
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluences of novel microwave drying on dissolution of new formulated naproxen sodium
Maha Al-Ali ac,
P. R. Selvakannanb and
Rajarathinam Parthasarathy*a
ac,
P. R. Selvakannanb and
Rajarathinam Parthasarathy*a
aChemical and Environmental Engineering, School of Engineering, RMIT University, Melbourne, Victoria 3001, Australia. E-mail: rajarathinam.parthasarathy@rmit.edu.au
bApplied Chemistry, School of Science, RMIT University, Melbourne, Victoria 3001, Australia
cChemical Engineering Department, Engineering College, Tikrit University, Iraq
First published on 1st May 2018
Abstract
Drying of a pharmaceutical composition is an important step during its processing, which can affect its quality attributes including its texture, dispersion of the drug within the formulation, drug dissolution kinetics and eventually the drug's efficacy. This study presents the influence of varying drying techniques on the textural properties of the wet granulated formulation consisting of the drug naproxen sodium (NapSod) during the drying process. A new pharmaceutical formulation consisting of the NapSod drug was prepared by wet granulation and dried by novel microwave drying (MW), freeze drying (FD), vacuum drying (VD), and convective drying (CD) techniques before being processed in the form of tablets. The dissolution rate of NapSod from the tablet was measured in gastric (pH = 1.3) and intestinal fluid (pH = 6.8) mediums. The drug release was found to be influenced by the specific surface area, size distribution and the crystalline structure of dried particles, which were found to vary with the type of drying technique used as confirmed by the results of XRD, FTIR, SEM and particle size analyses. This study shows that using microwave technique to dry pharmaceutical granules containing a polar drug, such as NapSod, is an efficient and economical process, which can maintain the drug release at an appropriate rate to realize its desired pharmaceutical effect.
A Introduction
Naproxen sodium is widely used as an anti-inflammatory drug and prescribed as a painkiller for rheumatoid arthritis and osteoarthritis.1 The oral intake of the drug provides fast relief from the pain without any undesirable side effects. However, the release or dissolution rate of the drug and the factors that influence them need to be optimized to obtain improved pharmaceutical effects from the drug.Drug release rate is primarily the rate of mass transfer of the drug from the tablet into the dissolution medium for facilitating suitable pharmacological activity.2,3 The release rate of drugs is usually measured using dissolution tests that measure the physiological variation of the dissolved drug in the dissolution medium. Studying the dissolution rate provides critical information on the drug's physical and chemical stabilities.4 The major factors that can influence the drug dissolution rate are those that influence the nature of the interaction of the drug with the dissolution medium and other drug constituents such as the polymer fillers and binders. The influencing factors include the pH and ionic strength of the dissolution medium, drug's ionic nature,5–7 drug's physical and chemical properties,8 drug-polymer structure9 and drug formulation.10 Many studies available in the literature have investigated the influence of the above-mentioned factors on the dissolution kinetics of drugs from tablets. For example, the pH of the dissolution media has been shown to have a significant impact on the release behavior of the Asacol drug.11 Bhise et al. showed that the pH of the medium has a strong influence on the breakup tendency of the organic compounds of drugs into ionic species, which promote the drug release.7
Poor solubility of orally-administrated drugs can affect the release of drugs and hence its absorption in blood vessels due to the resulting low concentration gradient.5 Therefore, many studies in the literature have investigated the ways to enhance the dissolution rate of poorly soluble drugs. A recent review of the pharmaceutical particle technologies reviewed different methods used in various studies to improve the drug solubility.12 The effect of adding 5 or 20% v/v ethanol to a phosphate buffer with a pH of 6.5 and a fasted-state simulated intestinal fluid with a pH of 6.5 on the apparent solubility and dissolution of poorly soluble drugs such as naproxen was studied.13 It was found that there is a nine-fold increase in the apparent solubility of naproxen in the phosphate buffer and up to 4.5 fold increase in the fasted-state simulated fluid.13 Phaechamud and Ritthidej have attempted to study the influences of some formulation variables such as the amount of drug loading and lactose on the tree-layered matrix tablets containing chitosan and xanthan gum to improve the drug dissolution.14 Although increasing the amount of drug loading did not affect the release of these tablets, the addition of more lactose decreased the release of the tree-layered matrix tablets.14 Similarly, lipid-based solid dispersion15 and complexation with 2-hydroxypropyl-l∼iclodextrin (2-HPI3CD)16 were used to enhance the dissolution of naproxen oral. Bansal et al. used polyvinylpyrrolidones (PVPK-30 & PVPK-90) in the formulation of the drug as a binder to improve the dissolution and solubility of naproxen sodium.1
In addition to varying the formulation of the drug, the drying technique used in the preparation of the drug has been recognized to have a significant influence on the drug dissolution rate. Drugs, including naproxen, are usually blended with fillers and binders and subjected to wet granulation to achieve a uniform dispersion of the drug within the pharmaceutical formulation. Therefore, drying these formulations is essential to remove the excess solvent. The rate of drying and the nature of drying technique affect the textural properties of drug including its particle size, particle aggregation, crystallinity/amorphous ratio, surface area, etc., which eventually affect the dissolution of the drug. Bhise et al. studied the impacts of tray drying and freeze-drying on the ionic interactions of chitosan–naproxen sodium complexes.17 However, there are not many studies in the literature investigating the influence of different drying techniques on the dissolution kinetics of dried NapSod formulations. To understand the role of drying, four different drying methods namely microwave drying (MW), freeze drying (FD), convective drying (CD) and vacuum drying (VD) were employed in this work to dry the NapSod formulation containing NapSod, povidone, and microcrystalline cellulose. The aim of this work is to investigate the influence of drying technique on the kinetics of dissolution of NapSod formulations in solutions having the pH of gastric and intestinal liquids. In particular, microwave drying is used first time to dry NapSod formulations because the MW induced heating of polar drugs and water is more efficient and economical compared to other energy-intensive drying methods. Scanning Electron Microscopy (SEM), X-ray diffraction and particle size analyses of dried materials were carried out to correlate the results of dissolution studies with the textural properties of drug resulting from different drying methods. This study shows that the MW maintains the dissolution rate and solubility of NapSod formulation while being an energy efficient and economical drying technique.
B Experimental
Materials
The drug used in our experiments was Naproxen Sodium powder (NapSod), which is also known as (S)-6-methoxy-α-methyl-2-naphthaleneacetic acid (CAS: 26159-34-2; purity (titration by HClO4): 98.0–102.0%). The other materials used include microcrystalline cellulose powder (MCC) (CAS: 9004-34-6) and povidone powder, polyvinylpyrrolidone, (PVP) (CAS: 9003-39-8) which were used as filler and binder, respectively. All these chemicals were purchased from Sigma-Aldrich. The other laboratory grade chemicals used in our experiments include sodium chloride, hydrochloric acid, sodium dihydrogen phosphate, and sodium hydroxide. Both mediums of the simulated gastric and intestinal buffer solutions were prepared according to the specifications in the International European Pharmacopeia EP, 5th Ed., which are shown in Table 2.18,19 The gastric fluid medium was prepared by dissolving 2 g of sodium chloride in 800 mL of deionized water, and its pH was adjusted to 1.3 using hydrochloric acid. The solution was then diluted with water to obtain 1000 mL of the buffer solution.18,19 Similarly, the intestinal medium was prepared by dissolving about 6.9 g of sodium dihydrogen phosphate and 0.9 g of sodium hydroxide in 800 mL of deionized water. Consequently, the pH of the solution was adjusted to 6.8 using sodium hydroxide. The solution was then diluted with water to obtain 1000 mL of buffer solution.Methods
| Method | Operation conditions | ||
|---|---|---|---|
| Temperature (°C) | Pressure (kPa) | Time (min) | |
| VD | 60 | 67.7 | 210 |
| CD | 100 | 101.3 | 72 |
| FD | −55 | 0.004 | 390 |
| MW | 70 | 101.3 | 4.8 |
SEM images of the sample were obtained using Philips XL30 scanning electron microscope equipped with tungsten filament source. Samples for SEM analysis were prepared by dispersing 2–3 mg of the powder on a double-sided adhesive carbon tab mounted on aluminum stubs and were gently shaken off to remove the excess powder. The samples were then sputter coated under vacuum with a ∼30 nm layer of Au. The scan was performed by applying the secondary electron detector of 3 nm resolution at 30 kV and an accelerating voltage of 5 kV.
Powder X-ray diffraction patterns of samples were determined using Bruker AXS D4 Endeavour diffractometer with CuKα radiation. Samples were placed on a specimen holder at room temperature, and XRD patterns were collected over a 2θ range of 5 to 90° and 1.5406 Å wavelength using CuKα1 radiation at 40 kV, 35 mA at a scanning rate of 0.02° min−1. From the XRD patterns of DDP, WG and dried particles from FD, VD, CD and MW, percentage crystallinity and percentage amorphous were calculated using Gaussian peak fitting with a linear background or Gaussian peaks method as per eqn (1) and (2):
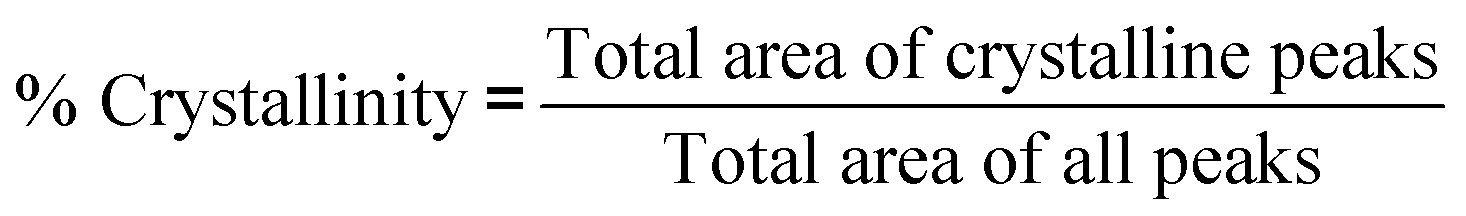 | (1) |
| % Amorphous = 100 − % Crystallinity | (2) |
Mastersizer (Malvern 3000, UK) that involves laser light scattering diffraction was used to measure the particle size distribution and the average specific surface area (m2 kg−1) distribution of DDP, WG, and dried particles. About 0.05 to 0.1 g of each powder sample was dispersed in a 100 mL vessel filled with acetone with a particle refractive index of 1.6 and an absorption index of 0.01. D50, the volume mean diameter, that represents the 50% of particles in the cumulative distribution, was used to denote the average particle size in μm. The average value of five scans of each run was used to determine the final result.
The porosity of tablets was calculated using eqn (3),21:
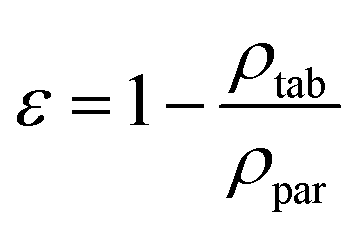 | (3) |
The dissolution rate tests in both the gastric and the intestinal mediums were performed in an agitated vessel equipped with a paddle running at 100 rpm. The solution volume used in the test was 1000 mL. All tests were conducted in triplicates at 37 ± 0.5 °C. During the test, the tablet was placed in a mesh basket and fully submerged in the solution. Samples were withdrawn every 5 minutes during the first 60 minutes and every hour for the next 9 hours. The samples collected were then filtered using a 45 μm PTFE membrane filter and then subjected to absorbance measurement by UV-vis spectrophotometer using a wavelength (λ) of 261 nm.23,24
C Results and discussion
Drug dissolution studies
The dissolution rate (Diss.R) of the tablets in (mg L−1 h) was calculated using the drug concentration change (mg L−1) in the dissolution medium within time interval Δti (h) in eqn (4).17,25 Eqn (5) was used to determine the cumulative drug content (DC.C%) at any given time or at the end of the experiment or drug release percentages (% release) of tablets at any given time in (mg mg−1).26
 | (4) |
 | (5) |
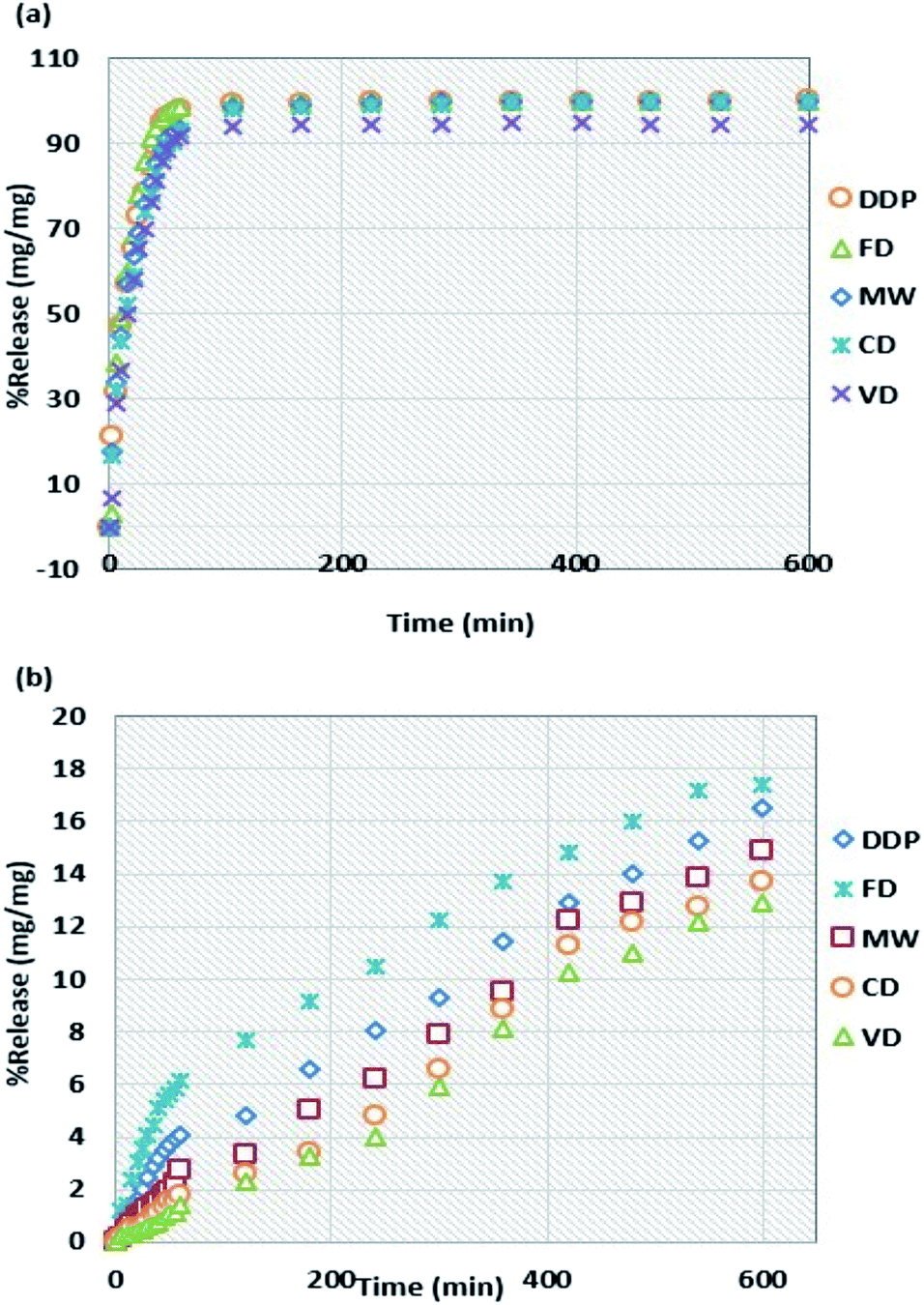 | ||
| Fig. 1 Release percentage of NapSod drug in (a) 6.8 pH medium and (b) 1.3 pH medium. Test duration: 10 h; all values shown are mean ± standard deviation. Standard deviation for (a) ± (3.64–4.94) and (b) ± (0.25–0.53); (n = 3). | ||
| Medium | pH | Composition | Weight (g) |
|---|---|---|---|
| Intestinal medium | 6.8 | KH2PO4 | 68.05 |
| NaOH | 8.96 | ||
| Deionized water | 1000 mL | ||
| Gastric medium | 1.3 | NaCl | 2 g |
| HCl | 70 g L−1 | ||
| Deionized water | 1000 mL |
Within the initial 60 minutes, the release percentages for the tablets dried by MW and CD in the intestinal medium have similar values of 93.4 and 93.1%, respectively while they have different release percentage values of 2.8 and 1.8%, respectively in the gastric medium. These results are consistent with the end-of-the-experiment cumulative drug content (DC.C) values shown in Table 3. Thus, the influence of drying methods on the release percentages of naproxen sodium in both gastric and intestinal mediums can be ranked in the following decreasing order: FD > DDP > MW > CD > VD.
| Tablet type | Gastric medium (mg mg−1) (pH = 1.3) | Intestinal medium (mg mg−1) (pH = 6.8) |
|---|---|---|
| DDP tablets | 16.54 ± 0.39 | 99.97 ± 4.94 |
| MW tablets | 14.93 ± 0.28 | 99.95 ± 4.73 |
| FD tablets | 17.42 ± 0.35 | 99.98 ± 3.59 |
| CD tablets | 13.75 ± 0.25 | 99.42 ± 4.31 |
| VD tablets | 12.88 ± 0.53 | 94.49 ± 3.64 |
Fig. 2a and b show the dissolution rates of tablets made of DDP and WG dried using the four drying methods as a function of time for intestinal and gastric mediums, respectively. The dissolution rate in the intestinal medium decreases sharply to low values after one hour for all samples and fluctuates around those values for the next 9 hours. The dissolution rate for DDP decreases from 3.28 mg L−1 min−1 after one hour to 0.040 mg L−1 min−1 after two hours. After two hours, the dissolution rates for tablets dried by CD, MW, FD, and VD are 0.210, 0.166, 0.030, and 0.025 mg L−1 min−1, respectively. After 10 hours, the dissolution rates for all samples reach values in the range of 0.003–0.008 mg L−1 min−1. Compared to the dissolution rates for the intestinal medium, the results for the gastric medium are much higher. After one hour, the dissolution rate for sample dried by FD decreases from 0.205 to 0.051 mg L−1 min−1 whereas the one for VD decreases from 0.047 to 0.030 mg L−1 min−1. The dissolution rates for samples dried by other drying methods lie in between those for FD and VD. The results during the next 9 hours for samples from all four drying methods fluctuate between the average rates of 0.041 to 0.046 mg L−1 min−1. The fluctuations in the dissolution rate results are not constant probably due to the slight variations of the solution temperature (±0.5 °C) during the test. From the results shown in Fig. 2b, it is observed that there were no significant changes in the dissolution rates in the gastric medium for samples obtained from all four drying methods after the 1st hr of the test.
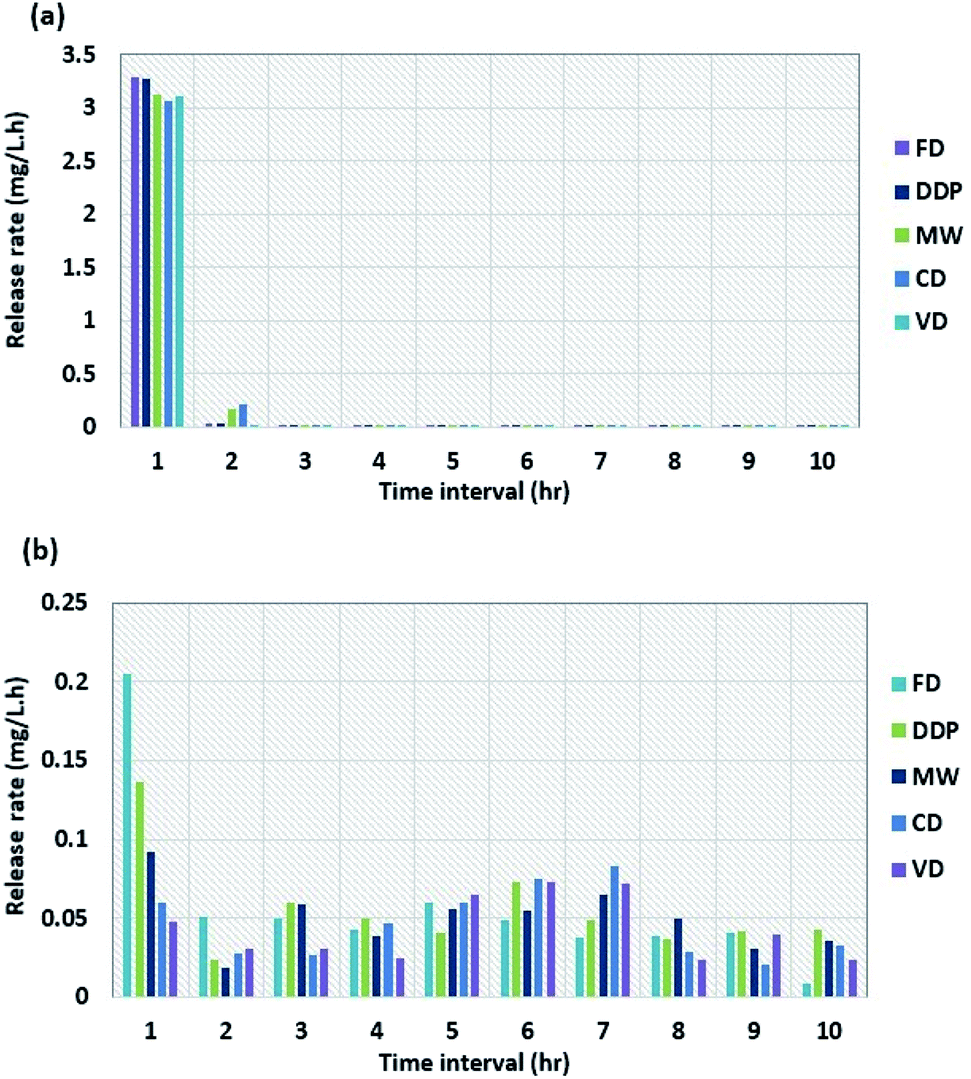 | ||
| Fig. 2 Dissolution rate of NapSod drug in (a) 6.8 pH medium and (b) 1.3 pH medium. Duration: 10 h; all values shown are mean ± standard deviation. Standard deviation for (a) ± (0.38–0.51), & (b) ± (0.02–0.06); (n = 3). | ||
SEM imaging of DDP and WG dried by the four drying methods
Scanning Electron Microscope (SEM) images of DDP, WG, and particles dried using the four drying methods are shown in Fig. (3) using the same scale. The SEM image for DDP shows that it was comprised of non-uniform semi-rectangular particles, needle-like and spherical particles of NapSod, MCC, and PVP, respectively with a mean particle size of 25.4 μm. The morphology of WG and dried particles are significantly different from that of DDP. The SEM image of WG shows that the wetting process has led to significant agglomeration of particles leading to an increase in the particle size. However, the removal of moisture during the drying process leads to the breakup of agglomerates and generation of smaller particles regardless of the drying technique used. Although the dried particles appear smaller than WG particles, their morphology is different from that of DDP. This can be ascribed to the change in the shape of particles of DDP due to the dissolution of sodium in NapSod in water during the wetting process to the extent that it cannot return to its original shape after drying. These changes in the morphology can influence the dissolution rate of the drugs as shown in the following sections. | ||
| Fig. 3 SEM images of DDP, WG, and the particles dried by FD, VD, CD, and MW – (scale bar = 500 μm). | ||
XRD profiles of the materials
The XRD (Powder X-Ray Diffraction analysis) patterns of DDP, WG and the particles dried using the four drying techniques are shown in Fig. 4. The XRD results provide insight into the crystallinity and the distribution of active components in the active matrix.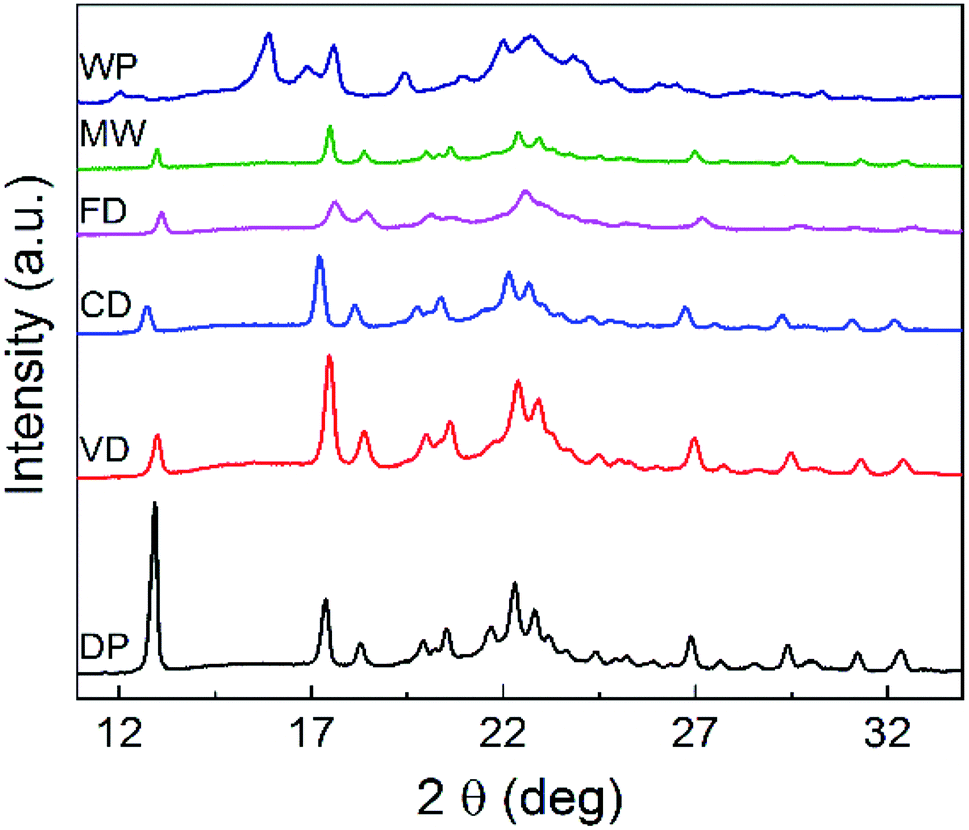 | ||
| Fig. 4 XRD patterns obtained for DDP, WG and the particles dried using the four drying techniques. | ||
XRD patterns for the granules dried by all four drying techniques are significantly different from the diffraction pattern of DDP. This difference is notable especially for the pack observed at 2θ = 12.9°. This peak appears to be shifted towards the right-hand side of the graph for the patterns of the particles dried by all four drying techniques. Moreover, the intensities of the peaks, as determined by their heights, are lower for samples from all drying methods compared to that for DDP. This shift in the position of the peak at 2θ = 12.9° and the variation in its intensity as measured by the peak height can be attributed to the greater level of dispersion of naproxen which eventually transforms the particles from the crystalline form into an amorphous form during the drying process. Thus, these observations confirm that crystallinity of materials dried by all four drying methods is significantly lower than that of DDP. These variations can, therefore, be expected to have an impact on the dissolution characteristics of the NapSod drug.28
In pharmaceutical applications, the correlations between drug bioavailability and drug particle size distribution and surface area are very important.29 The particle size of materials such as powders, suspensions, emulsions, and aerosols has significant influences on the drug characteristics, quality, and performance. The content uniformity, dissolution, and absorption rate are also significantly affected by the particle size of the active ingredient.30 To investigate how the drying methods used in this work influence the particle size and thereby the dissolution rate of the material, particle size measurements were carried out for DDP, and dried samples using Mastersizer analyzer and the results are shown in Table (4). The results of % crystallinity and % amorphous obtained from XRD analysis are included in Table (4). D50 values for particles dried by all four drying techniques are higher than that of DDP. D50 value of particles dried by MW (= 30.3 μm) is slightly higher than that of DDP (= 25.4 μm) while D50 of particles from VD (= 59.2 μm) is more than two times higher than that of DDP. There is no significant difference between the specific surface area values of DDP (= 363.8 m2 kg−1) and that of particles dried by MW (= 333.0 m2 kg−1). However, the specific surface area values for particles subjected to other three drying methods are significantly lower than that of DDP. The most remarkable decrease in the specific surface area occurs for particles dried by VD, which is 194.2 m2 kg−1. As the average size and specific surface area of particles decrease with drying, it can be expected that they will influence the release rate of the drug. The following sections will discuss the changes in the drug release rate as a function of drying technique.
The differences in the dissolution rate of tablets observed for different drying methods can be attributed to the differences in the particle size of the dried particles.30,31 Smaller particles are known to have higher solubility and tendency to dissolve readily than larger particles. DDP has the smallest average particle size (D50) of 25.4 μm followed by particles dried by MW with 30.3 μm, which suggests that particle dried by MW will have a dissolution rate closer to that of DDP. D50 values for samples from FD and CD are close to each other with 40.9 to 44.1 μm, respectively. D50 values of particles from FD and CD are higher than that of DDP which explain their lower dissolution rates, especially that for particles from CD. The dissolution rate of particles from FD is lower than that of DDP but is greater than that for particles from CD. It could be attributed to the higher percentage of the amorphous structure of tablet prepared using particles from FD which leads to higher percentage release as explained in the following section. However, the lower dissolution rate observed for particles from VD is mainly due to their higher D50 value (= 59.2 μm). The results obtained in this work agree well with the dissolution rate results obtained for the coenzyme Q10, which exhibits increased dissolution and diffusion rates when particle size decreases.32
Another factor that has an important effect on the drug release is the particle surface area. According to Noyes–Whitney equation (eqn (6)), the particle surface area is directly proportional to the dissolution rate.12
 | (6) |
| Sample name | DDP | MW | FD | CD | VD |
|---|---|---|---|---|---|
| a Values are mean ± standard deviation (0.4–0.6); n = 5.b Values are mean ± standard deviation (2.5–3.5); n = 5.c Tablet diameter = 13 mm ± 0.01. | |||||
| Particle sizea D50 (μm) | 25.4 | 30.3 | 40.9 | 44.1 | 59.2 |
| Particle specific surface areab (m2 kg−1) | 363.8 | 333.0 | 260.0 | 242.6 | 194.2 |
| % Crystallinity | 63.7 | 47.2 | 40.9 | 47.5 | 52.4 |
| % Amorphous | 36.3 | 52.8 | 59.1 | 52.5 | 47.6 |
| Tablet thicknessc | 2.67 | 2.36 | 2.43 | 2.33 | 2.30 |
| Total porosityc | 0.165 | 0.131 | 0.115 | 0.120 | 0.152 |
The above results may be explained using the crystallinity of particles because it is known to play a significant role in the drug release. The pharmaceutical formulation of tablets consisting a small percentage of crystalline materials and a large percentage of amorphous materials tends to have improved dissolution rate and solubility of the drug.1,28 The results shown in Table 4 indicate that the crystalline materials percentages for the tablets prepared by the four drying methods are lower than that of DDP. The amorphous nature of the tablet is higher especially for samples subjected to FD and MW, which are 59.1 and 52.8%, respectively compared to 36.3% for DDP. The relationship between the extent of transformation from the crystalline to an amorphous structure and the dissolution rate could be the reason for the highest and the next highest dissolution rates exhibited by samples from FD and MW, respectively.
Among the four different drying processes used, MW is determined to have the highest drying efficiency because it is shorter in duration and energy efficient. Therefore, MW was considered as the preferred drying process and studied in detail. Moreover, the mechanism of drying of the formulated NapSod drug is different for MW. MW decreases the average particle size and particle specific surface area and increases the amorphous structure of particles. These changes occur because MW depends on the rapid heating and quick evaporation of water from the sample. This mechanism could transform the crystalline materials into amorphous form, which subsequently could increase the dissolution rate of materials because of higher Gibbs free energy of amorphous structure.12,28 The crystalline percentage of samples for MW decreases from 63.7 to 47.2% as can be seen from the results shown in Table 4, which explains its high dissolution rate and solubility. Hence, the release percentage of tablets prepared by MW is influenced not only by the changes in particles size and the specific surface area but also by its crystalline structure. Table 5 shows the molar solubility of DDP and tablets dried by MW and other drying techniques in the intestinal and gastric mediums. The samples from MW exhibit the second highest molar solubilities with 43.459 and 8.488 mg mol L−1 in intestinal and gastric mediums, respectively after the samples from FD whose solubilities are 43.472 and 8.833 mg mol L−1, respectively. However, the samples from VD have the lowest solubilities in both intestinal and gastric mediums with 41.882 and 8.167 mg mol L−1, respectively. The solubilities of samples from all four drying methods have a dependency on particle size which is similar to that for release %. Smaller particles have a higher solubility than larger particles. The decrease in the particle size and the increase in the amorphous structure of samples after drying are the important factors that affect the solubility of the drug.1,9,28 Yu (2001) reported that the solubility of solids increased by increasing the amorphous state of the solids as these solids are physically and chemically not stable.33 Also, the solubility of a drug-polymer system was found to be enhanced with the increase in the extent of the amorphous structure,34 while the drugs with higher levels of the amorphous structure have higher dissolution rate because of their higher Gibbs free energy than those with the crystalline structure.12 Thus, the increase in the levels of the amorphous structure and the drastic decrease in the levels of the crystalline forms of the dried particles promote the solubility and the percentage release of the NapSod drug.28 In the present work, XRD pattern of the particles dried by MW shows clearly the transformation of crystalline components into amorphous forms, thereby providing further evidence to enhanced solubility of samples from MW (Fig. 4). On the other hand, the solubilities of NapSod tablets in gastric and intestinal mediums are found to be different. The solubility is found to be higher in the intestinal medium than that in the gastric medium. The reason is that naproxen is a weak acid drug with pKa of 4.15,27 which indicates a low solubility in the acidic medium with a pH of 1.3 and high solubility in the basic medium with a pH of 6.8. These results are very consistent with those of aceclofenac and ibuprofen drugs, which have pKa of 4.7 and 4.6, respectively, which have a strong solubility in a basic medium with a pH of 6.8 and poor solubility in an acidic medium with a pH of 1.2.35 Thus, the solubility of NapSod is proportional to its release in the intestinal and gastric mediums, because of its weak solubility in acidic mediums.5
Overall, wet formulation of NapSod requires a suitable drying process to enhance its dissolution. A comparison of the drying temperature and time, and the resulting mean particle size for the four drying methods used in this work is shown in Fig. 5. FD and MW led to similar results of enhanced dissolution. However, FD is energy intensive in terms of cooling, as it removes the water vapor at −55 °C. Moreover, it takes more than six hours to obtain similar drying efficiency. In contrast, MW is efficient, rapid, less energy intensive, but it produces the pharmaceutical composition that has desirable dissolution efficiency. The rapid and efficient MW drying is partly associated with the permanent dipole moment of the NapSod and water because MW acts on molecules that have a permanent dipole moment. When compared to CD and VD, heating in MW occurs from the moisture and the drug itself. Based on the above discussions it can be concluded that MW drying of the wet formulation of NapSod provides better dissolution of the drug than other drying processes.
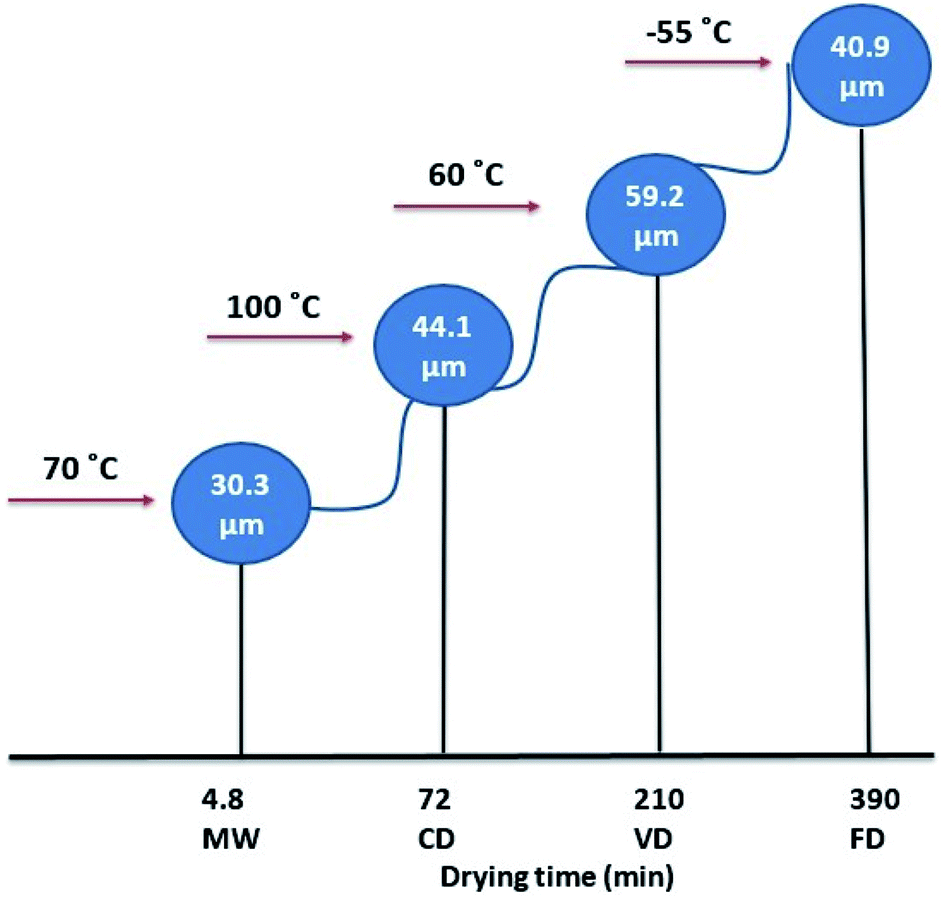 | ||
| Fig. 5 Comparison of drying temperature, drying time and particle size for four drying methods used in this work. | ||
Conclusions
To summarize, this study demonstrated the role of drying techniques used to remove the moisture in modifying the textural properties of the pharmaceutical composition and dissolution kinetics of a drug. Naproxen sodium was chosen as the drug and a novel wet formulation with an appropriate filler and binder was made. The wet granulated material was dried using four different drying methods namely freeze drying (FD), vacuum-drying (VD), convective drying (CD) and microwave drying (MW). The influences of the aforementioned drying techniques on the release and solubility of NapSod tablets in the gastric and intestinal mediums were studied and compared. The release rate of NapSod was rapid in the intestinal medium and slow in the gastric medium. The particle size, particle specific surface area, and the amorphous structure of drug particle were found to be the factors that control the dissolution of NapSod tablets significantly. Among the drug samples dried by the four drying methods, the one dried by MW was found to have the second highest dissolution rate due to its smaller particle size, large particle specific surface area and a high percentage of amorphous structure. The solubilities of samples from all four drying methods were found to be proportional to the dissolution rate. This study demonstrated clearly that the drying method plays a critical role in the dissolution of the drug, particularly MW which maintained the release and solubility of the NapSod drug. In addition, MW is more economical compared to other methods based on energy efficiency and drying time, which makes it a potential and promising drying technique in the production of powder-based pharmaceuticals.Conflicts of interest
We authors declare that “There are no conflicts to declare.”Acknowledgements
The authors would like to acknowledge the facilities and the technical assistance of the Australian Microscopy & Microanalysis Research Facility at RMIT University, the laboratory of Applied Chemistry School at RMIT University and Chemical Engineering Laboratory at RMIT University. Financial support from the Iraqi Ministry of Higher Education and Scientific Research is sincerely acknowledged.References
- P. Bansal, K. Haribhakti, V. Subramanian and F. Plakogiannis, Drug Dev. Ind. Pharm., 1994, 20, 2151–2156 CrossRef CAS.
- G. Singhvi and M. Singh, Int. J. Pharm. Sci. Res., 2011, 2, 77–84 Search PubMed.
- H. K. Shaikh, R. Kshirsagar and S. Patil, WJPPS, 2015, 4, 324–338 CAS.
- K. Singh, Asian J. Pharm., 2016, 10(4), S623–S627 CAS.
- K. S. Bhise, R. S. Dhumal, B. Chauhan, A. Paradkar and S. S. Kadam, AAPS PharmSciTech, 2007, 8, E110–E118 CrossRef PubMed.
- K. Asare-Addo, B. R. Conway, H. Larhrib, M. Levina, A. R. Rajabi-Siahboomi, J. Tetteh, J. Boateng and A. Nokhodchi, Colloids Surf., B, 2013, 111, 384–391 CrossRef CAS PubMed.
- K. S. Bhise, R. S. Dhumal, B. Shailesh, A. R. Paradkar and S. S. Kadam, AAPS PharmSciTech, 2010, 11, 226–232 CrossRef CAS PubMed.
- C. G. Wilson and P. J. Crowley, Controlled release in oral drug delivery, Springer, 2011 Search PubMed.
- Ö. B. Ünlüer, L. Genç, Ş. G. Kahyaoğlu and A. Ersöz, J. Pharm. Pharmacol., 2014, 2, 527–533 Search PubMed.
- S. C. Chattaraj and S. K. Das, Drug Dev. Ind. Pharm., 1996, 22, 555–559 CrossRef CAS.
- J. A. Spencer, Z. Gao, T. Moore, L. F. Buhse, P. F. Taday, D. A. Newnham, Y. Shen, A. Portieri and A. Husain, J. Pharm. Sci., 2008, 97, 1543–1550 CrossRef CAS PubMed.
- P. Khadka, J. Ro, H. Kim, I. Kim, J. T. Kim, H. Kim, J. M. Cho, G. Yun and J. Lee, Asian J. Pharm. Sci., 2014, 9, 304–316 CrossRef.
- J. H. Fagerberg, Y. Al-Tikriti, G. Ragnarsson and C. A. Bergström, Mol. Pharmaceutics, 2012, 9, 1942–1952 CrossRef CAS PubMed.
- T. Phaechamud and G. C. Ritthidej, AAPS PharmSciTech, 2008, 9, 870–877 CrossRef CAS PubMed.
- V. Nagabandi, A. K. Chandragiri, S. Thota and P. Katakam, J. Pharm. Sci. Res., 2014, 6, 78–82 Search PubMed.
- B.-J. Lee and J.-R. Lee, Arch. Pharmacal Res., 1995, 18, 22–26 CrossRef CAS.
- K. S. Bhise, R. S. Dhumal, A. R. Paradkar and S. S. Kadam, AAPS PharmSciTech, 2008, 9, 1–12 CrossRef CAS PubMed.
- W. H. Organization, 1994.
- M. Berteli, E. Rodier and A. Marsaioli Jr, Braz. J. Chem. Eng., 2009, 26, 317–329 CrossRef CAS.
- P. Di Martino, L. Malaj, R. Censi and S. Martelli, J. Pharm. Sci., 2008, 97, 5263–5273 CrossRef CAS PubMed.
- A. Juban, C. Nouguier-Lehon, S. Briancon, T. Hoc and F. Puel, Int. J. Pharm., 2015, 490, 438–445 CrossRef CAS PubMed.
- K. Hajera and Z. Zaheer, Int. J. PharmTech Res., 2012, 4, 661–668 Search PubMed.
- W. McMinn, C. McLoughlin and T. Magee, Drying Technol., 2005, 23, 513–532 CrossRef CAS.
- W. H. Organization, The international pharmacopoeia, World Health Organization, 2006 Search PubMed.
- K. Čerpnjak, A. Zvonar, F. Vrečer and M. Gašperlin, Int. J. Pharm., 2015, 485, 215–228 CrossRef PubMed.
- K. Adibkia, M. Barzegar-Jalali, H. Maheri-Esfanjani, S. Ghanbarzadeh, J. Shokri, A. Sabzevari and Y. Javadzadeh, Powder Technol., 2013, 246, 448–455 CrossRef CAS.
- H. Sevelius, R. Runkel, E. Segre and S. Bloomfield, Br. J. Clin. Pharmacol., 1980, 10, 259–263 CrossRef CAS PubMed.
- M. Dixit, P. K. Kulkarni and R. N. Charyulu, World J. Pharm. Pharm. Sci., 2015, 4, 715–725 CAS.
- P. Sharma, S. Ganta and S. Garg, Scale-Up and Postapproval Changes (SUPAC) Regulations, Wiley Online Library, 2008 Search PubMed.
- K. Harun, M. Mistafa and S. Alisa, Int. J. Pharm. Pharm. Sci., 2013, 5, 775–779 Search PubMed.
- S. C. Gad, Pharmaceutical manufacturing handbook: production and processes, John Wiley & Sons, 2008 Search PubMed.
- J. Sun, F. Wang, Y. Sui, Z. She, W. Zhai, C. Wang and Y. Deng, Int. J. Nanomed., 2012, 7, 5733 CAS.
- L. Yu, Adv. Drug Delivery Rev., 2001, 48, 27–42 CrossRef CAS PubMed.
- M. M. Knopp, L. Tajber, Y. Tian, N. E. Olesen, D. S. Jones, A. Kozyra, K. Löbmann, K. Paluch, C. M. Brennan and R. Holm, Mol. Pharmaceutics, 2015, 12, 3408–3419 CrossRef CAS PubMed.
- K. R. Chu, E. Lee, S. H. Jeong and E.-S. Park, Arch. Pharmacal Res., 2012, 35, 1187–1195 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |