a single unpaired electron in a low-spin 4 d5 configuration for a Ru3+ ion in an octahedral environment.32 This value is lower than the predicted value of 2.10 B.M. This lowering may be due to the presence of ligand fields of lower symmetry, or extensive electron delocalization in the species.33 Therefore, the magnetic moment value gives an impression that the ruthenium(III) complex has been found in the (+3) oxidation state.
is ≤1.0. The decrease in β value is also associated with a reduction in the effective positive charge of the metal ion and with an increasing tendency to reduction to the lower oxidation state.31 These results suggest the presence of strong covalent bonding between the Pip–H donor and Ru3+ ions.30 The magnetic susceptibility of [Ru(Pip–H)3(Cl)3]·6H2O complex (1) was measured using a Gouy magnetic balance at room temperature. The ruthenium(III) complex being d5 (low spin), S = 1/2 has a paramagnetic character. The magnetic moment of the ruthenium(III) Pip–H complex is 1.90 B.M., which confirms the exhibition of a single unpaired electron in a low-spin 4 d5 configuration for a Ru3+ ion in an octahedral environment.32 This value is lower than the predicted value of 2.10 B.M. This lowering may be due to the presence of ligand fields of lower symmetry, or extensive electron delocalization in the species.33 Therefore, the magnetic moment value gives an impression that the ruthenium(III) complex has been found in the (+3) oxidation state.
The electronic spectrum of [pt(Pip–NH4)2(Cl)4] complex (2) shows that charge transfer transitions may interfere and prevent the observation of all the expected bands.34 The distinct bands at 326 and 354 nm are assignable to a combination of metal ligand charge transfer (M → LCT) and d–d transition band. The other weak band at 389 nm is attributed to a combination of N → Pt(IV) metal charge transfer (Lπ → MCT) and d–d transition bands. The Pt(IV) complex 2 is found to be diamagnetic in character, so the platinum(IV) complex must have octahedral geometry. The Pt(IV) is a d6 system and four bands are expected due to 1A1g → 3T1g, 1A1g → 3T2g, 1A1g → 1T1g and 1A1g → 1T2g transitions. The shift to lower frequency after complexation is due to the binding between the Pt(IV) ion and the nitrogen atom of the piperazyl ring.
The iridium(III) complex 3 is diamagnetic in character. The UV-vis spectrum of [Ir(Pip–H)3(Cl)3]·6H2O shows bands in the range of 26525–30675 cm−1 and 34246–34602 cm−1. These two regions can be deduced from the ground state 1A1g and exist in the expected ranges35 for two spin-allowed transitions 1A1g → 1T1g (ν1) and 1A1g → 1T2g (ν2). The ratio ν2/ν1 in the Ir(III) complex 3 is 1.12, which is agreement with the octahedral geometry for iridium(III) complexes in the literature.36 The 1A1g → 1T1g (ν1) and 1A1g → 1T2g (ν2) transitions have been used to calculate the ligand field parameters 10Dq and nephelauxetic (B) based on the following equations:31
The outcome data of ν2/ν1, 10Dq, B, C and β parameters are in agreement with other iridium(III) complexes in previous studies.35,36 The B value is about 34% of the free iridium ion (660 cm−1) in the Ir(III) complexes, which refers to the overlap with significant covalency in the metal ligand σ-bond.
3.4 1H-NMR spectra
1H-NMR spectra of Pip–H and [pt(Pip–NH4)2(Cl)4] complex (2) have been recorded in d6-DMSO. The assignment of each signal has been achieved by comparing the 1H-NMR spectrum of the Pt(IV) complex (Fig. 5) to that of the Pip–H free drug ligand and these spectra have been reported in the literature.37 The assignable results are shown in Table 5. The proton-NMR spectrum of the platinum(IV) complex showed a set of signals which were almost identical to those of Pip–H, while shifts occurred particularly at the piperazinyl protons ((3.341(4H2a,2a′: piperazinyl group) and 4.060, 4.095(4H2b,2b′: piperazinyl group)) and the singlet proton of the –NH piperazinyl group (9.253(1H11: –NH piperazinyl group)). The singlet peak at δ = 7.345 ppm was assigned to eight protons of two –NH4 groups which existed instead of the deprotonated carboxylic group. The spectrum showed a singlet peak at δ = 4.347 ppm for –CH2 protons and a singlet at δ = 8.992 ppm for the two protons of the pyrimidine and quinolone moieties at positions H4 and H7 (Table 5). No broad weak band for the acidic proton at δ = 11.00 ppm can be seen in the spectrum of complex 2, indicating that this moiety is deprotonated by the –NH4 basic ion, so Pip–H acts as a uni-dentate deprotonated ligand bound to the target metal ion through the piperazinyl nitrogen and completed coordination sphere by chlorine atoms.38
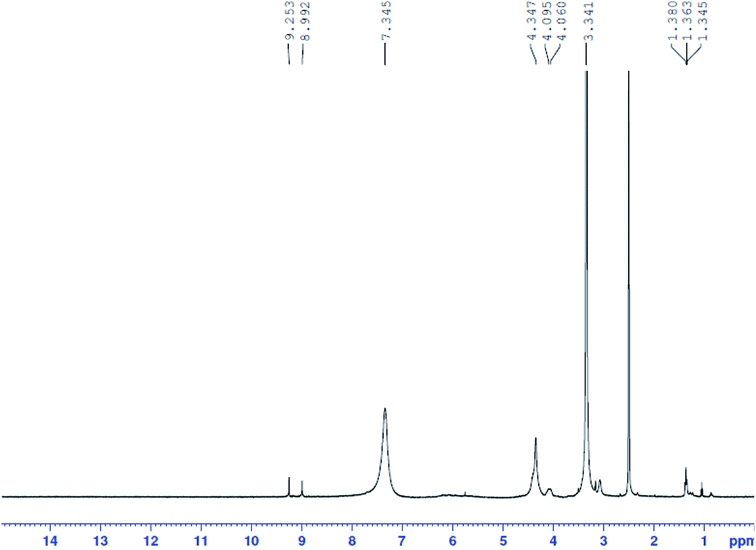 |
| | Fig. 5 1H-NMR spectrum of Pt(IV) complex 2. | |
Table 5 1H-NMR data of Pip–H and its Pt(IV) complex 2
| Compounds |
H-NMR (δ, ppm) |
| Pip–H |
1.36(3H8b: 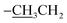 ), 3.14–3.25(4H2a,2a′: piperazinyl group), 3.86(4H2b,2b′: piperazinyl group), 4.39(2H8a: ), 3.14–3.25(4H2a,2a′: piperazinyl group), 3.86(4H2b,2b′: piperazinyl group), 4.39(2H8a: 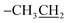 ), 8.90(2H4,7: pyrimidine and quinolone groups); 9.19(1H11: –NH piperazinyl group) ), 8.90(2H4,7: pyrimidine and quinolone groups); 9.19(1H11: –NH piperazinyl group) |
| Complex 2 |
1.363–1.380(3H8b: 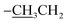 ), 3.341(4H2a,2a′: piperazinyl group), 4.060, 4.095(4H2b,2b′: piperazinyl group), 4.347(2H8a: ), 3.341(4H2a,2a′: piperazinyl group), 4.060, 4.095(4H2b,2b′: piperazinyl group), 4.347(2H8a: 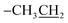 ), 8.992(2H4,7: pyrimidine and quinolone groups); 9.253(1H11: –NH piperazinyl group); 7.345(8H: 2NH4 group) ), 8.992(2H4,7: pyrimidine and quinolone groups); 9.253(1H11: –NH piperazinyl group); 7.345(8H: 2NH4 group) |
3.5 Thermal analyses and thermodynamic parameters
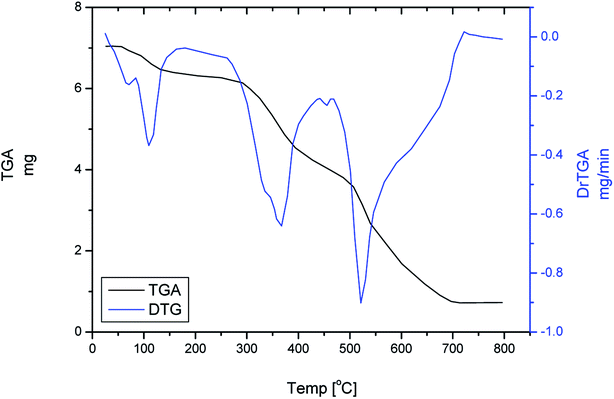
Thermogravimetric (TG) and differential thermogravimetric (DTG) analyses were carried out to identify the formulas of Ru(III), Pt(IV) and Ir(III) Pip–H complexes 1–3 reported in this study. TGA and DTG thermograms are performed under N2 flow, as shown in Fig. 6a–c. Table 6 includes the maximum temperature values, DTGmax °C−1, and the mass losses of the thermal decomposition steps for each synthetic complex. The thermal data led us to find the number of crystallized water molecules outside the coordination sphere for Pip–H complexes 1–3.
 |
| | Fig. 6 TGA and DTG of [Ru(Pip–H)3(Cl)3]·6H2O complex 1. | |
Table 6 Thermodynamic parameters of the thermal decomposition of Ru(III), Pt(IV) and Ir(III) Pip–H complexes 1–3
| Complex |
Stage |
Method |
Parameter |
r |
| E (J mol−1) |
A (s−1) |
ΔS (J mol−1 K−1) |
ΔH (J mol−1) |
ΔG (J mol−1) |
| 1 |
2nd |
CR |
7.35 × 104 |
3.24 × 103 |
−1.84 × 102 |
6.83 × 104 |
1.84 × 105 |
0.9804 |
| HM |
8.32 × 104 |
4.95 × 104 |
−1.61 × 102 |
7.79 × 104 |
1.80 × 105 |
0.9677 |
| 2 |
2nd |
CR |
1.56 × 105 |
3.74 × 1010 |
−4.88 × 101 |
1.51 × 105 |
1.82 × 105 |
0.9394 |
| HM |
1.56 × 105 |
6.81 × 1010 |
−4.39 × 101 |
1.51 × 105 |
1.79 × 105 |
0.9314 |
| 3 |
2nd |
CR |
3.18 × 104 |
3.62 × 10−1 |
−2.61 × 102 |
2.59 × 104 |
2.12 × 105 |
0.9781 |
| HM |
4.46 × 104 |
4.71 × 100 |
−2.39 × 102 |
3.86 × 104 |
2.10 × 105 |
0.9829 |
The thermal decomposition of [Ru(Pip–H)3(Cl)3]·6H2O complex (1) proceeds with approximately three main degradation steps (Fig. 6). The first stage occurs at a maximum temperature of 103 °C with weight loss of 9.863% (theoretical 8.810%) due to dehydrated 6H2O crystallization water molecules. Therefore, it could be associated with changes in the lattice structures. While the second and third stages occur at two different maximum temperatures of 345 and 554 °C. The weight loss associated with these stages is 80.584%, which is very close to the theoretical value of 81.637% corresponding to the loss of three Pip–H molecules. The final thermal product obtained at 800 °C is RuO (found 9.553%; theoretical 9.553%).
The thermal degradation of [pt(Pip–NH4)2(Cl)4] complex (2) takes place with three degradation stages. This complex is anhydrous, so the first stage of decomposition occurs at a high maximum temperature of 258 °C. Therefore, the complex starts to decompose at the first DTGmax and continuously decomposes through the second and third stages, with a total weight loss of 80.475%, corresponding to the loss of two Pip–NH4 molecules. Theoretically, the loss of these molecules corresponds to a weight loss of 80.045%. The weight found for the residue (Pt metal) after final decomposition is 19.525% (theoretical, 19.955%) giving an actual total weight loss in agreement with our calculated total weight loss value of 80.475%.
The thermal decomposition of [Ru(Pip–H)3(Cl)3]·6H2O complex (3) proceeds with three main degradation steps. The first stage of the decomposition occurs at a maximum temperature of 67 °C and is accompanied by a weight loss of 8.295% corresponding to the loss of the six uncoordinated water molecules. Theoretically, the loss of these molecules corresponds to a weight loss of 8.203%, which agrees with the experimental result. The second and third decomposition stages occur at maximum temperatures of 317 and 587 °C, respectively. The weight loss at these steps is 73.224% associated with the loss of three Pip–H molecules. The theoretical weight loss value is 74.767%. The weight found for the residue after decomposition is 18.481%, giving an actual total weight loss in agreement with our calculated total weight loss value of 17.030%. The final thermal decomposition product is IrO2.
Three different methods are used for the evaluation of the kinetic parameters: the Freeman and Carroll (FC) differential method,39 the Horowitz and Metzger (HM) approximation method40 and the Coats and Redfern (CR) integral method41. The kinetic parameters were evaluated by using two of these methods: the Horowitz and Metzger (HM) approximation method40 and the Coats and Redfern (CR) integral method,41 and the data are listed in Table 6. The satisfactory values of correlation coefficients (∼1) in all cases indicate good agreement with the experimental data and the values of the kinetic parameters are reasonable and in good agreement.
Table 6 can be used to make some points:
(i) The higher values of activation energies Pt(IV) (156 kJ mol−1) > Ru(III) (78.35 kJ mol−1) > Ir(III) (38.20 kJ mol−1) lead to the thermal stability of the Pip–H complexes.
(ii) The thermodynamic data resulting from the two methods are in harmony with each other.
(iii) The correlation coefficients (r) of the Arrhenius plots of the thermal decomposition steps were found to lie in the range 0.98–0.93, showing a good fit with the linear function.
(iv) It is clear that the process of thermal decomposition of the complexes is non-spontaneous (ΔS* = negative value); i.e., the complexes are thermally stable.
3.6 X-ray powder diffractions
The X-ray solid powder diffractions of [M(L)n(Cl)x]·yH2O ((1) M = Ru3+, L: Pip–H, n = 3, x = 3, y = 6; (2) M = Pt4+, L: Pip–NH4, n = 2, x = 4, y = 0 and (3) M = Ir3+, L: Pip–H, n = 3, x = 3, y = 6) complexes 1–3 are shown in Fig. 7. These diffraction patterns are semi-crystalline in nature. The particle size (D) of Pip–H complexes 1–3 was estimated from the major diffraction patterns of the respective complex using the Debye–Scherrer formula (eqn (1)):42| |
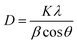 | (1) |
where λ is the wavelength of the X-ray (1.5418 Å) for Cu Kα radiation, K is a constant taken to be 0.94, β is the full width at half maximum (FWHM) of the prominent intensity peak (100% relative intensity peak), and θ is a peak position. The grain sizes using the Debye–Scherrer formula were found to be 28, 22 and 14 nm for Ru(III), Pt(IV) and Ir(III) complexes, respectively. The lower grain size can be discussed according to the increase in Pip–H chelates around the metal ions (ratio 1:3 or 1:2).43 The collected data of XRD such as 2θ, intensities and d-spacing are listed in Table 7. The dislocation density (δ) was evaluated from eqn (2)44 and is listed in Table 7; it indicates the formation of high-quality complexes.| |
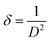 | (2) |
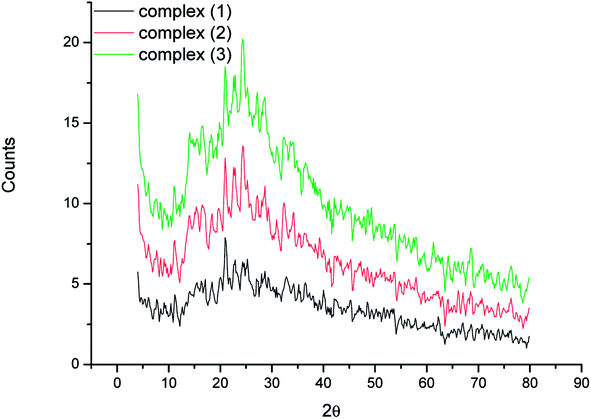 |
| | Fig. 7 X-ray powder diffraction patterns of [M(L)n(Cl)x]·yH2O ((1) M = Ru3+, L: Pip–H, n = 3, x = 3, y = 6; (2) M = Pt4+, L: Pip–NH4, n = 2, x = 4, y = 0 and (3) M = Ir3+, L: Pip–H, n = 3, x = 3, y = 6) complexes 1–3. | |
Table 7 The XRD collected data of crystallite sizes (D), dislocation density (δ), 2θ, intensities, and d-spacing
| Complex |
D (nm) |
δ (1012 lin m−2) |
2 theta |
Intensity |
d-Spacing |
| Complex 1 |
28 |
0.001 |
21 |
100 |
4.2088 |
| Complex 2 |
22 |
0.002 |
24 |
100 |
3.6395 |
| Complex 3 |
14 |
0.005 |
24 |
100 |
3.6395 |
The XRD of the Ru(III) complex has three significant peaks at 36.47, 45.35, and 48.41° due to the presence of the (002), (100), and (101) planes of ruthenium metal.45 The XRD patterns due to metallic platinum (JCPDS PDF card no. 04-0802)46 are present at 41.07, 51.16, and 66.00° for the (111), (200), and (220) planes, respectively. XRD patterns of the synthesized iridium metal in situ of the Ir(III) complex agreed well with standard card JCPDS no. 06-0598 for bulk iridium at 2θ = 41.75, 49.09, and 68.57°. The considerably broadened contours of the peaks resulted from the ultrasmall grain sizes of iridium metal.47
3.7 SEM and TEM morphology
The scanning electron microscope images are used here to investigate the surface morphology and particle size of the Pip–H complexes 1–3, revealing a crystalline, regular, and glassy structure. Layers in the micrograph reveal that the system contains atoms in a well-defined pattern; thus the reactants have reacted completely to form a clear homogenous compound. In general, the SEM photograph shows single phase formation with well-defined grain-like shape and particle size in the range of 1 mm to 20 μm. The SEM images of the Pip–H complexes 1–3 are depicted in Fig. 8a–c.
 |
| | Fig. 8 SEM images of [M(L)n(Cl)x]·yH2O ((A) M = Ru3+, L: Pip–H, n = 3, x = 3, y = 6; (B) M = Pt4+, L: Pip–NH4, n = 2, x = 4, y = 0 and (C) M = Ir3+, L: Pip–H, n = 3, x = 3, y = 6) complexes 1–3. | |
TEM photos of Pip–H complexes 1–3 (Fig. 9a–c) confirm the presence of spherical or semispherical NPs, which appear as dark spots.
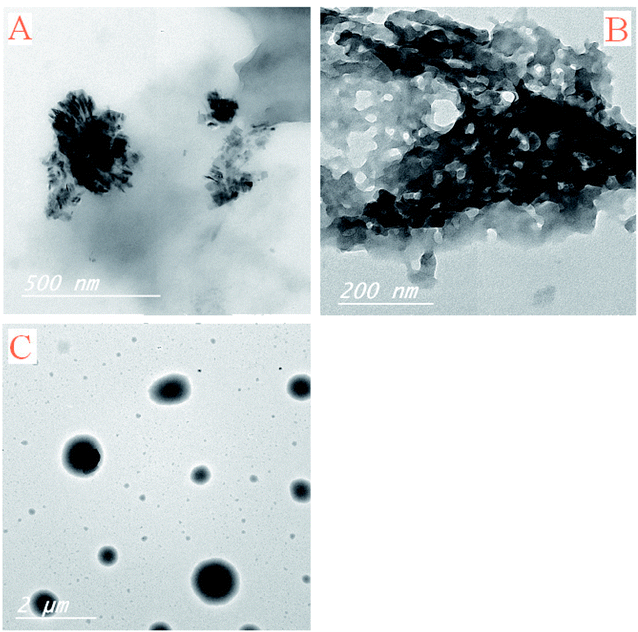 |
| | Fig. 9 TEM images of [M(L)n(Cl)x]·yH2O ((A) M = Ru3+, L: Pip–H, n = 3, x = 3, y = 6; (B) M = Pt4+, L: Pip–NH4, n = 2, x = 4, y = 0 and (C) M = Ir3+, L: Pip–H, n = 3, x = 3, y = 6) complexes 1–3. | |
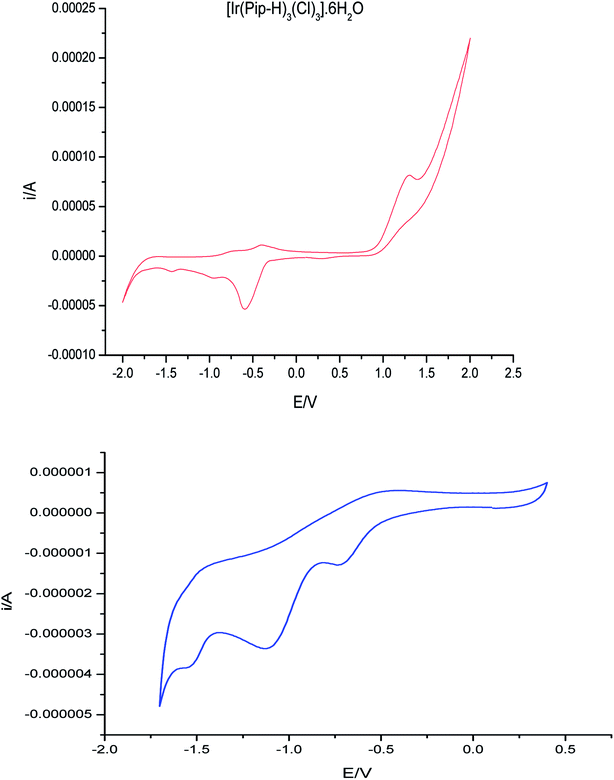
3.8 Redox behavior of [Ir(Pip–H)3(Cl)3]·6H2O
Cyclic voltammograms (CVs) of the complex [Ir(Pip–H)3(Cl)3]·6H2O in (Bu)4N+·BF4−–DMSO solution at 100 mV s−1 vs. Ag/AgCl electrode are shown in Fig. 10. One well-defined anodic peak at 1.25 V is assigned to the Ir3+/Ir4+ electrode couple. In the reverse scan, three well-defined cathodic peaks at −0.6, −1.1 and −1.5 V were noticed and were safely assigned to the couples Ir4+/Ir3+, Ir3+/Ir2+ and the ligand centre.48 The potential–potential difference (ΔEp = Ep,a − Ep,c) was < 90 mV, revealing the quasi-reversible nature of the reduction process. The irreversible nature of the couple Ir4+/Ir3+ was also confirmed from the observed increase in ΔEp on increasing the scan rate. Thus, the couple Rh4+/Rh3+ is most likely to be irreversible metal-based reduction.48,49
 |
| | Fig. 10 CV of (Ir(Pip-H)3(Cl)3]·6H2O in (Bu)4N+·BF4−–DMSO solution at 100 mV s−1 vs. Ag/AgCl, voltage. | |
The plot of ip,c vs. ν1/2 was linear (R2 = 0.986) (Fig. 11) reflecting the diffusion-controlled feature of the electrochemical process.48,49
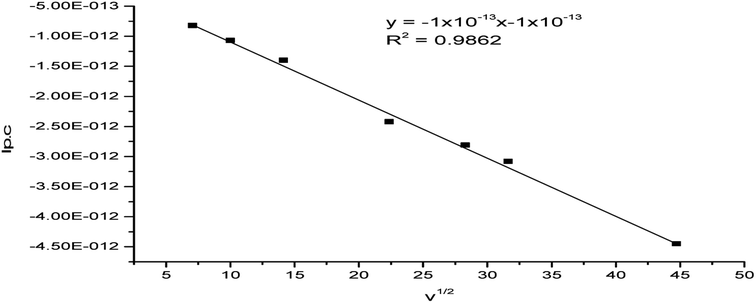 |
| | Fig. 11 Plot of ip,c vs. the square root of the scan rate for the (Ir(Pip-H)3(Cl)3]·6H2O complex in (Bu)4N+·BF4−–DMSO solution at the Pt electrode vs. Ag/AgCl reference electrode. | |
The variation in the current function (ip,c/ν1/2) with the scan rate (50–2000 mV s−1) was also studied. The plot of the current function (ip,c/ν1/2) vs. the scan rate (ν) increased linearly at a low sweep rate (Fig. 12). Thus, the reduction process of the complex favors the EE-type mechanism.50 The product of this reduction step undergoes a very rapid follow-up chemical reaction and the redox processes are of the EE type.49,50 The observed behavior may possibly be explained by considering that the protonation reaction is very fast or virtually complete. The irreversibility and most often quasi-reversibility is attributed to the adsorption onto the electrode surface.
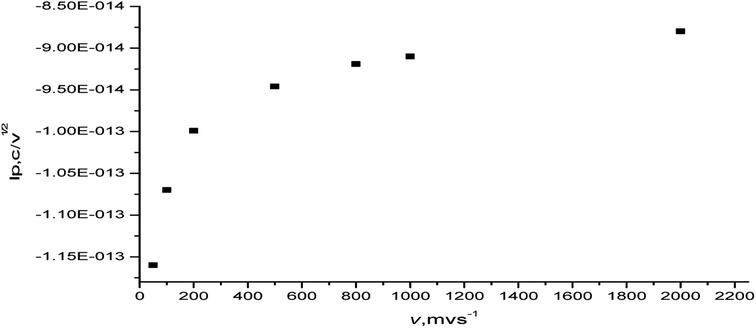 |
| | Fig. 12 Plot of current function (ip,c/ν1/2) vs. the scan rate of the [Ir(PipH)3(Cl)3]·6H2O complex in (Bu)4N+·BF4−–DMSO at the Pt electrode vs. Ag/AgCl electrode. | |
3.9 Biological studies
3.9.1 Antibacterial assessments. The antibacterial efficiency of pipemidic acid complexes (Pip–H) with Ru3+, Pt4+ and Ir3+ was assessed in vitro against four kinds of bacteria G(+) (Staphylococcus epidermidis and Staphylococcus aureus) and G(−) (Klebsiella and Escherichia coli). Inhibition zone diameters of the test samples are summarized in Table 8. The ruthenium(III) complex has a higher antibacterial efficiency than augmentin or unasyn, standard drugs against Klebsiella. Also, both iridium and platinum(IV) complexes have more antibacterial inhibition than the unasyn standard drug against Klebsiella. The Ru3+ and Ir3+ complexes have more antibacterial efficiency than the unasyn standard drug against Escherichia coli bacteria. The ruthenium(III), platinum(IV) and iridium(III) complexes have no inhibition against Staphylococcus epidermidis or Staphylococcus aureus, comparable with the unasyn and augmentin standard drugs. The +ve charge on the metal ions is partially shared with the donation sites in the chelates and there is π-electron delocalization over the whole chelate ring. This leads to increases in the lipophilic character of the metal chelate and favors its permeation through the lipoid layers of the bacterial membranes.51 The increased activity of the metal complexes can be explained on the basis of chelation theory.52 It is known that chelation tends to make the ligand act as a more powerful and potent bactericidal agent, killing more of the bacteria than the ligand. There are other factors which also increase the activity, such as solubility, conductivity, and the bond length between the metal and the ligand, which may explain the values shown in the present case.53
Table 8 Inhibition zone diameter (mm mg−1 sample) of (IrIII, PtIV and RuIII) pipemidic acid complexes against some kinds of bacteriaa
| Sample |
Inhibition zone diameter (mm mg−1 sample) |
| Klebsiella (G−) |
Escherichia coli (G−) |
Staphylococcus epidermidis (G+) |
Staphylococcus aureus (G+) |
| (−)G: gram reaction. Solvent: DMSO. |
| Control: DMSO |
0.0 |
0.0 |
0.0 |
0.0 |
| Standard |
Augmentin |
0.5 |
0.3 |
1.0 |
0.4 |
| Unasyn |
0.2 |
0.1 |
1.0 |
0.2 |
| Ir(III) complex |
0.5 |
0.2 |
0.0 |
0.0 |
| Pt(IV) complex |
0.3 |
0.0 |
0.0 |
0.0 |
| Ru(III) complex |
0.7 |
0.3 |
0.2 |
0.0 |
3.9.2 Anticancer assessments
In vitro cytotoxicity assessments of the pipemidic acid complexes of Ru3+, Pt4+ and Ir3+ metal ions comparable with two commonly anticancer drugs (doxorubicin and cisplatin) were carried out against the human breast cancer (MCF-7) tumor cell line. The inhibitory activity data assessed based on the calculation of the inhibitory concentration IC50 are listed in Table 9. The IC50 of the platinum(IV) complex shows that it is more effective than ruthenium(III) and iridium(III) complexes against the MCF-7 cell line.
Table 9 Inhibitory activities against MCF-7 cell lines for the pipemidic acid, complexes of (IrIII, PtIV and RuIII) and (doxorubicin and cisplatin) standard drugs
| Sample conc. (μg mL−1) |
Viability (%) |
| Doxorubicin |
Cisplatin |
Ir(III) complex |
Pt(IV) complex |
Ru(III) complex |
| 500 |
1.51 |
3.72 |
8.63 |
12.94 |
20.45 |
| 250 |
2.36 |
4.98 |
16.79 |
23.18 |
32.78 |
| 125 |
3.21 |
7.83 |
24.82 |
30.97 |
46.29 |
| 62.5 |
5.07 |
14.68 |
39.46 |
43.52 |
71.56 |
| 31.25 |
6.93 |
23.79 |
56.89 |
67.45 |
86.31 |
| 15.6 |
15.46 |
34.62 |
74.04 |
82.13 |
94.20 |
| 7.8 |
19.89 |
46.71 |
90.64 |
96.58 |
98.76 |
| 3.9 |
24.98 |
52.85 |
98.15 |
99.71 |
100 |
| 2 |
31.69 |
61.74 |
100 |
100 |
100 |
| 0 |
100 |
100 |
100 |
100 |
100 |
| IC50 (μg mL−1) |
0.03 |
0.5 |
1.8 |
1.2 |
5.9 |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by the Deanship of Scientific Research at “Princess Nourah bint Abdulrahman University”, through the Research Groups Program No. RGP-1438-0001.
References
- I. Fonseca, S. Martinex-Carrera and S. Garcia-Blanco, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1618–1621 CrossRef.
- M. A. Toscano, A. Serrao, B. Ventimiglia, V. Colosi, M. Pennisi, R. Morgana and T. Bevilacqua, Miner. Urol., 1982, 34, 257–260 Search PubMed.
- S. Babić, A. J. M. Horvat, D. M. Pavlović and M. Kaštelan-Macan, Trends Anal. Chem., 2007, 26, 1043–1061 CrossRef.
- M. Shimizu, Y. Takase, S. Nakamura, H. Katae and A. Minami, Antimicrob. Agents Chemother., 1976, 9, 569–574 CrossRef PubMed.
- E. K. Efthimiadou, Y. Sanakis, N. Katsaros, A. Karaliota and G. Psomas, Polyhedron, 2007, 26, 1148–1158 CrossRef.
- H. M. Refaat and D. A. N. El-Din, J. Mol. Struct., 2018, 1163, 103–113 CrossRef.
- A. Barmpa, O. Frousiou, S. Kalogiannis, F. Perdih, I. Turel and G. Psomas, Polyhedron, 2018, 145, 166–175 CrossRef.
- I. Turel, N. Bukovec and E. Farkas, Polyhedron, 1996, 15, 269 CrossRef.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef PubMed.
- J. M. Hill, Anticancer Res., 1982, 2, 173–186 Search PubMed.
- M. N. Patel, H. N. Joshi and C. R. Patel, J. Chem. Sci., 2014, 126(3), 737–749 CrossRef.
- A. R. Shaikh, R. Giridhar, F. Megraud and M. R. Yadav, Acta Pharm., 2009, 59, 259 Search PubMed.
- F. A. I. Al-Khodir and M. S. Refat, J. Pharm. Sci. Innovation, 2015, 10, 335–347 CrossRef.
- A. Rusu, G. Hancu, A. C. Munteanu and V. Uivarosi, J. Organomet. Chem., 2017, 839, 19–30 CrossRef.
- R. Joshi, S. K. Yadav, N. Pandey, H. Mishra, R. Tilak and S. Pokharia, J. Mol. Struct., 2018, 1167, 44–56 CrossRef.
- F. A. Al-Saif, K. A. Alibrahim, J. A. Alfurhood and M. S. Refat, J. Mol. Liq., 2018, 249, 438–453 CrossRef.
- P. F. Selwood, Magnetochemistry, Wiley (Interscience), N.Y., 2nd edn, 1956 Search PubMed.
- T. A. Yousef, G. M. Abu El-Reash and R. M. El Morshedy, J. Mol. Struct., 2013, 1045, 145 CrossRef.
- M. H. Helal, S. A. El-Awdan, M. A. Salem, T. A. Abd-elaziz, Y. A. Moahamed, A. A. El-Sherif and G. A. M. Mohamed, Spectrochim. Acta, Part A, 2015, 135, 764 CrossRef PubMed.
- Hyper chem. Version 7.51 Hyper cube, INC Search PubMed.
- R. Gupta, R. K. Saxena, P. Chatarvedi and J. S. Virdi, J. Appl. Bacteriol., 1995, 78, 378 CrossRef PubMed.
- T. Mosmann and J. Immunol, Methods, 1983, 65, 55–63 Search PubMed.
- S. M. Gomha, S. M. Riyadh, E. A. Mahmmoud and M. M. Elaasser, Heterocycles, 2015, 91(6), 1227–1243 CrossRef.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81 CrossRef.
- E. K. Efthimiadou, N. Katsaros, A. Karaliota and G. Psomas, Inorg. Chim. Acta, 2007, 360, 4093 CrossRef.
- L. Yang, W. Li, D. Tao, Y. Li and X. Yang, Synth. React. Inorg. Met.-Org. Chem., 1999, 29(8), 1585 CrossRef.
- A. Beteringhe, C. Racuciu, C. Balan, E. Stoican and L. Patron, Adv. Mater. Res., 2013, 787, 236 Search PubMed; Z. Bikadi, L. Demko and E. Hazai, Arch. Biochem. Biophys., 2007, 461, 225 CrossRef PubMed.
- G. Venkatachalam, S. Maheswaran and R. Ramesh, Indian J. Chem., 2005, 44(4), 705 Search PubMed.
- Y. Tanabe and S. Sugano, J. Phys. Soc. Jpn., 1954, 9, 766 CrossRef.
- S. Chandra, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 1992, 22(10), 1565 CrossRef.
- A. B. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 1968 Search PubMed.
- B. N. Figgis, Introduction to Ligand Field Theory, Interscience Publishers, New York, 1st edn, 1966, p. 287 Search PubMed.
- F. A. El-Saied, R. M. El-Bahnasawy, M. Abdel-Azeem and A. K. El-Sawaf, Polyhedron, 1994, 13(11), 1781 CrossRef.
- D. X. West, M. S. Lockwood, A. E. Liberta, X. Chen and R. D. Willet, Transition Met. Chem., 1993, 18, 221 CrossRef.
- C. Preti and G. Tosi, Transition Met. Chem., 1978, 3, 17 CrossRef.
- F. Bertelli, C. Preti and G. Tosi, J. Inorg. Nucl. Chem., 1975, 37, 1421 CrossRef.
- E. K. Efthimiadou, Y. Sanakis, N. Katsaros, A. Karaliota and G. Psomas, Polyhedron, 2007, 26, 1148 CrossRef.
- M. Badea, R. Olar, D. Marinescu, V. Uivarosi, T. O. Nicolescu and D. Iacob, J. Therm. Anal. Calorim., 2010, 99, 829 CrossRef.
- E. S. Freeman and B. Carroll, J. Phys. Chem., 1958, 62, 91 Search PubMed.
- H. H. Horowitz and G. Metzger, Anal. Chem., 1963, 35, 1464 CrossRef.
- A. W. Coats and J. P. Redfern, Nature, 1964, 201, 68 CrossRef.
- B. D. Cullity, Elements of X-ray Diffraction, Addison-Wesley, Reading, MA, 1972, p. 102 Search PubMed.
- M. Salavati-Niasari, F. Mohandes, F. Davar, M. Mazaheri, M. Monemzadeh and N. Yavarinia, Inorg. Chim. Acta, 2009, 362(10), 3691 CrossRef.
- S. Velumani, X. Mathew and P. J. Sebastian, Sol. Energy Mater. Sol. Cells, 2003, 76, 359 CrossRef.
- J. P. Singh, T. Karabacak, P. Morrow, S. Pimanpang, T.-M. Lu and G.-C. Wang, J. Nanosci. Nanotechnol., 2007, 7, 2192 CrossRef PubMed.
- S. Krehula and S. Musić, Croat. Chem. Acta, 2011, 84(4), 465 CrossRef.
- T. Zhang, S.-C. Li, W. Zhu, J. Ke, J.-W. Yu, Z.-P. Zhang, L.-X. Dai, J. Gu and Y.-W. Zhang, Surf. Sci., 2016, 648, 319 CrossRef.
- B. S. Parajon-Costa, A. C. Gonzalez-Baro and E. J. Baran, Z. Anorg. Allg. Chem., 2002, 628, 1419 CrossRef.
- M. L. P. Santos, I. A. Bagatin, E. M. Pereira and A. M. D. Ferreira, J. Chem. J. Chem. Soc., Dalton Trans., 2001, 6, 838 RSC.
- A. J. Bard and L. R. Faulkner, Electrochemical
Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- Z. Chohan and S. Mushtaq, Pak. J. Pharm. Sci., 2000, 13(1), 21–27 Search PubMed.
- S. K. Sengupta, O. P. Pandey, B. K. Srivastava and V. K. Sharma, Transition Met. Chem., 1998, 23(4), 349–353 CrossRef.
- S. Chandra, S. Raizada, M. Tyagi and A. Gautam, Bioinorg. Chem. Appl., 2007, (1), 51483 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bc,
Y. M. Moustafad and
Moamen S. Refat
bc,
Y. M. Moustafad and
Moamen S. Refat



 is ≤1.0. The decrease in β value is also associated with a reduction in the effective positive charge of the metal ion and with an increasing tendency to reduction to the lower oxidation state.31 These results suggest the presence of strong covalent bonding between the Pip–H donor and Ru3+ ions.30 The magnetic susceptibility of [Ru(Pip–H)3(Cl)3]·6H2O complex (1) was measured using a Gouy magnetic balance at room temperature. The ruthenium(III) complex being d5 (low spin), S = 1/2 has a paramagnetic character. The magnetic moment of the ruthenium(III) Pip–H complex is 1.90 B.M., which confirms the exhibition of a single unpaired electron in a low-spin 4 d5 configuration for a Ru3+ ion in an octahedral environment.32 This value is lower than the predicted value of 2.10 B.M. This lowering may be due to the presence of ligand fields of lower symmetry, or extensive electron delocalization in the species.33 Therefore, the magnetic moment value gives an impression that the ruthenium(III) complex has been found in the (+3) oxidation state.
is ≤1.0. The decrease in β value is also associated with a reduction in the effective positive charge of the metal ion and with an increasing tendency to reduction to the lower oxidation state.31 These results suggest the presence of strong covalent bonding between the Pip–H donor and Ru3+ ions.30 The magnetic susceptibility of [Ru(Pip–H)3(Cl)3]·6H2O complex (1) was measured using a Gouy magnetic balance at room temperature. The ruthenium(III) complex being d5 (low spin), S = 1/2 has a paramagnetic character. The magnetic moment of the ruthenium(III) Pip–H complex is 1.90 B.M., which confirms the exhibition of a single unpaired electron in a low-spin 4 d5 configuration for a Ru3+ ion in an octahedral environment.32 This value is lower than the predicted value of 2.10 B.M. This lowering may be due to the presence of ligand fields of lower symmetry, or extensive electron delocalization in the species.33 Therefore, the magnetic moment value gives an impression that the ruthenium(III) complex has been found in the (+3) oxidation state.


 ), 3.14–3.25(4H2a,2a′: piperazinyl group), 3.86(4H2b,2b′: piperazinyl group), 4.39(2H8a:
), 3.14–3.25(4H2a,2a′: piperazinyl group), 3.86(4H2b,2b′: piperazinyl group), 4.39(2H8a:  ), 8.90(2H4,7: pyrimidine and quinolone groups); 9.19(1H11: –NH piperazinyl group)
), 8.90(2H4,7: pyrimidine and quinolone groups); 9.19(1H11: –NH piperazinyl group) ), 3.341(4H2a,2a′: piperazinyl group), 4.060, 4.095(4H2b,2b′: piperazinyl group), 4.347(2H8a:
), 3.341(4H2a,2a′: piperazinyl group), 4.060, 4.095(4H2b,2b′: piperazinyl group), 4.347(2H8a:  ), 8.992(2H4,7: pyrimidine and quinolone groups); 9.253(1H11: –NH piperazinyl group); 7.345(8H: 2NH4 group)
), 8.992(2H4,7: pyrimidine and quinolone groups); 9.253(1H11: –NH piperazinyl group); 7.345(8H: 2NH4 group)







