
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed intramolecular C–H arylation of 2-halo-N-Boc-N-arylbenzamides for the synthesis of N–H phenanthridinones†
Quan-Fang
Hu
 ,
Tian-Tao
Gao
,
Yao-Jie
Shi
,
Qian
Lei
and
Luo-Ting
Yu
*
,
Tian-Tao
Gao
,
Yao-Jie
Shi
,
Qian
Lei
and
Luo-Ting
Yu
*
State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, West China Medical School, Sichuan University and Collaborative Innovation Center, Chengdu 610041, China. E-mail: yuluot@scu.edu.cn; Fax: +86-28-85164060; Tel: +86-28-85164063
First published on 13th April 2018
Abstract
A palladium catalyzed synthesis of N–H phenanthridinones was developed via C–H arylation. The protocol gives phenanthridinones regioselectively by one-pot reaction without deprotection. It exhibits broad substrate scope and affords targets in up to 95% yields. Importantly, it could be applied for the less reactive o-chlorobenzamides.
Introduction
Palladium-catalyzed direct functionalization of C–H bonds has become one of the most efficient and environmentally friendly mild procedures for building carbon–carbon bonds.1 It holds a special place among various types of palladium-catalyzed coupling reactions. C–H direct arylation is its typical application, which is catalyzed by Pd(0) or Pd(II) with phosphine ligands operating under a Pd(0)/Pd(II) or Pd(II)/Pd(IV) catalytic cycle with or without the assistance of base.2 This coupling strategy has been well developed in terms of regioselectivity and efficiency; the tolerance of diverse functional groups makes the C–H arylation particularly versatile for organic synthesis. The starting materials may be easily achieved and thus this strategy has been used for building a variety of aromatic and heteroaromatic systems.3Phenanthridinones are important structural units found in many natural products and pharmaceuticals that exhibit wide range of biological activities.4 A variety of novel synthetic approaches have been developed to the synthesis of the phenanthridinone cores and related lactams, most of them are based on palladium catalyzed biaryl coupling by the regioselective C–H bond activation (Scheme 1a, synthesis of N-alkyl phenanthridinones).5 Although they are effective for synthesis of N-alkyl phenanthridinones, the obvious drawbacks were observed when applied to N–H phenanthridinones.5e,6 Their scope of substrates is quite limited and high reaction temperature is necessary. To the best of our knowledge, only the yields with iodo-substituted anilides are reported. Usually a complex mixture of side-products is observed, decreasing the yield of desired product.6a,6b This is possibly resulted from the coordination of nitrogen to palladium in the presence of the NH free amide.5e Earlier reported efficient protocols for such analog were mediated by potassium tert-butoxide7 or photochemistry.8,9 However, those procedures are not regioselective with substituted anilides due to radical cyclization mechanism (Scheme 1b, radical pathway for synthesis of N–H phenanthridinones), limiting application of substrate scopes.7 So based on those disadvantages researchers reported utilization of protection groups which could be easily cleaved after the ring closure. Sandro Cacchi and co-workers5e developed N-benzyl derivatives of N-benzoyl-o-iodoanilides which can be converted into the corresponding phenanthridinones in good to high yields, but needed one more deprotection step in TFA. François Tillequin et al.6d developed Boc as protection and leaving group, however, one equivalent of Pd catalyst and two equivalents of phosphine ligands were involved, and only 26% yield was obtained.
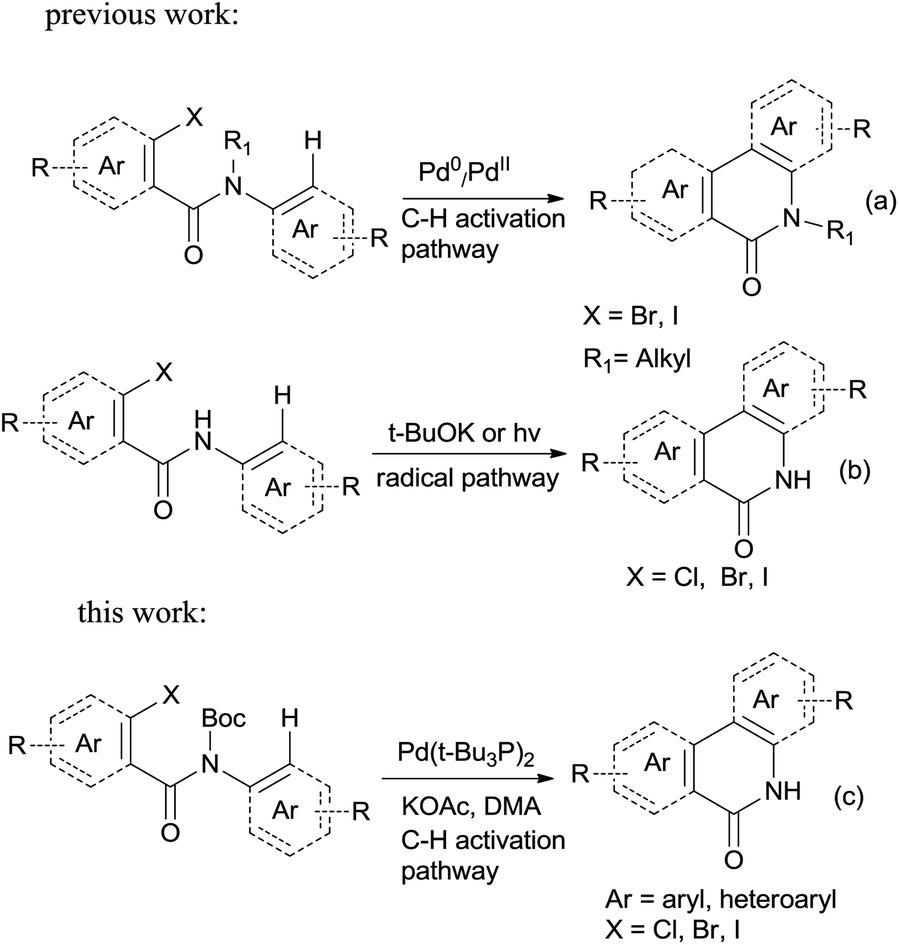 | ||
| Scheme 1 Synthesis of phenanthridinones via intramolecular cyclization. | ||
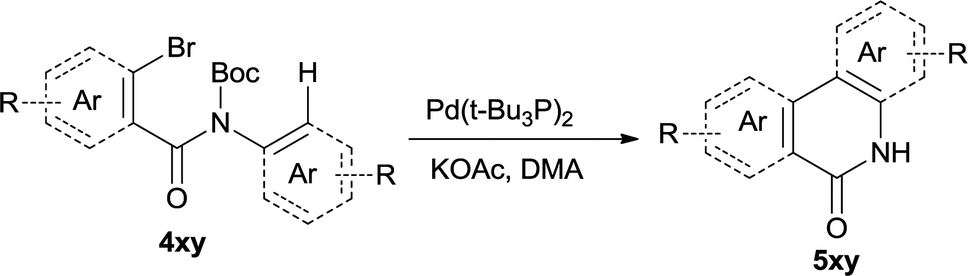
In this account it is important to develop an operationally simple catalyst system for direct intramolecular arylation processes exhibiting broad scope for aryl chlorides, bromides, and iodides. Here we disclosed a palladium catalyzed one pot C–H activation protocol, cyclization and decarboxylation of N-Boc protected o-halobenzamides to produce phenanthridinones directly, which gave good to excellent yields as high as 95% (Scheme 1c, synthesis of N–H phenanthridinones by C–H activation from Boc protected amide). It can be easily scaled up even with increasing of yield. The reaction was promoted by Pd(t-Bu3P)2 combining together with KOAc, a convenient, commercial available catalyst inexpensive, but highly efficient to produce a broad range of phenanthridinones.
Results and discussion
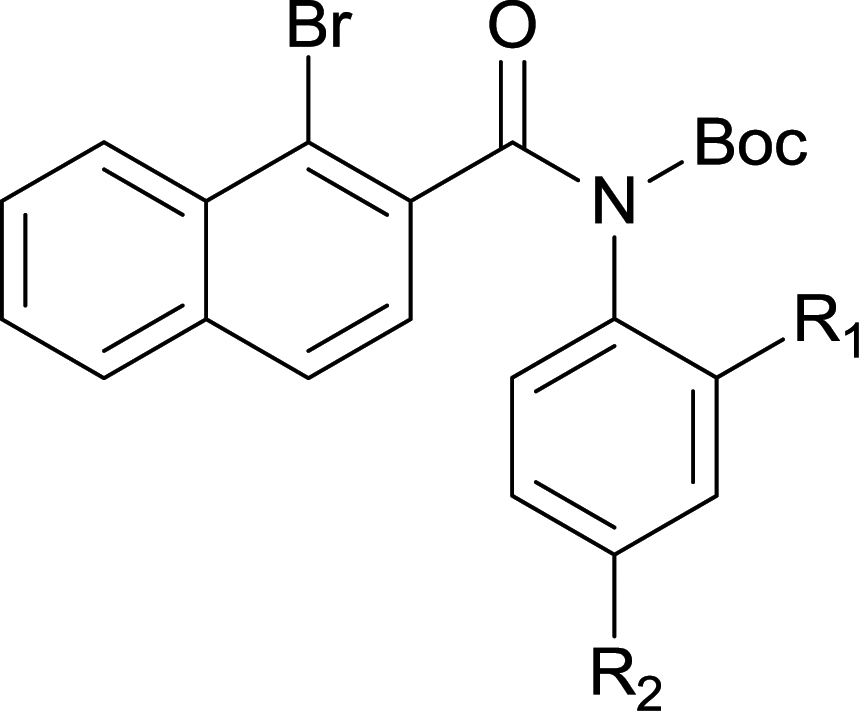
The synthesis of the amide starting material 4xy for this investigation is shown in Scheme 2. Ortho-halogen substituted arylcarboxylic acid was easily converted to acyl chloride, which reacted with aniline to give amide 3xy. Then under catalytic amount of DMAP in DCM, 3xy was almost quantitatively converted to 4xy.10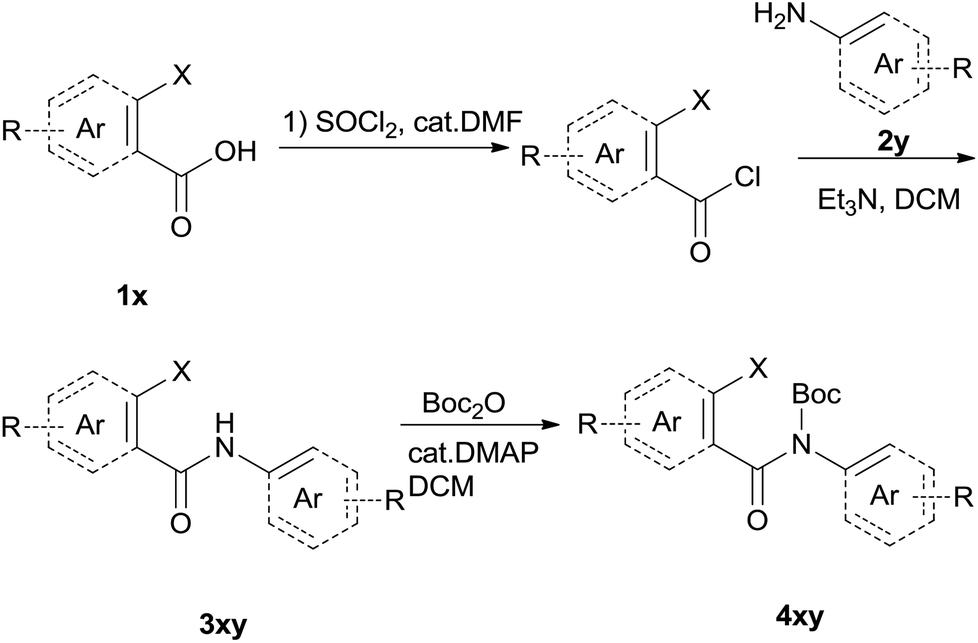 | ||
| Scheme 2 Synthesis of cyclization substrates. | ||
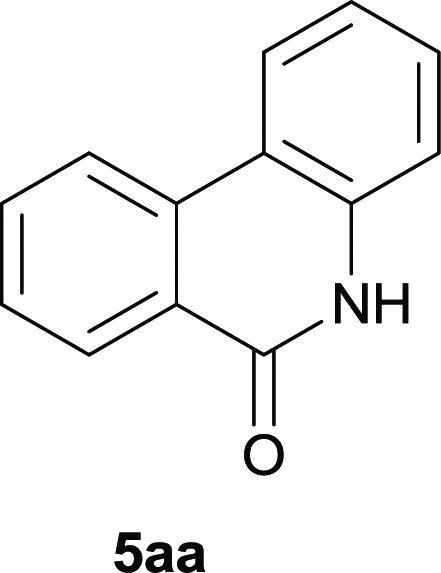
In our initial investigation to optimize cyclization conditions of 4xy, we selected 4aa as starting point for screening, which is briefly summarized in Table 1. The C–H direct arylation is strongly dependent on the catalyst, the solvent and the base. Firstly we tried most popular coupling catalyst/base system (entries 1–3),11 but obtained low yields; Pd(OAc)2/Bu3P/Ag2CO3 system12 gave better yield (38%). After that, we tried Pd(PCy3)2 under various basic conditions, and found the yield can be increased to 48% when KOAc was used. Speculating that different ligand could be used to fine-tune the reaction due to its electronic and steric particularities,2d we further screened Pd(t-Bu3P)2 catalyst13 with different bases and finally identified Pd(t-Bu3P)2/KOAc was the best combination (entry 11, 72% yield). Other bases with different cation or anion were inferior (entries 9, 10). When the optimized condition was utilized with non-Boc protected substrate 3aa (entry 12), the strategy was not effective, suggesting that unprotected NH completely inhibited the reaction, presumably resulted by coordination of nitrogen to palladium in the presence of the NH free amide.5e,14
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst/ligand (mol%) | Base (equiv.) | Subs. conv. (%) | Yieldb (%) |
| a Reaction conditions: 4aa (0.5 mmol), Pd catalyst and base in DMA (5 ml) were heated at 120 °C for 90 min under Ar atmosphere. b Yield determined by LCMS. c 3aa was used as substrate. | ||||
| 1 | Pd(PPh3)4 (5) | K2CO3 (2) | 100 | 22 |
| 2 | Pd(OAc)2/PCy3·HBF4 (5/10) | Cs2CO3 (2) | 100 | 33 |
| 3 | Pd(OAc)2/Bu3P (5/10) | Ag2CO3 (2) | 89 | 38 |
| 4 | Pd(PCy3)2 (5) | Cs2CO3 (2) | 97 | 35 |
| 5 | Pd(PCy3)2 (5) | K2CO3 | 98 | 31 |
| 6 | Pd(PCy3)2 (5) | AgOAc (4) | 83 | 20 |
| 7 | Pd(PCy3)2 (5) | K3PO4 (2) | 88 | 11 |
| 8 | Pd(PCy3)2 (5) | KOAc (4) | 89 | 48 |
| 9 | Pd(t-Bu3P)2 (5) | Cs2CO3 (2) | 98 | 38 |
| 10 | Pd(t-Bu3P)2 (5) | AgOAc (4) | 82 | 23 |
| 11 | Pd(t-Bu3P)2 (5) | KOAc (4) | 100 | 72 |
| 12c | Pd(t-Bu3P)2 (5) | KOAc (4) | 18 | Trace |
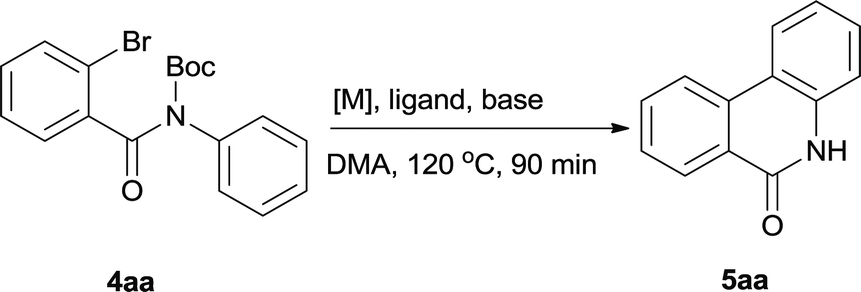
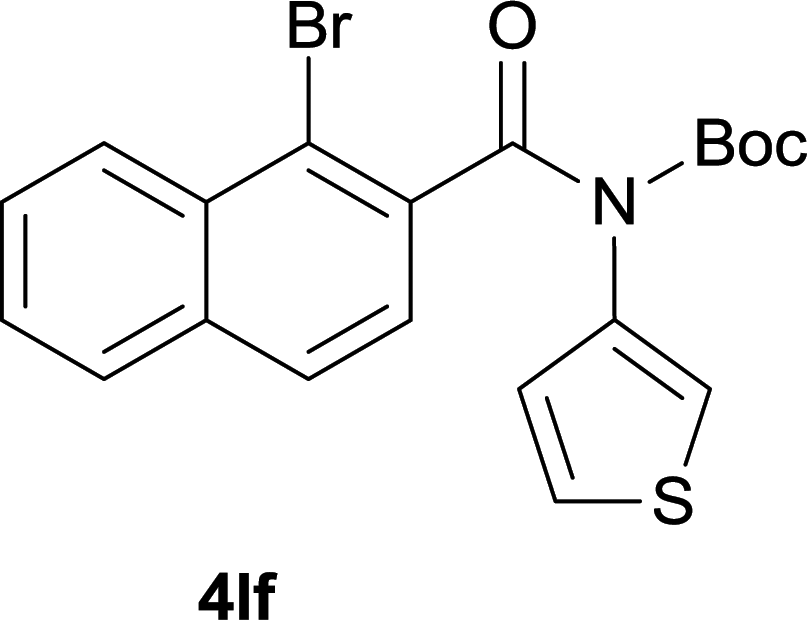
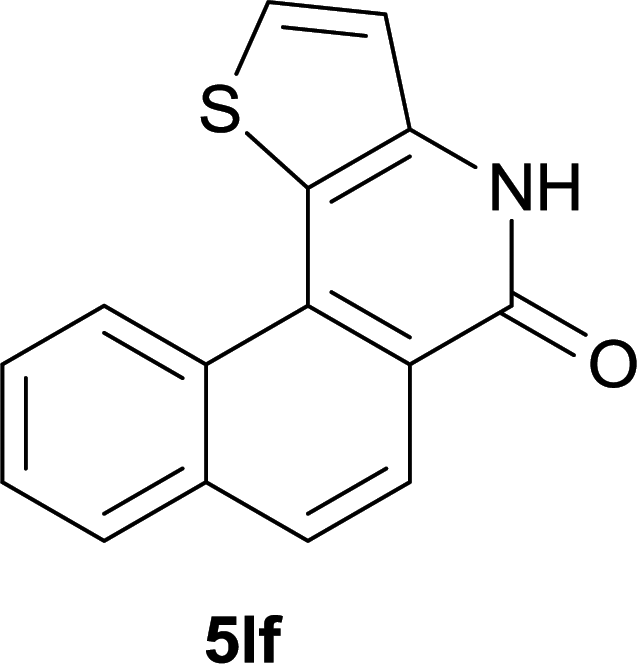
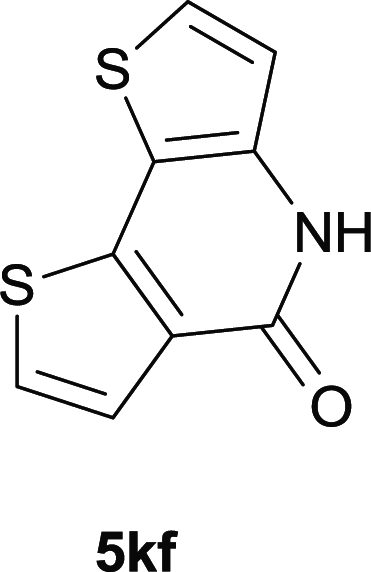
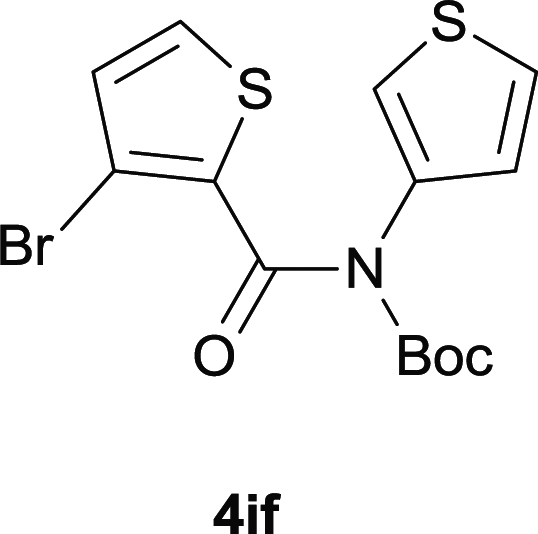
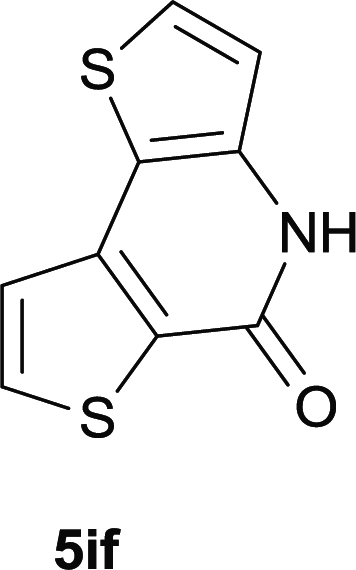
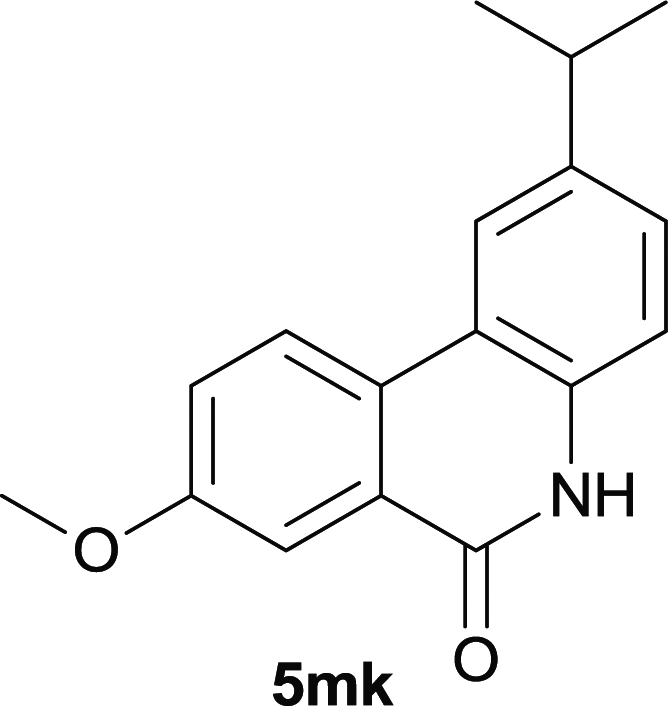
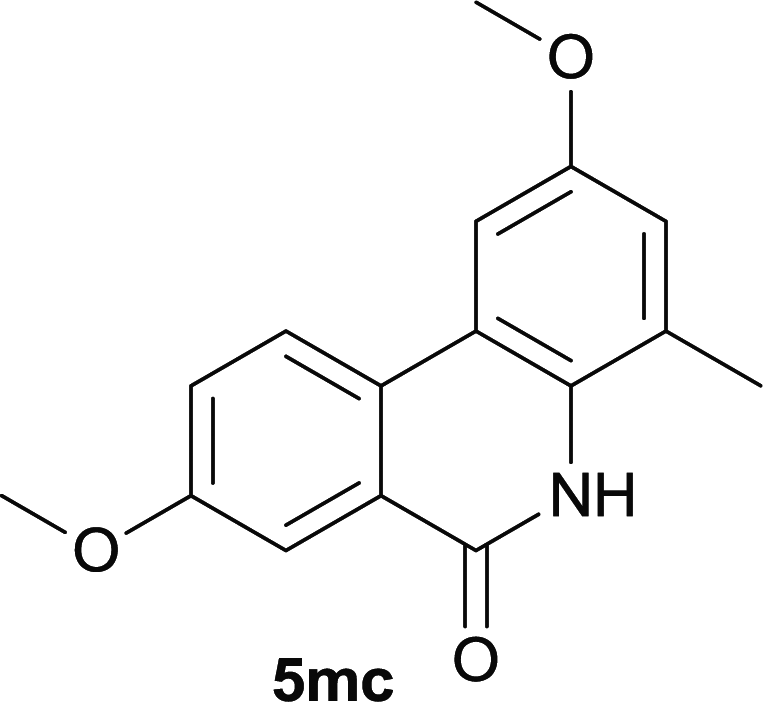
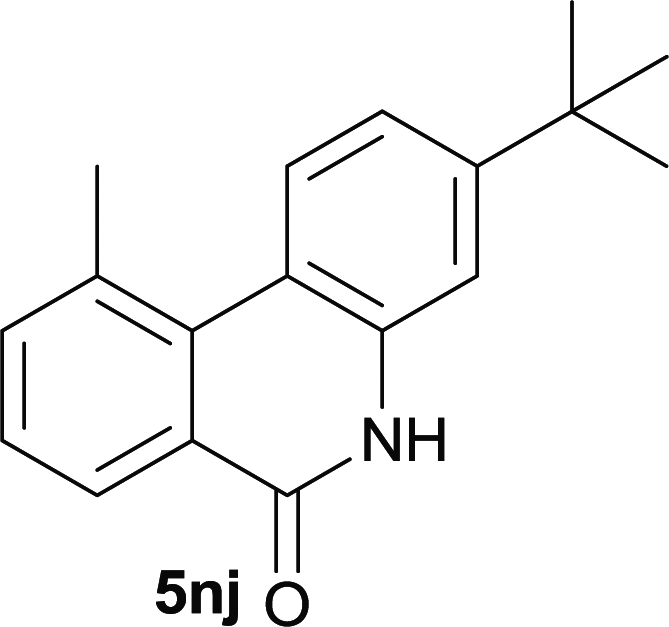
After optimization of the reaction condition, we attempted to extend the scope and generality of the intramolecular coupling as shown in Table 2. Various aryl acids and aryl amines were examined. Electron-rich groups (e.g. Me and OMe) either on the acid partner or on the aniline partner could smoothly undergo cyclization-decarboxylation which exclusively provided the desired products in 81–92% yields (entries 1–5). Notably, naphthyl-based substrates 4lb and 4lg also gave good to excellent yields (75–95%). 4lg gave lower yield due to slight electron deficiency. When this protocol was subjected to strong electron-deficient substrate such as cyano group (5jh, entry 8), the yield reduced sharply. To explore the cyclization position with respect to meta-substituted anilines, we tested meta-isopropyl aniline (entry 9). The reaction gave para and ortho-cyclization mixtures (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); However when tert-butyl analine was tested (entry 10), only para-cyclization was produced, presumably due to stronger steric hindrance. It was worth noting that the 3-amino thiophene also gave moderate to high yields (61–93%, entries 11–14), suggesting it's high C–H activation activity and regioselectivity. NMR confirmed the coupling is at second position. The reaction scope is not limited to benzoic acids, thiophene-carboxylic acids could also go cyclization smoothly (entries 15–20) with aniline or aminothiophene. Similar to early Pd-catalyzated protocol,15 we found that pyridine ring was not an effective acid partner under our reaction condition (entries 21–23). To further evaluation the protocol's scaling up efficiency, we tested 4lb/4af/4kc as substrates for gram scale synthesis. To our delight, the yields increased up from 91%/84%/83% to 95%/93%/91% respectively.
:1); However when tert-butyl analine was tested (entry 10), only para-cyclization was produced, presumably due to stronger steric hindrance. It was worth noting that the 3-amino thiophene also gave moderate to high yields (61–93%, entries 11–14), suggesting it's high C–H activation activity and regioselectivity. NMR confirmed the coupling is at second position. The reaction scope is not limited to benzoic acids, thiophene-carboxylic acids could also go cyclization smoothly (entries 15–20) with aniline or aminothiophene. Similar to early Pd-catalyzated protocol,15 we found that pyridine ring was not an effective acid partner under our reaction condition (entries 21–23). To further evaluation the protocol's scaling up efficiency, we tested 4lb/4af/4kc as substrates for gram scale synthesis. To our delight, the yields increased up from 91%/84%/83% to 95%/93%/91% respectively.
|
|
|||
|---|---|---|---|
| Entry | 4xy | 5xy | Yieldb [%] |
| a Reaction conditions: 1 mmol of substrate, 4 equiv. of KOAc, and 5% equiv. of Pd (t-Bu3P)2 in 10 ml DMA were heated at 120 °C for 2 h under Ar atmosphere. b Isolated yield. c Reaction conducted at 20 mmol scale. d Reacted at 135 °C. | |||
|
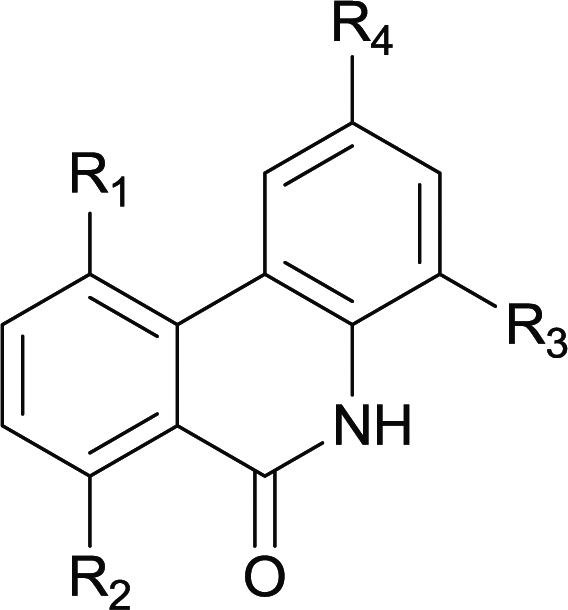
|
||
| 1 | 4ab R1 = R2 = R3 = H R4 = OMe | 5ab R1 = R2 = R3 = H R4 = OMe | 88 |
| 2 | 4db R1 = Me R2 = R3 = H R4 = OMe | 5db R1 = Me R2 = R3 = H R4 = OMe | 81 |
| 3 | 4dc R1 = Me R2 = H R3 = Me R4 = OMe | 5dc R1 = Me R2 = H R3 = Me R4 = OMe | 87 |
| 4 | 4ec R1 = H R2 = OMe R3 = Me R4 = OMe | 5ec R1 = H R2 = OMe R3 = Me R4 = OMe | 92 |
| 5 | 4hd R1 = H R2 = F R3 = Cl R4 = OMe | 5hd R1 = H R2 = F R3 = Cl R4 = OMe | 89 |
|
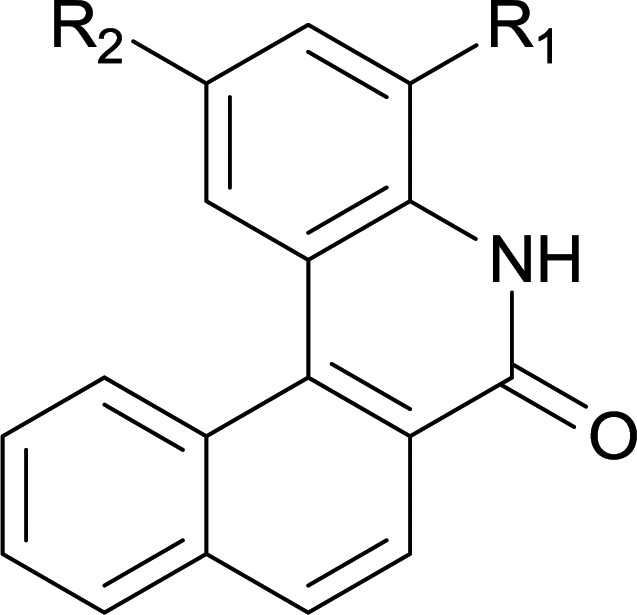
|
||
| 6 | 4lb R1 = H R2 = OMe | 5lb R1 = H R2 = OMe | 91/95c |
| 7 | 4lg R1 = Cl R2 = H | 5lg R1 = Cl R2 = H | 75 |
| 8 |
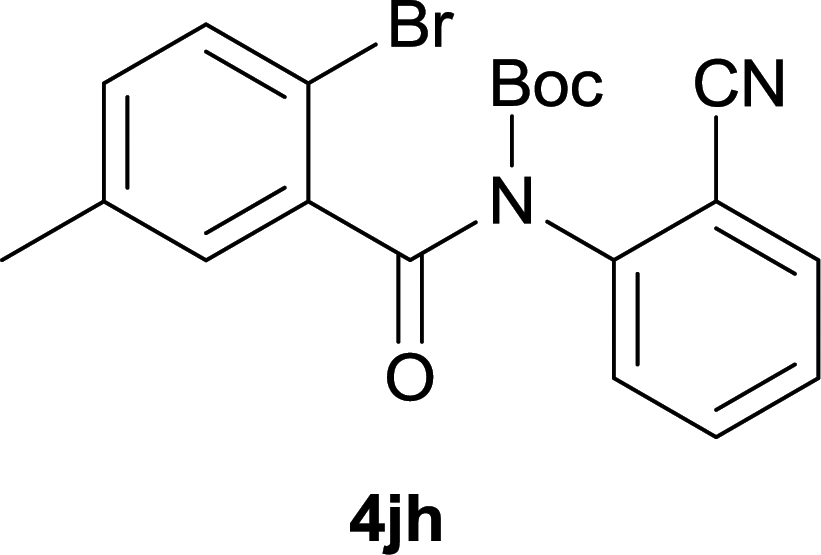
|
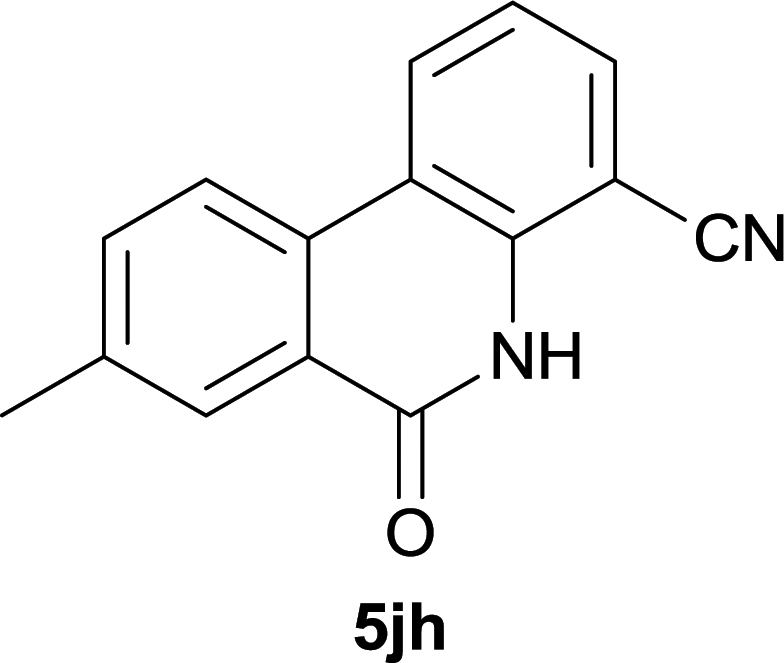
|
37d |
| 9 |
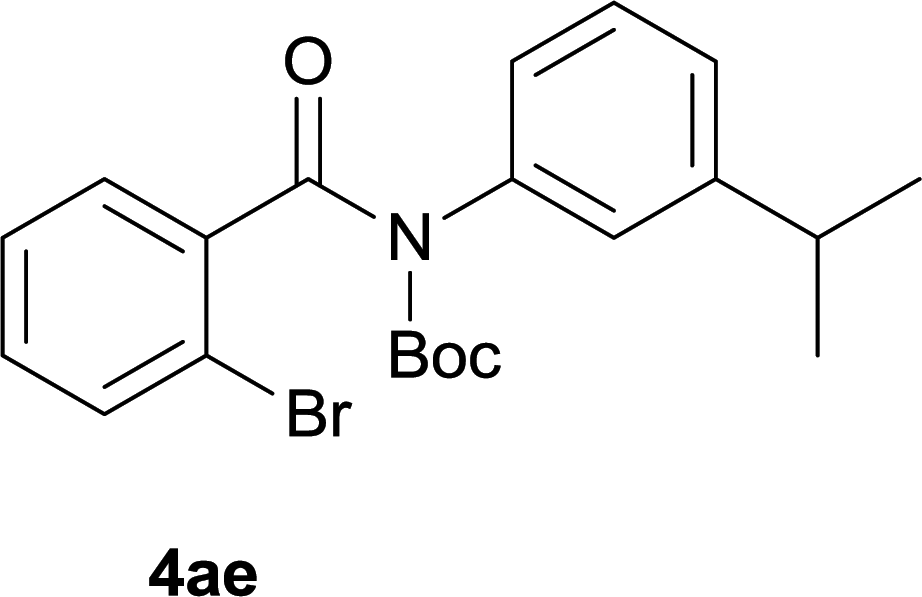
|
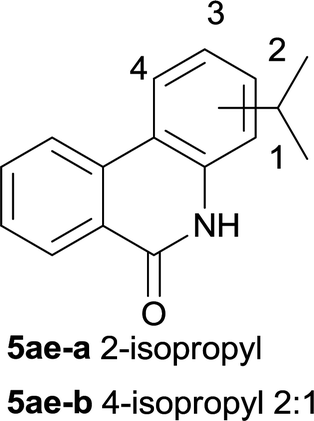
|
78 |
| 10 |
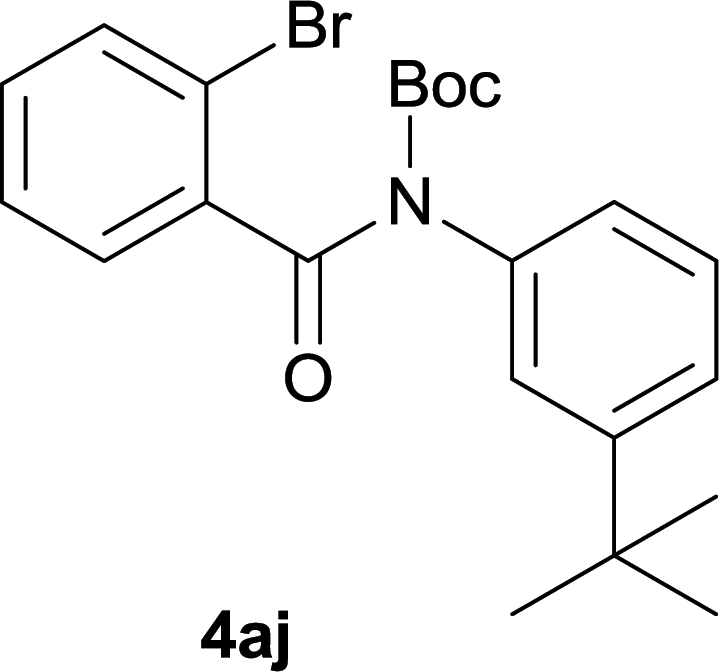
|
|
82 |
|
|
||
| 11 | 4af R1 = R2 = H | 5af R1 = R2 = H | 84/93c |
| 12 | 4df R1 = Me R2 = H | 5df R1 = Me R2 = H | 71 |
| 13 | 4ef R1 = H R2 = OMe | 5ef R1 = H R2 = OMe | 61 |
| 14 |
|
|
91 |
|
|
||
| 15 | 4kc R1 = Me R2 = OMe | 5kc R1 = Me R2 = OMe | 83/91c |
| 16 | 4kg R1 =Cl R2 = H | 5kg R1 =Cl R2 = H | 71 |
| 17 |
|
|
84 |
| 18 |
|
|
85 |
|

|
||
| 19 | 4fb R1 = H R2 = OMe | 5fb R1 = H R2 = OMe | 62 |
| 20 | 4fc R1 = Me R2 = OMe | 5fc R1 = Me R2 = OMe | 58 |
|
|
||
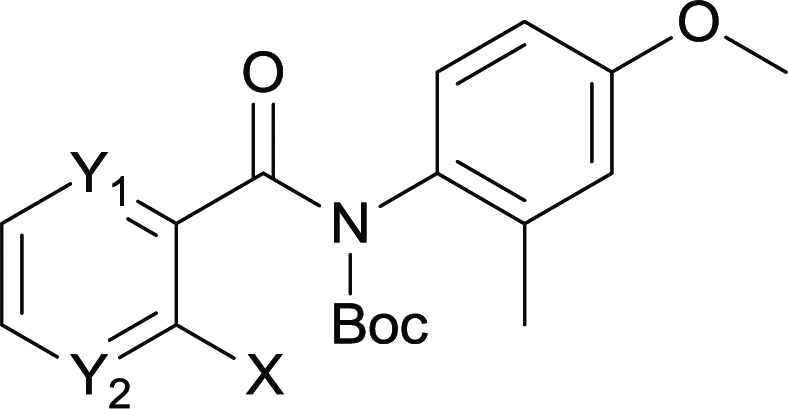
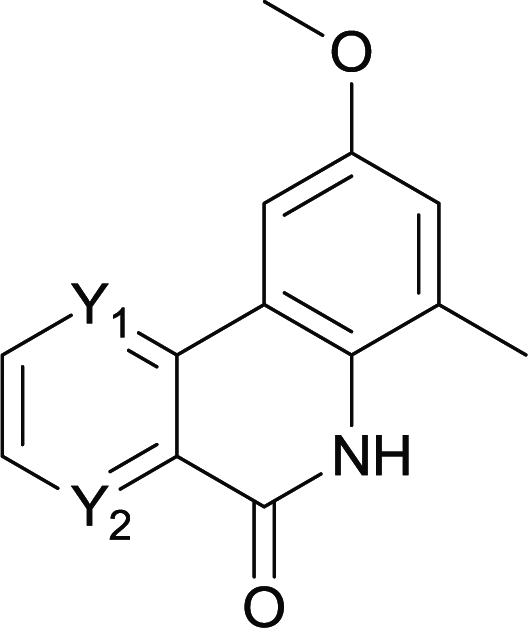
| 21 | 4rc Y1 = C Y2 = N X = Br | 5rc Y1 = C Y2 = N | 0 |
| 22 | 4sc Y1 = N Y2 = C X = Br | 5sc Y1 = N Y2 = C | 0 |
| 23 | 4tc Y1 = N Y2 = C X = I | 5tc Y1 = N Y2 = C | 0 |
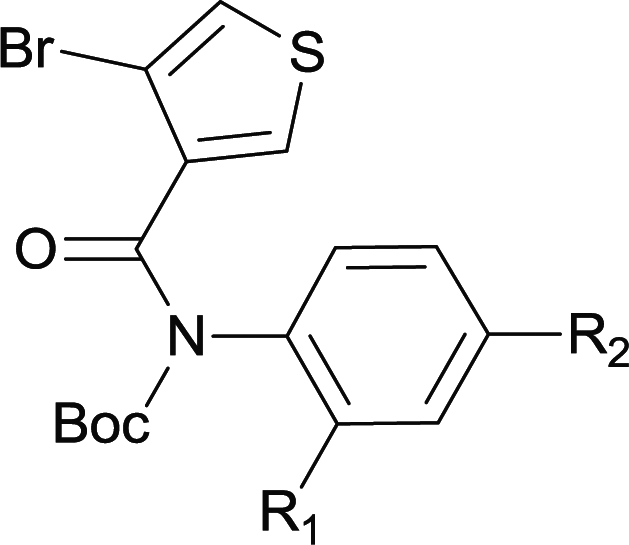
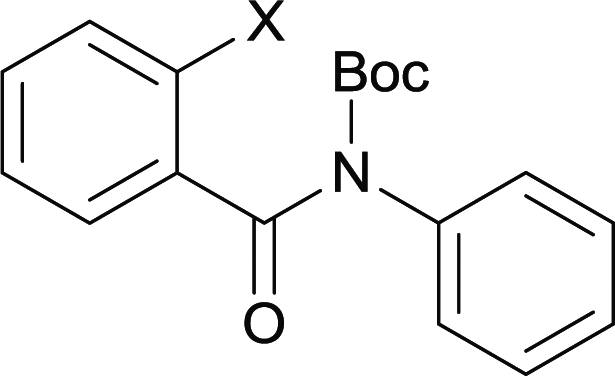
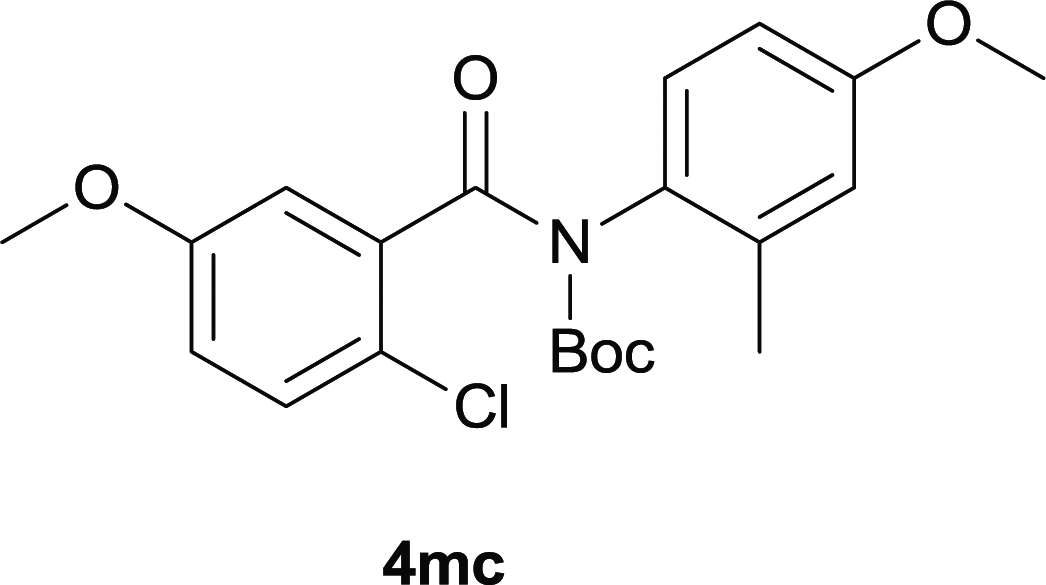
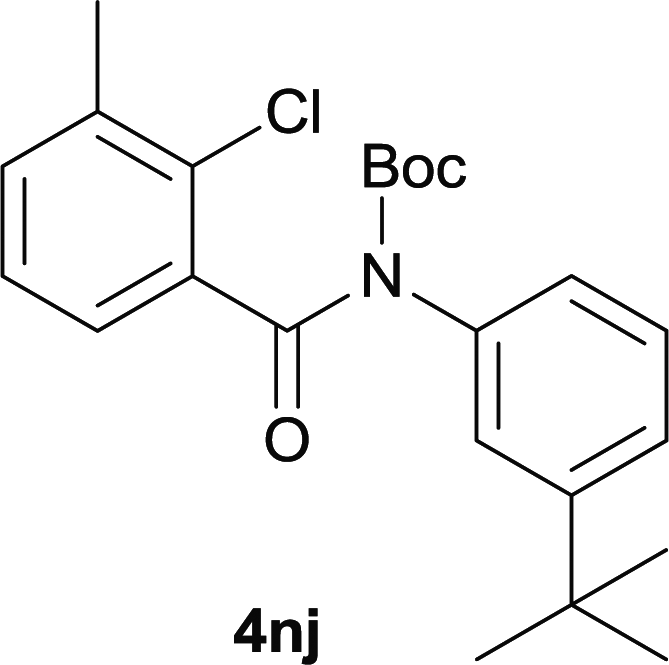
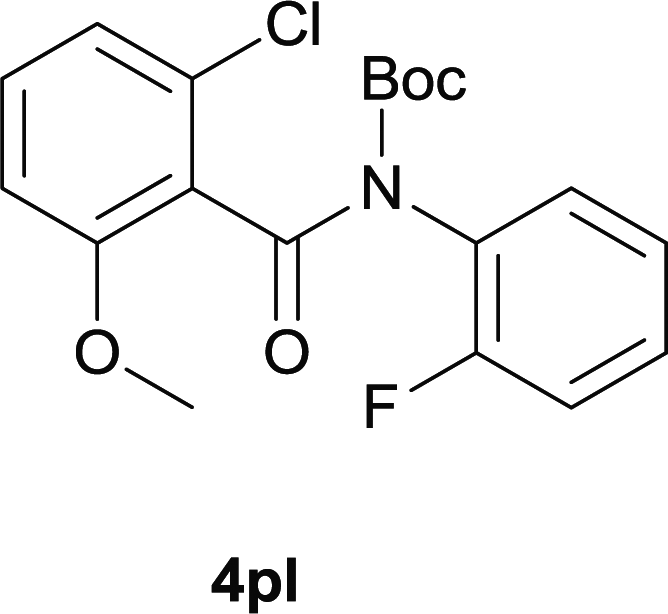
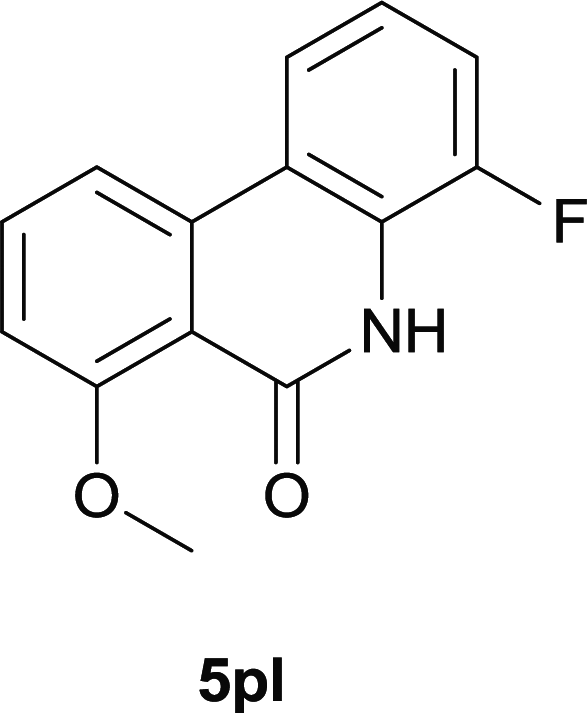
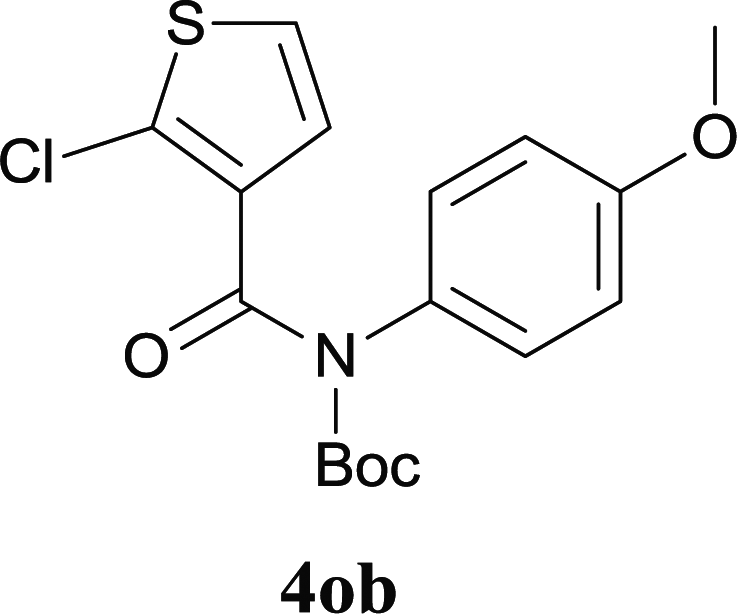
Our above results showed that diverse functional groups, including F, Cl, CN, OMe, alkyl and a variety of aryl carboxylic acids and aryl amines were well tolerated. It leads us to speculate the reaction scope is not limited to bromo-substituted acid, but can also be extended to chloro-substituted acid. So we tested the activity of halogens in Table 3. Firstly, we selected the synthesis of 5aa as example (entries 1a to 1c). When iodide and bromide were tested, the reaction can be finished at 120 °C, given 77% and 66% yield respectively. When chloride was utilized, the yield decreased to moderate (45%) and must be conducted at higher temperature. After that, we tested electron-donating substitutes on either carboxylic moiety or aniline moiety (entries 2–5) and found yield can be as high as 78%; instead electron-withdrawing substitutes were negative to the reaction (entry 6, 38% yield). When the reaction was extended to strong electron withdrawing substituents, the yield decreased significantly even with iodide (entry 9). Chloro-thiophene carboxylic acid also delivered the corresponding products without a problem (entries 7, 8). Considering both chlorine and bromine were active as acid partner, we subjected 4uc for the reaction. Just as we predicted, the reaction turned to be quite messy and yielded no desired target (entry 10).
|
|
|||
|---|---|---|---|
| Entry | 4xy | 5xy | Yieldb [%] |
| a Reaction conditions: 1 mmol of substrate, 4 equiv. of KOAc, and 5% equiv. of (t-Bu3P)2Pd in 10 ml DMA were heated at 135 °C for 2 h under Ar atmosphere. b Isolated yield. c Reacted at 120 °C. | |||
|
|
||
| 1a | 4aa1 X = I | 77c | |
| 1b | 4aa2 X = Br | 66c | |
| 1c | 4aa3 X = Cl | 45 | |
| 2 |
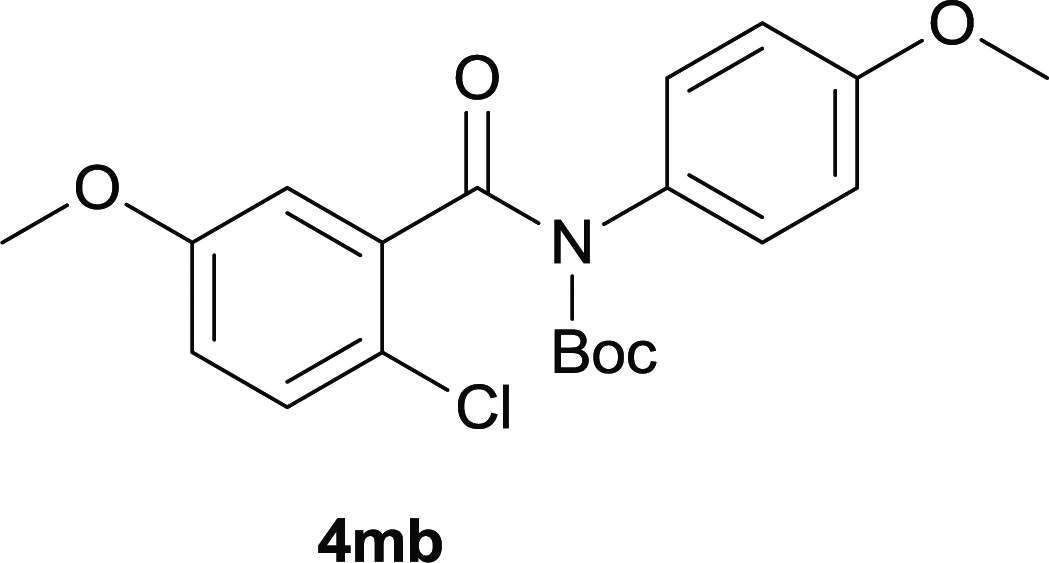
|
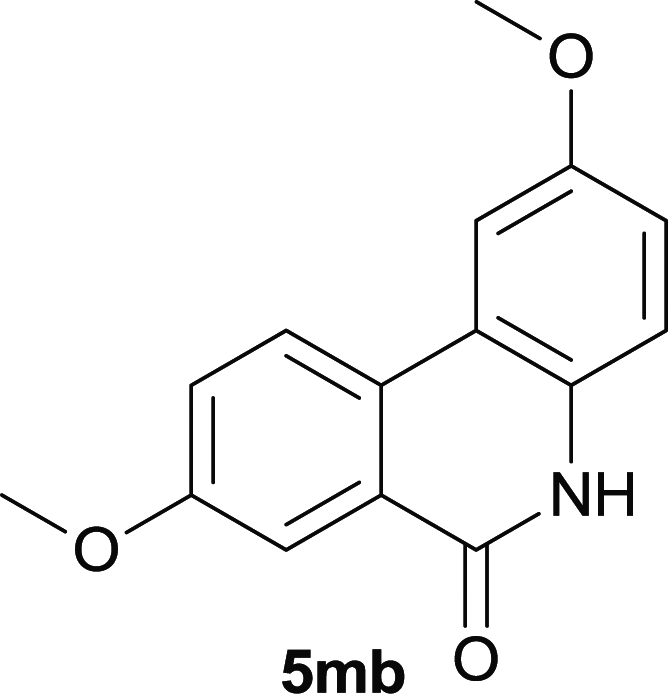
|
68 |
| 3 |
|
|
45 |
| 4 |
|
|
67 |
| 5 |
|
|
78 |
| 6 |
|
|
38 |
| 7 |
|
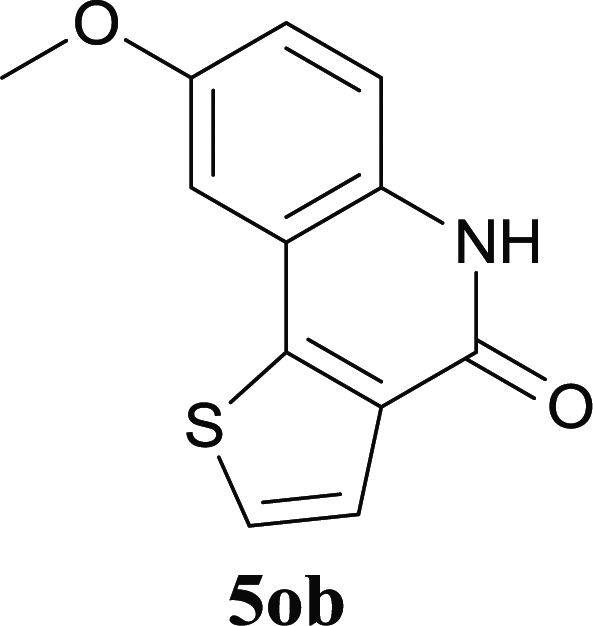
|
53 |
| 8 |
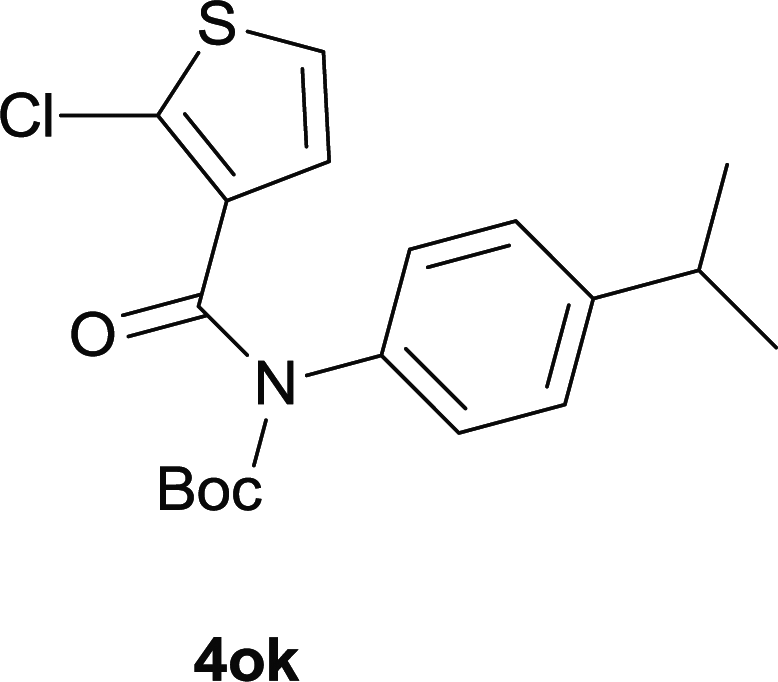
|
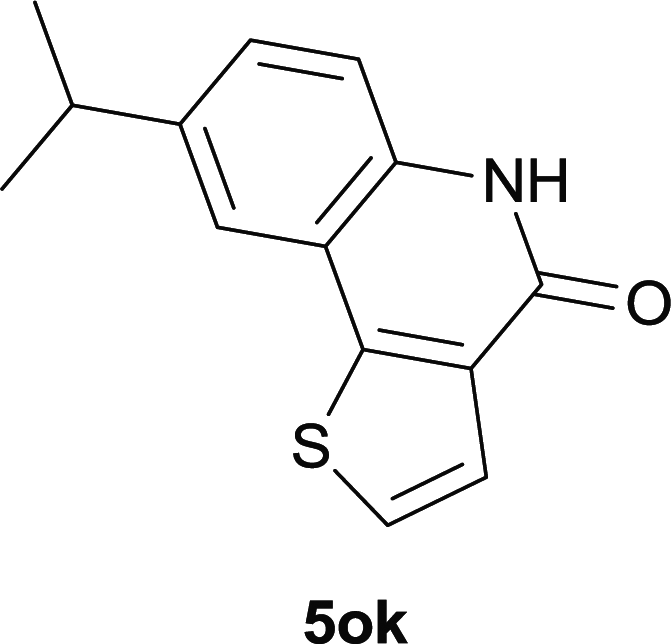
|
51 |
| 9 |
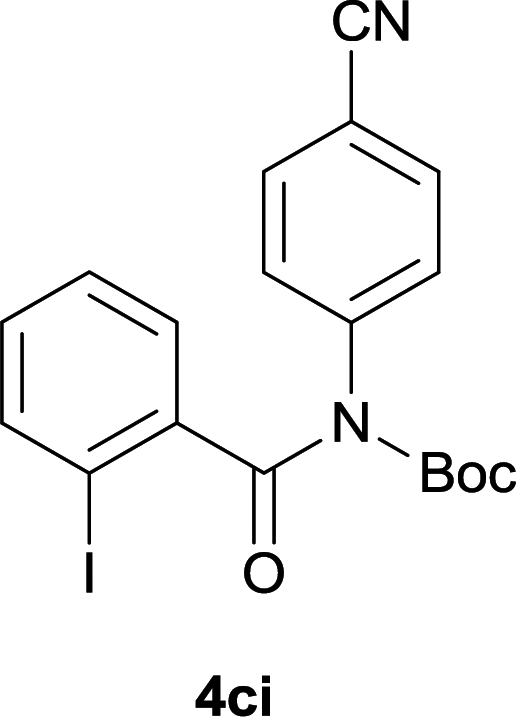
|
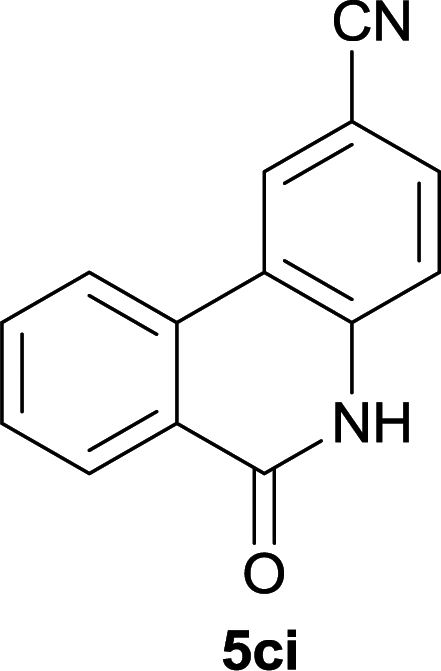
|
40 |
| 10 |
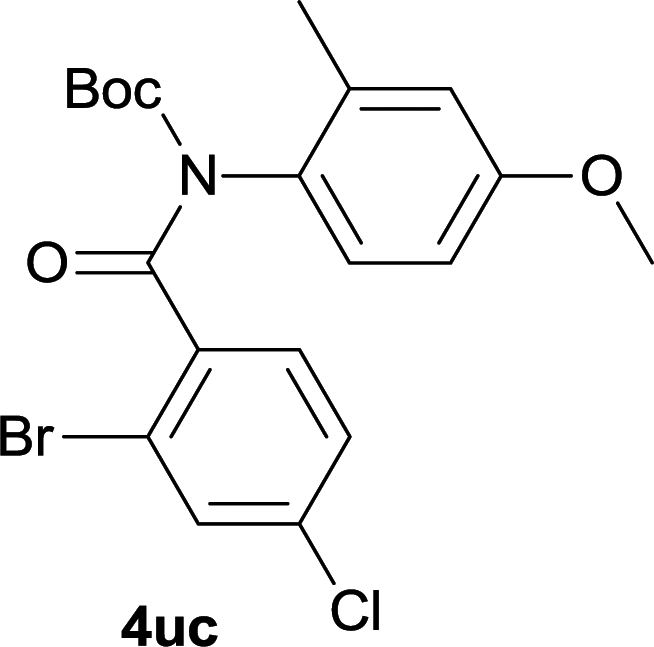
|
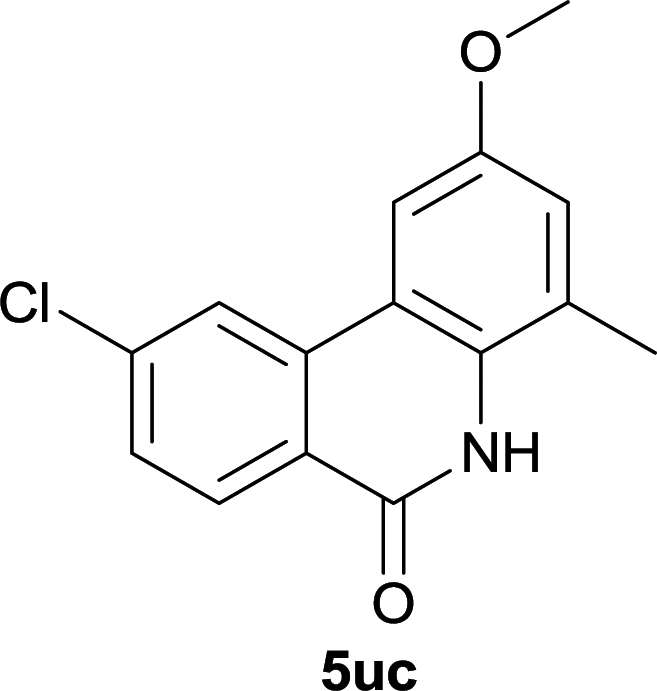
|
0 |
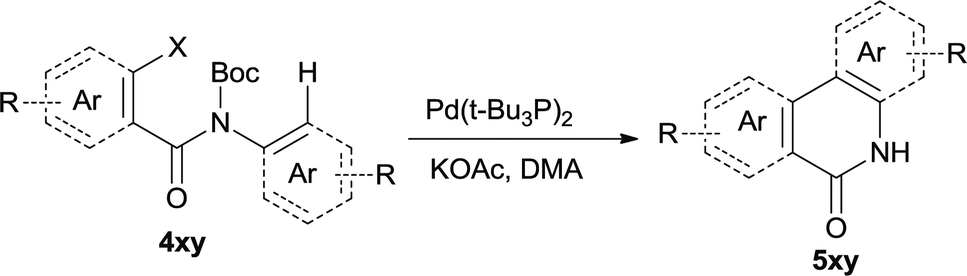
Next we examined the effect of catalyst loading to further optimize the reaction condition. Substrates 4lb and 4af were selected as examples which are shown in Table 4. Firstly we tested 4lb, when the catalyst loading increased from 5 mol% to 6 mol%, the influence was insignificant. However, when it was reduced to 4 mol%, the starting material remained and the yield was reduced by 13%; and only 60% yield was obtained when decreased further to 3 mol%; the same situation was observed with substrate 4af. Based on research below, 5 mol% catalyst was established as an optimal catalyst loading.
| Entry | Substrate | Pd(t-Bu3P)2 | Subs. conv. (%) | Yield (%) of 5b |
|---|---|---|---|---|
| a Reaction conditions: 1 mmol of substate, 4 equiv. of KOAc, Pd catalyst and 10 ml DMA were heated at 120 °C for 2 h under Ar atmosphere. b Isolated yield. | ||||
| 1 | 4lb | 6% | 100 | 93 |
| 2 | 4lb | 5% | 100 | 91 |
| 3 | 4lb | 4% | 94 | 78 |
| 4 | 4lb | 3% | 89 | 60 |
| 5 | 4af | 5% | 100 | 84 |
| 6 | 4af | 4% | 93 | 61 |
| 7 | 4af | 3% | 82 | 52 |
The directly cleavage of Boc was left for investigation for further exploring of reaction mechanism. We tested another protection group acetyl and used it for cyclization. However acetyl group is extremely unstable and only trace cyclization product was produced. It leaded us to suspect the stability of Boc on such conformationally rigid tricyclic system. So we re-protected the final target 5ec with Boc again and then heated it in the reaction system at same condition without Pd catalyst (Table 5). To our delight, 26% 5ec was produced and 69% 6ec remained. This result revealed that Boc was a fragile spot however just heating the substrate couldn't result full decomposition. Pd catalyst should have played a critical role in catalytic cycle for complete cleavage.
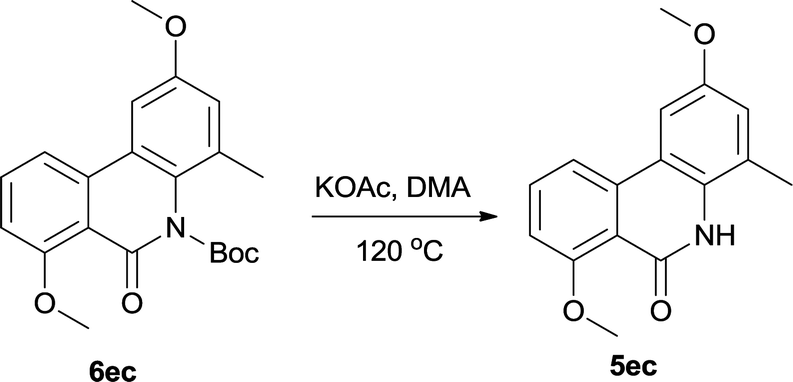
Although additional data are needed to establish the mechanism, the fact that Pd catalyst loading influences the reaction significantly and the base KOAc gives the best yield, Boc group is some extend of fragile suggests their critical roles. It leads us to propose a plausible Pd(0)/Pd(II) catalytic cycle2 (Scheme 3). A proton abstraction mechanism previously proposed by Echavarren and Maseras,16 was found to explain our reaction outcomes: (1) the anionic ligand KOAc is directly involved in C–H bond cleaving; (2) the anion ligand must bind to the catalyst, but not block the catalytic cycle by competitive occupation of vacant coordination sites;17 (3) the arene which interacts with the catalyst weakly must compete for binding to the arylpalladium(II) intermediate with the excess anionic ligand.18 So initially oxidative addition of polarized Ph–Br bond to the Pd(0) catalyst forms a highly electrophilic arylpalladium intermediate I,18,19 which exchange with KOAc to produce intermediate II,18,20 followed by phosphine dissociation. Secondly Pd(II) interacts with arene at the ortho position of aniline in a C–H activation manner which forms intermediate III.20,21 Here HOAc plays a critical role in the stabilized coordination intermediate.21,22 Next is irreversible deprotonation: Pd(II) inserts into the C–H bond of the arene to give cyclobiarylpalladium intermediate IV,20–22 accompanied by elimination of HOAc. Under the effect of produced HOAc, Pd presumably kicks off Boc and coordinates to nitrogen to give intermediate V, which finally undergoes reductive elimination to provide the desired product and regenerates the Pd(0) catalyst.16,20,22
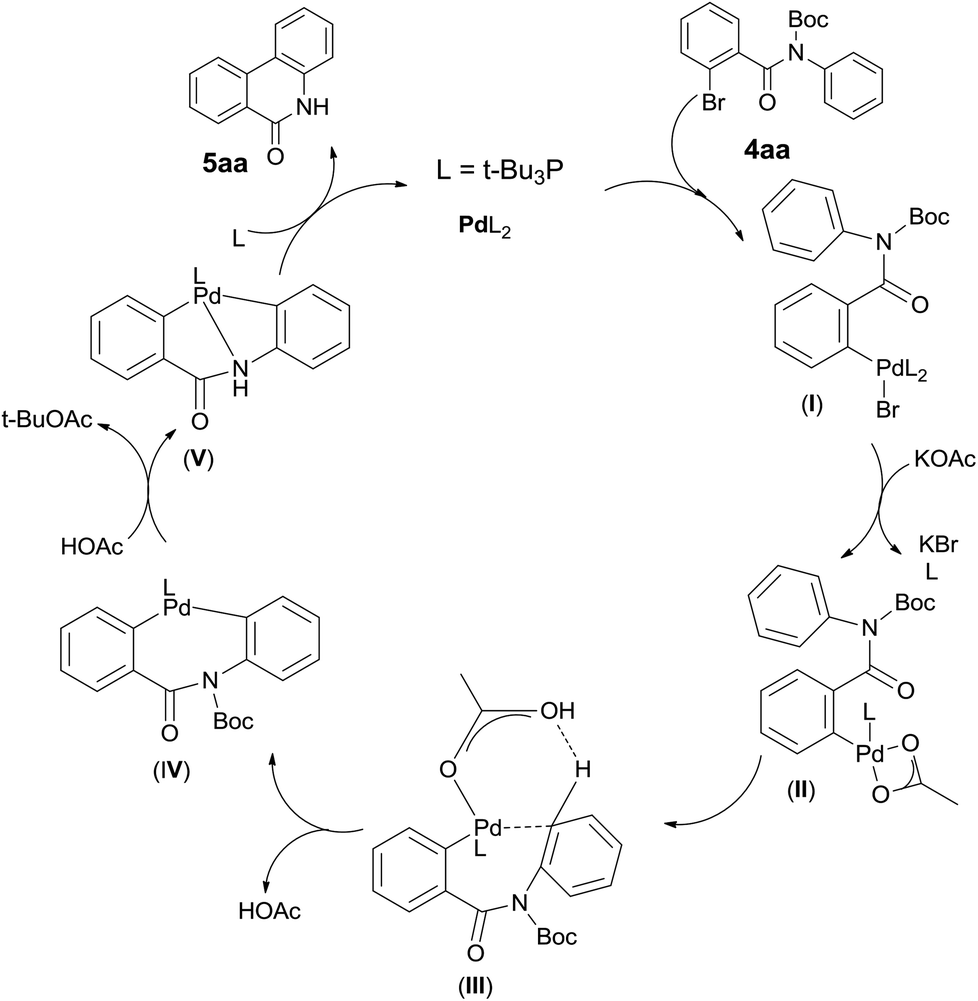 | ||
| Scheme 3 Proposed mechanism for the phenanthridinone synthesis. | ||
Conclusions
In summary, we have developed a palladium catalyzed C–H arylation method for synthesis of N–H phenanthridinones from Boc protected diaryl amide. There are four advantages of using Boc as protection group: (1) the protection stage gives quantitive yield and just simple workup produces pure product; (2) the protection group is stable to some extent compared with acetyl group; (3) the coupling stage directly gives de-protection final targets, additional de-proctection is unnecessary; (3) in contrast to the system of radical pathway without protection reported by Bhakuni et al., the regioisomer is not a concern, only a single desired target was obtained. The catalyst Pd(t-Bu3P)2 is commercial available, inexpensive and the catalyst loading is reasonable; a second phosphine ligand is not required compared to published method.6d This practical and convenient method can be easily scaled up, applicable for versatile substrates and produces high yields. Most important of all, it could be applied for the less reactive o-chlorobenzamides which greatly extend the substrates scope.Experimental
General experimental details
Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. The 1H and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer at 25 °C using DMSO-d6 or CDCl3 as the solvent. Chemical shifts (δ) are reported in ppm relative to Me4Si (internal standard), coupling constants (J) are reported in hertz, and peak multiplicity are reported as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), or br s (broad singlet). High resolution mass analysis is performed on a Waters Q-TOF Premier mass spectrometer with electron spray ionization (ESI). Thin layer chromatography (TLC) was performed on 0.20 mm silica gel F-254 plates (Qingdao Haiyang Chemical, China). Visualization of TLC was accomplished with UV light and/or aqueous potassium permanganate or I2 in silica gel. Column chromatography was performed using silica gel 60 of 300–400 mesh (Qingdao Haiyang Chemical, China).General procedure for synthesis of 4xy10,23
To suspension of 1x (5 mmol) in SOCl2 (5 ml) was added with 2 drops of DMF. The mixture was heated under reflux for 3 h. Then the mixture was concentrated and diluted with DCM and concentrated, then diluted and concentrated again to give light brown semi-solid. The residue was dissolved in anhydrous DCM (10 ml), added dropwise to a mixture of 2y (5 mmol) and Et3N (12.5 mmol) in DCM (10 ml). The resulting suspension was stirred for another 1 h after addition finished. The mixture was diluted with water, washed with 1 N HCl, 2 N NaOH and brine successively. The organic layer was dried over Na2SO4, filtered and concentrated to give the crude amide 3xy which was used for next step directly.To the above crude 3xy was added anhydrous DCM (20 ml), DMAP (0.05 eq., 0.25 mmol), and Boc2O (1.2 eq., 6 mmol). The suspension was stirred until the bubbling was not observed which indication completion of the reaction. TLC showed a less polar product which was also confirmed by LCMS. The mixture was concentrated, diluted with hexane and water. After stirring for a couple of minutes a precipitate deposited. The solid was filtered, washed with water and hexane, dried under vacuum to give off-white solid 4xy.
General procedure for synthesis of product 5 (Tables 2 and 3)
To a 50 ml two necked flask equipped with a thermometer was added 4xy (1 mmol), DMA (10 ml), KOAc (4 eq., 4 mmol), Pd(t-Bu3P)2 (0.05 mmol, 0.05 eq.). The mixture was heated under argon at 120–135 °C for 2 h. It was diluted with H2O (30 ml), filtered and the solid was washed with H2O and ethanol successively. The crude product was dissolved in DCM/MeOH (5:1), filtered through celite to remove catalyst residue, then concentrated and re-crystallized from hexane/ethyl acetate to give almost pure compound 5xy.
Most of the final compound can be purified by crystallization easily, for those compounds which has good solubility, a short silica gel column purification is necessary.
Representative gram scale procedure for synthesis of 5
To a three necked 500 ml flask equipped with a thermometer was added 4lb (9.13 g, 20 mmol, 1 eq.), DMA (200 ml), anhydrous KOAc (azeotroped with toluene prior to use) (7.85 g, 80 mmol, 4 eq.), Pd(t-Bu3P)2 (511.05 mg, 1 mmol, 0.05 eq.). The mixture was heated under argon at 120 °C for 2 h. The mixture was diluted with H2O (30 ml) to precipitate the product, filtered and the solid was washed with H2O and ethanol successively. The crude product was dissolved in DCM/MeOH (5:1), filtered through celite to remove catalyst residue, then concentrated and re-crystallized from hexane/ethyl acetate to give pure compound 5lb (5.23 g, 19 mmol, 95% yield).
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, DMSO-d6): δ 11.69 (s, 1H), 8.52 (d, J = 8.0 Hz, 1H), 8.40 (d, J = 8.0 Hz, 1H), 8.33 (d, J = 7.6 Hz, 1H), 7.86 (t, J = 7.2 Hz, 1H), 7.65 (t, J = 7.4 Hz, 1H), 7.50 (t, J = 7.4 Hz, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.27 (t, J = 7.4 Hz, 1H). 13C NMR (100 MHz, DMSO-d6): δ 160.8, 136.5, 134.2, 132.8, 129.6, 127.9, 127.5, 125.7, 123.2, 122.6, 122.3, 117.5, 116.1. ESI-HRMS: calculated for C13H9NNaO [M + Na]+ 218.0576, found 218.0576.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, CDCl3): δ 11.23 (s, 1H), 8.61 (d, J = 7.6 Hz, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.83–7.79 (m, 1H), 7.68–7.62 (m, 2H), 7.38 (d, J = 8.8 Hz, 1H), 7.14–7.11 (m, 1H), 3.93 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 154.8, 134.1, 132.5, 130.7, 127.9, 127.5, 125.9, 123.0, 118.3, 117.7, 117.3, 106.2, 55.6; HRMS (ESI) m/z calcd for C14H11NNaO2 [M + Na]+ 248.0682, found 248.0690.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, DMSO-d6): δ 11.60 (s, 1H), 8.46 (d, J = 8.0 Hz, 1H), 8.31–8.29 (m, 2H), 7.83 (t, J = 7.6 Hz, 1H), 7.61 (t, J = 7.4 Hz, 1H), 7.23 (s, 1H), 7.17 (d, J = 8.4 Hz, 1H), 2.93–3.00 (m, 1H), 1.25 (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6): δ 160.9, 150.2, 136.6, 134.3, 132.7, 127.4, 125.3, 123.2, 122.4, 120.9, 115.6, 113.4, 33.3, 23.6; HRMS (ESI) m/z calcd for C16H15NNaO [M + Na]+ 260.1046, found 260.1033.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, CDCl3): δ 10.37 (s, 1H), 8.56 (d, J = 7.6 Hz, 1H), 8.24 (d, J = 8.0 Hz, 1H), 7.71 (t, J = 7.6 Hz, 1H), 7.55 (t, J = 6.8 Hz, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.28 (d, J = 7.6 Hz, 1H), 7.11 (d, J = 7.6 Hz, 1H), 3.98–3.93 (m, 1H), 1.39 (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6): δ 160.4, 147.0, 137.2, 134.4, 131.9, 128.8, 127.6, 127.2, 127.0, 121.0, 116.1, 113.8, 30.0, 24.5; HRMS (ESI) m/z calcd for C16H15NNaO [M + Na]+ 260.1046, found 260.1046.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 12.01 (s, 1H), 8.27 (d, J = 7.6 Hz, 1H), 7.79–7.75 (m, 3H), 7.54–7.50 (m, 1H), 7.06 (d, J = 5.2 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 161.4, 138.4, 133.1, 133.0, 128.2, 127.7, 126.3, 123.6, 122.4, 117.9, 115.0; HRMS (ESI) m/z calcd for C11H7NNaOS [M + Na]+ 224.0141, found 224.0137.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, CDCl3): δ 10.99 (s, 1H), 8.51 (d, J = 8.0 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 8.17 (d, J = 8.4 Hz, 1H), 7.74–7.69 (m, 1H), 7.54–7.50 (m, 1H), 7.33–7.27 (m, 2H), 1.35 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 163.4, 153.3, 136.0, 135.0, 132.8, 128.2, 127.4, 125.5, 122.6, 121.9, 120.7, 116.2, 113.4, 35.0, 31.2; HRMS (ESI) m/z calcd for C17H18NO [M + H]+ 252.1383, found 252.1395.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 12.07 (s, 1H), 8.98 (d, J = 1.6 Hz, 1H), 8.65 (d, J = 8.0 Hz, 1H), 8.34–8.32 (m, 1H), 7.93–7.88 (m, 2H), 7.75–7.71 (m, 1H), 7.48 (d, J = 8.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 160.9, 139.7, 133.2, 132.9, 132.4, 129.0, 128.6, 127.4, 125.7, 123.2, 119.1, 118.2, 117.1, 104.5; HRMS (ESI) m/z calcd for C14H8N2NaO [M + Na]+ 243.0529, found 243.0516.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, DMSO-d6): δ 11.59 (s, 1H), 8.32 (d, J = 7.6 Hz, 1H), 7.94 (d, J = 1.6 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.55 (t, J = 7.8 Hz, 1H), 7.36 (d, J = 8.8 Hz, 1H), 7.19–7.16 (m, 1H), 3.85 (s, 3H), 2.97 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.4, 153.8, 136.9, 135.1, 133.0, 131.0, 127.5, 127.3, 126.1, 119.4, 117.1, 116.1, 111.3, 55.3, 25.4; HRMS (ESI) m/z calcd for C15H13NNaO2 [M + Na]+ 262.0838, found 262.0842.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, DMSO-d6): δ 8.77 (s, 1H), 8.51 (d, J = 7.6 Hz, 1H), 7.86 (s, 1H), 7.63 (d, J = 6.8 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 6.98 (s, 1H), 3.89 (s, 3H), 2.99 (s, 3H), 2.51 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.9, 153.2, 137.1, 135.1, 133.6, 129.2, 127.4, 127.3, 126.1, 125.2, 119.5, 117.6, 109.2, 55.3, 25.6, 18.2; HRMS (ESI) m/z calcd for C16H15NNaO2 [M + Na]+ 276.0995, found 276.1007.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 12.10 (s, 1H), 8.25 (d, J = 8.0 Hz, 1H), 7.90 (d, J = 5.2 Hz, 1H), 7.68 (d, J = 7.2 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.15 (d, J = 5.6 Hz, 1H), 2.75 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 161.3, 138.2, 134.7, 132.6, 132.1, 128.6, 126.3, 125.6, 124.6, 117.2, 113.9, 22.0; HRMS (ESI) m/z calcd for C12H9NNaOS [M + Na]+ 238.0297, found 238.0318.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 10.08 (s, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.73 (t, J = 8.2 Hz, 1H), 7.63 (d, J = 2.4 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 2.0 Hz, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 2.42 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.9, 159.3, 153.9, 137.2, 133.4, 129.7, 125.0, 119.2, 117.8, 114.9, 114.7, 110.8, 104.4, 56.0, 55.5, 17.5; HRMS (ESI) m/z calcd for C16H16NO3 [M + H]+ 270.1125, found 270.1118.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 11.63 (s, 1H), 7.73 (d, J = 5.2 Hz, 1H), 7.65 (t, J = 8.2 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 7.03 (d, J = 8.4 Hz, 1H), 6.96 (d, J = 5.2 Hz, 1H). 3.86 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 161.3, 160.2, 138.9, 135.6, 134.0, 127.9, 117.5, 114.6, 114.3, 112.5, 108.9, 55.8; HRMS (ESI) m/z calcd for C12H9NNaO2S [M + Na]+ 254.0246, found 254.0257.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 11.06 (s, 1H), 8.49–8.47 (m, 2H), 7.66 (d, J = 2.8 Hz, 1H), 7.22 (d, J = 8.8 Hz, 1H), 7.03–7.00 (m, 1H), 3.84 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 157.7, 154.7, 136.3, 130.9, 130.2, 129.9, 119.6, 117.3, 117.2, 116.1, 107.5, 55.6; HRMS (ESI) m/z calcd for C12H9NNaO2S [M + Na]+ 254.0246, found 254.0246.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 10.08 (s, 1H), 8.52 (d, J = 2.0 Hz, 1H), 8.47 (s, 1H), 7.52 (s, 1H), 6.89 (s, 1H), 3.82 (s, 3H), 2.40 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 158.1, 154.3, 136.6, 130.7, 130.0, 128.5, 125.8, 119.7, 117.5, 117.3, 105.3, 55.4, 17.9; HRMS (ESI) m/z calcd for C13H11NNaO2S [M + Na]+ 268.0403, found 268.0398.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, DMSO-d6): δ 10.50 (s, 1H), 8.43 (d, J = 8.0 Hz, 1H), 7.89–7.85 (m, 2H), 7.45 (dd, J = 11.6, 8.4 Hz, 1H), 7.36 (d, J = 2.0 Hz, 1H); 3.89 (s. 3H); 13C NMR was not obtained due to low solubility; HRMS (ESI) m/z calcd for C14H9ClFNNaO2 [M + Na]+ 300.0198, found 300.0208.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 12.23 (s, 1H), 8.17 (d, J = 4.0 Hz, 1H), 7.79 (d, J = 4.8 Hz, 1H), 7.61 (d, J = 4.4 Hz, 1H), 7.11 (d, J = 4.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 156.0, 139.9, 135.3, 127.5, 127.1, 122.6, 117.4, 113.3; HRMS (ESI) m/z calcd for C9H5NNaOS2 [M + Na]+ 229.9705, found 229.9718.
:1, DCM:MeOH); 1H NMR (400 MHz, CDCl3): δ 8.93 (s, 1H), 8.42 (d, J = 8.0 Hz, 1H), 8.36 (s, 1H), 8.18 (d, J = 8.0 Hz, 1H), 7.75–7.68 (m, 2H), 7.36 (t, J = 7.8 Hz, 1H), 2.57 (s, 3H); 13C NMR was not obtained due to low solubility; HRMS (ESI) m/z calcd for C15H10N2NaO [M + Na]+ 257.0685, found 257.0696.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 10.73 (s, 1H), 7.78 (d, J = 5.6 Hz, 1H), 7.60 (d, J = 5.2 Hz, 1H), 7.11 (d, J = 2.4 Hz, 1H), 7.01 (d, J = 2.0 Hz, 1H), 3.83 (s, 3H), 2.46 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 158.1, 154.1, 145.8, 131.1, 129.0, 126.7, 126.4, 125.3, 119.3, 116.8, 103.1, 55.4, 17.8; HRMS (ESI) m/z calcd for C13H11NNaO2S [M + Na]+ 268.0403, found 268.0411.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 12.15 (s, 1H), 7.79 (d, J = 5.2 Hz, 1H), 7.62 (d, J = 5.2 Hz, 1H), 7.54 (d, J = 4.8 Hz, 1H), 7.09 (d, J = 5.2 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 158.4, 142.1, 139.4, 128.5, 127.6, 125.3, 124.1, 117.6, 112.2; HRMS (ESI) m/z calcd for C9H6NOS2 [M + H]+ 207.9885, found 207.9880.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 10.91 (s, 1H), 7.88–7.86 (m, 2H), 7.66–7.63 (m, 2H), 7.27 (t, J = 7.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6): δ 157.9, 145.3, 132.5, 131.5, 129.4, 128.0, 125.3, 123.1, 122.6, 119.3, 117.9. ESI-HRMS: calculated for C11H6ClNNaOS [M + Na]+ 257.9751, found 257.9739.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, DMSO-d6): δ 11.85 (s, 1H), 9.01 (d, J = 7.2 Hz, 1H), 8.33 (d, J = 8.8 Hz, 1H), 8.17–8.08 (m, 3H), 7.80–7.78 (m, 2H), 7.47 (d, J = 8.8 Hz, 1H), 7.28–7.25 (m, 1H), 3.90 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.4, 154.2, 135.7, 132.5, 131.5, 129.0, 128.6, 128.4, 128.1, 127.4, 126.9, 124.9, 122.9, 118.1, 117.4, 117.2, 111.1, 55.5; HRMS (ESI) m/z calcd for C18H14NO2 [M + H]+ 276.1019, found 276.1028.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 12.55 (s, 1H), 8.89 (d, J = 8.4 Hz, 1H), 8.39 (d, J = 8.8 Hz, 1H), 8.17 (d, J = 7.6 Hz, 1H), 8.04–8.01 (m, 2H), 7.90 (t, J = 7.4 Hz, 1H), 7.83 (t, J = 7.4 Hz, 1H), 7.26 (d, J = 5.6 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 161.2, 140.1, 135.1, 131.8, 129.4, 129.2, 128.4, 127.6, 127.1, 126.8, 125.5, 124.1, 122.1, 117.4, 112.8; HRMS (ESI) m/z calcd for C15H9NNaO [M + Na]+ 274.0297, found 274.0314.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, DMSO-d6): δ 11.08 (s, 1H), 8.89–8.87 (m, 1H), 8.58 (d, J = 8.0 Hz, 1H), 8.33 (d, J = 8.4 Hz, 1H), 8.20–8.15 (m, 2H), 7.83–7.74 (m, 3H), 7.38 (t, J = 8.0 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 156.2, 131.3, 128.2, 127.7, 124.3, 123.9, 123.7, 123.1, 122.2, 122.0, 121.9, 119.6, 118.0, 117.0, 115.1, 114.9, 113.6; HRMS (ESI) m/z calcd for C17H11ClNO [M + H]+ 276.1019, found 276.1026.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, DMSO-d6): δ 11.58 (s, 1H), 8.49 (d, J = 9.2 Hz, 1H), 7.80–7.76 (m, 2H), 7.45–7.42 (m, 1H), 7.28 (d, J = 8.8 Hz, 1H), 7.08 (dd, J = 8.8, 2.8 Hz, 1H), 3.92 (s, 3H), 3.86 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.1, 159.0, 154.9, 129.6, 127.5, 127.3, 124.9, 121.3, 118.5, 117.1, 116.5, 108.7, 105.7, 55.6, 55.4; HRMS (ESI) m/z calcd for C15H13NNaO3 [M + Na]+ 278.0788, found 278.0797.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, CDCl3): δ 8.66 (s, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.86 (d, J = 2.8 Hz, 1H), 7.40 (d, J = 2.4 Hz, 1H), 7.31 (dd, J = 8.8, 2.8 Hz, 1H), 6.84 (d, J = 2.0 Hz, 1H), 3.90 (s, 3H), 3.83 (s, 3H), 2.40 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.5, 159.0, 154.3, 128.0, 127.8, 127.0, 125.7, 125.2, 121.7, 118.5, 117.9, 108.4, 103.5, 55.5, 17.7; HRMS (ESI) m/z calcd for C16H15NNaO3 [M + Na]+ 292.0944, found 292.0954.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, CDCl3): δ 11.16 (s, 1H), 8.27 (d, J = 9.2 Hz, 1H), 8.03 (d, J = 2.0 Hz, 1H), 7.99 (s, 1H), 7.42–7.35 (m, 3H), 4.02 (s, 3H), 3.10–3.03 (m, 1H), 1.36 (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 159.3, 143.5, 133.1, 128.6, 127.0, 123.8, 122.6, 119.6, 118.5, 116.7, 108.7, 55.7, 34.1, 24.3; HRMS (ESI) m/z calcd for C17H17NNaO2 [M + Na]+ 290.1151, found 290.1147.
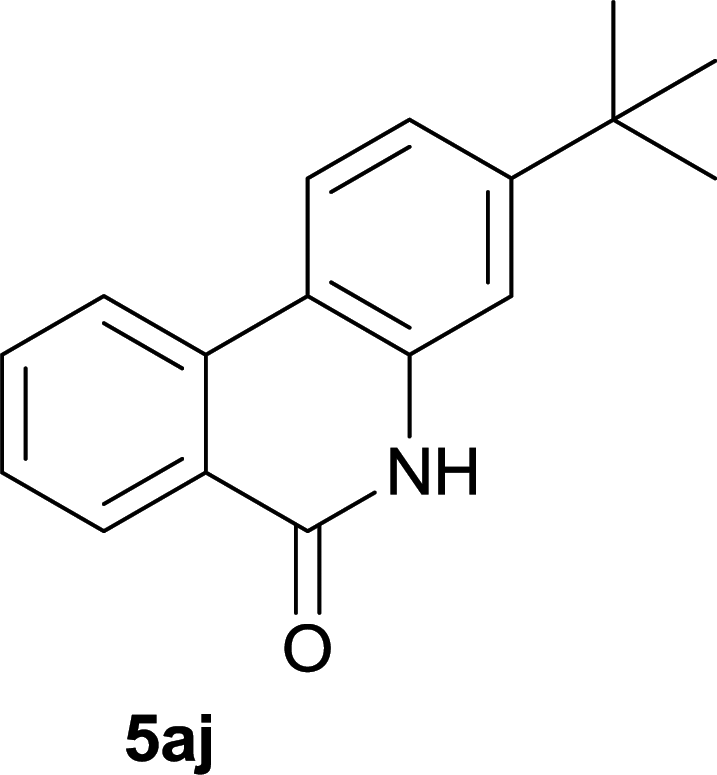
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, DMSO-d6): δ 11.58 (s, 1H), 8.39 (d, J = 8.8 Hz, 1H), 8.29 (d, J = 7.2 Hz, 1H), 7.69 (d, J = 7.2 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.45 (d, J = 1.6 Hz, 1H), 7.32 (dd, J = 8.8, 2.0 Hz, 1H), 2.92 (s, 3H), 1.34 (s, 9H); 13C NMR (100 MHz, DMSO-d6): δ 161.0, 151.5, 136.9, 136.8, 134.9, 133.2, 127.1, 126.8, 126.0, 119.3, 116.4, 112.7, 34.4, 30.8, 25.5; HRMS (ESI) m/z calcd for C18H19NNaO [M + Na]+ 288.1359, found 288.1370.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 11.63 (s, 1H), 7.79 (d, J = 5.2 Hz, 1H), 7.58 (d, J = 5.2 Hz, 1H), 7.36 (d, J = 8.8 Hz, 1H), 7.26 (d, J = 2.4 Hz, 1H), 7.14 (dd, J = 8.8, 2.8 Hz, 1H), 3.85 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 157.7, 154.6, 145.1, 131.4, 130.4, 126.7, 125.3, 118.2, 117.6, 116.8, 105.2, 55.6; HRMS (ESI) m/z calcd for C12H9NNaO2S [M + Na]+ 254.0246, found 254.0252.
:1, DCM:MeOH); 1H NMR (400 MHz, DMSO-d6): δ 11.67 (s, 1H), 7.77 (d, J = 5.2 Hz, 1H), 7.63 (d, J = 2.8 Hz, 1H), 7.58 (d, J = 5.2 Hz, 1H), 7.42–7.35 (m, 2H), 3.04–2.97 (m, 1H), 1.26 (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6): δ 158.1, 145.6, 142.7, 134.4, 131.1, 127.9, 126.4, 125.3, 120.3, 116.3, 116.1, 32.9, 23.9; HRMS (ESI) m/z calcd for C14H13NNaOS [M + Na]+ 266.0610, found 266.0641.
:1, petroleum ether:EtOAc); 1H NMR (400 MHz, DMSO-d6): δ 11.12 (s, 1H), 8.14 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.78 (t, J = 8.2 Hz, 1H), 7.38–7.34 (m, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.20–7.15 (m, 1H), 3.90 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 161.0, 159.0, 150.0, 147.6, 136.4, 133.9, 125.8, 125.7, 121.4, 121.3, 119.5, 114.9, 114.8, 114.7, 111.5, 56.0; HRMS (ESI) m/z calcd for C14H10FNNaO2 [M + Na]+ 266.0588, found 266.0599.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Chengdu ChemPartner Co., Ltd. for technical, reagents and analysis support. We also thank Lihua Zhou and Professor Lijuan Chen's team of State Key Laboratory of Biotherapy (Sichuan University) for NMR and HRMS measurements.Notes and references
- (a) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (b) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540–548 RSC; (c) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef CAS; (d) L.-C. Campeau and K. Fagnou, Chem. Commun., 2006, 1253–1264 RSC; (e) R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17–117 CrossRef CAS; (f) I. Hussain and T. Singh, Adv. Synth. Catal., 2014, 356, 1661–1696 CrossRef CAS; (g) D. D. Vachhani, A. Sharma and E. V. Eycken, J. Org. Chem., 2012, 77, 8768–8774 CrossRef CAS.
- (a) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS; (b) L. Théveau, C. Schneider, C. Fruit and C. Hoarau, ChemCatChem, 2016, 8, 3183–3194 CrossRef; (c) M. A. Campo, Q. Huang, T. Yao, Q. Tian and R. C. Larock, J. Am. Chem. Soc., 2003, 125, 11506–11507 CrossRef CAS; (d) N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972–4973 CrossRef CAS; (e) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS; (f) Y. Jaiswal, Y. Kumar, R. Thakur, J. Pal, R. Subramanian and A. Kumar, J. Org. Chem., 2016, 81, 12499–12505 CrossRef CAS.
- (a) R. Srinivasan, A. Dey, N. S. Nagarajan, R. S. Kumaran, T. Gandhi and D. Maiti, Chem. Commun., 2017, 53, 11709–11712 RSC; (b) B. Wang, W. A. Nack, G. He, S.-Y. Zhang and G. Chen, Chem. Sci., 2014, 5, 3952–3957 RSC; (c) W. A. Nack, B. Wang, X. Wu, R. Jiao, G. He and G. Chen, Org. Chem. Front., 2016, 3, 561–564 RSC; (d) Y. Mu, X. Tan, Y. Zhang, X. Jing and Z. Shi, Org. Chem. Front., 2016, 3, 380–384 RSC; (e) H. Y. Fu, L. Chen and H. Doucet, J. Org. Chem., 2012, 77, 4473–4478 CrossRef CAS; (f) S. E. Kazzouli, J. Koubachi, N. E. Brahmi and G. Guillaumet, RSC Adv., 2015, 5, 15292–15327 RSC; (g) A. A. Ruch, S. Handa, F. Kong, V. N. Nesterov, D. R. Pahls, T. R. Cundari and L. M. Slaughter, Org. Biomol. Chem., 2016, 14, 8123–8140 RSC; (h) S.-Y. Chang, P.-H. Lin and C.-Y. Liu, RSC Adv., 2014, 4, 35868–35878 RSC.
- (a) M. Nakamura, A. Aoyama, M. T. A. Salim, M. Okamoto, M. Baba, H. Miyachi, Y. Hashimoto and H. Aoyama, Bioorg. Med. Chem., 2010, 18, 2402–2411 CrossRef CAS; (b) H. Aoyama, K. Sugita, M. Nakamura, A. Aoyama, M. T. A. Salim, M. Okamoto, M. Baba and Y. Hashimoto, Bioorg. Med. Chem., 2011, 19, 2675–2687 CrossRef CAS; (c) J. D. Koružnjak, M. Grdiša, N. Slade, B. Zamola, K. Pavelić and G. Karminski-Zamola, J. Med. Chem., 2003, 46, 4516–4524 CrossRef; (d) Z. Chen and X. Wang, Org. Biomol. Chem., 2017, 15, 5790–5796 RSC; (e) R. L. Dow, T. T. Chou, B. M. Bechle, C. Goddard and E. R. Larson, J. Med. Chem., 1994, 37, 2224–2231 CrossRef CAS; (f) M. Moser, X. Sun and T. Hudlicky, Org. Lett., 2005, 7, 5669–5672 CrossRef CAS PubMed.
- (a) R. Ferraccioli, D. Carenzi, O. Rombolà and M. Catellani, Org. Lett., 2004, 6, 4759–4762 CrossRef CAS; (b) T. Furuta, Y. Kitamura, A. Hashimoto, S. Fujii, K. Tanaka and T. Kan, Org. Lett., 2007, 9, 183–186 CrossRef CAS; (c) N. Conde, F. Churruca, R. SanMartin, M. T. Herrero and E. Domínguez, Adv. Synth. Catal., 2015, 357, 1525–1531 CrossRef CAS; (d) J. Cao, L. Qian, F. Lu, J. Zhang, Y. Feng, X. Qiu, H.-L. Yip and L. Ding, Chem. Commun., 2015, 51, 11830–11833 RSC; (e) R. Bernini, S. Cacchi, G. Fabrizi and A. Sferrazza, Synthesis, 2008, 729–738 CAS.
- (a) J. V. Suárez-Meneses, A. Oukhrib, M. Gouygou, M. Urrutigoïty, J.-C. Daran, A. Cordero-Vargas, M. C. Ortega-Alfaro and J. G. López-Cortés, Dalton Trans., 2016, 45, 9621–9630 RSC; (b) J. V. Suárez-Meneses, E. Bonilla-Reyes, E. A. Blé-González, M. C. Ortega-Alfaro, R. A. Toscano, A. Cordero-Vargas and J. G. López-Cortés, Tetrahedron, 2014, 70, 1422–1430 CrossRef; (c) K. C. Majumdar, B. Chattopadhyay and S. Nath, Tetrahedron Lett., 2008, 49, 1609–1612 CrossRef CAS; (d) S. Prado, S. Michel, F. Tillequin and M. Koch, Lect. Heterocycl. Chem., 2006, 43, 1261–1264 CrossRef CAS.
- (a) D. S. Roman, Y. Takahashi and A. B. Charette, Org. Lett., 2011, 13, 3242–3245 CrossRef CAS; (b) B. S. Bhakuni, A. Kumar, S. J. Balkrishna, J. A. Sheikh, S. Konar and S. Kumar, Org. Lett., 2012, 14, 2838–2841 CrossRef CAS.
- I. Jarak, M. Kralj, L. Šuman, G. Pavlović, J. Dogan, I. Piantanida, M. Žinić, K. Pavelić and G. Karminski-Zamola, J. Med. Chem., 2005, 48, 2346–2360 CrossRef CAS.
- M. Aleksić, B. Bertoša, R. Nhili, L. Uzelac, I. Jarak, S. Depauw, M.-H. David-Cordonnier, M. Kralj, S. Tomić and G. Karminski-Zamola, J. Med. Chem., 2012, 55, 5044–5060 CrossRef.
- (a) M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590–11593 CrossRef CAS; (b) A. Aoyama, H. Aoyama, K. Dodo, M. Makishima, Y. Hashimoto and H. Miyachi, Heterocycles, 2008, 76, 137–142 CrossRef CAS.
- Y. Nishiyama, M. Nakamura, T. Misawa, M. Nakagomi, M. Makishima, M. Ishikawa and Y. Hashimoto, Bioorg. Med. Chem., 2014, 22, 2799–2808 CrossRef CAS.
- A.-D. Yapi, N. Desbois, J.-M. Chezal, O. Chavignon, J.-C. Teulade, A. Valentin and Y. Blache, Eur. J. Med. Chem., 2010, 45, 2854–2859 CrossRef CAS.
- (a) J. Ye, Z. Shi, T. Sperger, Y. Yasukawa, C. Kingston, F. Schoenebeck and M. Lautens, Nat. Chem., 2017, 9, 361–368 CrossRef CAS PubMed; (b) D. Heijnen, F. Tosi, C. Vila, M. C. A. Stuart, P. H. Elsinga, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 3354–3359 CrossRef CAS; (c) K. Matsumoto, M. Yoshida and M. Shindo, Angew. Chem., Int. Ed., 2016, 55, 5272–5276 CrossRef CAS; (d) P. Li, L. Wang, L. Zhang and G.-W. Wang, Adv. Synth. Catal., 2012, 354, 1307–1318 CrossRef CAS.
- (a) J. Alonso, N. Halland, M. Nazaré, O. R'Kyek, M. Urmann and A. Lindenschmidt, Eur. J. Org. Chem., 2011, 234–237 CrossRef CAS; (b) O. Kitagawa, M. Takahashi, M. Yoshikawa and T. Taguchi, J. Am. Chem. Soc., 2005, 127, 3676–3677 CrossRef CAS.
- V. Polshettiwar, P. Hesemann and J. J. E. Moreau, Tetrahedron, 2007, 63, 6784–6790 CrossRef CAS.
- D. García-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066–1067 CrossRef.
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS.
- M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS.
- L.-C. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581–590 CrossRef CAS PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- H.-Y. Sun, S. I. Gorelsky, D. R. Stuart, L.-C. Campeau and K. Fagnou, J. Org. Chem., 2010, 75, 8180–8189 CrossRef CAS PubMed.
- N. Yang, L. Dong, Z. Su and C. Hu, RSC Adv., 2013, 3, 20772–20781 RSC.
- J. Robertson, M. J. Palframan, S. A. Shea, K. Tchabanenko, W. P. Unsworth and C. Winters, Tetrahedron, 2008, 64, 11896–11907 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02099j |
| This journal is © The Royal Society of Chemistry 2018 |