
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHierarchically porous N-doped carbon derived from supramolecular assembled polypyrrole as a high performance supercapacitor electrode material†
Li Lai‡
,
Yu Zhao‡,
Shu Ying,
Lanlan Li,
Zhong Ma and
Lijia Pan *
*
National Laboratory of Microstructures, School of Electronic Science and Engineering, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, P. R. China. E-mail: ljpan@nju.edu.cn
First published on 22nd May 2018
Abstract
Rationally designed precursors of N-doped carbon are crucial for high performance carbon materials of supercapacitor electrodes. Herein, we report a scalable preparation of hierarchically structured N-doped carbon of micro/meso porous nanofiber morphology by using a supramolecular assembled polypyrrole as the precursor. The influences of the dose of supramolecular dopant on final products after carbonization and sequential chemical activation were investigated. The interconnected nanofiber backbone allows better electron transport and the optimized hierarchically porous structure of the material exhibits a large specific surface area of 2113.2 m2 g−1. The N content of the carbon is as high as 6.49 atom%, which is favorable to improve the supercapacitive performance via additional reversible redox reaction over pure carbon. The hierarchically porous N-doped carbon electrode delivered an outstanding specific capacitance of 435.6 F g−1 at 0.5 A g−1, significantly higher than that of the control sample derived from undoped polypyrrole samples. Moreover, the capacitance retention is as high as 96.1% after 5000 cycles. This precursor's structural control route is readily applicable to various conducting polymers, and provides a methodology to design carbon materials with advanced structure for developing high-performance supercapacitor electrode materials.
Introduction
Supercapacitors have been regarded as one of the most promising sustainable energy storage technologies with potential to alleviate the growing energy demand in the face of the continuous depletion of fossil fuels and the release of excessive greenhouse gasses. This is because they have intriguing characteristics of high power density, extraordinary cycle life, and high energy efficiency.1,2 Generally, the amount of charge stored by the electric double layer (EDL) does not meet the growing performance demands. An alternative way to enhance the specific capacitance is to utilize faradaic reactions by employing pseudocapacitive materials,3 such as conducting polymers,4,5 doped carbon materials,6,7 transition metal oxides (TMOs),8,9 metal organic frameworks (MOFs),10,11 etc. Among them, carbon materials are outstanding because of their high surface area,12 abundant low dimensional morphologies, and superb physical/chemical robustness (which prolongs the electrode lifespan to tens of thousands of cycles).13,14 However, traditional carbon materials, including activated carbon,12 carbon nanotubes,15 and graphene,16 suffer from low specific capacitance due to their low electronic conductivity, large pore tortuosity, lack of pseudocapacitance, or severe aggregation.Several strategies were developed to address this problem, including forming composites with conducting polymers17,18 and TMOs,19,20 or doping carbon with heteroatoms,6,7 but each of them has their own limitations. For example, conducting polymers are electrochemically unstable during the cycling process,19 TMOs are either too expensive or hardly conductive, and their side reactions would put a drag on cycling stability too.19,21 Doping carbon with B, N, or S elements is usually adopted by either a carefully controlled chemical vapour deposition (CVD) process which is complicated,7 or hydrothermal reaction using graphene oxide as the building blocks,6 however, their specific surface area was dramatically decreased by random stacking of individual 2D sheets.20,22 Although some specially designed 3D porous graphene networks show good pore connectivity,23,24 their large-scale fabrication still remains a challenge. Carbonizing with precursors of biomass (e.g., chicken egg whites,25 straw,26 and catkin27) or conducting polymers is also a practical approach for carbon doping while suitable for large-scale and low-cost fabrication. However, the structures of biomass cannot be tuned once the type of the biomass is chosen, and the products usually contain a large portion of impurities.28 As for carbonizing with conducting polymer precursors, Bao et al. reported a 3D hierarchically porous graphitic carbon framework with a specific capacitance of 225 F g−1 at 0.5 A g−1, and Tan et al. reported highly doped carbon nanobelts with a specific capacitance of 458 F g−1 at 0.2 A g−1.28,29 Although these studies demonstrate the potential of polymer derived carbon, they focused more on conditions of carbonization or chemical activation rather than structure control. As is known, conducting polymers excel for flexibility of structure control compared with biomass.30,31 Thus, it is necessary to investigate the carbonized materials by tuning the morphology of the conducting polymer precursors to improve their performance as supercapacitor electrode materials.
Herein, we report a scalable and low temperature preparation of hierarchically porous N-doped carbon (HPNC) from a structure-tunable precursor of the conductive polymer polypyrrole (PPy) based on a dopant enabled supramolecular assembly approach we reported previously.32 Our strategy requires no sacrificial templates and PPy self-assembled into nanostructures with gradual changes, which can be readily achieved with the assistance of different doses of dopant copper phthalocyanine-3,4′,4′′,4′′′-tetrasulfonic acid tetrasodium salt (CuPcTs). During 800 °C carbonization under nitrogen flow, the CuPcTs with higher thermal stability and rigid conjugated framework of PPy help to maintain the interconnected nanofiber structure and form the macro pore architecture of N-doped carbons (NCs). The chemical activation of NCs at 700 °C with potassium hydroxide (KOH) produces HPNCs and renders the HPNCs micro/mesopores on its surface as extra pseudocapacitance sites and simultaneously enlarging the specific surface area to 2113.2 m2 g−1 with 1.69 cm3 g−1 pore volume. The relative low activation temperature preserves much N content to provide pseudocapacitance. Due to the synergetic effect of structure optimization and redox active N content, the supercapacitive performance of our HPNC makes a considerable progress (up to 435.6 F g−1 at 0.5 A g−1) in comparison with the ordinary PPy derived carbon materials, for example, 179 F g−1 at 0.5 A g−1 reported by Manyala et al., and 182 F g−1 at low scan rates reported by Shrestha and Mustain.33,34
Results and discussion
Preparation of HPNCs
Preparation process of the HPNCs is illustrated in Fig. 1. The PPy precursors were facilely synthesized by mixing a pyrrole solution in isopropanol, and an aqueous solution of an oxidative initiator and CuPcTs. As the polymerization of pyrrole progressed, the blue (the color of the CuPcTs) solution turned into black (the color of PPy) hydrogel quickly, losing its fluidic properties in approximately 2 min. After the PPy precursors were rinsed and dried, there are only minor mass differences among the undoped PPy and doped PPys synthesized with 0.008 mmol, 0.016 mmol, 0.032 mmol CuPcTs from 2 ml solution B (see Experimental section), indicating nearly the same amount of PPy in each sample. This is because the amount of pyrrole is always 1.2 mmol in 1 ml solution A for each sample, and CuPcTs does not affect the amount of PPy. It can be observed from the scanning electron microscopic (SEM) image (Fig. S1, ESI†) that the PPys assembles into different morphologies in the presence of different dose of CuPcTs. In the beginning, the PPy consists of agglomerated granular particles, and then the particles start to separate with slow width shrinkage and length stretch (Fig. S1a and c†). With the growing amount of CuPcTs, the PPy appears as interconnected nanofibers (Fig. S1d–f†). Besides, the nanofibers become thinner (ranged approximately from 40 to 200 nm), longer, and denser. It had been demonstrated that the rigid disc shape of the CuPcTs is in favor of the alignment of PPy to from a 1D nanostructure by electrostatic interaction between the sulfonic acid functional groups and the pyrrolic N.32 Specifically, the relatively large size of the disc-shaped CuPcTs may sterically hinder the formation of the side chains and cross-linked structure between PPy main chains.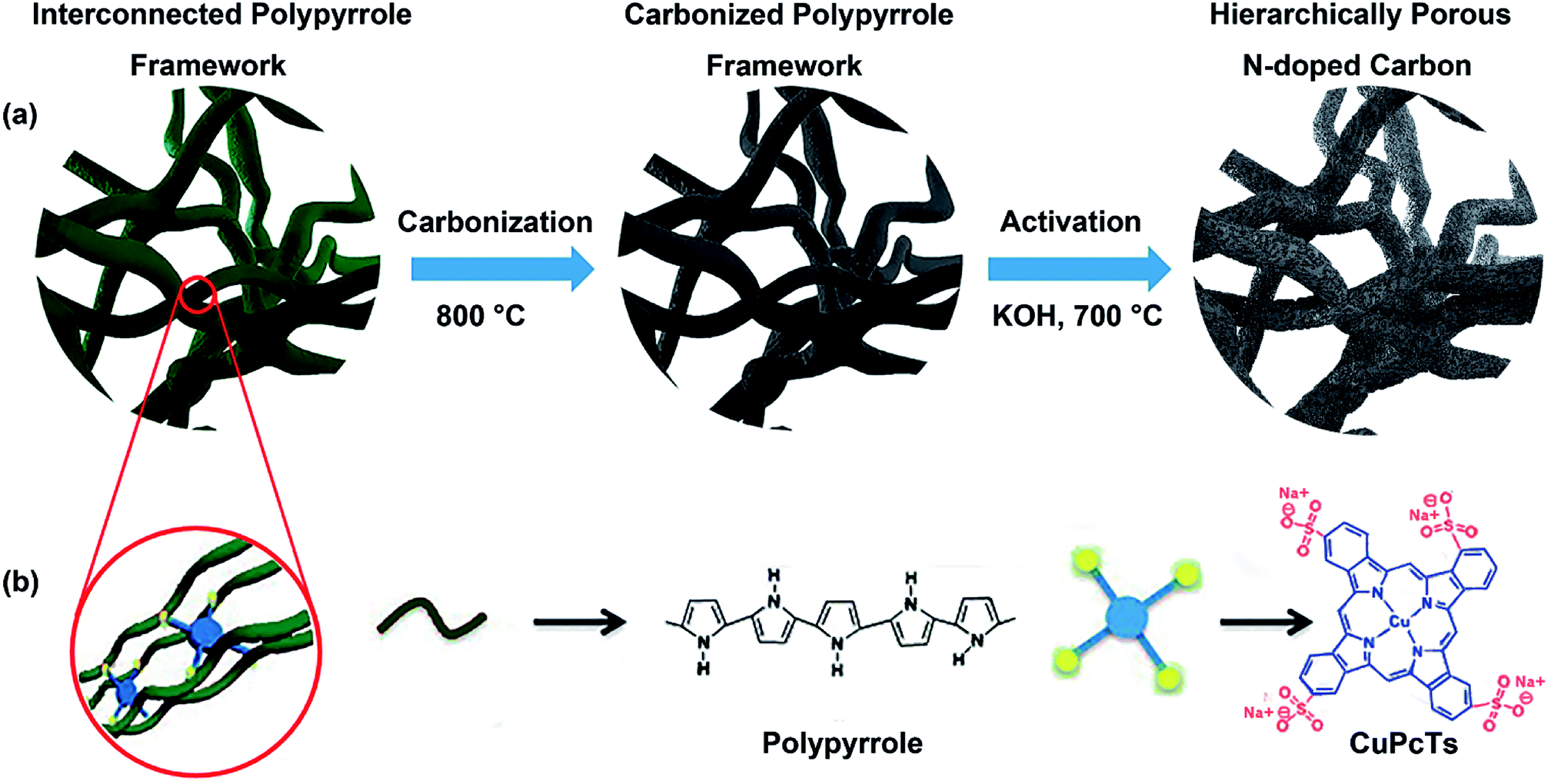 | ||
| Fig. 1 (a) Schematic of the fabrication process of the HPNCs. (b) Illustration of the controlled synthesis of the CuPcTs doped PPy hydrogel and the molecular structures of PPy and CuPcTs. The CuPcTs act to align the PPy chains into 1D nanostructure through a steric effect. | ||
In subsequent pyrolysis, CuPcTs starts its pyrolysis at around 490 °C, and decomposes further at around 630 °C according to the thermogravimetric analysis (TGA) (Fig. S2, ESI†). Thus, the weight of CuPcTs doped PPy decreases slower than the undoped PPy at a lower temperature, and then surpasses the undoped PPy at around 550 °C. Indeed, the high degradation temperature of CuPcTs effectively prevents pores and the 1D structure from collapsing, which results about 50 wt% carbon yield. Further chemical activation with KOH by a heat treatment increases the porosity and surface area. A lower activation temperature (700 °C) was selected to prevent substantial N content lose during chemical activation. A typical overall HPNC yield (vs. mass of PPy) of about 21 wt% can be achieved, higher than common carbon yield (about 8 wt%) of commercial ACs made from activation of biomass under similar condition.35,36
Physical and chemical characterization of the HPNCs
The HPNCs exactly inherit the backbone of their PPy precursor, showing hierarchical architectures (Fig. 2 and 3). Compared with the agglomerated particles (Fig. 2a–c), this type of interconnected nanofiber morphology (primary structure) is ample of macropores (>50 nm), which not only avoids surface area decreasing due to aggregation but also shows good pore connectivity, facilitating the permeation of ions (Fig. 2d–f). Close examination of the HPNC backbone by transmission electron microscopy (TEM) (Fig. 3a, and S3a and b, ESI†) reveals a porous surface morphology (secondary structure) swarming with micropores (0–2 nm) and mesopores (2–50 nm). High magnification TEM image (Fig. 3b, and S3c and d, ESI†) displays structurally defective microporous carbons with local aromatic ordering, suggesting the existence of both amorphous and graphitized carbon of the HPNCs. In addition, there are no heterogeneous structure from peripheral edges to inner axis of the nanofiber except for the pores. Therefore, it can be inferred that the CuPcTs disperses uniformly in the pristine PPy precursor instead of concentrating in confined areas and it degrades completely after successive carbonization and activation. | ||
| Fig. 2 SEM images of HPNCs, whose precursor are PPy without (a) CuPcTs, and PPy with (b) 0.002 mmol, (c) 0.004 mmol, (d) 0.008 mmol, (e) 0.016 mmol, (f) 0.032 mmol CuPcTs, respectively. Samples in (a), (d), (e), and (f) are HPNC-1, HPNC-2, HPNC-3, and HPNC-4, respectively. | ||
 | ||
| Fig. 3 TEM images of HPNCs (a) revealing the microporous surface, and (b) with a typical lateral dimension of a few nanometers. | ||
Given that the parameters of surface area and porosity play an important role in determining the electrochemical characteristics of a material by means of ion accommodation and facilitating ions transportation, nitrogen adsorption/desorption isotherm was applied to evaluate the NC and HPNC samples. The nitrogen adsorption/desorption isotherms, pore width distributions of the samples by density functional theory (DFT) of HPNCs are shown in Fig. 4a and b, respectively. The HPNCs exhibit similar nitrogen adsorption/desorption isotherm patterns and similar pore width distributions. There are two rapid nitrogen uptakes in the isotherm curves. The first (when P/Po < 0.1) can be ascribe to micropores generated by chemical activation, while the second (when P/Po > 0.9.) is due to larger macropores originated from interconnected nanofiber structure. Followed the first rapid uptake is a slower nitrogen adsorption until the curves reach a plateau when the relative pressure is around 0.5. This slow uptake is considered to be the process of nitrogen filling mesopores that are probably inherited from intrinsic rough PPy surface or generated by activation. The DFT pore size distributions derived from the nitrogen adsorption branches confirm the presence of micropores and mesopores. There are several peaks when the pore width is less than 2 nm, and the mesopores lies in range 2–6 nm (Fig. 4b). That the micropores and mesopores adsorb much more nitrogen than the macropores reveals their abundance in the HPNCs (Table 1). Such hierarchical pore structure can help to shorten the ion diffusion path and these ample micro/meso pores can offer a substantial amount of redox sites. Generally, the HPNC-3 absorbed the most nitrogen volume (up to 1.69 cm3 g−1), followed by HPNC-2, HPNC-4 and HPNC-1 in a decreasing order (Table 1). In view of structure tuning, the conversion from particle to fiber morphology and the reduced diameter from HPNC-1 to HPNC-3 causes the increase of surface area for chemical activation to generate micro/meso pores. Indeed, the BET surface area of NCs increases from 9.1 m2 g−1 (NC-1) to 46.5 m2 g−1 (NC-2) to 315.9 m2 g−1 (NC-3), proving the influence of morphology change on surface area directly and indicating both micro and meso pores are created during activation. Correspondently, the BET surface areas are 1686.7 m2 g−1 (HPNC-1), 1864.1 m2 g−1 (HPNC-2), and 2113.2 m2 g−1 (HPNC-3). When it comes to HPNC-4, the diameter of nanofiber stops to decrease, but the density of the nanofibers increases, leading to severe overlap between nanofibers (NC-4, 290.0 m2 g−1). So, a drop (HPNC-4, 1789.8 m2 g−1) of surface area was observed.
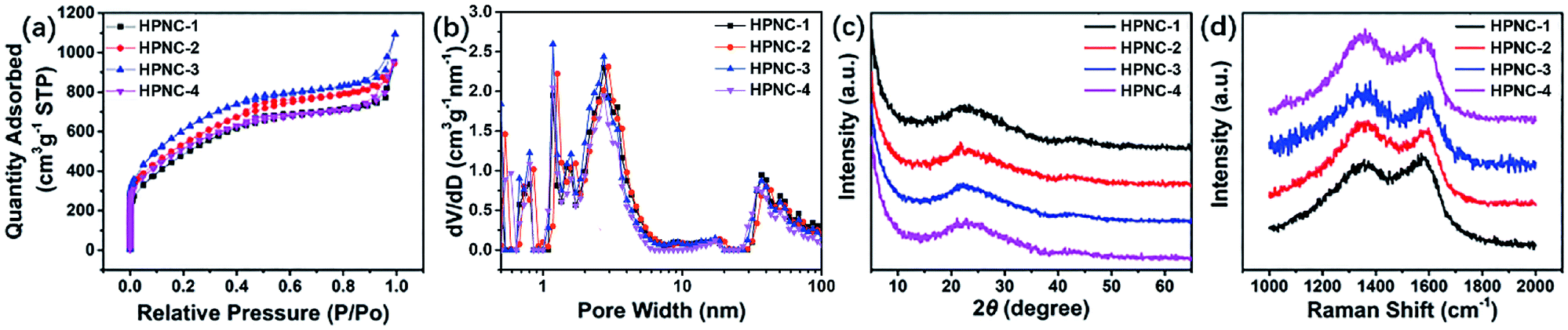 | ||
| Fig. 4 (a) Nitrogen adsorption/desorption isotherms, (b) DFT pore width distributions, (c) XRD curves, and (d) Raman spectra of the HPNCs, respectively. | ||
| Samples | SBET (m2 g−1) | Vtotal (cm3 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | Vmacro (cm3 g−1) |
|---|---|---|---|---|---|
| HPNC-1 | 1686.7 | 1.48 | 0.67 | 0.50 | 0.31 |
| HPNC-2 | 1864.1 | 1.60 | 0.83 | 0.51 | 0.28 |
| HPNC-3 | 2113.2 | 1.69 | 0.86 | 0.52 | 0.31 |
| HPNC-4 | 1789.8 | 1.47 | 0.72 | 0.48 | 0.27 |
Powder X-ray diffraction (XRD) analysis and Raman spectroscopy were used to further investigate the graphitization degree of the HPNCs. In Fig. 4c, all samples shares similar features that two peaks centred at 22.8° and 43.5° representing (002) and (100) diffraction. The (002) diffraction has a broad width, and the (100) diffraction is relatively low in intensity. These suggests amorphous carbon structure.26,37 In Fig. 4d, there are two prominent peaks at 1356 cm−1 (D band) and 1589 cm−1 (G band). The D band is related to disordered carbon, while the G band is a feature of graphitic carbon. The former can accommodate electrolyte ions and the latter improves conductivity. The average ID/IG intensity ratio of HPNC-1, HPNC-2, HPNC-3, and HPNC-4 are calculated to be 0.96, 1.06, 1.08, and 1.08, respectively. These suggests a balance between the abundant micro/meso pores caused by chemical activation and low resistance because of graphitic carbon.38,39 The slightly higher ration of HPNC-2, HPNC-3, and HPNC-4 is consistent with their higher surface area, confirming that the nanofiber morphology is benefit for creating more micro/meso pores.
To further understand the intrinsic chemical features, we conducted X-ray photoelectron spectroscopy (XPS) on the NCs and HPNCs. Since the samples were treated at the same carbonization and activation condition, they show similar surface elemental composition. The HPNCs consisted of dominant C (84.41–86.25 atom%), sample O (7.27–9.54 atom%), and N (5.29–6.49 atom%) spices (Table 2). Only negligible Cu (0.53 atom%) and S (0.27–0.30 atom%) species were found in HPNC-3 and HPNC-4, indicating that the dopant CuPcTs does not caused a substantial influence on the near surface contents of the HPNCs. One reason is that the dose of CuPcTs (no more than 0.032 mmol) is relatively a small compared with pyrrole (1.2 mmol). Cu and S species are also found in NC-3 and NC-4 (Table S1, ESI†), and they are slightly higher than in HPNC-3 and HPNC-4. Therefore, the pyrolysis of CuPcTs and the final purification with 2 M HCl may further minimize the possible remnants. In NCs, the O species ranges from 2.54 atom% to 3.61 atom%, suggesting the majority of O species of HPNCs is introduced during KOH activation. In Fig. 5 and Fig. S4 (ESI†), the fitted XPS peaks for N 1s are deconvoluted into multiple bands, which are attributed to the pyridinic N (397.5 eV), the graphitic N (399.7 eV), and N oxide (403.0 eV), respectively.40,41 The integration of peak areas provides the relative abundance of each type of N species, which is also summarized in Table 2 for HPNCs and Table S1 for NCs (ESI†). The main difference between HPNCs and NCs in N species is that HPNCs has less N in regardless of its form, confirming that the activation decreases N species. In HPNCs, the pyridinic N (19.3–30.0 atom% for total N) originated from structure confinement, namely, the irregular edges of the nanofiber structure and the surface micro/meso pores. The graphitic N predominates (58.1–76.7 atom% for total N) due to its highest thermal stability. The N oxide (3.9–12.8 atom% for total N) can be ascribed to the oxidizing environment of the activation process. These N species is expected favorable for enhancing pseudocapacitance in view of their redox sites, especially in the form of pyridinic N and N oxide. By close comparison of the four samples, the relative integration area of pyridinic N peaks maximized in HPNC-3 and the integration area of N oxides peaks also starts to saturate in HPNC-3, while the graphitic N peaks becomes weaker. This is because the nanofiber structure, especially with thinner diameter, would induce more carbon edges during chemical activation to generate pyridinic N and N oxide.
| Samples | Elemental composition (atom%) | ||||
|---|---|---|---|---|---|
| C | N (pyridinic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :graphitic:oxidized) :graphitic:oxidized) |
O | S | Cu | |
| HPNC-1 | 85.17 | 5.29 (1.02:4.06:0.21) |
9.54 | 0 | 0 |
| HPNC-2 | 86.07 | 5.42 (1.58:3.34:0.50) |
8.51 | 0 | 0 |
| HPNC-3 | 86.25 | 6.49 (1.95:3.77:0.77) |
7.00 | 0.26 | 0 |
| HPNC-4 | 84.41 | 6.33 (1.53:3.99:0.79) |
8.42 | 0.31 | 0.53 |
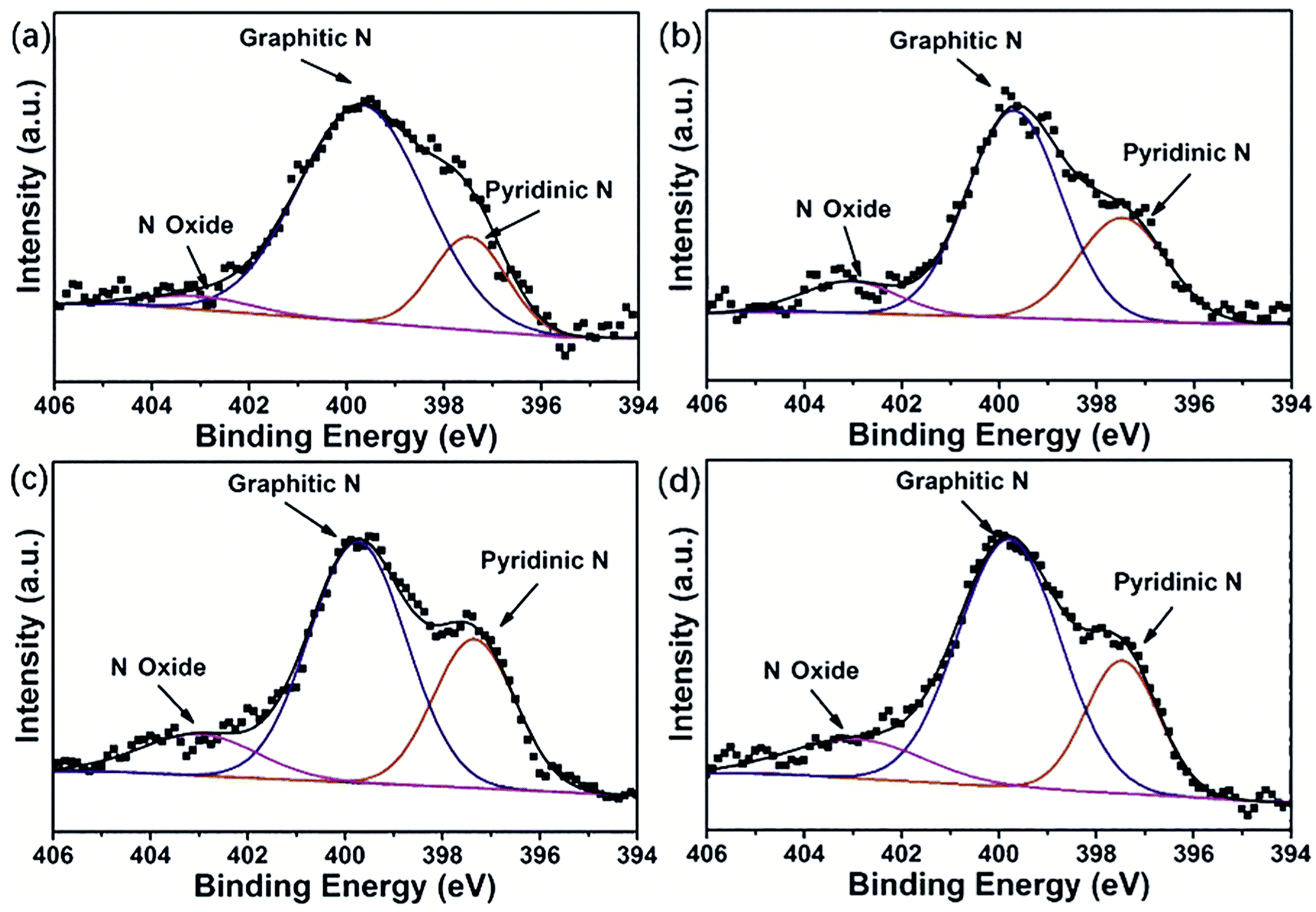 | ||
| Fig. 5 N 1s detail spectra of (a) HPNC-1, (b) HPNC-2, (c) HPNC-3, and (d) HPNC-4 conducted by XPS analysis. | ||
Electrochemical performances of HPNC electrodes
To evaluate the electrochemical characteristics of the HPNCs, cyclic voltammetry (CV), galvanostatic charge/discharge (GCD), and electrochemical impedance spectroscopy (EIS) tests were carried out in a conventional three-electrode system with the electrodes immersed in 1 M H2SO4 electrolyte. Fig. 6a and b display typical CV and GCD curves. (See Fig. S5, ESI† for more curves of each HPNC sample). The CV curves show rectangular-like shapes at the scan rates varied from 20 to 200 mV s−1. This is in consistence with the nearly symmetric triangular GCD curves at current densities ranging from 0.5 to 10 A g−1 and the nearly vertical shape of the obtained EIS curves at lower frequencies (Fig. S7, ESI†). The above shapes and symmetry features indicates rapid EDL charging/discharging kinetics. Additionally, there are two obvious peaks at approximately 0.35 V and 0.45 V in the CV curves (Fig. 6a and S5, ESI†), and they are observable even when the scan rate increased to 100 mV s−1. They evidently indicate the reversible redox reactions, which also demonstrates a rapid kinetics during the charge/discharge cycle. | ||
| Fig. 6 (a) CV curves of HPNC-3 at various scan rates. (b) GCD curves of HPNC-3 at various current density. (c) and (d) CV and GCD curves of HPNCs at 20 mV s−1 and 0.5 A g−1, respectively. (e) Gravimetric capacitance at different charge/discharge current densities. (f) Cycling stability (inset, the GCD curves between 4981th and 5000th cycle at 10 A g−1). | ||
In Fig. 6c and d, the enhanced current density and the prolonged time from HPNC-1 to HPNC-2 to HPNC-3 for a charge/discharge cycle indicated growing specific capacitance. The specific capacitance growth is calculated to be approximately 55.9% from HPNC-1 to HPNC-3, with 279.4 F g−1, 308.1 F g−1, and 435.6 F g−1 at 0.5 A g−1 in detail (Fig. 6d). However, an unanticipated decrement came up to HPNC-4, whose specific capacitance is 379.1 F g−1 at 0.5 A g−1 in spite of its lowest equivalent series resistance (ESR). As is mentioned before, the density of the nanofibers shows an increasing tendency from HPNC-2 to HPNC-4. Giving this factor, it can be ascribed to the extensive overlap between nanofibers (Fig. 2f, and S1f, ESI†), which consumes a lot of surface area (2113.2 m2 g−1 for HPNC-3 and 1789.8 m2 g−1 for HPNC-4). Another possible unfavorable factor is the residue of CuPcTs after carbonization and chemical activation, which is not rivalled to the carbonized PPy in providing capacitance.
For comparison, we also conducted electrochemical analysis on the intermediate products, namely the unactivated samples derived from PPy (see Fig. S6, ESI† for all CV and GCD curves of each NC sample). At the scan rate of 20 mV s−1, the four CV curves also show rectangular-like shapes with two peaks (Fig. 7a). However, the peaks are relatively less intense than they are in the HPNCs and they shift left by about 0.05 V (0.3 V and 0.4 V for NCs vs. 0.35 V and 0.45 V for HPNCs). These can be ascribed to the much smaller surface area and different surface chemical environment. The BET surface area of NC-1, NC-2, NC-3, and NC-4 are 9.1 m2 g−1, 46.5 m2 g−1, 315.9 m2 g−1, and 290.0 m2 g−1, respectively. In NCs, scarce O species (no more than 3.61 atom%) are detected. These two factors suggest a small quantity of active sites on the NCs surfaces, leading to weaker redox peaks and peak shift towards lower potential. At the current density of 0.5 A g−1, the discharging time first increases, maximizes at NC-3, and then decreases (Fig. 7b). This is nearly exactly the same changing trend showed in HPNCs, but the increment (120.2%) from NC-1 to HPNC-1 is much larger than the other three pair of samples (no more than 32.9%, Fig. 7c). The reason is that the specific surface area ratio SHPNC/SNC is much larger in samples derived from undoped or low CuPcTs doped PPy than highly CuPcTs doped ones. For example, SHPNC/SNC is up to 185.35 for HPNC-1 vs. NC-1, while only 6.17 for HPNC-4 vs. NC-4. The SHPNC/SNC values suggest that the activation process can greatly enlarge the surface area, but diminish the surface area differences between activated samples. As a result, the specific capacitance increment between HPNC-1 and HPNC-2 is not as prominent as the increment between NC-1 and NC-2. Therefore, the smaller surface area the unactivated carbon is, the larger capacitance activation contributes. That is to say, the surface area is the leading factor for small surface area carbon. Anyway, the large specific capacitance increment of the four pair of samples indicates that activation does enhance the performance of HPNCs substantially.
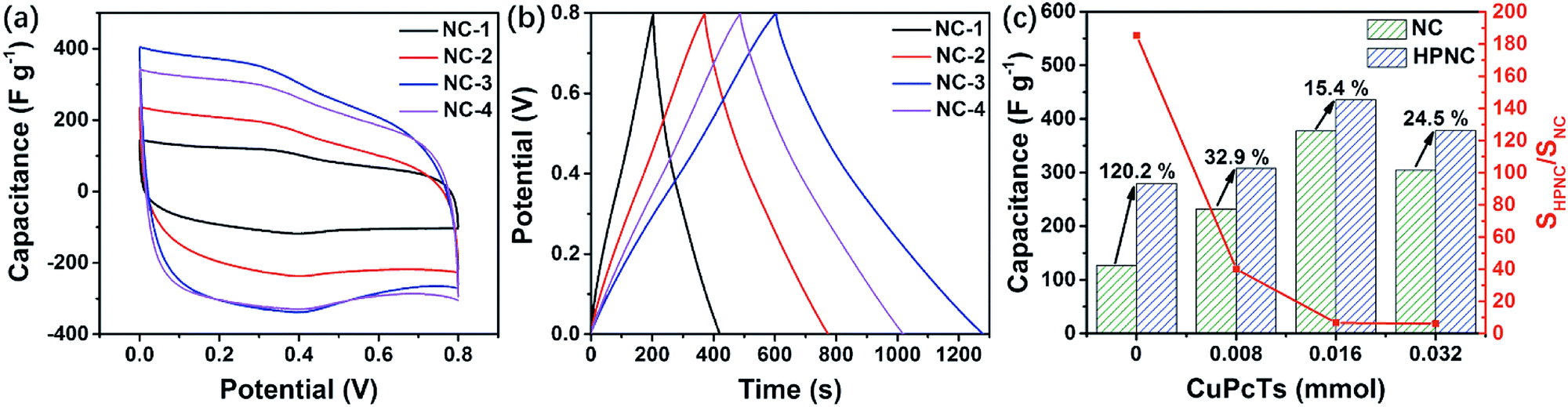 | ||
| Fig. 7 (a) and (b) CV and GCD curves of NCs at 20 mV s−1 and 0.5 A g−1, respectively. (c) Specific capacitance and surface area comparison between unactivated (NCs) and activated carbon derived (HPNCs) from PPy. | ||
Moreover, the capacitance retention of HPNCs are all above 72% even the current density reached 10 A g−1, and the specific capacitance of HPNC-3 retains 343.8 F g−1 at 10 A g−1. It is the best rate performance (78.9% capacitance retention) except HPNC-1 (84.3% capacitance retention) (Fig. 6e). A cyclic test of 5000 cycles at 10 A g−1 shows only a negligible drop (3.9%), indicating great cycling stability (Fig. 6f).
Discussion
Based on the characterization and the electrochemical performance, the excellent supercapacitive performance can be ascribed to several aspects as the following: (1) when the morphology gradually turns from granular particles (HPNC-1) to interconnected nanofibers (HPNC-2) without obvious size difference, the elimination of aggregation increases the surface area. (2) As the diameter of nanofiber decreases by more than a half (from 140–200 nm of HPNC-2 to 40–60 nm of HPNC-3), the relative surface area of each single fiber increases with respect to its volume. Therefore, the utilization of HPNC is substantially improved, especially the utilization of N content. Nevertheless, the non-graphitic N increment (1.23 atom% in HPNC-1, 2.08 atom% in HPNC-2, 2.72 atom% in HPNC-3, and 2.32 atom% in HPNC-4 as summarized in Table 1), resulting from the growing portion of exposed micro/meso pore edges, would supply more redox active sites. These factors lead to the substantial specific capacitance growth from HPNC-2 to HPNC-3 and such performance of specific capacitance up to 435.6 F g−1 at 0.5 A g−1 is superior or comparable for carbon material derived from biomass to polymer precursors as shown in Table S2 (ESI†). (3) Among various morphologies, the 1D nanofiber structure is also intrinsically advantageous to electron transport. Although the nanofibers become thinner from HPNC-2 to HPNC-4, they become much longer and well intertwined. Such densely intertwined network successfully improves the electronic conductivity. The ESRs extracted from high frequency (10 kHz) data confirms this point (Fig. S7 inset, ESI†), which also ensures efficient charge transfer and high chemical stability. Synergized with the hierarchically porous structure for fast electrode/electrolyte interface kinetics, the HPNC can provides high rate capability and impressively high cycling stability. Hence, the HPNC can be regarded as a promising candidate material for high performance supercapacitors, due to its high specific capacitance and outstanding stability.Conclusions
In summary, we have successfully developed a scalable approach to prepare hierarchically porous N-doped carbon derived from CuPcTs assembled PPy using carbonization and sequential chemical activation. Particularly, the obtained hierarchically porous interconnected nanofiber architecture brings more efficient electron transfer, better pore connectivity and higher surface area along with increased non-graphitic N content by further rational optimization of PPy morphology prior to the heat treatments. These properties render the HPNCs specific capacitance as high as 435.6 F g−1 at 0.5 A g−1 without sacrificing the extraordinary cyclic stability. This work presents a new route in tuning the morphology of carbon material, which is benefit for electrochemical performance enhancement and illuminating for other tunable precursors. Besides, the scalability of the fabrication process and the substitutability with cheaper supramolecular dopant with multiple sulfonic acid functional groups, in view of the underlying assembling mechanism, would make this approach potential for commercialization.Experimental section
Synthesis of the doped PPy
Typically, 84 μl (1.2 mmol) pyrrole (99%, Aldrich) was dissolved in 1 ml isopropanol (denoted as solution A). Dopant solution was prepared by mixing 0.0327 g (0.032 mmol) CuPcTs in 2 ml DI water. 0.274 g (1.2 mmol) mass of persulfate (Aldrich) was added to the dopant solution (denoted as solution B). After cooling solution A and B to 4 °C, they were mixed quickly. The product was purified by immersion in ethanol (12 h) and DI water (24 h) sequentially to remove excessive reagents and by-products. Then it was dried in vacuum at 60 °C.Preparation of the HPNCs
The as-synthesized CuPcTs doped PPy was first calcined at 800 °C for 2 h under nitrogen. The product after carbonization was added to KOH aqueous solution (carbonized PPy and KOH mass ratio, 1:3) for 12 h immersion, after which it was dried. The followed process was a heat treatment at 700 °C for 1 h under nitrogen. After activation, the yield was cleaned using 2 M HCl and washed for several times with DI water until pH = 7. Finally, the sample was dried again in vacuum at 60 °C. In this work, 0 mmol, 0.008 mmol, 0.016 mmol, and 0.032 mmol CuPcTs were used in the initial synthesis step, while other conditions keep unchanged. The unactivated samples derived from PPy were designated NC-1, NC-2, NC-3, and NC-4, respectively. The corresponding final obtained HPNCs were designated HPNC-1, HPNC-2, HPNC-3, and HPNC-4, respectively.
Material characterization
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were carried out using an JSM-7000F and a JEM-200CX, respectively. Thermogravimetric analysis (TGA) was performed in a temperature range of 20–750 °C under nitrogen atmosphere with a heating rate of 10 °C min−1 using a Pyris 1 DSC thermal analyzer. Powder X-ray diffraction (XRD) analysis was performed in a X'TRA with Cu Kα radiation from 3 to 65° at a step size of 0.02° to examine the crystalline morphology. Raman spectra of the HPNCs were analysed using a JY HR800. X-ray photoelectron spectroscopy (XPS) was conducted on PHI 5000 Versa Probe. Nitrogen adsorption–desorption isotherms were obtained using an ASAP 2020 at 77 K.Electrochemical measurement
The electrochemical measurements were performed in a three-electrode system using 1 M H2SO4 aqueous as electrolyte on a CHI660C electrochemical instrument. The working electrodes were prepared using 1 cm × 2 cm carbon cloth sheets as current collector. The carbon cloth sheets were immersed in 8 M HNO3 for 24 h before putting into use. The as-prepared HPNC and isopropanol were ultrasonicated for 0.5 h to form homogeneously dispersed solution at a concentration of 5 mg ml−1. The solution was drop-casted onto a carbon cloth sheet covering an area of 1 cm2, and then it was dried at 80 °C for 2 h. Each carbon cloth sheet was weighed before and after coverage of the HPNC to calculate the weight of the HPNC. Cyclic voltammetry (CV) and galvanostatic charge/discharge (GCD) measurements were carried out in a potential window from 0 to 0.8 V. The electrochemical impedance spectroscopy (EIS) was carried out in frequency ranged from 0.01 Hz to 10 KHz with a voltage amplitude of 5 mV. The cyclic stability was tested at a current density of 10 A g−1 for 5000 charging/discharging cycles. The specific capacitance of the HPNC on working electrode can be calculated from the discharge process according to the equation:
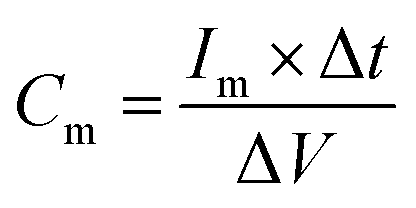 | (1) |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support from the Chinese National Key Fundamental Research Project (2017YFA0206302), the National Natural Science Foundation of China (61674078, 61229401, and 41401257), and the Natural Science Foundation of Jiangsu Province (BK20130055, BK20141054).Notes and references
- A. González, E. Goikolea, J. A. Barrena and R. Mysyk, Renewable Sustainable Energy Rev., 2016, 58, 1189–1206 CrossRef.
- H. Chen, T. N. Cong, W. Yang, C. Tan, Y. Li and Y. Ding, Prog. Nat. Sci., 2009, 19, 291–312 CrossRef.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P. L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 16070 CrossRef.
- F. Zhao, Y. Shi, L. Pan and G. Yu, Acc. Chem. Res., 2017, 50, 1734–1743 CrossRef PubMed.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef.
- Z. S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Müllen, Adv. Mater., 2012, 24, 5130–5135 CrossRef PubMed.
- T. Lin, I.-W. Chen, F. Liu, C. Yang, H. Bi, F. Xu and F. Huang, Science, 2015, 350, 1508–1513 CrossRef PubMed.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef PubMed.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- Y. Zhao, Z. Song, X. Li, Q. Sun, N. Cheng, S. Lawes and X. Sun, Energy Storage Materials, 2016, 2, 35–62 CrossRef.
- L. Wang, X. Feng, L. Ren, Q. Piao, J. Zhong, Y. Wang, H. Li, Y. Chen and B. Wang, J. Am. Chem. Soc., 2015, 137, 4920–4923 CrossRef PubMed.
- J. Gamby, P. L. Taberna, P. Simon, J. F. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109–116 CrossRef.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41–53 Search PubMed.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef PubMed.
- M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9, 1872–1876 CrossRef PubMed.
- Z. Tan, G. Chen and Y. Zhu, Nanocarbons Adv. Energy Storage, 2015, 1, 211–225 Search PubMed.
- Y. G. Wang, H. Q. Li and Y. Y. Xia, Adv. Mater., 2006, 18, 2619–2623 CrossRef.
- S. Biswas and L. T. Drzal, Chem. Mater., 2010, 22, 5667–5671 CrossRef.
- G. Yu, L. Hu, N. Liu, H. Wang, M. Vosgueritchian, Y. Yang, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 4438–4442 CrossRef PubMed.
- L. Peng, X. Peng, B. Liu, C. Wu, Y. Xie and G. Yu, Nano Lett., 2013, 13, 2151–2157 CrossRef PubMed.
- X. Zhu, Y. Zhu, S. Murali, M. D. Stoller and R. S. Ruoff, ACS Nano, 2011, 5, 3333–3338 CrossRef PubMed.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2013, 7, 174–182 CrossRef PubMed.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CrossRef PubMed.
- X. Yang, C. Cheng, Y. Wang, L. Qiu and D. Li, Science, 2013, 341, 534–537 CrossRef PubMed.
- Z. Li, Z. Xu, H. Wang, J. Ding, B. Zahiri, C. M. B. Holt, X. Tan and D. Mitlin, Energy Environ. Sci., 2014, 7, 1708–1718 Search PubMed.
- Q. Xie, R. Bao, A. Zheng, Y. Zhang, S. Wu, C. Xie and P. Zhao, ACS Sustainable Chem. Eng., 2016, 4, 1422–1430 CrossRef.
- Y. Li, G. Wang, T. Wei, Z. Fan and P. Yan, Nano Energy, 2016, 19, 165–175 CrossRef.
- J. W. F. To, Z. Chen, H. Yao, J. He, K. Kim, H. H. Chou, L. Pan, J. Wilcox, Y. Cui and Z. Bao, ACS Cent. Sci., 2015, 1, 68–76 CrossRef PubMed.
- C. Su, C. Pei, B. Wu, J. Qian and Y. Tan, Small, 2017, 13, 1–12 Search PubMed.
- Y. Shi, L. Pan, B. Liu, Y. Wang, Y. Cui, Z. Bao and G. Yu, J. Mater. Chem. A, 2014, 2, 6086–6091 Search PubMed.
- L. Pan, L. Pu, Y. Shi, S. Song, Z. Xu, R. Zhang and Y. Zheng, Adv. Mater., 2007, 19, 461–464 CrossRef.
- Y. Wang, Y. Shi, L. Pan, Y. Ding, Y. Zhao, Y. Li, Y. Shi and G. Yu, Nano Lett., 2015, 15, 7736–7741 CrossRef PubMed.
- S. Shrestha and W. E. Mustain, J. Electrochem. Soc., 2010, 157, B1665 CrossRef.
- A. Bello, F. Barzegar, M. J. Madito, D. Y. Momodu, A. A. Khaleed, T. M. Masikhwa, J. K. Dangbegnon and N. Manyala, Electrochim. Acta, 2016, 213, 107–114 CrossRef.
- J. A. Conesa, M. Sakurai and M. J. Antal, Carbon, 2000, 38, 839–848 CrossRef.
- X. Dai and M. J. Antal, Ind. Eng. Chem. Res., 1999, 38, 3386–3395 CrossRef.
- G. Ma, Q. Yang, K. Sun, H. Peng, F. Ran, X. Zhao and Z. Lei, Bioresour. Technol., 2015, 197, 137–142 CrossRef PubMed.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef PubMed.
- X. Huang, S. Kim, M. S. Heo, J. E. Kim, H. Suh and I. Kim, Langmuir, 2013, 29, 12266–12274 CrossRef PubMed.
- F. Su, C. K. Poh, J. S. Chen, G. Xu, D. Wang, Q. Li, J. Lin and X. W. Lou, Energy Environ. Sci., 2011, 4, 717–724 Search PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02110d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |