
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manipulating extracellular tumour pH: an effective target for cancer therapy
Guanyu Hao
,
Zhi Ping Xu
* and
Li Li
 *
*
Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, Queensland, Australia 4072. E-mail: l.li2@uq.edu.au; gordonxu@uq.edu.au
First published on 19th June 2018
Abstract
The pH in tumour cells and the tumour microenvironment has played important roles in cancer development and treatment. It was thought that both the extracellular and intracellular pH values in tumours are acidic and lower than in normal cells. However, recent progress in the measurement of pH in tumour tissue has disclosed that the intracellular pH (pHi) of cancer cells is neutral or even mildly alkaline compared to normal tissue cells. This review article has summarized the recent advancement in the measurement pHi and extracellular pH (pHe) in cancer cells, and the effect of pHi and pHe on proliferation, migration and biological functions of cancer cells. This paper has also elaborated recent treatment strategies to manipulate pHi and pHe for cancer treatment. Based on the recent progress in pHi and pHe manipulation in cancer treatment, we have proposed potential nanoparticle-based strategies to manipulate pHi and pHe to effectively treat cancer.
 Guanyu Hao | Guanyu Hao obtained his BS degree in Chemical Biology from Xiamen University (Xiamen, China) in 2015 and Master degree in Biotechnology from The University of Queensland. He has been working with Dr Li Li and Professor Zhi Ping Xu at Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland since January 2017. His current research focuses on the design and development of alkaline nanoparticle-based platforms for cancer imaging and therapy. |
 Zhi Ping Xu | Professor Zhi Ping Xu received his PhD degree from National University of Singapore in 2001. After his postdoctoral fellowship at University of North Texas, he joined the Australian Institute for Bioengineering and Nanotechnology, the University of Queensland as a research fellow in 2004. Currently, he is a senior group leader at Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland. He is an expert in anionic clay materials, i.e. layered double hydroxides (LDH) and related materials (CaP) for drug/gene/vaccine delivery to treat tumour and infectious diseases, as well as imaging for bioanalysis and diagnosis. |
 Li Li | Dr Li Li is currently an Advance Queensland Research Fellow (Mid) at Australian Institute for Bioengineering and Nanotechnology, the University of Queensland. She received her PhD in Chemical Engineering from China University of Petroleum in 2007. She has broad research experience in developing novel functional nanomaterials and nanocomposites for biomedical applications. Currently, her research focuses on design and engineering of functional nanoparticles for drug/gene delivery, sustainable release and oral vaccine delivery for cancer treatment and animal health management. |
1. Introduction
Cancer is one of the most severe diseases in the world. According to statistics, in total 8.8 million people died from cancer in 2015, accounting for 17% of the total deaths.1,2 Researchers have made great efforts to understand the pathogenesis and properties of cancer in order to develop effective treatments for clinic application. As known, extracellular and intracellular pHs in tissues affect the function of the cells and play an important role in cancer development and treatment. As reported, the extracellular pH (pHe) affects the proliferation of human T cells and the expression of the interleukin-2 receptor.3It is widely accepted that the pHe of cancer cells is more acidic than normal cells.4–6 Generally, pHe values of the normal tissues (brain tissues, subcutaneous tissues, etc.) are in the rage of 7.2–7.5. However, pHe of tumour cells is mildly acidic in the range of 6.4–7.0. Since Warburg et al. first reported the abnormal anaerobic glycolysis in tumour cells, they measured the glucose and lactic acid in tumor veins and found more lactic acid and less glucose on the tumour tissue than on the normal tissue due to the fermentation process in the tumour side, which may affect pHe and pHi in tumour.7 Consequently, it has been assumed that pHe and pHi in cancer cells should be more acidic than those in normal cells during 1930s to 1980s.7,8
With the progress on sensing technologies, several techniques have been developed to measure pHi and pHe in cancer cells including pH-sensitive nuclear magnetic resonance spectroscopy (MRS), positron emission tomography (PET) radiotracers, magnetic resonance imaging (MRI) and optical imaging (Optics).9 It has been found that pHi in cancer cells is actually mildly alkaline or near neutral, similar to normal cells.10,11 These new findings subvert the traditional assumption that the pHi in cancer cells is more acidic than normal cells. More extracellular acidity and more intracellular alkalinity means a smaller ratio of pHe/pHi.
Subsequently, researchers have investigated the mechanisms of pH controls in cancer cells and microenvironments. Numerous membrane transporters across tumour cells have been found for pH hemeostasis in cancer cells, and further been used to manipulate pHe and pHi.2,12–17 These novel strategies have been developed to control the pHe/pHi ratio in cancer microenvironments and cells to induce apoptosis of cancer cells, improving the treatment efficiency.18
In this review, we have summarized the recent progress on the studies of pHe and pHi in tumour tissues and their corresponding normal tissues. Then, we have further outlined the mechanisms of pHe/pHi maintenance in cancer cells and the developed therapeutics to manipulate the pHe/pHi in cancer tissues. In the outlook, the potentials of new strategies using state-of-art nanotechnology to manipulate the pHe/pHi in cancer tissues have been proposed for cancer treatment.
2. pHe/pHi in tumour tissues versus normal tissues
2.1. Technologies for in vivo pH measurement and their accuracies
Several approaches for the measurement of pHe and pHi in tumour have been developed including pH-sensitive electrodes (POT), chemical exchange saturation transfer MRI (CEST-MRI), MRS, PET, MRI, and Optics.4,6,19–33 Table 1 summarized some basic information of four major technologies for in vivo pH measurement.| Technology | First use | Accuracy (pH unit) | Mechanism | Major Advantage | Reference |
|---|---|---|---|---|---|
| POT | 1950s | ±0.1–0.2 pH | Use of pH-sensitive electrodes with tip diameters ranging from 0.5 pm to 2 mm | Electrodes can be directly controlled by hand and the results can be easily read | 4 and 6 |
| PET | 1970s | ±0.08 pH | Based on the presence of pH-dependent biologically active molecule | High sensitivity (nM–pM level detected) | 23 and 24 |
| MRS | 1980s | ±0.06 pH | Based on the pH-dependent chemical shift of the resonance frequency | Real-time observation of multiple metabolites | 25 and 26 |
| MRI | 1990s | ±0.1 pH | Based on the pH-dependent relaxation agent, hyperpolarized 13C-labelled agent, and/or proton-electron double resonance imaging | Visible, concentration-independent | 19–22 and 27–29 |
| Optics | 2000s | ±1.5% (±0.1 pH) | Based on the specificity of fluorescence probes and pH sensitivity of their emission lifetime | Non-invasive, Independent to the concentration of agent and intensity of the excitation light | 31 and 32 |
| CEST-MRI | 2000s | ±0.01 pH | Based on the agents that are capable of exchanging protons with the surrounding water molecules, lead to the continuous buildup of magnetic saturation of water, resulting in extremely enhanced sensitivity | Very low concentration extremely high sensitivity | 21 and 33 |
Although several novel MRI and optical imaging agents (probes) have been developed and applied for in vivo pH measurement, there is not adequate data of the pH values measured by the same MRI or Optics method for comparable analysis. Thus, for the consistency of the comparison, the pH measured by POT or MRS were collected and compared in Section 2.2 and 2.3.
2.2. Extracellular pH (pHe)
According to the literature reports, pHe of eight types of tumour tissues and the corresponding normal tissues has been summarized in Fig. 1. These data were selected based on the measurements using pH-sensitive electrodes.4,6,34–39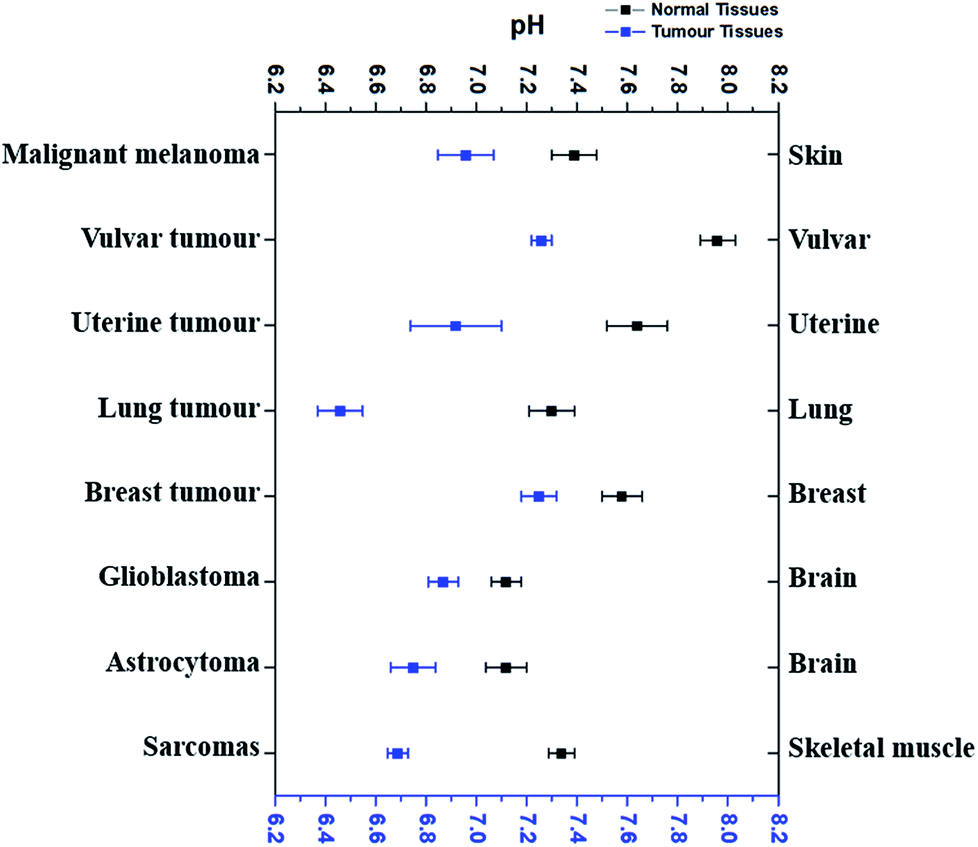 | ||
| Fig. 1 The comparison of average extracellular pH values of different tumours with normal tissues. Blue dots refer to the average extracellular pH of some cancer tissues, while black dots the average extracellular pH of corresponding normal tissues. All dots (average pHe ± SEM) referred to the average extracellular pH of a specific kind of cancer or normal tissues listed. Data were taken from several different sources (ref. 4, 6 and 34–39), which were given in the text correspondingly. | ||
As shown in Fig. 1, pHe of cancer cells is 0.3–0.7 pH unit lower than that of corresponding normal cells. For example, malignant melanoma tissues have an average pHe of 6.96 while the average pHe in normal skin cells is 7.39,4 which is 0.43 difference. The average pHe in vulvar tumours is 7.26, 0.7 pH unit less than in normal vulvar tissues (with an average pHe of 7.96).6 Uterine tumour tissues also have a lower average pHe (6.92) than normal uterus, whose average pHe is 7.64.6 Although the average pHe of two kinds of brain tumours is slightly different, they are both more acidic than normal brain tissues.38 Similar results have also been observed in other tissues, such as lung,34 breast,39 and skeletal muscle.35–37 Thus, it is very clear that most cancer cells usually have a more acidic pHe than their corresponding normal cells, and the differences vary from 0.3–0.7.
Warburg et al. proposed that tumour cells used glycolysis rather than oxidative phosphorylation to acquire energy, even in the presence of oxygen.7 Excess anaerobic glycolysis has been considered as the major reason for the extracellular acidity of tumour tissues.10,40 For most animal cells, there are two different pathways for glucose metabolism, i.e. aerobic and anaerobic glycolysis. The detailed processes of glucose metabolism in the cells have been briefly outlined in Fig. 2. There are two possible pathways for glucose metabolism in the cells: aerobic and anaerobic pathway. Generally, one glucose molecule is metabolized to two pyruvate molecules, producing two ATP molecules as the energy. In the aerobic pathway, two pyruvate molecules react with CoA-SH and form acetyl-CoA by releasing CO2. Subsequently, the produced acetyl-CoA undergoes the citric acid cycle, finally degrading into CO2 and producing 30 ATP molecules. In the anaerobic process, two pyruvate molecules transfer into two lactate molecules with the assistance of lactate dehydrogenase, but this transfer only produce 2 ATP. The overall reactions of these two ways are briefly expressed as follows:
| C6H12O6 (D-glucose) → 6CO2 + 6H2O + 38ATP (aerobic) |
| C6H12O6 (D-glucose) → 2C3H6O3 (lactate) + 2ATP (anaerobic) |
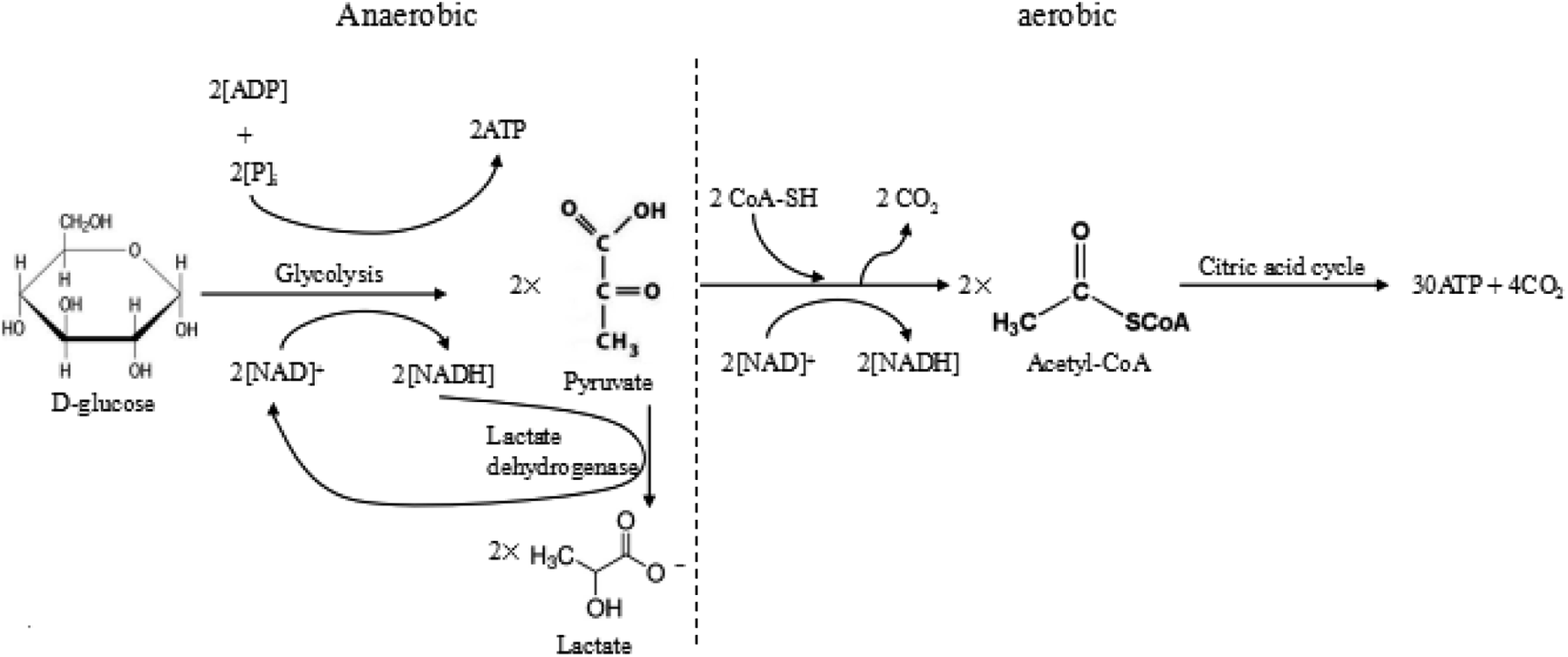 | ||
| Fig. 2 The anaerobic and aerobic pathways of glycolysis. | ||
In the normal cells, most glucose is fully metabolized to produce carbon dioxide, water and the energy via the aerobic pathway. However, in the tumour cells, the glucose is mostly metabolized through the anaerobic pathway, which produces a large amount of lactate and releases limited energy due to a high level of pyruvate and hypoxia in the tumour environment. During the process, the tumour growth requires a large amount of energy compared to the normal tissue, which produces more CO2 and lactic ions in tumour. The produced CO2 was excreted extracellularly, resulting in the acidic condition in the tumour microenvironment, i.e. 0.3–0.7 pH units lower than the average pHe of normal tissues.
2.3. Intracellular pHi: acidic or not?
Interestingly, pHi of cancer cells is not acidic, not as postulated previously. Since the 1980's, more research outcomes have demonstrated that pHi of cancer cells is around neutral and even mildly alkaline.10,11 Fig. 3 has displayed the pHi of six kinds of tumour tissues and their corresponding normal tissues collected from MRS method.11,41–45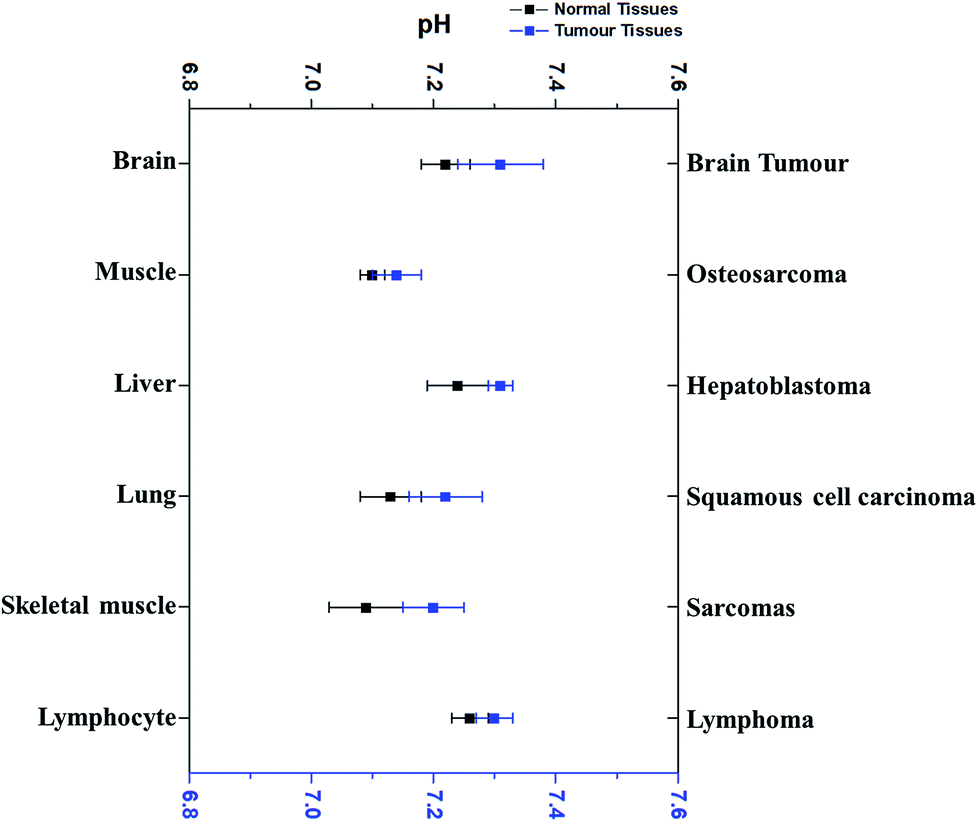 | ||
| Fig. 3 The comparison of the average intracellular pH values of different tumours with that in the corresponding normal cells. Blue dots refer to the average intracellular pH of tumour cells, while black dots represent the average intracellular pH of corresponding normal cells. All dots (average ± SEM) refer to the average intracellular pH of a specific kind of cancer or normal cells listed. Data were taken from different sources (ref. 11 and 41–45). | ||
Very surprisingly, the average pHi of these tumour cells is slightly higher than that in their corresponding normal cells, although the difference is less than 0.1 pH unit and not significant. For example, the average pHi of brain tumours is 7.31, slightly higher than normal brain cells (7.24).11,42,46 Redmond et al. reported that the intracellular environment of osteosarcoma cells is also mildly more alkaline than in normal cells.43 Furthermore, this weak alkalinity of the intracellular environment in tumour cells has also been discovered in many other types of tumours, such as hepatoblastoma11 and squamous cell carcinoma.44,45 These evidences thus clearly indicate that pHi of tumour cells is near neutron or even more alkaline. Thus, the discrepancy of pHe and pHi in tumour cells is much larger than in normal tissues.
2.4. How cancer cells maintain their unbalanced pHe/pHi ratio?
For most cells, the maintenance of neutral (or mild alkaline) pHi is achieved by transporting respiratory end-products (such as CO2 and lactate) across the cell membrane. When the extracellular concentration of acidic respiratory end-products is lower than intracellular, the excess CO2 can passively across the cell membrane by diffusion.47 However, in most cases, the CO2 and lactate generated from glucose metabolise is accumulated in extracellular tumour site due to low blood flow rate, resulting in development of acidic microenvironments in tumour.48,49 In this situation, the release of CO2 and lactate in microenvironments mainly relies on numerous special membrane proteins, such as carbonic anhydrase enzymes (CA2, CA9 and CA12). More relevant pH regulators are listed in Table 2 and discussed in Section 3. Overall, the maintenance of pHe and pHi is based on passive diffusion and active membrane transporters. Table 2 briefly summarizes some major pH regulators in tumours and their main functions, including anion exchangers (SLC4A1, SLC4A2, and SLC4A3), proton transporter vacuolar ATPase (V-ATPase), mono-carboxylate transporters (MCT1, MCT2, MCT3, and MCT4), sodium ion based chloride/bicarbonate exchanger (SLC4A8) and Na+/H+ exchanger 1 (SLC9A1).50–59| Name | Description | Function | Reference |
|---|---|---|---|
| SLC4A1 | Anion exchangers | Transport HCO3− out of cancer cells | 53 and 54 |
| SLC4A2 | |||
| SLC4A3 | |||
| SLC4A7 | Sodium bicarbonate cotransporters | Mediate the coupled movement of sodium and bicarbonate ions across the plasma membrane | 55 |
| SLC4A8 | Sodium ion-based chloride/bicarbonate | Transport Cl− out of tumour cells and simultaneously import HCO3− into cancer cells powdered by Na+ | 56 |
| SLC9A1 | Na+/H+ exchanger 1 | Transport intracellular produced H+ to the extracellular environment, and import Na+ at the same time | 56 |
| MCT1 | Monocarboxylate transporters | Transport (both inside to outside and outside to inside) the products of glycolysis (such lactic acid and other monocarboxylates) | 57 and 58 |
| MCT2 | |||
| MCT3 | |||
| MCT4 | |||
| V-ATPase | Proton transporter vacuolar ATPase | A proton pump on the membrane of tumour cells, responsible for the stransportation of H+ between intracellular and extracellular plasma | 59 |
In the last two decades, several complicated mechanisms have been revealed about how cancer cells maintain the alkaline pHi and acidic pHe.60–65 Among them, the mechanism for the import of weak bases (e.g. bicarbonate) and the extrusion of weak acids (e.g. CO2, H2CO3, and lactate) with the assistance of proteins in tumour cell membrane has been clearly demonstrated.66 Apart from this, the intracellular protons have been pumped out of tumour cells in three different ways, including direct discharge from the cells, exchange with other extracellular cations (e.g. Na+), and extrusion by the vacuolar ATPase.56,67
3. The effect of pHe and pHi on tumour activity
As discussed above, the difference of pHe and pHi in tumour cells is much larger than in normal cells. The maintenance of pHe and pHi in the tumour mainly relies on some specific proton pumps and intracellular buffer systems.10,68,69 For instance, the balance of HCO3−/CO32− buffer system in tumour is administrated by carbonic anhydrase enzymes CA2, CA9 and CA12.12,70,71 Besides, the Na+/H+ buffer system is manipulated by Na+/H+ exchangers, such as SLC9A1.72 The regulation of pHe and pHi depends on the synergic effect of all of these pumps and buffer systems.It is known that even the little change of pHe/pHi ratio may severely affect many biological and chemical processes in the cells, and eventually result in the proliferation and aggressiveness of cancer cells.60 For example, the incubation of melanoma in the acidic environment can significantly enhance its metastasis, aggressiveness and migratory activity in vitro.73 Martinez-Zaguilan reported that C8161 and A375P cells were cultured in acidic medium (pH 6.8) for 3 weeks and then transferred to the membrane invasion culture system (MICS) chambers.73 They found that C8161 cells and A375P cells treated in acidic medium have significantly enhanced migration and invasion, as shown in Fig. 4. Moellering et al. also reported that acidic-treated C8161 cells cultured in normal medium (pH 7.4) showed higher aggressiveness than those cultured in acidic environment (low pH group) and control (native group), as shown in Fig. 5.74 The C8161 cells cultured in lower pH medium (6.7) has shown the inhibition of the cell invasion, indicating less aggressiveness. These results have demonstrated that the regulation of pHe and pHi ratio in the tumour is highly important for metastasis, aggressiveness and migratory activity. Fine control of pHe and pHi in tumour may improve the cancer treatment.
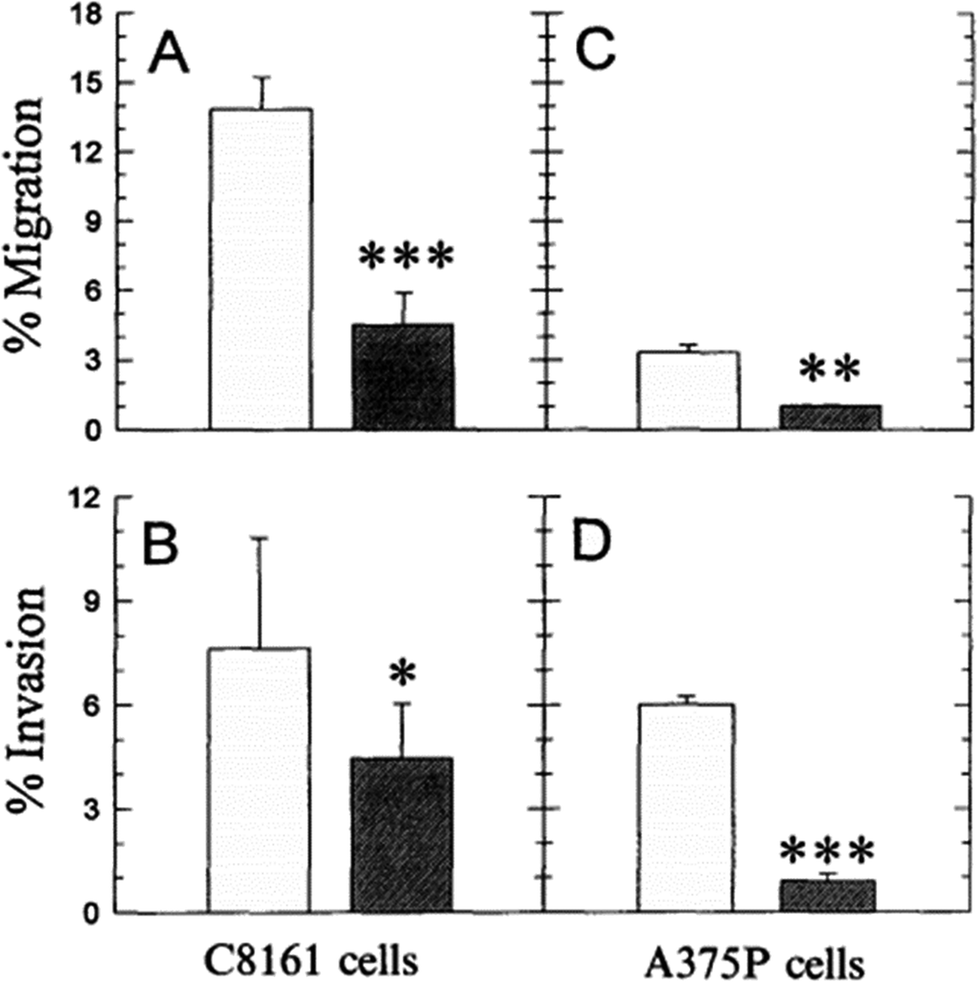 | ||
| Fig. 4 Comparison of the migration and invasion of C8161 and A375P cells treated in standard and acid media. The migration and invasion properties of cells treated in acidic medium (pH 6.8) were drawn in white bar, while black bar referred to the value of cells cultured in standard medium (pH 7.4). Data analysis was performed using Student's t-test: *P < 0.01; **P < 0.005; ***P < 0.001. This figure is adapted from ref. 73 with permission from Kluwer Academic Publishers. | ||
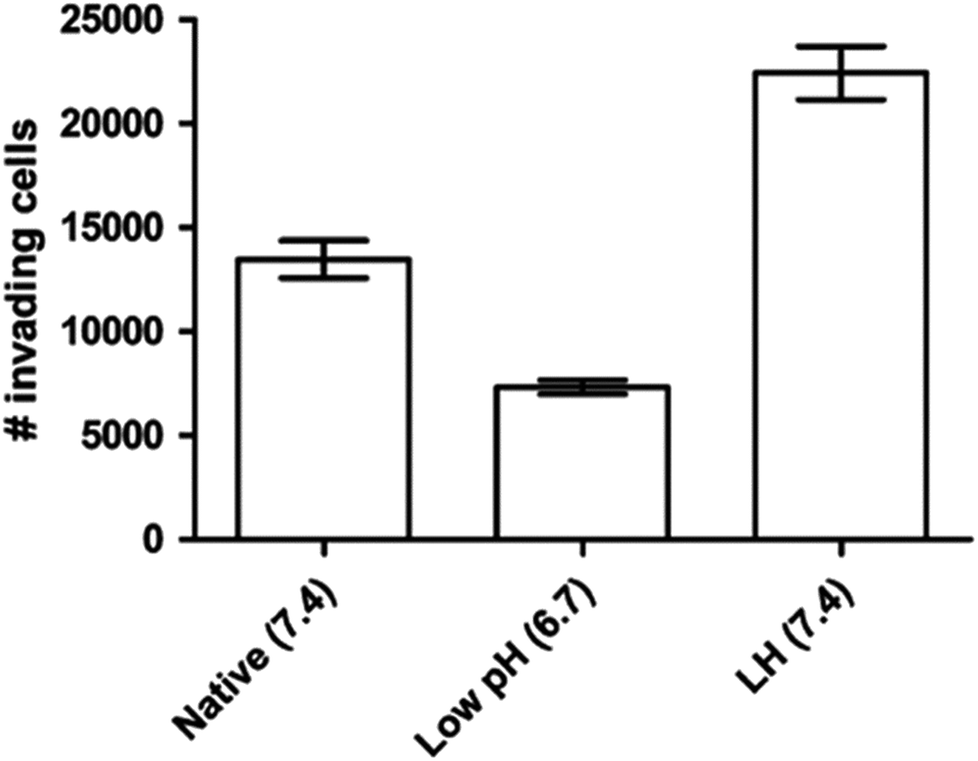 | ||
| Fig. 5 Invasion of different C8161 phenotypes. Representative invasion assay results for C8161 phenotypes assayed in their respective media. Native group meant the cells were incubated in normal medium. Low pH group represented the cells cultured in acidic medium (pH 6.7). LH group meant the cells were cultured in acidic medium for 1 month and then transferred into normal medium before the experiment. This figure is reproduced from ref. 74 with permission from Springer Netherlands. | ||
Furthermore, the slight change of pHe and pHi may also disorder the function of some proteins (such as tenascin and fibronectin), particularly in cancer cells.75,76 For example, mild change of environmental pH by 0.7 pH unit dramatically affected the RNA alternative slicing. The major expression of tenascin-C (TN-C) isoforms was 8 kb TN mRNA in human skin fibroblasts at pH 7.4, while 6 kb TN mRNA isoform was the majority of TN-C expression at pH 6.7 (see Fig. 6).
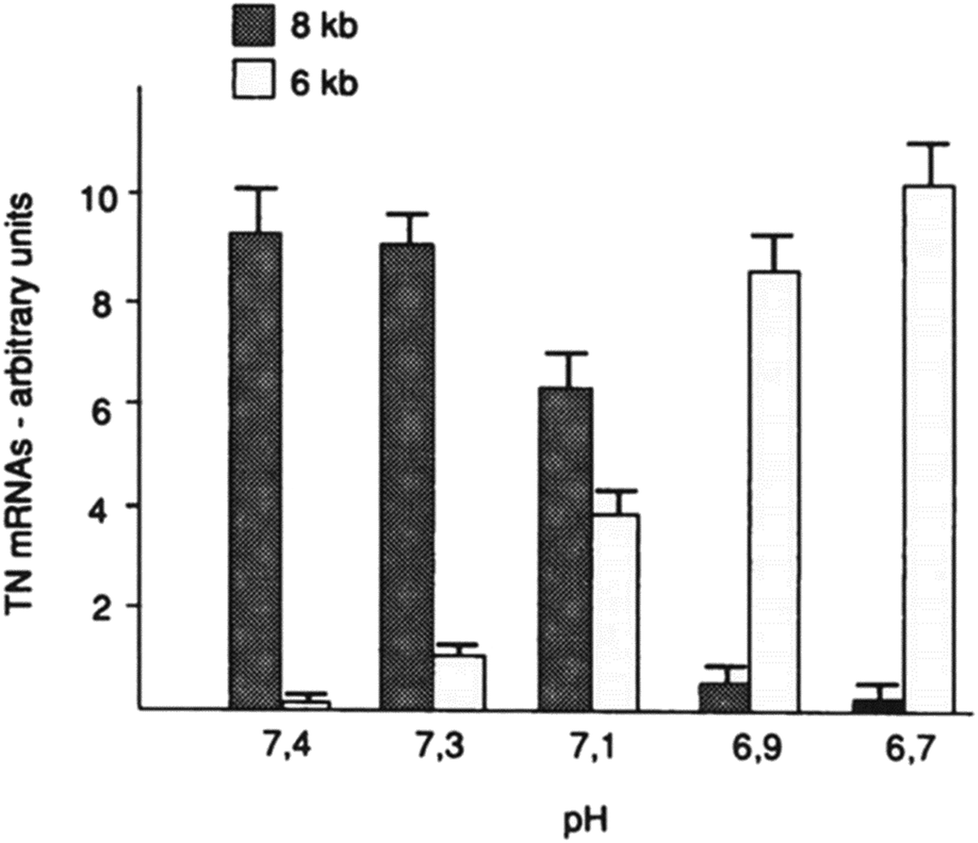 | ||
| Fig. 6 Effect of environmental pH on the RNA alternative slicing of TN-C mRNA isoforms in human skin fibroblasts. Skin fibroblasts were incubated in DMEM medium at different pH for 4 days, and the expressed TN-C mRNA amounts were derived from northern blot analyses and shown in arbitrary units. This figure is adapted from ref. 75 with permission from American Society for Biochemistry and Molecular Biology. | ||
Tumour microenvironment triggers the tumour heterogeneity during the cancer development. It is well known that acidic condition and hypoxia are important characteristics in the tumour microenvironment. The homeostasis of pHe and pHi is very important for all kinds of cells. As discussed above, compared with normal cells, cancer cells have a more acidic pHe and more alkaline pHi, suggesting that the pH homeostasis regulation of tumour tissues may be more complex and involve in more proteins and buffer systems. The pH environment may influence the growth and function of the cells in two main ways. On the one hand, the 0.1 alteration in the ratio of pHe/pHi may affect many essential biochemical processes in the cell metabolism system, such as ATP synthesis, cell proliferation, aggressiveness, migration and diffusion, and the function of some membrane proteins.60 On the other hand, the tiny disturbance of pHe may activate the mechanism of alternative splicing of constituents in extracellular matrix to produce isoform of tenascin and fibronectin, which specifically occur in cancer cells rather than in normal cells.75,76 Although the isoforms of these alternatively spliced proteins do not involve in the manipulation of tumour's pHe/pHi ratio, they may provide binding sites for antigen-based cancer therapy.18
4. Strategies to manipulate the pHe/pHi ratio
As discussed above, the small change in pHe/pHi ratio of tumour cells may disturb many biological functions, including proliferation, aggressiveness, and migration. This relationship demonstrates that adjusting the pHe/pHi ratio in the tumour tissues may halt cancer progress or even completely inhibit cancer growth. In recent years, several approaches have been developed to manipulate pHe/pHi ratio for cancer treatment. These approaches can be classified as direct manipulation and indirect manipulation. Direct manipulation is to regulate the pHe/pHi ratio of tumour cells by using acidic/alkaline drugs and indirect manipulation is based on operating the pH regulators of tumour cells.4.1. Direct manipulation using small molecule drugs
The drugs for direct manipulation are mainly small molecular substances (such as bicarbonates). This approach is to directly increase pHe of tumour tissues to the normal level (0.3–0.7 pH unit). It can be achieved by oral administration of alkaline agents or even by simple adjustment of diet habit.The alkaline agents include sodium bicarbonate and trisodium citrate.77 In practice, it seems difficult to maintain the mildly alkaline microenvironment near tumour tissues via oral administration, as a high dose and continuous intake of the alkaline substrate is required. Based on the breast cancer study, White et al. investigated the exact daily dose of sodium bicarbonate needed for breast cancer treatment.78 The calculated daily dose for a normal human (with 70 kg weight) would be 31.75 g sodium carbonate or 32.5 g trisodium citrate.79 Another example is the Tris–base buffer to inhibit tumour progression and metastasis.80 The size of the pancreatic tumour in the mouse model was significantly decreased after 200 mM of Tris–buffer treatment. Based on their data, the daily dose for the mice can be calculated as 18.2 g of Tris–base buffer per kg, equivalents to 31 g of Tris–base intake per day for an adult (70 kg). Although it is possible for a cancer patient to intake more than 30 g alkaline agents (such as sodium carbonate or trisodium citrate) with daily drinking water, it would be more efficient to deliver alkaline agent to the tumour tissues rather than to the whole body. A recent non-randomized controlled study investigated the efficacy of local infusion of alkaline agent.81 Researchers found that there was a 6.4-fold difference of geometric mean of viable tumour residues (VTR) when the hepatocellular carcinoma patients were treated with transarterial chemoembolization (TACE) accompanied with or without locally infusing bicarbonate (LIB) into tumour (Table 3). Such a local administration may be a better strategy for anticancer therapy.
| TACE (n = 27) | TACE + LIB (n = 30) | P value | |
|---|---|---|---|
| Crude VTR | 45.1% (30.3–67.0%) | 7.1% (4.4–11.5%) | <0.0001 |
| Multivariable adjusted VTR | 45.6% (28.9–72.0%) | 7.1% (4.6–10.9%) | <0.0001 |
The adjusted diet could be low in protein but high in potassium and/or magnesium.82–84 It has been proved that potassium can effectively neutralize mineral acidity and even mildly alkaline pH of urine via KHCO3 generation or glutamine sparing.85 The pHi may be altered by a large change of the intake of potassium due to its fundamental physiologic and metabolic importance.85 Based on another big data analysis (based on more than 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cases), the risk of suffering from pancreatic cancer decreased by 18% for each 100 mg increase of magnesium intake per day by men on the continuous scale.86 These results may provide a diet-based way to manipulate the pH environment in vivo and assist cancer treatment.
000 cases), the risk of suffering from pancreatic cancer decreased by 18% for each 100 mg increase of magnesium intake per day by men on the continuous scale.86 These results may provide a diet-based way to manipulate the pH environment in vivo and assist cancer treatment.
4.2. Indirect manipulation: proton pump inhibitors
The second alternative strategy to administrate the pHe/pHi ratio is to inhibit the functional proton pumps. It is well known that the maintenance of high pHe/pHi ratio in tumour tissues relies on many proton regulators (pumps) on the cell membrane. Most of these proton pumps on the tumour cell membrane have a few specific isoforms that do not exist on the normal cell surface. Thus these isoforms may provide some specific target sites for cancer therapy. Once these functional proton pumps are inhibited, the pH balancing system of tumour cells may be disordered and the pHe/pHi ratio may increase. The abnormal proton transportation and change of the pHe/pHi ratio may affect the behaviour of tumour cells. Recent research reports have demonstrated that the inhibition of proton regulators have suppressed the proliferation and promoted the programmed cell death in some tumour cell lines.87–92 For example, treatment with proton pump inhibitors led to the induction of apoptosis in many types of gastric cancer cells, which involves in the regulation of tumour pH.89 Besides, the inhibition of proton extrusion by Na+/H+ exchanger inhibitors72 or V-ATPase inhibitors93 may make cancer cells susceptible or vulnerable. Now a few proton pump inhibitor drugs have been used in the clinical stage. Table 4 lists some inhibitors and their target proton pumps.90,94–102 As seen in Table 4, the current inhibitor drugs mainly focus on two major pH regulators (V-ATPase and SLC9A1) and only one of these drugs, cariporide, has been successfully developed to phase III clinical trial.| Inhibitors drugs | Identification site | Function & description | Reference |
|---|---|---|---|
| Omeprazole, esomeprazole | V-ATPase | Can be activated in the slightly acidic environment, and then inhibit V-ATPase via covalent interaction. Work on V-ATPase at high dose | 94 and 95 |
| Bafilomycin | V-ATPase | Commonly inhibits V-ATPase (not selective for tumour cells) with high cell toxicity | 96 and 97 |
| Diuretic amiloride | SLC9A1 | Inhibits NHE-1 with unacceptable high concentration | 98 |
| EIPA (derivative of amiloride) | SLC9A1 | 200 times stronger than amiloride, has not used in clinical trial yet | 98 and 99 |
| Cariporide | SLC9A1 | Decrease the intracellular pH of cancer cells. Has been developed to the third stage of clinical trial | 90 and 100–102 |
Interestingly, decreasing pHi may increase hyperthermia efficacy (over 42 °C) and the programming cell death response to TNF (tumour necrosis factor) induced by apoptosis ligand, also known as TRAIL.96,103–105 For example, both bafilomycine A1 (an inhibitor of V-ATPase) and EIPA (an inhibitor of the Na+/H+ exchanger) increased the thermo-sensitivity of the AsPC-1 tumours (grown in nude mice) by individually mildly decreasing pHi, and the thermo-sensitivity was markedly enhanced by the sharp decrease in pHi, resulting from the synergetic effect of the combination of these two therapies.96
Fig. 7 outlines the functions of some specific pH regulators and relatively main inhibitors for tumour cells. The functions of these ion exchangers and proton pumps, and their main inhibitors have been described in Tables 3 and 4 The overall process of ion exchangers is the cellular intake of HCO3− and cellular exhaust of CO2 and Cl−, which both lead to pHe decrease and pHi increase. Proton pumps (or more exactly Na+/H+ exchangers) directly exchange intracellular H+ with extracellular Na+.
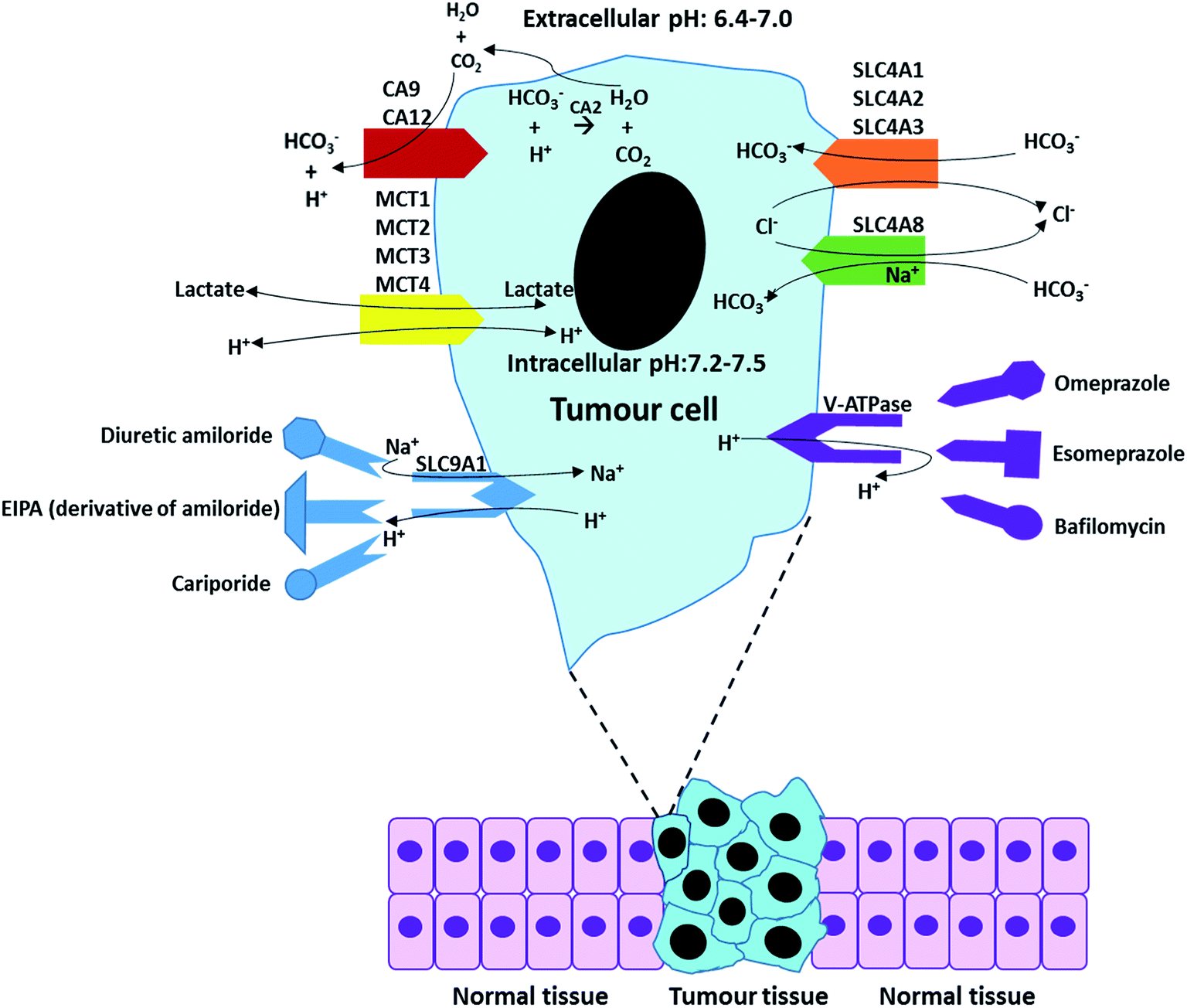 | ||
| Fig. 7 A summary of the regulation processes in a tumour cell and some “manipulators (inhibitors)”. | ||
To conclude, more targeting sites and relevant inhibitors need to be explored in order to more efficiently manipulate pHe/pHi in tumour cells for potential and effective cancer therapy.
4.3. Alternative methods to manipulate the pHe/pHi ratio
Several research reports have showed that the apoptosis of tumour cells can be boosted by the adequately large decrease of their pHi.87–92 One way to achieve the reduction of pHi in tumour cells is to promote cancer glycolysis to the utmost extent by maximizing the glucose supplement. The extremely high rate of glycolysis may break the capacity of proton pumps in tumour cells, which means that tumour cells cannot timely transport acidic metabolites (such as H+, H2CO3, lactate etc.) outside and hence decreases pHi. For example, a very high glycolysis rate was observed in human melanoma cells (cultured in the medium containing high amount of glucose) when DNP, an uncoupling agent, was added.106The programmed cell death can also be activated by the sharp reduction of pHi, finally leading to cell death.102 This may result from several different mechanisms, one of which is the reduction of glycolysis metabolism.107 For example, the enzymatic functions of hexokinase, one of the vital enzymes for the maintenance of the high level of glycolysis metabolism in tumours cells, was strongly inhibited (activity decreased from 82 ± 3.2% to 31.2 ± 5.7) with the sharp decrease (from 8 to 6 respectively) of pHi in SNB-19 glioma cells.108
The tumour glycolysis can be promoted by inhibiting the production of mitochondrial ATP, which requires some specific inhibitors. meta-Iodobenzylguanidine is one of the inhibitors of mitochondrial complex 1, acting as proton extrusion inhibitors (or hyperglycemia) and then decreasing pHi in cancer cells.100,109–111 However, this drug is normally used as a radioiodine therapy agent, and the dose used for radioiodine therapy is not high enough to perform a strong inhibition on proton transportation. Dinitrophenol (DNP), a new type of chemotherapeutic drugs, has also shown a remarkable enhancement in glycolysis with the increase of blood pressure at a low dose. It has been reported that mM-level DNP can inhibit the proliferation of cancer cells and lead to apoptosis in the human pulmonary adenocarcinoma Calu-6 cell line.112
Overall, even though there are some drugs (such as meta-iodobenzylguanidine and DNP) that have shown their ability to decrease pHi by boosting the glycolysis rate in tumour cells, the hyperglycemia-reliable mechanism restricts the feasibility of this cancer therapy strategy.
5. Conclusions and future prospective
In this review, pHe and pHi in tumour cells have been summarized and the ways to manipulate cellular pH in cancer cells have been discussed. It is clear that tumour cells have a more acidic pHe (0.3–0.7 lower) than normal cells, and pHi in tumour cells is neutral or even more alkaline than that in normal cells. The abnormally high ratio of pHe/pHi in tumour cells is due to the high rate of glycolysis in tumour cells, which produces numerous acidic products (such as H2CO3 and CO2). The maintenance of pHe/pHi relies on several special proton pumps on tumour cell membranes, such as SLC9A1 and V-ATPase. Then the mechanisms of these proton pumps are discussed and two potential pH manipulating strategies are presented, including direct manipulation by delivering small molecule drugs and indirect manipulation sing proton pump inhibitors.It has been demonstrated that treatment of cancers (halting its proliferation, aggressiveness and even inducing programmed cell death) is very possible by manipulating pHe/pHi ratio in tumour. A future potential method is to combine 2 or 3 inhibitors so that pHe/pHi can be well controlled, which may significantly enhance the efficacy of the cancer treatment.
The other future approach to manipulating the pHe/pHi ratio for cancer treatment is to use functional nanoparticle delivery systems to efficiently transport the known inhibitors. Compared to small molecular inhibitors, nanoparticles could have more advantages. For example, nanoparticles can be accumulated around tumour tissues through enhanced permeability and retention effect (EPR effect).113 Of course, inhibitor-loaded nanoparticles can be further functionalized with target ligands, which may enhance the accumulation in the tumour tissues and manipulate the pHe/pHi ratio.
Another potential way to direct pH manipulation can be achieved by target delivery of alkaline nanoparticles to the tumour tissues by virtue of EPR effect. Thus, accumulated alkaline nanoparticles neutralize the extracellular acids and efficiently increase pHe. Moreover, some alkaline nanoparticles can be modified as a carrier for delivering anticancer drugs to more efficiently treat cancers.114,115
Conflicts of interest
There are no conflicts to declare.References
- H. Wang, M. Naghavi, C. Allen, R. Barber, A. Carter, D. Casey, F. Charlson, A. Chen, M. Coates and M. Coggeshall, Lancet, 2016, 388, 1459–1544 CrossRef.
- A. B. Thomsen, S. Kim, F. Aalbaek, C. Aalkjaer and E. Boedtkjer, J. Cereb. Blood Flow Metab., 2014, 34, 161–168 CrossRef PubMed.
- K. S. Carswell and E. T. Papoutsakis, J. Immunother., 2000, 23, 669–674 CrossRef PubMed.
- B. S. Ashby, Lancet, 1966, 288, 312–315 CrossRef.
- K. Engin, D. Leeper, J. Cater, A. Thistlethwaite, L. Tupchong and J. McFarlane, Int. J. Hyperthermia, 1995, 11, 211–216 CrossRef PubMed.
- J. Naeslund and K. E. Swenson, Acta Obstet. Gynecol. Scand., 1953, 32, 359–367 CrossRef PubMed.
- O. Warburg, F. Wind and E. Negelein, J. Gen. Physiol., 1927, 8, 519–530 CrossRef PubMed.
- J. Griffiths, Br. J. Cancer, 1991, 64, 425 CrossRef PubMed.
- X. Zhang, Y. Lin and R. J. Gillies, J. Nucl. Med., 2010, 51, 1167–1170 CrossRef PubMed.
- P. Vaupel, F. Kallinowski and P. Okunieff, Cancer Res., 1989, 49, 6449–6465 Search PubMed.
- R. Oberhaensli, P. Bore, R. Rampling, D. Hilton-Jones, L. Hands and G. Radda, Lancet, 1986, 328, 8–11 CrossRef.
- P. Swietach, S. Patiar, C. T. Supuran, A. L. Harris and R. D. Vaughan-Jones, J. Biol. Chem., 2009, 284, 20299–20310 CrossRef PubMed.
- W. F. Boron and P. De Weer, J. Gen. Physiol., 1976, 67, 91–112 CrossRef PubMed.
- A. B. Thomsen, S. Kim, F. Aalbaek, C. Aalkjaer and E. Boedtkjer, J. Cereb. Blood Flow Metab., 2014, 34, 161–168 CrossRef PubMed.
- E. Boedtkjer, L. Bunch and S. F. Pedersen, Curr. Pharm. Des., 2012, 18, 1345–1371 CrossRef PubMed.
- S. C. Kong, A. Gianuzzo, I. Novak and S. F. Pedersen, Biochem. Cell Biol., 2014, 92, 449–459 CrossRef PubMed.
- E. Boedtkjer and C. Aalkjaer, Frontiers in Physiology, 2013, 4, 388 CrossRef PubMed.
- D. Neri and C. T. Supuran, Nat. Rev. Drug Discovery, 2011, 10, 767–777 CrossRef PubMed.
- S. Düwel, C. Hundshammer, M. Gersch, B. Feuerecker, K. Steiger, A. Buck, A. Walch, A. Haase, S. J. Glaser, M. Schwaiger and F. Schilling, Nat. Commun., 2017, 8, 15126 CrossRef PubMed.
- A. Samouilov, O. V. Efimova, A. A. Bobko, Z. Sun, S. Petryakov, T. D. Eubank, D. G. Trofimov, I. A. Kirilyuk, I. A. Grigor’ev, W. Takahashi, J. L. Zweier and V. V. Khramtsov, Anal. Chem., 2014, 86, 1045–1052 CrossRef PubMed.
- V. R. Sheth, Y. Li, L. Q. Chen, C. M. Howison, C. A. Flask and M. D. Pagel, Magn. Reson. Med., 2012, 67, 760–768 CrossRef PubMed.
- F. A. Gallagher, M. I. Kettunen, S. E. Day, D. E. Hu, J. H. Ardenkjær-Larsen, R. Zandt, P. R. Jensen, M. Karlsson, K. Golman, M. H. Lerche and K. M. Brindle, Nature, 2008, 453, 940–943 CrossRef PubMed.
- D. Rottenberg, J. Ginos, K. Kearfott, L. Junck and D. Bigner, Ann. Neurol., 1984, 15, 98–102 CrossRef.
- R. A. Hawkins and M. E. Phelps, Cancer Metastasis Rev., 1988, 7, 119–142 CrossRef PubMed.
- J. K. Roberts, N. Wade-Jardetzky and O. Jardetsky, Biochemistry, 1981, 20, 5389–5394 CrossRef PubMed.
- J. W. Prichard, J. R. Alger, K. L. Behar, O. Petroff and R. G. Shulman, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 2748–2751 CrossRef.
- S. Aime, S. G. Crich, M. Botta, G. Giovenzana, G. Palmisano and M. Sisti, Chem. Commun., 1999, 1577–1578 RSC.
- S. Aime, F. Fedeli, A. Sanino and E. Terreno, J. Am. Chem. Soc., 2006, 128, 11326–11327 CrossRef PubMed.
- S. Zhang, K. Wu and A. D. Sherry, Angew. Chem., Int. Ed., 1999, 38, 3192–3194 CrossRef PubMed.
- X. Zhang, G. Martinez, M. Garcia-Martin, M. Woods, D. Sherry and R. Gillies, Proceedings of the 16th Annual Meeting of ISMRM, Toronto, Ontario, Canada, 2008 Search PubMed.
- M. Hassan, J. Riley, V. Chernomordik, P. Smith, R. Pursley, S. B. Lee, J. Capala and G. Amir H, Mol. Imaging, 2007, 6, 229–236 CrossRef PubMed.
- I. Gannot, I. Ron, F. Hekmat, V. Chernomordik and A. Gandjbakhche, Lasers Surg. Med., 2004, 35, 342–348 CrossRef PubMed.
- B. Wu, G. Warnock, M. Zaiss, C. Lin, M. Chen, Z. Zhou, L. Mu, D. Nanz, R. Tuura and G. Delso, EJNMMI Physics, 2016, 3, 19 CrossRef PubMed.
- T. E. Mürdter, G. Friedel, J. T. Backman, M. McClellan, M. Schick, M. Gerken, K. Bosslet, P. Fritz, H. Toomes and H. K. Kroemer, J. Pharmacol. Exp. Ther., 2002, 301, 223–228 CrossRef.
- A. J. Thistlethwaite, D. B. Leeper, D. J. Moylan and R. E. Nerlinger, Int. J. Radiat. Oncol., Biol., Phys., 1985, 11, 1647–1652 CrossRef.
- H. Laks, J. R. Dmochowski and N. P. Couch, Ann. Surg., 1976, 183, 193 CrossRef PubMed.
- J. R. Piñeyro, J. Pediatr. Surg., 1979, 14, 77–79 CrossRef.
- F. Pampus, Acta Neurochir., 1963, 11, 305–318 CrossRef PubMed.
- A. Van den Berg, J. Wike-Hooley, A. Van Den Berg-Block, J. Van der Zee and H. Reinhold, Eur. J. Cancer Clin. Oncol., 1982, 18, 457–462 CrossRef PubMed.
- L. E. Gerweck and K. Seetharaman, Cancer Res., 1996, 56, 1194–1198 Search PubMed.
- H. D. Sostman, H. C. Charles, S. Rockwell, K. Leopold, C. Beam, D. Madwed, M. Dewhirst, G. Cofer, D. Moore and R. Burn, Radiology, 1990, 176, 837–843 CrossRef PubMed.
- B. Hubesch, D. Sappey-Marinier, K. Roth, D. Meyerhoff, G. Matson and M. Weiner, Radiology, 1990, 174, 401–409 CrossRef PubMed.
- O. Redmond, J. Stack, P. Dervan, B. Hurson, D. Carney and J. Ennis, Radiology, 1989, 172, 811–815 CrossRef PubMed.
- S. C. Wood and K. E. Schaefer, J. Appl. Physiol., 1978, 45, 115–118 CrossRef PubMed.
- T. Ng, A. Majors, S. Vijayakumar, N. Baldwin, F. Thomas, I. Koumoundouros, M. Taylor, S. Grundfest, T. Meaney and R. Tubbs, Radiology, 1989, 170, 875–878 CrossRef PubMed.
- H. D. Sostman, D. M. Prescott, M. W. Dewhirst, R. K. Dodge, D. E. Thrall, R. L. Page, J. A. Tucker, J. M. Harrelson, G. Reece and K. A. Leopold, Radiology, 1994, 190, 269–275 CrossRef PubMed.
- A. Hulikova, A. L. Harris, R. D. Vaughan-Jones and P. Swietach, Curr. Pharm. Des., 2012, 18, 1331–1337 CrossRef PubMed.
- R. K. Jain and K. Ward-Hartley, IEEE Trans. Sonics Ultrason., 1984, 31, 504–525 Search PubMed.
- P. W. Vaupel, S. Frinak and H. I. Bicher, Cancer Res., 1981, 41, 2008–2013 Search PubMed.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef PubMed.
- C. T. Supuran, Bioorg. Med. Chem. Lett., 2010, 20, 3467–3474 CrossRef PubMed.
- J. Pastorek, S. Pastoreková, I. Callebaut, J. P. Mornon, V. Zelník, R. Opavský, M. Zat'ovicová, S. Liao, D. Portetelle and E. J. Stanbridge, Oncogene, 1994, 9, 2877–2888 Search PubMed.
- D. Sterling, N. J. Brown, C. T. Supuran and J. R. Casey, Am. J. Physiol.: Cell Physiol., 2002, 283, C1522–C1529 CrossRef PubMed.
- P. E. Morgan, C. T. Supuran and J. R. Casey, Mol. Membr. Biol., 2004, 21, 423–433 CrossRef PubMed.
- M. Soleimani and C. E. Burnham, Kidney Int., 2000, 57, 371–384 CrossRef PubMed.
- J. Pouysségur, F. Dayan and N. M. Mazure, Nature, 2006, 441, 437–443 CrossRef PubMed.
- A. P. Halestrap and N. T. Price, Biochem. J., 1999, 343, 281–299 CrossRef PubMed.
- B. E. Enerson and L. R. Drewes, J. Pharm. Sci., 2003, 92, 1531–1544 CrossRef PubMed.
- M. Pérez-Sayáns, J. M. Somoza-Martín, F. Barros-Angueira, J. M. Rey and A. García-García, Cancer Treat. Rev., 2009, 35, 707–713 CrossRef PubMed.
- J. S. Fang, R. D. Gillies and R. A. Gatenby, Semin. Cancer Biol., 2008, 18, 330–337 CrossRef PubMed.
- P. Sonveaux, F. Végran, T. Schroeder, M. C. Wergin, J. Verrax, Z. N. Rabbani, C. J. De Saedeleer, K. M. Kennedy, C. Diepart and B. F. Jordan, J. Clin. Invest., 2008, 118, 3930–3942 Search PubMed.
- V. Huber, A. De Milito, S. Harguindey, S. J. Reshkin, M. L. Wahl, C. Rauch, A. Chiesi, J. Pouysségur, R. A. Gatenby and L. Rivoltini, J. Transl. Med., 2010, 8, 57 CrossRef PubMed.
- C. C. Wykoff, N. J. Beasley, P. H. Watson, K. J. Turner, J. Pastorek, A. Sibtain, G. D. Wilson, H. Turley, K. L. Talks and P. H. Maxwell, Cancer Res., 2000, 60, 7075–7083 Search PubMed.
- P. Swietach, S. Wigfield, P. Cobden, C. T. Supuran, A. L. Harris and R. D. Vaughan-Jones, J. Biol. Chem., 2008, 283, 20473–20483 CrossRef PubMed.
- P. Swietach, S. Wigfield, C. T. Supuran, A. L. Harris and R. D. Vaughan-Jones, BJU Int., 2008, 101, 22–24 CrossRef PubMed.
- P. Ebbesen, E. O. Pettersen, T. A. Gorr, G. Jobst, K. Williams, J. Kieninger, R. H. Wenger, S. Pastorekova, L. Dubois and P. Lambin, J. Enzyme Inhib. Med. Chem., 2009, 24, 1–39 CrossRef PubMed.
- G. Kroemer and J. Pouyssegur, Cancer Cell, 2008, 13, 472–482 CrossRef PubMed.
- R. J. Gillies, N. Raghunand, M. L. Garcia-Martin and R. A. Gatenby, IEEE Eng. Med. Biol. Mag., 2004, 23, 57–64 CrossRef PubMed.
- A. S. Silva, J. A. Yunes, R. J. Gillies and R. A. Gatenby, Cancer Res., 2009, 69, 2677–2684 CrossRef PubMed.
- T. H. Maren, Physiol. Rev., 1967, 47, 595–781 CrossRef PubMed.
- E. Švastová, A. Hulíková, M. Rafajová, M. Zat'ovičová, A. Gibadulinová, A. Casini, A. Cecchi, A. Scozzafava, C. T. Supuran and J. r. Pastorek, FEBS Lett., 2004, 577, 439–445 CrossRef PubMed.
- S. Wakabayashi, M. Shigekawa and J. Pouyssegur, Physiol. Rev., 1997, 77, 51–74 CrossRef PubMed.
- R. Martinez-Zaguilan, E. A. Seftor, R. E. Seftor, Y.-W. Chu, R. J. Gillies and M. J. Hendrix, Clin. Exp. Metastasis, 1996, 14, 176–186 CrossRef PubMed.
- R. E. Moellering, K. C. Black, C. Krishnamurty, B. K. Baggett, P. Stafford, M. Rain, R. A. Gatenby and R. J. Gillies, Clin. Exp. Metastasis, 2008, 25, 411–425 CrossRef PubMed.
- L. Borsi, E. Balza, B. Gaggero, G. Allemanni and L. Zardi, J. Biol. Chem., 1995, 270, 6243–6245 CrossRef PubMed.
- L. Borsi, G. Allemanni, B. Gaggero and L. Zardi, Int. J. Cancer, 1996, 66, 632–635 CrossRef PubMed.
- B. Requena, M. Zabala, P. Padial and B. Feriche, J. Strength Cond. Res., 2005, 19, 213–224 Search PubMed.
- C. R. White and R. S. Seymour, J. Exp. Biol., 2005, 208, 1611–1619 CrossRef PubMed.
- M. F. McCarty and J. Whitaker, Altern. Med. Rev., 2010, 15, 264–272 Search PubMed.
- A. Ibrahim-Hashim, D. Abrahams, P. M. Enriquez-Navas, K. Luddy, R. A. Gatenby and R. J. Gillies, Cancer Med., 2017, 6, 1720–1729 CrossRef PubMed.
- M. Chao, H. Wu, K. Jin, B. Li, J. Wu, G. Zhang, G. Yang and X. Hu, eLife, 2016, 5, e15691 CrossRef PubMed.
- K. L. Tucker, M. T. Hannan, H. Chen, L. A. Cupples, P. W. Wilson and D. P. Kiel, Am. J. Clin. Nutr., 1999, 69, 727–736 CrossRef PubMed.
- C. Demigné, H. Sabboh, C. Rémésy and P. Meneton, J. Nutr., 2004, 134, 2903–2906 CrossRef PubMed.
- A. Sebastian, L. A. Frassetto, D. E. Sellmeyer and R. C. Morris, Semin. Nephrol., 2006, 26, 447–453 CrossRef PubMed.
- T. Remer, Seminars in Dialysis, 2000, 13, 221–226 CrossRef PubMed.
- E. Molina-Montes, P. A. Wark, M. J. Sánchez, T. Norat, P. Jakszyn, L. Luján-Barroso, D. S. Michaud, F. Crowe, N. Allen and K. T. Khaw, Int. J. Cancer, 2012, 131, E1134–E1147 CrossRef PubMed.
- M. Yamagata and I. Tannock, Br. J. Cancer, 1996, 73, 1328–1334 CrossRef PubMed.
- A. De Milito, E. Iessi, M. Logozzi, F. Lozupone, M. Spada, M. L. Marino, C. Federici, M. Perdicchio, P. Matarrese and L. Lugini, Cancer Res., 2007, 67, 5408–5417 CrossRef PubMed.
- M. Yeo, D. K. Kim, H. J. Park, S. W. Cho, J. Y. Cheong and K. J. Lee, Cancer Sci., 2008, 99, 185 Search PubMed.
- A. Di Sario, E. Bendia, A. Omenetti, S. De Minicis, M. Marzioni, H. Kleemann, C. Candelaresi, S. Saccomanno, G. Alpini and A. Benedetti, Dig. Liver Dis., 2007, 39, 60–69 CrossRef PubMed.
- T. Morimura, K. Fujita, M. Akita, M. Nagashima and A. Satomi, Pediatric Surgery International, 2008, 24, 1087–1094 CrossRef PubMed.
- R. Supino, A. I. Scovassi, A. C. Croce, L. D. Bo, E. Favini, A. Corbelli, C. Farina, P. Misiano and F. Zunino, Ann. N. Y. Acad. Sci., 2009, 1171, 606–616 CrossRef PubMed.
- M. E. Finbow and M. A. Harrison, Biochem. J., 1997, 324, 697 CrossRef PubMed.
- F. Luciani, M. Spada, A. De Milito, A. Molinari, L. Rivoltini, A. Montinaro, M. Marra, L. Lugini, M. Logozzi and F. Lozupone, J. Natl. Cancer Inst., 2004, 96, 1702–1713 CrossRef PubMed.
- E. Carlsson, Scand. J. Gastroenterol., 1985, 20, 23–35 CrossRef.
- Y. Hayashi, K. Katayama, T. Togawa, T. Kimura and A. Yamaguchi, Int. J. Hyperthermia, 2006, 22, 275–285 CrossRef PubMed.
- Y. C. Wu, W. K. K. Wu, Y. Li, L. Yu, Z. J. Li, C. C. M. Wong, H. T. Li, J. J. Y. Sung and C. H. Cho, Biochem. Biophys. Res. Commun., 2009, 382, 451–456 CrossRef PubMed.
- R. Maidorn, E. Cragoe and I. Tannock, Br. J. Cancer, 1993, 67, 297–303 CrossRef PubMed.
- B. Masereel, L. Pochet and D. Laeckmann, Eur. J. Med. Chem., 2003, 38, 547–554 CrossRef PubMed.
- R. Zhou, N. Bansal, D. B. Leeper, S. Pickup and J. D. Glickson, Academic Radiology, 2001, 8, 571–582 CrossRef PubMed.
- P. Wong, C. Lee and I. F. Tannock, Clin. Cancer Res., 2005, 11, 3553–3557 CrossRef PubMed.
- P. Wong, H. Kleemann and I. Tannock, Br. J. Cancer, 2002, 87, 238–245 CrossRef PubMed.
- J. C. Lyons, G. E. Kim and C. W. Song, Radiat. Res., 1992, 129, 79–87 CrossRef PubMed.
- C. W. Song, J. C. Lyons, R. J. Griffin, C. M. Makepeace and E. J. Cragoe, Cancer Res., 1993, 53, 1599–1601 Search PubMed.
- Y. L. Cho, K. S. Lee, S. J. Lee, S. Namkoong, Y. M. Kim, H. Lee, K. S. Ha, J. A. Han, Y. G. Kwon and Y. M. Kim, Biochem. Biophys. Res. Commun., 2005, 326, 752–758 CrossRef PubMed.
- R. Burd, P. R. Wachsberger, J. E. Biaglow, M. L. Wahl, I. Lee and D. B. Leeper, Cancer Res., 2001, 61, 5630–5635 Search PubMed.
- S. Oudard, F. Poirson, L. Miccoli, Y. Bourgeois, A. Vassault, M. Poisson, H. Magdelénat, B. Dutrillaux and M. F. Poupon, Int. J. Cancer, 1995, 62, 216–222 CrossRef PubMed.
- L. Miccoli, S. Oudard, F. Sureau, F. Poirson, B. Dutrillaux and M. F. Poupon, Biochem. J., 1996, 313, 957 CrossRef PubMed.
- D. Prescott, H. Charles, H. D. Sostman, R. Page, D. Thrall, D. Moore, J. Oleson and M. Dewhirst, Int. J. Hyperthermia, 1993, 9, 745–754 CrossRef PubMed.
- W. Mueller-Klieser, S. Walenta, D. Kelleher, H. Dinh, E. Marx and P. Vaupel, Int. J. Hyperthermia, 1996, 12, 501–511 CrossRef PubMed.
- N. S. Al-Waili, G. J. Butler, J. Beale, R. W. Hamilton, B. Y. Lee and P. Lucas, Med. Sci. Monit., 2005, 11, RA279–RA289 Search PubMed.
- Y. H. Han, S. W. Kim, S. H. Kim, S. Z. Kim and W. H. Park, Toxicol. in Vitro, 2008, 22, 659–670 CrossRef PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 Search PubMed.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1–20 CrossRef PubMed.
- Z. P. Xu, G. S. Stevenson, C. Q. Lu, G. Q. Lu, P. F. Bartlett and P. P. Gray, J. Am. Chem. Soc., 2006, 128, 36–37 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |