
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHemoglobin-driven iron-directed assembly of gold nanoparticles†
Jacquelyn G. Egan,
Nicole Drossis,
Iraklii I. Ebralidze ,
Holly M. Fruehwald,
Nadia O. Laschuk,
Jade Poisson,
Hendrick W. de Haan‡
and
Olena V. Zenkina*
,
Holly M. Fruehwald,
Nadia O. Laschuk,
Jade Poisson,
Hendrick W. de Haan‡
and
Olena V. Zenkina*
Faculty of Science, University of Ontario Institute of Technology, 2000 Simcoe Street North, Oshawa, ON L1H 7K4, Canada. E-mail: olena.zenkina@uoit.ca; Web: http://faculty.uoit.ca/zenkina
First published on 26th April 2018
Abstract
The ability to form complex 3D architectures using nanoparticles (NPs) as the building blocks and complex macromolecules that direct these assemblies remains a challenging objective for nanotechnology. Here we report results in which the partial substitution of classical Turkevich citrate-capped gold NPs by a novel, heteroaromatic ligand (L) results in NPs able to form coordination-driven assemblies mediated by free or protein-bound iron ions. The morphology of these assemblies can be tuned depending on the source of iron. To prove the concept, classical citrate and novel NPs were reacted with iron-containing protein hemoglobin (Hb). To diminish the influence of possible electrostatic interactions of native Hb and gold NPs, the reaction was performed at the isoelectric point of Hb. Moreover, thiol groups of Hb were protected with p-quinone to exclude thiol–gold bond formation. As expected, citrate-capped gold NPs are well dispersed in functionalized Hb, while L-functionalized NPs form assemblies. The blue shift of the Soret band of the functionalized Hb, when reacted with novel NPs, unambiguously confirms the coordination of a NP-anchored heteroaromatic ligand with the heme moiety of Hb. Coarse-grained molecular dynamics of this system were performed to gain information about aggregation dynamics and kinetics of iron- and hemoglobin-templated assemblies of L–NPs. A multi-scale simulation approach was employed to extend this model to longer time scales. The application of this model towards novel coordination-based assemblies can become a powerful tool for the development of new nanomaterials.
Introduction
Gold nanoparticles (Au NPs) are at the core of current and emerging nanotechnologies. Within the area of nanomaterials, Au NPs are widely employed as a tunable core for novel nanostructures for applications in materials science,1 molecular transport,2 catalysis,3 and energy conversion.4 Additionally, solutions of Au NPs have several useful optical properties and are used as the functional element in a wide range of applications including chemical logic gates,5 analytical chemistry of extracellular vesicles,6 and biomedical sensing such as cancer detection.7 Citrate-stabilized gold nanoparticles (C–Au NPs), initially developed by Turkevich et al.,8 have a number of desirable properties including high stability in aqueous solutions, availability of uniform quasi-spherical shape particles of up to ∼200 nm,9 and high biocompatibility.10 These characteristics lead to C–Au NPs being used in diverse biomedical applications.11 However, as the nanoparticle travels in the body through different compartments with different composition and pH, initially adsorbed moieties are progressively replaced by proteins of higher affinity.12 As a result, incorporation of functional organic moieties via stronger (thiol13 or carbene-based14,15) bonding is preferred. There is thus a need for functionalized Au NPs exhibiting strong attachment of the ligand to the NP and the attachment of species in solution to the functional end of the ligand – which may drive self-assembly. In this work, we introduce an L-receptor that attaches to gold nanoparticles and demonstrate that this receptor allows the formation of clusters of Au NPs driven by free iron ions or hemoglobin.Incorporation of functional thiols into C–Au NPs via citrate-to-thiol partial ligand exchange16 is a promising strategy for functionalization and well-tuning of interfacial properties of NPs. Functionalization of citrate-stabilized Au NPs with 11-mercaptoundecanoic acid results in Au NPs able to react with heavy metals to form cross-linked agglomerates.17 Metal-directed association of Au NPs was used last decade for Pb, Cd, and Hg sensing in aqueous solutions17,18 and for preparation of metallo-supramolecular assemblies of gold clusters.19 Agglomeration of NPs can lead to the formation of either amorphous aggregates or self-assembled periodic arrays in two or three dimensions, NPs clusters20 or even superlattices.21
In contrast to the use of metals, controlled NPs assembly using high molecular weight molecules of biological origin such as lipids,6,13 DNA,22 and especially proteins23,24 remains a challenging task for nanobiotechnology.25 Many biomolecule-directed assemblies of Au NPs with a certain level of NPs organization employ quite rigid molecular templates to control the process. Liu and Yan report nanomaterials that were scaffolded by self-assembling triangular DNA origami structures leading to a programmable DNA-directed positioning of gold nanorods.26 Jin and Raines employed collagen-related synthetic peptides that pre-form fibers, followed by NPs plating to yield metallic nanowires.27 McMillan and Trent report on the design of special protein templates crystallized into 2-D hollow double-ring structures called chaperonins that selectively bind and organize gold NPs into arrays.28 There have also been several studies that achieve directed self-assembly of NPs driven by “soft” matter objects in the solution that involve interactions between well-defined binding sites of coupling partners.29 A benefit of this approach is the precise control of inter-particle distances thus granting enhanced collective properties of the resulting materials. However, the majority of biomolecule directed assemblies employ electrostatic interactions and weak non-covalent forces to guide the assemblies. Thus, electrostatic interactions between coiled coil peptides,30 weak-non-covalent binding between artificial repeat protein pairs29 or DNA pairs,26 and interactions between biotin–streptavidin pairs31 have been used to design colloidal assemblies in solution. In addition, dynamics simulations of coarse-grained models of a DNA molecule and positively charged nanoparticle have been recently performed as an important step for understanding the motion of molecules and colloids and control of their interactions.32 Moreover, recent progress in modelling of polymers self-organization into soft patchy nanoparticles, the shape and size of which can be fine-tuned either by adjusting the amphiphilicity of the polymer or by thermal stimuli,33 has a great potential for developing soft-matter based materials. Finally, modelling the influence of directional and isotropic interactions on self-assembly in a system of patchy particles34 helps to quantify both the mass distribution and the shape for all clusters and thus opens the door for a possible directing of these assemblies.
The use of proteins and NPs both with well-defined reacting sides and ability to react via covalent bond formation are less common, yet highly desired because it allows much better control for the architecture of the resulting nanomaterials. Very limited literature examples include the design of the hierarchical assembly of protein nanocages with Au NPs that utilize metal complex formation was reported by Li and Wang.35 In more detail, Au NPs were functionalized by ligands containing nickel(II) complex, while the protein nanocage was bearing polyhistidine tag on the external surface. As a result, the protein nanocage controllably assembles onto the Au NPs via the polyhistidine–Ni2+ affinity.
In this work we use hemoglobin (Hb) to drive self-assembly of the Au NPs via covalent interactions with the L-receptor. Previous works have reported the adsorption of Hb onto metal oxide nanoparticles36,37 such as silica, zirconia, titania, and on Au NPs.38 The Hb–NP binding mechanism was postulated to arise from mainly hydrophobic forces and hydrogen bonding; direct interaction between the Au NPs and heme groups was not found and interactions between Au NPs and Hb thiol groups were not considered.38 These studies did not explore self-assembly of NPs driven by Hb. However, Hayashi has developed a protein bearing a heme pocket39 that, when combined with heme-functionalized Au NPs, leads to the formation of hemoprotein–nanoparticle conjugates due to heme–heme pocket interaction.40 This is again a non-covalent binding interaction. In contrast to that work, our design uses the heme moieties (Hb has definite tertiary structure and contains four globin chains each associated with one molecule of iron heme) for the creation of better-directed NPs assemblies. High binding affinities of Hb for heteroaromatic amines is well documented41 and used in Hb-like proteins determination.42
In this manuscript we use strong metal to ligand coordinative forces as a powerful tool to self-assemble Au NPs. To begin, we introduce the novel L-receptor that drives this process. As a preliminary step, we then demonstrate self-assembly of the L–Au NPs when iron ions are added to the solution. Aggregation of the nanoparticles is characterized by UV-vis, TEM, and cryo-TEM measurements. Following from this result, we proceed to demonstrate self-assembly driven by hemoglobin with no free iron ions. To study the dynamics of aggregation and cluster morphology in more detail, we introduce a coarse-grained model of the system and perform molecular dynamics simulations based on this model. Simulations employing a multi-scale approach are also introduced such that longer time-scales can be accessed. Comparison between the simulation and experimental results indicate that this multi-scale methodology provides a powerful method to study the dynamics of the system and thus complement the experimental measurements.
Experimental
Materials
Trisodium citrate, 1,10-phenanthroline, KCl, and KBr were purchased from Alfa Aesar. Lipoic acid, N,N′-dicyclohexylcarbodiimide (DCC), ammonium acetate, 2-pyridinecarboxaldehyde, glacial acetic acid, ammonia, sulfuric acid, nitric acid, hexanes, hemoglobin from bovine blood, and chloroauric acid were purchased from Sigma-Aldrich. All reagents were used without further purification.Instrumentation
Nuclear Magnetic Resonance (NMR) spectra were measured on a Bruker Avance™III HD NanoBay 400 MHz spectrophotometer. UV-visible spectra were measured using an Agilent Technologies Cary 60 UV-visible spectrometer. TEM images were taken using a Hitachi H7500 Transmission Electron Microscope (TEM). Cryo-TEM images were recorded using JEOL 2010 instrument at the Canadian Centre for Electron Microscopy, Hamilton, ON. To perform cryo-TEM measurements, a droplet of the solution was placed on the grid, the excess fluid was blotted using a filter paper to prepare a thin layer of the sample solution. The specimen was vitrified by immediate transferring under liquid N2 into the cryo-TEM chamber using a cryogenic holder and transfer station. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo Scientific K-Alpha spectrophotometer equipped with an Al Kα (1486.6 eV) X-ray source. The binding energies were referenced to the Au 4f7/2 peak at 84.0 eV.Synthesis of the ligand
Compound 1, (Scheme 1) 2-(pyridin-2-yl)-1H-imidazo[4,5-f][1,10]phenanthroline, was synthesized according to published procedure.43 The method of preparing compound L, 2-(pyridin-2-yl)-1H-imidazo[4,5-f][1,10]phenanthrolin-1-yl-5-(1,2-dithiolan-3-yl)pentanoate was optimized from the closely related literature procedure.44 In the round bottom flask 0.99 g (8.5 mmol) of N-hydroxysuccinimide was mixed with 1.48 g (7.1 mmol) of lipoic acid in 20 mL of THF. The solution was stirred for 10 minutes, then 1.87 g of N,N′-dicyclohexylcarbodiimide (DCC) was added. The reaction was stirred vigorously for 22 hours, after which the precipitate was filtered off. Then 2.06 g (6.9 mmol) of 2-(2-pyridine)imidazo[4,5,f]-1,10-phenanthroline, 1 was added to the filtrate and the reaction mixture was stirred for 72 hours. All volatiles were removed in vacuum and resulting sticky dark solid was re-dissolved in 25 mL of dichloromethane (DCM). After liquid extraction (DCM/DI water × 2 times, and DCM/brine) all volatiles were removed in vacuum resulting in brown-red solid of L. Resulting solid was washed multiple times by hexanes (5 × 100 mL) then sonicated with the solution of hexanes (3 × 100 mL). Compound L was isolated as dark-vine powder in 70% yield, 2.58 g.1 1H NMR (400 MHz, CDCl3): δ 9.35 (d, J = 5.0 Hz, 1H), 9.31 (d, J = 6.1 Hz, 1H), 9.11 (d, J = 8.0 Hz, 1H), 8.91 (d, J = 4.6 Hz, 1H), 8.86 (d, J = 8.1 Hz, 1H), 8.49 (d, J = 8.1 Hz, 1H), 7.99 (t, J = 7.7 Hz, 1H), 7.86 (ddd, J = 15.8, 7.9, 4.3 Hz, 2H), 7.54 (t, J = 5.6 Hz, 1H), 3.49 (m, J = 10.4 Hz, 1H), 1.93 (d, J = 11.7 Hz, 2H), 1.69 (dt, J = 13.3, 3.6 Hz, 2H), 1.59 (dt, J = 13.0, 4.3 Hz, 2H), 1.33 (m, 2H), 1.28–1.14 (m, 2H), 1.13–0.85 (m, 2H). 13C{1H} NMR (101 MHz, CDCl3): δ 162.4 (Cq), 156.8 (Cq), 150.6, 150.1, 149.2 (Cq), 145.6, 144.2 (Cq), 137.3, 134.8 (Cq), 132.2 (Cq), 129.5, 125.7, 123.93, 123.90, 123.2 (Cq), 118.2 (Cq), 49.2 (S–CH–), 33.9 (CH2), 30.9, 29.7, 29.3, 25.6 (CH2), 24.9 (CH2). FTIR: 2928.20 (CH2 asymmetric), 1709.90 (carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1433 (CH2 bend alkane), 1211.93 (C–N), 1023.17, 819.86 (C–H), 740.97 (C–H). MS, m/z: calc. for L + CH3CN + Na 549.15; found 549.15.
O), 1433 (CH2 bend alkane), 1211.93 (C–N), 1023.17, 819.86 (C–H), 740.97 (C–H). MS, m/z: calc. for L + CH3CN + Na 549.15; found 549.15.
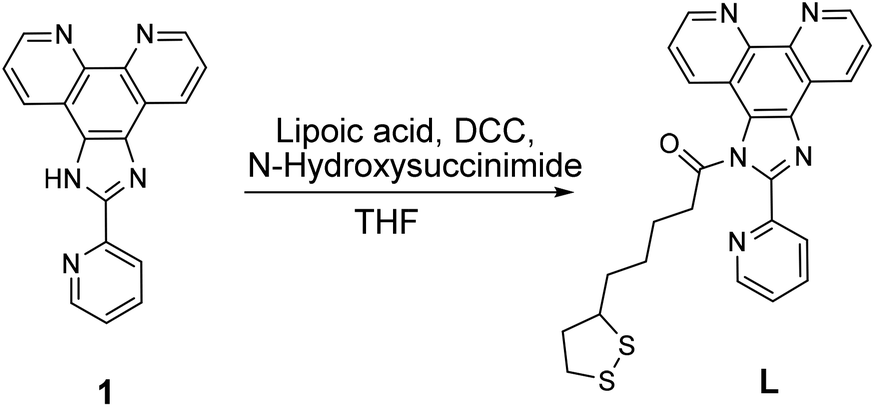 | ||
| Scheme 1 Synthesis of alpha-lipoic acid derivative of 2-(2-pyridine)imidazo[4,5,f]-1,10-phenanthroline, L. | ||
Preparation of C–Au NPs
The gold nanoparticles in aqueous medium were synthesized by the citrate reduction method using a slightly modified Turkevich method. Briefly, to a hot (100 °C) solution of HAuCl4 (5 mg, 14.7 μmol) in DI water (50 mL), a solution of sodium citrate (17 mg, 65.9 μmol) in DI water (1.5 mL) was swiftly ejected under vigorous stirring. The reaction mixture was refluxed for 30 minutes under a constant rate of stirring until solution became a red colour.Preparation of gold nanoparticles functionalized by L, L–Au NP
Compound L (2.4 mg, 4.8 μmol) was dissolved in 19.5 mL of 1,4-dioxane. After stirring for two hours to ensure complete solubility, an aliquot (5 mL) of this solution was added dropwise under vigorous stirring to the initial aqueous solution of C–Au NPs (2.5 mL) to give a final concentration of 83 ppm of L in the solution. The reaction mixture was stirred at ambient conditions for 10 min. During this time, the NPs solution experience a colour change from red to purple. The final solution was then washed with diethyl ether (5 mL). The aqueous phase containing L–Au NPs was collected, stored in the refrigerator at +4 °C, and used without future purification.The reaction of Au NP with Fe ions
A solution of Fe3+ or Fe2+ in DI water (750 ppm) was portionwise added to the corresponding L– or C–Au NP solution with vigorous stirring. Fe(ClO4)2·6H2O and Fe(ClO4)3·6H2O were used as Fe2+ and Fe3+ sources, respectively. The reactions were monitored by UV-vis. The complete Au NPs precipitation occurs when the overall concentration of Fe3+ or Fe2+ in the solution reached 20 ppm. Caution: while we have experienced no problems in working with perchlorate compounds, they are potentially explosive. Perchlorate salts must be handled in small amounts, and with special care.45The reaction of L–Au NPs with hemoglobin
A solution of hemoglobin (1 mg of hemoglobin in 1 mL of phosphate buffer pH = 6.8) was portionwise added to the corresponding C– or L–Au NP solution (7.5 mL) with vigorous stirring.Hemoglobin functionalization
Thiol residues in Hb were functionalized via classical Michael addition of thiols to p-quinone.46 A stock 10 mM solution of naphthoquinone (NQ) was made by dissolving 7.9 mg of NQ in 5 mL of phosphate buffer. An aliquot of NQ stock solution (300 μL) was added to a solution of hemoglobin (1 mg) in phosphate buffer (2.7 mL) with vigorous stirring. The mixture was stirred for 30 min, the unreacted NQ was extracted with toluene (2 × 1.5 mL) and the aqueous phase was lyophilized. The obtained functionalized hemoglobin (fHb) was stored at −20 °C and used without further purification.Results and discussion
To achieve assembly by iron coordination, a novel ligand referred to as receptor L was developed. Lipoic acid, a compound able to graft to gold by both sulfur ends, is often used for functionalization of gold surfaces47 and Au NPs.48 Reacting lipoic acid with compound 1 that contains N-aromatic coordinating moieties (Scheme 1) yields receptor L, which contains 2 sulphurs at one end and 2 nitrogens in close proximity at the other end. Further treatment of C–Au NPs with L leads to a partial substitution of citrate molecules on Au NPs surface by L leading to L–Au NPs (Fig. 1A).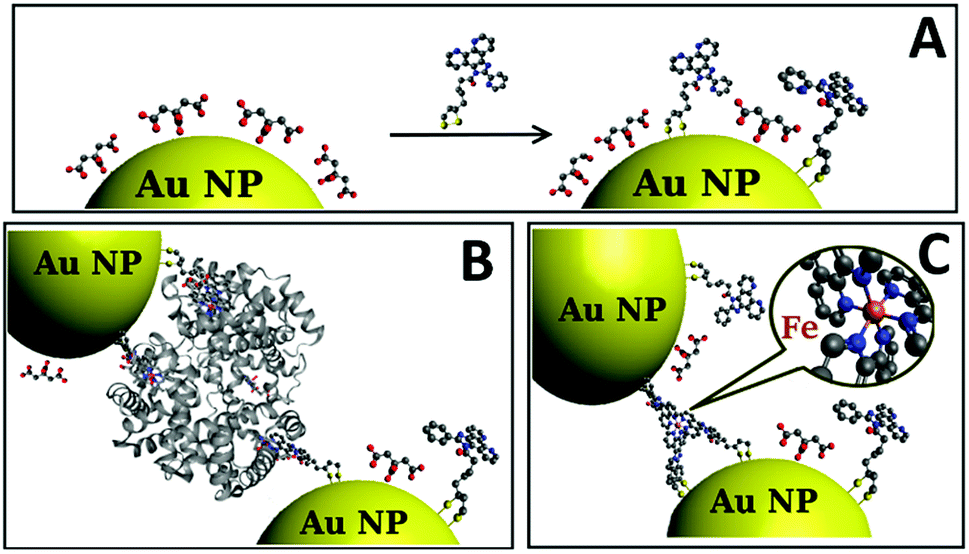 | ||
| Fig. 1 (A) Schematic representation of partial citrate substitution by L resulting in the formation of C–Au NPs. Schematic representation of NPs iron-directed assembly: (B) in presence of hemoglobin showing ligand to heme iron coordination, and (C) in presence of iron ions. | ||
Fig. 1B and C schematically represent the assembly of L–Au NPs. The ligand L anchored to the NP surface forms a complex with iron in heme and “free” iron ions, respectively.
Fig. 2 illustrates a representative X-ray photoelectron spectra of C–Au NPs reacted with L demonstrating that incubation of the initial NPs with L does result in ligand incorporation. A single S2s peak is observed at 227.7 eV that matches to previously reported for thiol group bonded to a metal surface.49 Neither “free” thiol nor oxidized sulfur species were found. A single N1s peak at 399.9 eV is consistent with N1s binding energy of 400.0 eV reported for similar aromatic amines.50 Peak area normalization between different elements using relative XPS sensitivity factors as determined by Scofield51 gives an S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N ratio equal to 2.0:4.8 that is close to expected stoichiometric ratio of 2.0:5.0. Deconvolution of the C1s spectrum using a Powell peak-fitting algorithm yields citrate carboxyl carbon atoms (288.7 eV), aromatic carbon atoms (286.6 eV), and aliphatic carbon atoms (285.0 eV). Analysis of the carbon atoms area ratio gives a citrate: L ratio of 1.9:1.0 confirming partial substitution. The L–Au NPs are thus still primarily coated with citrate thereby maintaining high solubility in water, but now are also partially coated by L receptors that are attached to the NPs via thiol–Au bonds and contain a heteroaromatic amine functional site at the NP-liquid interphase (Fig. 1A).
:N ratio equal to 2.0:4.8 that is close to expected stoichiometric ratio of 2.0:5.0. Deconvolution of the C1s spectrum using a Powell peak-fitting algorithm yields citrate carboxyl carbon atoms (288.7 eV), aromatic carbon atoms (286.6 eV), and aliphatic carbon atoms (285.0 eV). Analysis of the carbon atoms area ratio gives a citrate: L ratio of 1.9:1.0 confirming partial substitution. The L–Au NPs are thus still primarily coated with citrate thereby maintaining high solubility in water, but now are also partially coated by L receptors that are attached to the NPs via thiol–Au bonds and contain a heteroaromatic amine functional site at the NP-liquid interphase (Fig. 1A).
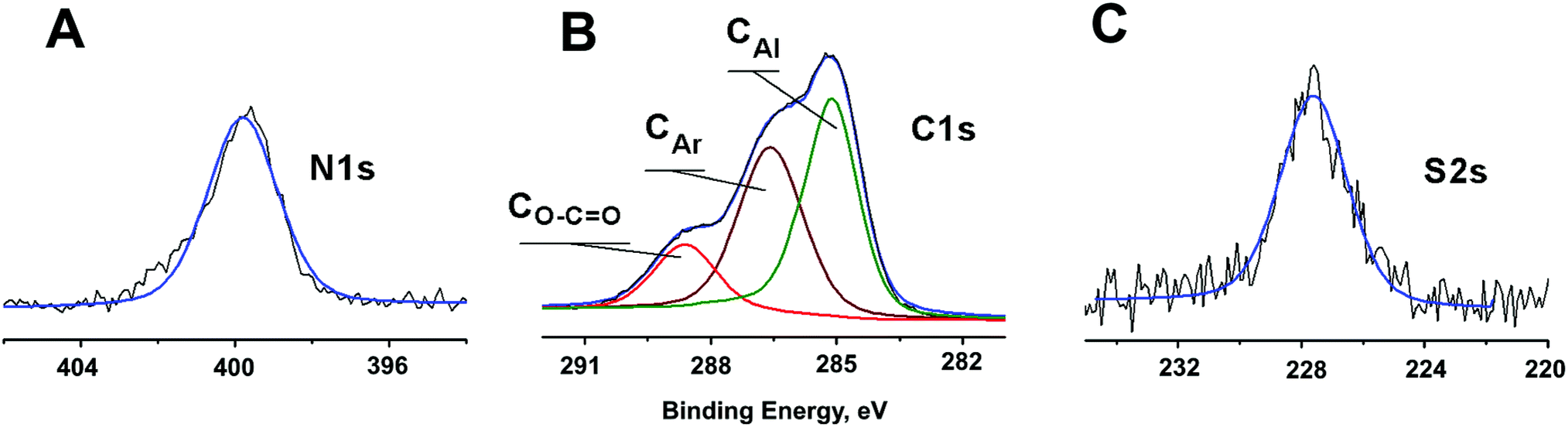 | ||
| Fig. 2 X-ray photoelectron spectra of L–Au NPs showing N1s, C1s, and S2s areas. The black line shows the experimental data, while the blue line is the overall fitted spectrum. C1s peak is deconvoluted: red traces represent the carbon atoms of the citrate carboxyl group O–CO, the brown traces correspond to aromatic (CAr) moiety, and the green traces line is associated with aliphatic (CAl) carbon. | ||
Before proceeding to self-assembly driven by iron containing protein, self-assembly of the L–Au NPs driven by iron ions was investigated. To this end, the behavior of C–Au NPs and L–Au NPs both with and without iron ions was investigated by UV-vis and TEM. The UV-vis curves for solutions of L–Au NPs with no iron ions can be seen in Fig. 3A and B (black line) and the curves for pure C–Au NPs is shown in Fig. 3C and D (black line). Comparing between them, the surface plasmon resonance (SPR) band shifts from 519 nm for initial C–Au NPs to 536 nm for L–Au NP. A shift to a higher wavelength of SPR indicates certain aggregation of L–Au NP over C–Au NPs even for solutions containing no iron ions. The L moieties are not charged compared to the citrate ions and thus partial substitution of citrate to L reduces the surface charge of the Au NPs and as a result, the repulsion forces between NPs. This effect appears to be strong enough to cause weak aggregation via hydrophobic interactions that drive clustering of the NPs to reduce interactions with water. This can be seen by comparing the TEM images shown in Fig. 4A and B.
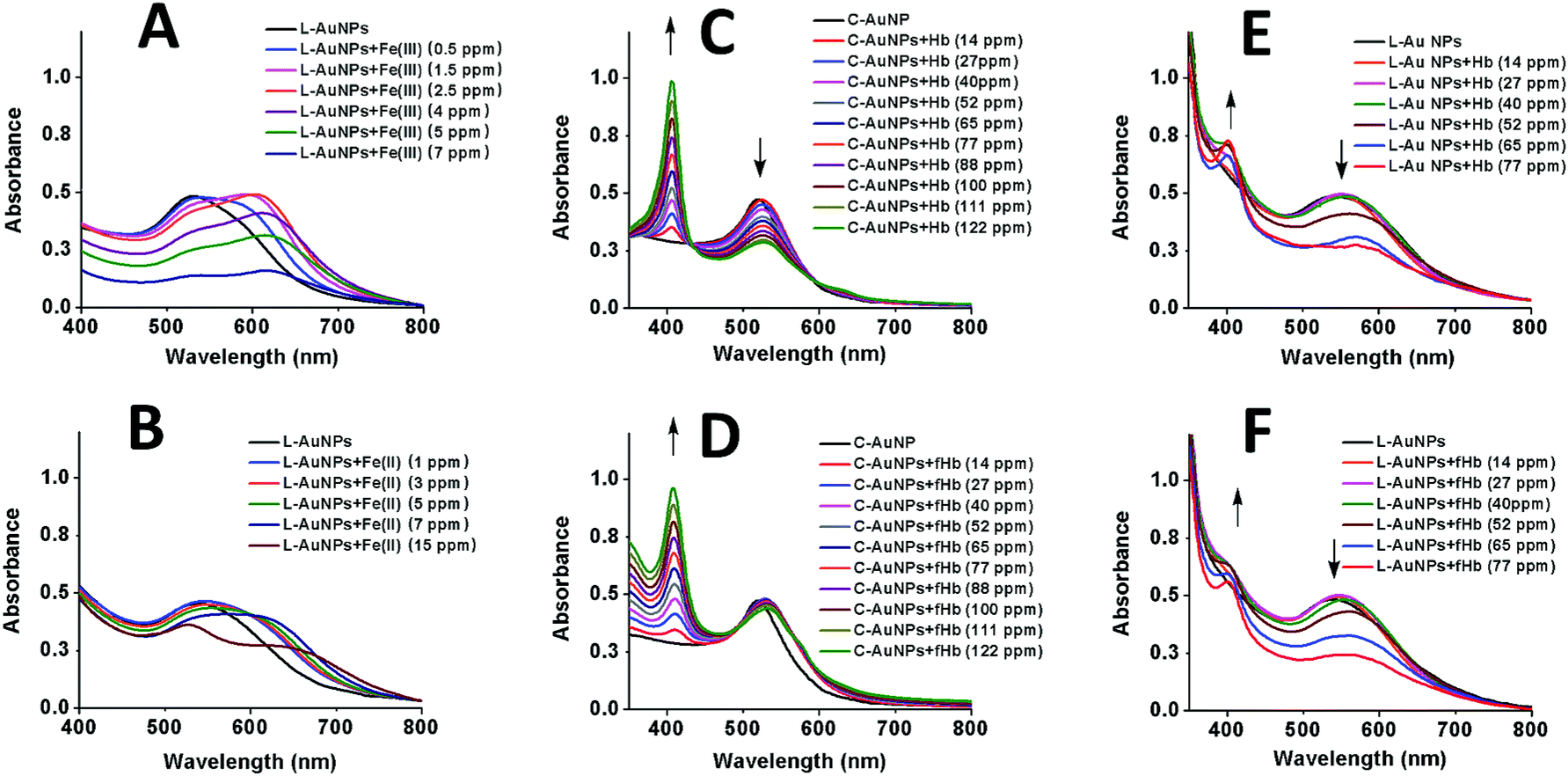 | ||
| Fig. 3 UV-vis spectra of (A) L–Au NPs solution upon reaction with Fe(III), (B) L–Au NP solution upon reaction with Fe(II), (C) C–Au NP upon reaction with Hb, (D) C–Au NP upon reaction with functionalized Hb (fHb), (E) L–Au NP solution upon reaction with Hb, and (F) L–Au NP solution upon reaction with fHb. | ||
 | ||
| Fig. 4 Representative TEM images of (A) C–Au NPs, (B) L–Au NPs, (C) C–Au NPs in presence of Hb (13 ppm), (D) C–Au NPs in presence of fHb (13 ppm), (E) C–Au NPs in presence of Fe(III), (F) C–Au NPs in presence of Fe(II), (G) L–Au NPs in presence of Hb (5 ppm), (H) L–Au NPs in presence of fHb (5 ppm), (I) L–Au NPs in presence of Fe(III), (J) L–Au NPs in presence of Fe(II), (K) L–Au NPs in presence of Hb (13 ppm), and (L) L–Au NPs in presence of fHb (13 ppm). Scale bar: 200 nm. | ||
To investigate self-assembly driven by iron ions, a range of concentrations of Fe(III) or Fe(II) was added to solutions of C–Au NPs and L–Au NPs. The change in the ionic strength of C–Au NPs solution after addition of up to 15 ppm of Fe(II) or Fe(III) ions neither changes the colour of the solution nor causes aggregation. This is confirmed by TEM images which show no clustering of C–Au NPs in presence of Fe(III) ions (Fig. 4E) or Fe(II) ions (Fig. 4F); instead the nanoparticles are spread out as for the pure C–Au NPs case (Fig. 4A).
The results are very different for the suspensions of L–Au NPs on the introduction of iron ions. If enough Fe(II) or Fe(III) was added to achieve a 1:1 ratio of iron ions to L receptors, all L-receptors would become a charged FeL complex that is grafted on the surface of NPs. This would generate a sufficient electrostatic repulsion between NPs to prevent aggregation and again yield stabilized NPs. However, lower concentrations should result in the sharing in iron ions via the formation of Fe(L)2 or Fe(L)3 motif of coordination thus driving the assembly of Au NPs.
As shown in Fig. 3A, the addition of Fe(III) solution to L–Au NP results in a red-shift and broadening of the SPR band. In fact, the solution of L–Au NPs visibly changes colour as Fe(III) is added with different colours corresponding to different concentrations of Fe(III) (see Fig. S7†). The assembly of L–Au NPs induces electric dipole–dipole interactions and coupling between the plasmons of neighboring particles, causing the colour change to blue. These results suggest that Fe(III) coordinates between N-aromatic units that were attached to different NPs thus causing agglomeration of the NPs. This is confirmed in Fig. 4I where the TEM image of L–Au NPs with Fe(III) displays a large cluster indicating strong aggregation.
Fig. 3B displays the UV-vis curves for the addition of increasing amounts of Fe(II) to the solution of L–Au NPs. Interestingly, the addition of Fe(II) results not only in broadening and significant red shift of the SPR band as seen upon the addition of Fe(III), but also the appearance of metal to ligand charge transfer (MLCT) band at 530 nm that is characteristic for similar complexes.52,53 The intensity of the MLCT band was used to establish the stoichiometry of L coordination with Fe(II) by means of continuous variation analysis procedure (Jobs plot shown in Fig. S3†). Examination of the Jobs plot suggests 3:1 ligand to metal binding, which is consistent with literature reports for structurally related complexes.54
The UV-vis curves (Fig. 3B) and the visible change in colour of the solution (Fig. S8†) indicate aggregation of L–Au NPs driven by Fe(II). TEM images confirm the formation of L–Au NPs assemblies in presence of Fe(II) ions as large aggregates are seen in Fig. 4J.
We have previously reported55 that the ethylenediaminetetraacetic acid (EDTA) removes the metal ions from the complexes formed on the surface of annealed TiO2– NP layers. In contrast, the addition of EDTA to the solution of dispersed Au NPs results in complex processes that include at least partial substitution of citric acid by EDTA and thus cannot be used for Au NPs recovery.
Having established the agglomeration of L–Au NPs via the coordination of iron ions, self-assembly driven by hemoglobin was explored. Hemoglobin has four iron-containing heme moieties. Each of these moieties consists of an iron ion that is strongly bound to a porphyrin cavity. The idea is thus that the amine groups on the L receptor will provide the final coordination to the iron ion in heme. High binding affinities of hemoglobin for heteroaromatic amines have been previously reported.41 Since there are four heme groups, iron ions from different groups may bind to L receptors on different gold nanoparticles thus driving self-assembly of the L–Au NPs.
Before testing this, the effect of hemoglobin on solutions of purely citrate coated NPs was explored. Note that for all hemoglobin experiments, no free iron ions were added to the solutions. As shown in Fig. 3C, adding increasing amounts of hemoglobin (Hb) to solutions of C–Au NPs results in the appearance of a characteristic Hb Soret absorption peak at 407 nm arising from the heme groups. There is also a small 7 nm red shift of the SPR peak that is due to the change in the local refractive index at the Au NP surface due to the reaction with Hb. The decrease in the intensity of the SPR band at 526 nm indicates NPs aggregation and this is confirmed by TEM images (Fig. 4C). Hence, Hb causes aggregation of C–Au NPs even though C–Au NPs do not contain the L receptor. This aggregation may be due to the thiol groups that are located in cysteine residues of Hb. In the same way that the thiols of the L-receptor bind to gold, cysteine thiol residues may stick to areas of the C–Au NP that are not protected by citrate molecules. This direct interaction between Hb and the Au NPs complicates the quantification of the efficacy our intended mechanism of self-assembly via the L-receptors. To remove this complicating factor, the Hb was reacted with p-quinone. This classical Michael addition reaction results in functionalized Hb (fHb) where the thiol groups are protected and not available for binding. Importantly, the addition of fHb to C–Au NPs results in the appearance of the distinctive Soret peak at 407 nm similarly to non-functionalized Hb. The SPR peak is 12 nm red shifted from that of initial C–Au NPs suggesting a different fHb–C–Au NPs binding mode. Interestingly, upon addition of fHb to a solution of C–Au NPs, the intensity of SPR band at 531 nm does not change significantly (Fig. 3D). This suggests that the Au NPs are no longer aggregating but instead fHb adsorbs on the C–Au NPs surface and stabilizes the NPs. The TEM images confirm this as Fig. 4D shows non-agglomerated, well-dispersed C–Au NPs in fHb. Comparison of the Hb and fHb results for C–Au NPs unambiguously confirms that the formation of thiol–gold bonds plays the major role in C–Au NPs aggregation via cysteine-induced assembly.
Fig. 3E and F display the UV-vis curves for the addition of the Hb and fHb to L–Au NPs respectively. In both cases, the Soret band is blue shifted to 401 nm suggesting that the L receptor interacts with the heme moieties of the Hb. As a strong electron donor, L provides electron density to a heme iron, which, in turn, donates it to the porphyrin macrocycle via metal to ligand π-backbonding. This results in an increased porphyrin π to π* energy separation causing the electronic absorptions to undergo a blue shift. As a result of the reaction, the SPR band of L–Au NPs red shifts to 550 nm and the colour intensity of the suspension diminishes. The redshift and the broadening of the SPR band observed in the absorption spectra indicate aggregation and thus the delocalization of the plasmon wave through adjacent L–Au NPs. The aggregation is confirmed by TEM images that show clustering of the L–Au NPs in presence of 5 ppm of Hb (Fig. 4G) and fHb (Fig. 4H). In both cases, the protein can be seen to act as a glue that joins the Au NPs together. For Hb, aggregation can be due to thiol groups on Hb binding to gold and via the coordination of heme irons by L receptors on the gold nanoparticles. Hence the L–Au NPs are enshrouded by Hb shell, forming protein corona that is stabilized by both L–heme and gold–thiol bonding. With both of these mechanisms, strong aggregation yielding dense clusters is observed. For the fHb case, direct interaction between fHb and the gold surface is eliminated and the only mechanism of aggregation is via the L receptors. The aggregation displayed in Fig. 4H thus confirms the self-assembly of L–Au NPs by using iron containing proteins as the template.
Increasing the concentrations, the reaction of L–Au NPs with excess (13 ppm) of Hb (Fig. 4K) or fHb (Fig. 4L) leads to the clusters of NPs that are fully encapsulated by the protein. Similar encapsulation of pre-associated C-NPs in a synthetic polymer was previously reported by Wu and Chen groups.56 They show that interactions between amphiphilic polymer solution with small clusters of hydrophobic Au NPs induce the formation of micelle type of self-assembled structures. The nature of our aggregates is thus quite different.
Images produced by regular TEM may reflect not only in-solution particle interactions but could be complicated by the precipitation and on-surface agglomeration processes taking place during the sample transfer to the TEM grid. To avoid these possible artifacts, we performed cryo-TEM measurements in which freshly-prepared solutions of NPs are frozen on the grid (Fig. 5). The cryo-TEM results confirm that for solutions containing only L–Au NPs, clusters containing mostly 2–3 NPs (Fig. 5A) are formed. Hence, the greater degree of association observed in the images produced by regular TEM (Fig. 4B) indeed should be assigned to processes taking place on the grid. The addition of Hb to L–Au NPs results in encapsulation of the NPs associates in Hb (Fig. 5B). In contrast, addition of fHb to the L–Au NPs initially results in the formation of clusters containing L–Au NPs associates and fHb ring-shaped units (Fig. 5C). The addition of Fe(II) ions to L–Au NPs results in immediate formation of coordination-based assemblies (Fig. 5D).
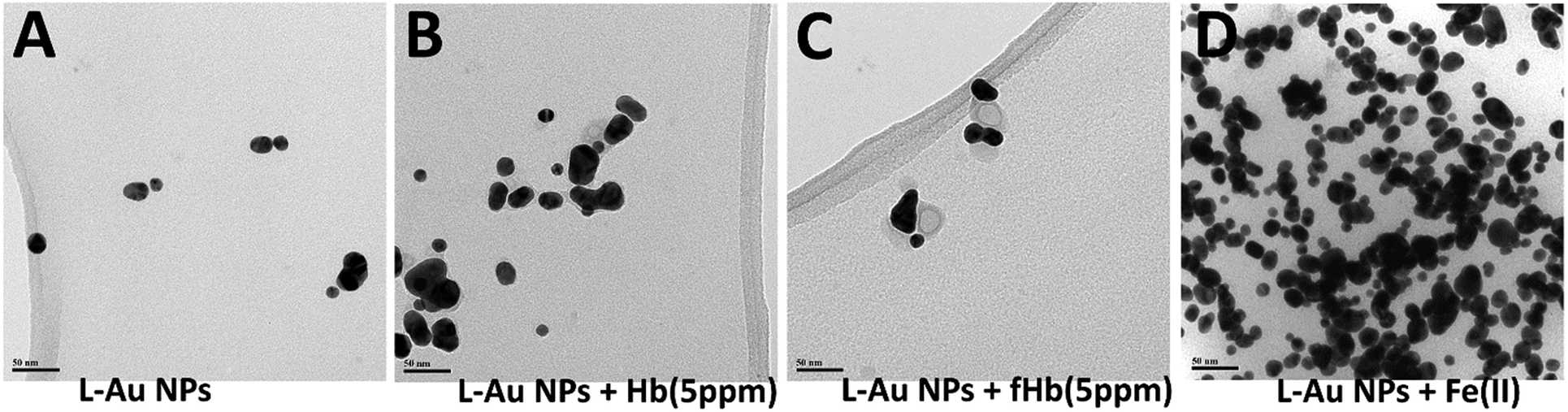 | ||
| Fig. 5 Representative cryo-TEM images of (A) L–Au NPs, (B) L–Au NPs in presence of Hb (5 ppm), (C) L–Au NPs in presence of fHb (5 ppm), (D) L–Au NPs in presence of Fe(II), scale bar: 50 nm. | ||
To gain insight into the details of the assembly of the NPs into clusters, coarse-grained Langevin dynamics simulation were performed in the HOOMD Blue simulation package57,58 using standard coarse-grained approaches.59,60 Simulations were performed at two levels of coarse-graining (see ESI† for details on the simulation approaches and video enhanced objects). At the more detailed level, Au NPs and L were explicitly modeled by spheres with L being bound to the surface of Au NPs. Iron was modeled implicitly by giving the L two states: iron bound and iron unbound. An attractive potential was implemented between iron-bound and iron-unbound L to mimic the sharing of iron between L that causes the clustering. With this approach, it was possible to simulate a system of 1000 NPs and 60000 L up to a time of 2.088 ms. A snapshot near the end of one of these simulations is shown in Fig. 6A.
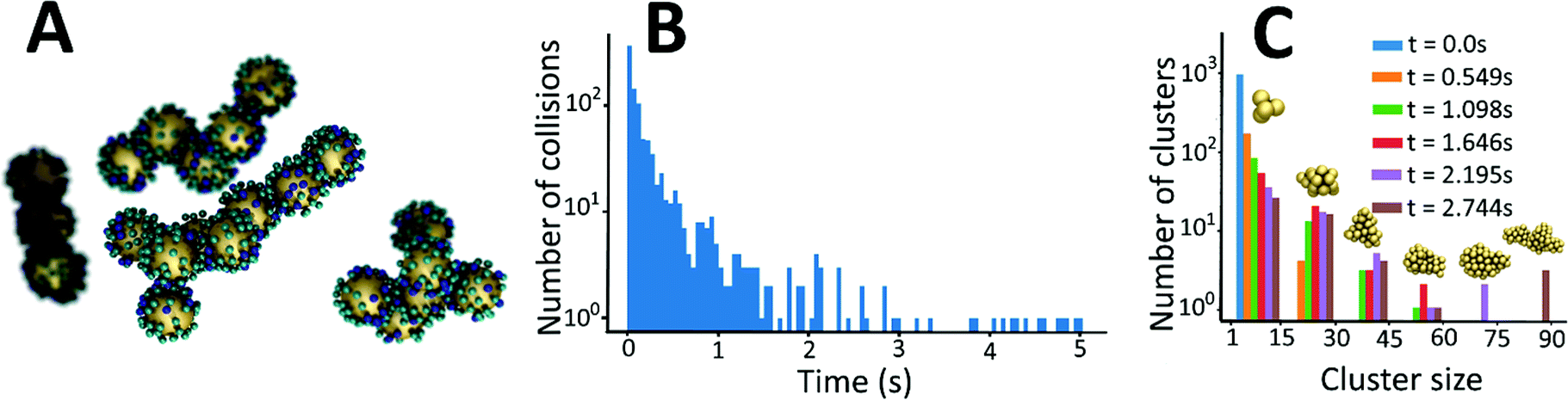 | ||
| Fig. 6 (A) A snapshot of calculated agglomerated nanostructures that are forming as a result of L–Au NPs interactions with iron ions obtained by coarse-grained computational model. Large yellow spheres represent gold nanoparticles (Au NPs); small blue spheres represent ligands, L, bonded to Au NPs and iron, small cyan spheres represent iron free ligands bonded to Au NPs. (B) Frequency of collisions between nanoparticles and/or clusters of nanoparticles as a function of time obtained from the simulations. (C) Histogram of the cluster size distribution at stages of the aggregation process. | ||
These simulations revealed a separation of time-scales that allowed for a more efficient, multi-scale approach. In brief, the assembly process is separated into a long time scale phase in which the clusters diffuse until a collision occurs and a short time scale collision where two clusters of NPs interact – often becoming a single cluster. This approach allowed for simulations up to 2.744 s and significant clustering was observed (Fig. 6C).
The simulations allow us to explore the dynamics of cluster formation. Fig. 6B displays the collision frequency (i.e. the number of collisions per unit time) as a function of time (up to a time of 5 s). Soon after the simulation starts, there is a high rate of collision between small clusters. As time progresses, the rate of collision declines quickly – approximately exponentially (linear on the semi-log plot). As small clusters merge, the average distance between clusters grows. Further, the larger clusters have a lower diffusion coefficient. Hence, it takes significantly longer for a collision to occur. However, this rapid decrease tapers off as time progresses and the collision frequency exhibits a long tail at medium to long times. That is, after the initial rapid local clustering, events become rare, but not vanishingly small as would be extrapolated from the short time behaviour.
Fig. 6C displays the distribution of cluster sizes at various times in the simulation. This data reinforces the picture suggested by the collision frequency data. Between t = 0 and t = 1.098 s, the system transitions between 1000 clusters of 1 Au NP to about a hundred small clusters (<15 gold NPs), about 11 clusters between 15-30 NPs, and only a few consisting of more than 30 NPs. For the subsequent time frames, there is a continual process in which there are 1 or 2 large clusters that continue grow in size while the smaller clusters sizes are depleted. The dynamics are thus quantified in two ways. At short times when only a limited number of cluster sizes are present, explicit reactions are examined; while at long times when the cluster sizes are very diverse, the mean cluster size as a function of time is examined.
Fig. 7A displays the number of single particles (NS) as well as dimers (ND), trimers (NT), tetramers (NQ), pentamers (NP), and hexamers (NH) as a function of time. There is a steady decline in the number of single Au NPs. At short times, collisions between two single particles are the dominant aggregation mechanism and there is a dramatic increase in the number of dimers. However, the number of dimers is depleted as higher order structures begin to form. There is thus a peak in the number of dimers at short times. Beyond this peak, the number of dimers is decreasing, but it is doing so at a slower rate than the number of single particles and the number of single particles drops below the number of dimers at longer times. Similar trends are seen for the higher order structures. The number of trimers also peaks at short times – but at a longer time than the peak in the number of dimers. In general, the peak corresponding to maximum density shifts to longer times and decreases in magnitude as the number of Au NPs in the cluster is increased. This behaviour is consistent with the coagulation equation developed by Smoluchowski.61–63
To examine the kinetics at short times, 20 independent simulations of length 0.056 s were conducted. The average number of dimers, trimers, tetramers, and pentamers at short times is shown at Fig. 7B. For the first 1.87 ms, the number of dimers is rapidly increasing but there are very few trimers (on average, less than half the simulations contain even 1 trimer). Hence, the kinetics are dominated by a single reaction
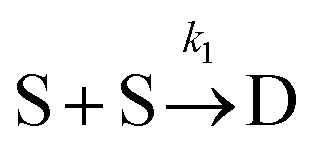
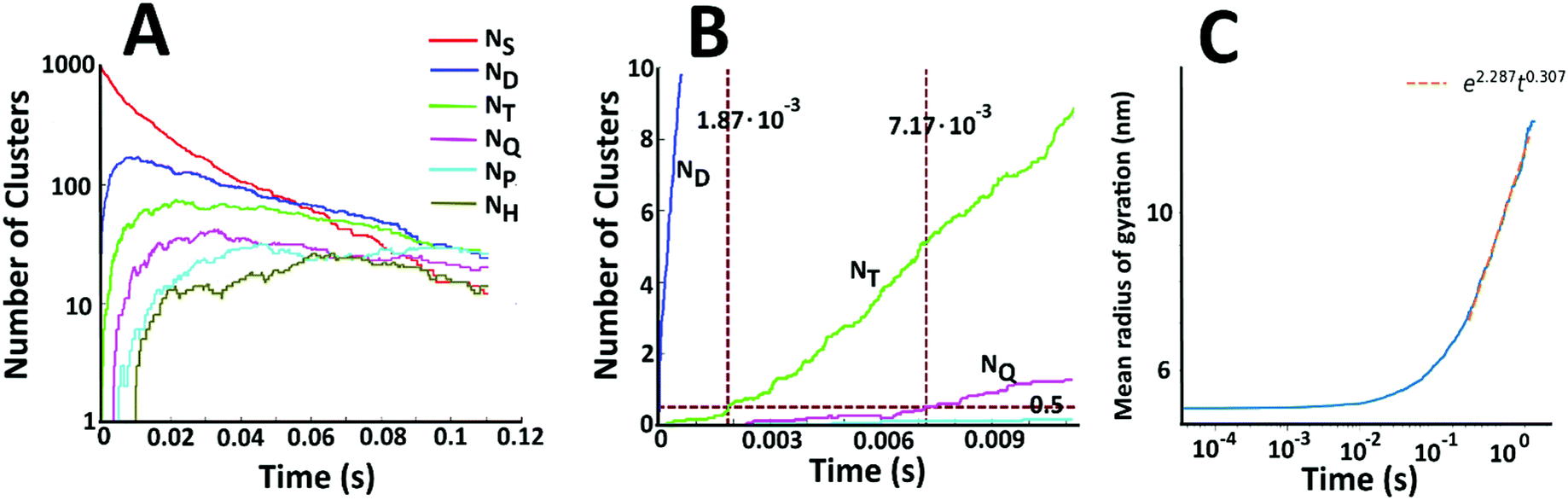 | ||
| Fig. 7 (A) Size evolution of gold aggregates (NS indicates primary (single) particles, ND dimers, NT trimers, NQ tetramers, NP pentamers, NH hexamers) in the solution containing iron ions. (B) Number of dimers, trimers, and tetramers at the early stages of aggregation. (C) The mean radius of gyration of the clusters as a function of time. | ||
Defining Ns as the number of single NPs, the rate of change in Ns is given by
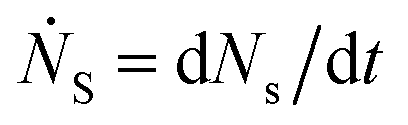
One can show that
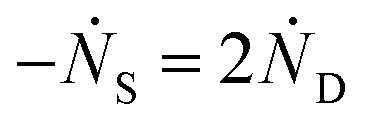
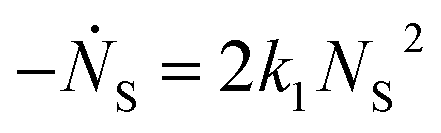
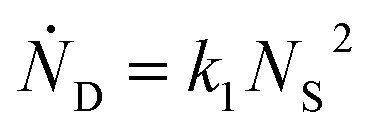
The value for k1 calculated from these expressions is identical up to the emergence of trimers and differs by only small amounts thereafter. Averaging over time and between the two methods of calculation yields k1 = 0.0143 part−1 s−1. Taking the simulation system size into account we obtain  It is shown in the ESI† that this rate constant does accurately model the decrease in number of single particles and increase in dimers.
It is shown in the ESI† that this rate constant does accurately model the decrease in number of single particles and increase in dimers.
Beyond 1.87 ms, the number of trimers begins to increase. However, the number of tetramers is negligible until 7.17 ms (Fig. 7B). Hence, during this time, the following two reactions are occurring:
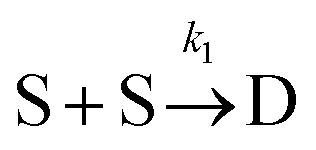
which yields:
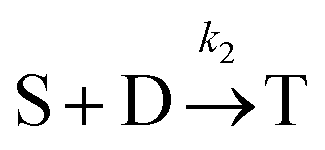
and rate constants given by the relations:
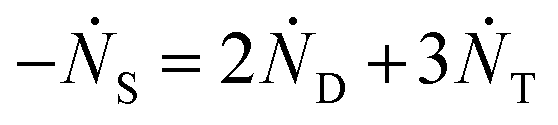
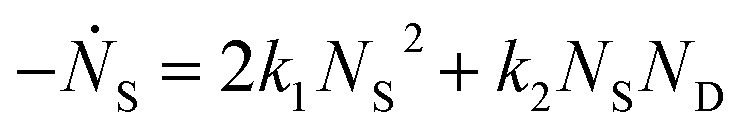
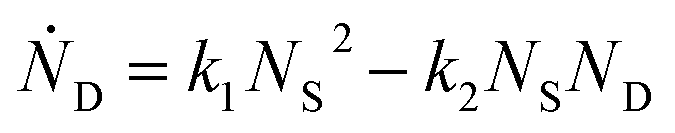
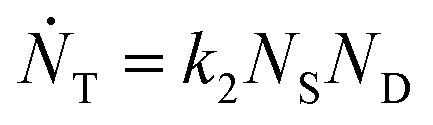
Averaging over this time window, we find k1 = 0.012 part−1 s−1 and k2 = 0.019 part−1 s−1 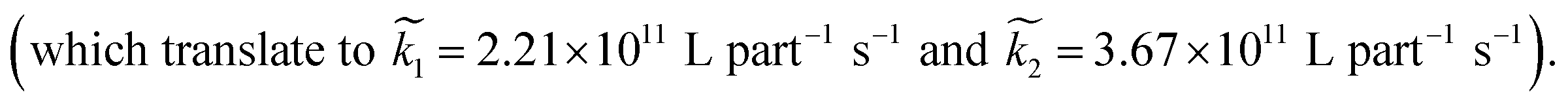 It is again shown in the ESI† that these rate constants accurately model the number of species. The value for k1 compares well across the time regions with a 15% difference easily being attributable to uncertainty in the simulation data. It is interesting to note that k2 is twice as large as k1. As the difference is between two single particles colliding or one single and a double, it is expected that these rate constants be of similar magnitude – as they are. However, the dimer is larger than a single particle and thus presents a bigger target, which increases the probability of a collision. On the other hand, the dimer also diffuses more slowly which slows down the reaction. The first effect appears to be stronger as the net result is a higher rate constant for a S–D reaction than a S–S reaction. Again, these results are consistent with the Smoluchowski63 coagulation equation.
It is again shown in the ESI† that these rate constants accurately model the number of species. The value for k1 compares well across the time regions with a 15% difference easily being attributable to uncertainty in the simulation data. It is interesting to note that k2 is twice as large as k1. As the difference is between two single particles colliding or one single and a double, it is expected that these rate constants be of similar magnitude – as they are. However, the dimer is larger than a single particle and thus presents a bigger target, which increases the probability of a collision. On the other hand, the dimer also diffuses more slowly which slows down the reaction. The first effect appears to be stronger as the net result is a higher rate constant for a S–D reaction than a S–S reaction. Again, these results are consistent with the Smoluchowski63 coagulation equation.
Beyond 7.17 ms, the number of tetramers starts to become non-negligible. However again note that very few pentamers have formed. Hence, there is a region where the kinetics are defined by the following reactions:
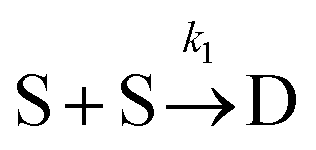
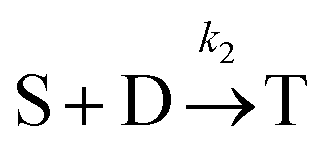
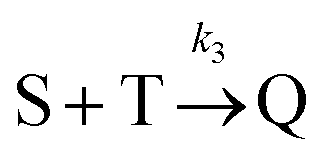
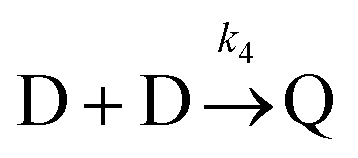
There are now two reactions that yield tetramers and this significantly complicates the analysis. Further exploration of the kinetics requires a great deal more simulation data and is the subject of future work.
As time progresses, the formation of larger clusters by the aggregation of smaller clusters becomes complex with many simultaneous processes. Hence, to characterize the growth of the clusters at long times, we instead calculate the mean radius of gyration, denoted by 〈Rg〉. Fig. 7C shows the mean radius of gyration of the clusters as a function of time. A linear fit on the log–log plot yields a scaling law of 〈Rg〉 = 9.84t0.31. Using this power law, we extrapolate to experimental times and calculate a predicted 〈Rg〉 of ∼320 nm at 24 hours. This compares well to the clusters resolved in the TEM images (Fig. 4I and J). The exponent of 0.31 compares well to previous reported values for similar aggregation processes that span between 0.4 and 0.5.64,65
We note that a likely source of any discrepancy are due to that in the current simulation large structures are able to relax and thus we observe more compact structures than those displayed in previous reports which tend to be more extended. Again, the simulation images compare well to the experimental TEM images indicating that continual relaxation of the aggregate structures is likely an important feature of the system. An extended analysis of the kinetics and the mean radius of gyration is the subject of further study.
Simulations were also performed for the system with hemoglobin. For this, a more detailed model that includes particles representing hemoglobin was used. Each hemoglobin contains four spheres harmonically bonded together, representing the heme sites. There is a weak attractive potential between heme sites and a strong attractive potential between the heme sites and L. Simulations were run up to a time of 0.02208 s and a snapshot of a representative cluster is shown in Fig. 8A (inset). The clusters bear a striking resemblance to the TEM images (Fig. 4 and 5) where the hemoglobin appears to act as a glue that binds the Au NPs together.
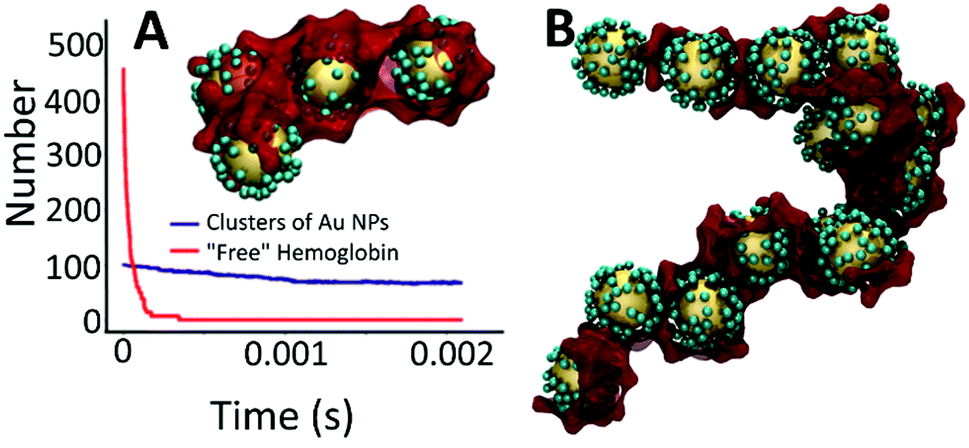 | ||
| Fig. 8 (A) Dynamics of association of hemoglobin centers to Au NP and small clusters of Au NP. Inset: a snapshot showing a representative calculated agglomerated nanostructure that is forming as a result of the interaction of L–Au NPs with hemoglobin obtained by coarse-grained simulations. Large yellow spheres represent Au NPs; small cyan spheres represent iron-free ligands bonded to Au NPs. Dark red colour represents hemoglobin protein acting as encapsulating agent interconnecting Au NPs into large clusters. Each hemoglobin center was modeled as four spheres harmonically bonded together to represent the heme sites. (B) Larger cluster obtained from simulations performed at higher densities of Au NPs. | ||
To explore this further, simulations were performed at a higher density to allow the formation of larger clusters. As shown in Fig. 8B, these simulations do generate larger structures thus demonstrating that hemoglobin not only mediates the formation of small clusters of L–Au NPs but also the aggregation of these small clusters in to larger structures. However, the structure displayed in Fig. 8B appears to be more extended than the relatively compact structures generated by the multi-scale simulations and the TEM images.
As only one of these simulations was performed, it is not known if this extended conformation is characteristic or not. Further, the impact that the artificially high density of L–Au NPs will have on the morphology of the resulting clusters has not been explored. These questions are the subject of further research, which requires further development of the modeling and simulation approach. To this end, we note that the hemoglobin simulations again reveal separable timescales. Fig. 8A shows the number of free hemoglobins and the number of clusters as a function of time for simulations performed at the experimental density of L–Au NPs. It is evident that the hemoglobin quickly associates with Au NP clusters on a much faster time scale than the aggregation of Au NPs. This again suggests a multiscale approach to yield efficient simulations to explore the formation of large L–Au NP clusters driven by hemoglobin.
Conclusions
We have shown that iron ion-mediated interactions can be utilized for assembly of Au NPs. The binding affinity of Au NPs is strongly dependent on the molecules grafted to the surface of NPs. While conventional citrate-stabilized Au NPs do not respond to the presence of up to 15 ppm of iron ions, partial substitution of citrate by a ligand able to complex iron ions, indeed, leads to Au NPs self-assembly in presence of less than 5 ppm of Fe(II) or Fe(III). Moreover, functionalized NPs can interact with iron centers of hemoglobin. To minimize the effect of electrostatic interactions, the behavior of Au NPs and hemoglobin were studied at the isoelectric point of hemoglobin (pH 6.8). To eliminate the impact of hemoglobin thiol groups on Au NPs assembly, Michael addition reaction was performed, protecting thiol groups. The assembly of Au NPs has been monitored via UV-vis and TEM techniques. The red shift of the surface plasmon absorption band of Au NPs and blue shift of Soret absorption peak of hemoglobin unambiguously confirms iron coordination by the surface-anchored ligands. A systematic and thorough analysis of the optical and structural characterization results show the formation of high-quality self-assembled metamaterial structures.Exploring the kinetics of NPs assembly induced by iron moieties (either iron ions of hemoglobin) by a multi-scale coarse-grained simulations gives insight into the mechanisms of particle assembly. The kinetics of the aggregation at short times displays interesting features such as the relative magnitude of the rate constants and peaks in the number of small clusters (e.g., dimers, trimers) at intermediate times. At longer times, the cluster morphology was characterized by the mean radius of gyration and a well defined power-law for the growth rate was obtained. The exponent compares well with previous work, but also indicates that continual relaxation of the clusters during aggregation may be a crucial element to capture in the simulations.
Going forward, development of the iron mediated self-assembly via the L-receptor presented in this work could yield novel materials based on NPs assembly – the design of which could be aided by the multi-scale modeling and simulation approach. In particular, we believe that applying proper 2D or 3D matrixes and well-tuning the number of binding sites will provide a means to tune the iron-mediated NPs assembly to achieve the protein mediated self-assembly of controlled, ordered structures for the development of novel nanomaterials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada through the Discovery Grants program (RGPIN-2016-05823, RGPIN-2014-06091) and University of Ontario Institute of Technology (UOIT). J. P. acknowledges NSERC Undergraduate Student Research Award. N. L. acknowledges NSERC Canada Graduate Scholarship – Master's Program. The authors acknowledge Dr Robert Temkin (Centre for the Neurobiology of Stress, University of Toronto Scarborough) and Dr Carmen Andrei (Canadian Centre for Electron Microscopy, University of MacMaster) for performing TEM and Cryo-TEM measurements.References
- J. Piella, N. G. Bastus and V. Puntes, Chem. Mater., 2016, 28, 1066–1075 CrossRef CAS.
- Y. Wang, O. Zeiri, M. Raula, B. Le Ouay, F. Stellacci and I. A. Weinstock, Nat. Nanotechnol., 2017, 12, 170–176 CrossRef CAS PubMed.
- E. Menumerov, R. A. Hughes and S. Neretina, Nano Lett., 2016, 16, 7791–7797 CrossRef CAS PubMed.
- N. Toshima, Pure Appl. Chem., 2013, 85, 437–451 CrossRef CAS.
- V. R. de la Rosa, Z. Y. Zhang, B. G. De Geest and R. Hoogenboom, Adv. Funct. Mater., 2015, 25, 2511–2519 CrossRef CAS.
- D. Maiolo, L. Paolini, G. Di Noto, A. Zendrini, D. Berti, P. Bergese and D. Ricotta, Anal. Chem., 2015, 87, 4168–4176 CrossRef CAS PubMed.
- P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nano Today, 2007, 2, 18–29 CrossRef.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- C. Freese, C. Uboldi, M. I. Gibson, R. E. Unger, B. B. Weksler, I. A. Romero, P. O. Couraud and C. J. Kirkpatrick, Part. Fibre Toxicol., 2012, 9, 23 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- V. Forest and J. Pourchez, Nano Today, 2016, 11, 700–703 CrossRef CAS.
- F. Wang, D. E. Curry and J. W. Liu, Langmuir, 2015, 31, 13271–13274 CrossRef CAS PubMed.
- Z. Li, K. Munro, I. I. Ebralize, M. R. Narouz, J. D. Padmos, H. Hao, C. M. Crudden and J. H. Horton, Langmuir, 2017, 33, 13936–13944 CrossRef CAS PubMed.
- R. W. Y. Man, C.-H. Li, M. W. A. MacLean, O. V. Zenkina, M. T. Zamora, L. N. Saunders, A. Rousina-Webb, M. Nambo and C. M. Crudden, J. Am. Chem. Soc., 2018, 140, 1576–1579 CrossRef CAS PubMed.
- J. W. Park and J. S. Shumaker-Parry, ACS Nano, 2015, 9, 1665–1682 CrossRef CAS PubMed.
- Y. J. Kim, R. C. Johnson and J. T. Hupp, Nano Lett., 2001, 1, 165–167 CrossRef CAS.
- Y. R. Kim, R. K. Mahajan, J. S. Kim and H. Kim, ACS Appl. Mater. Interfaces, 2010, 2, 292–295 CAS.
- R. K. Pandey, C. Chakraborty, U. Rana, S. Moriyama and M. Higuchi, J. Mater. Chem. A, 2016, 4, 4398–4401 CAS.
- J. K. Stolarczyk, A. Deak and D. F. Brougham, Adv. Mater., 2016, 28, 5400–5424 CrossRef CAS PubMed.
- M. Boterashvili, T. Shirman, R. Popovitz-Biro, Q. Wen, M. Lahav and M. E. van der Boom, Chem. Commun., 2016, 52, 8079–8082 RSC.
- E. W. Gehrels, W. B. Rogers and V. N. Manoharan, Soft Matter, 2018, 14, 969–984 RSC.
- J. M. Slocik and R. R. Naik, Curr. Opin. Biotechnol., 2017, 46, 7–13 CrossRef CAS PubMed.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- S. Pal, Z. Deng, H. Wang, S. Zou, Y. Liu and H. Yan, J. Am. Chem. Soc., 2011, 133, 17606–17609 CrossRef CAS PubMed.
- D. Gottlieb, S. A. Morin, S. Jin and R. T. Raines, J. Mater. Chem., 2008, 18, 3865 RSC.
- R. A. McMillan, C. D. Paavola, J. Howard, S. L. Chan, N. J. Zaluzec and J. D. Trent, Nat. Mater., 2002, 1, 247 CrossRef CAS PubMed.
- K. L. Gurunatha, A. C. Fournier, A. Urvoas, M. Valerio-Lepiniec, V. Marchi, P. Minard and E. Dujardin, ACS Nano, 2016, 10, 3176–3185 CrossRef CAS PubMed.
- S. C. Wagner, M. Roskamp, H. Colfen, C. Bottcher, S. Schlecht and B. Koksch, Org. Biomol. Chem., 2009, 7, 46–51 CAS.
- K. K. Caswell, J. N. Wilson, U. H. Bunz and C. J. Murphy, J. Am. Chem. Soc., 2003, 125, 13914–13915 CrossRef CAS PubMed.
- S. Park, H. Joo and J. S. Kim, Soft Matter, 2018, 14, 817–825 RSC.
- I. C. Garlea, E. Bianchi, B. Capone, L. Rovigatti and C. N. Likos, Curr. Opin. Colloid Interface Sci., 2017, 30, 1–7 CrossRef.
- D. J. Audus, F. W. Starr and J. F. Douglas, Soft Matter, 2018, 14, 1622–1630 RSC.
- L. Ma, F. Li, T. Fang, J. Zhang and Q. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 11024–11031 CAS.
- A. Kondo and H. Fukuda, J. Colloid Interface Sci., 1998, 198, 34–41 CrossRef CAS.
- A. Kondo and J. Mihara, J. Colloid Interface Sci., 1996, 177, 214–221 CrossRef CAS PubMed.
- W. Yang, L. Sun, J. Weng, L. Chen and Q. Zhang, IET Nanobiotechnol., 2012, 6, 26–32 CrossRef CAS PubMed.
- H. Kitagishi, K. Oohora, H. Yamaguchi, H. Sato, T. Matsuo, A. Harada and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 10326–10327 CrossRef CAS PubMed.
- A. Onoda, Y. Ueya, T. Sakamoto, T. Uematsu and T. Hayashi, Chem. Commun., 2010, 46, 9107–9109 RSC.
- J. J. Stephanos, S. A. Farina and A. W. Addison, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1996, 1295, 209–221 CrossRef.
- O. V. Kosmachevskaya and A. F. Topunov, Appl. Biochem. Microbiol., 2007, 43, 313–319 CrossRef CAS.
- X. L. Wang, W. Y. Zheng, H. Y. Lin, G. C. Liu, Y. Q. Chen and J. N. Fang, Tetrahedron Lett., 2009, 50, 1536–1538 CrossRef CAS.
- A. Detsi, D. Bouloumbasi, K. C. Prousis, M. Koufaki, G. Athanasellis, G. Melagraki, A. Afantitis, O. Igglessi-Markopoulou, C. Kontogiorgis and D. J. Hadjipavlou-Litina, J. Med. Chem., 2007, 50, 2450–2458 CrossRef CAS PubMed.
- I. I. Ebralidze, G. Leitus, L. J. W. Shimon and R. Neumann, Inorg. Chim. Acta, 2009, 362, 4760–4766 CrossRef CAS.
- J. S. Yadav, T. Swamy, B. V. S. Reddy and D. K. Rao, J. Mol. Catal. A: Chem., 2007, 274, 116–119 CrossRef CAS.
- K. Amini, I. I. Ebralidze, N. W. C. Chan and H. B. Kraatz, Anal. Methods, 2016, 8, 7623–7631 RSC.
- S. Roux, B. Garcia, J. L. Bridot, M. Salome, C. Marquette, L. Lemelle, P. Gillet, L. Blum, P. Perriat and O. Tillement, Langmuir, 2005, 21, 2526–2536 CrossRef CAS PubMed.
- M. A. Hanif, I. I. Ebralidze and J. H. Horton, Appl. Surf. Sci., 2013, 280, 836–844 CrossRef CAS.
- I. I. Ebralidze, M. Hanif, R. Arjumand, A. A. Azmi, D. Dixon, N. M. Cann, C. M. Crudden and J. H. Horton, J. Phys. Chem. C, 2012, 116, 4217–4223 CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- S. Shankar, M. Lahav and M. E. van der Boom, J. Am. Chem. Soc., 2015, 137, 4050–4053 CrossRef CAS PubMed.
- N. O. Laschuk, I. I. Ebralidze, D. Spasyuk and O. V. Zenkina, Eur. J. Inorg. Chem., 2016, 3530–3535 CrossRef CAS.
- Mudasir, K. Wijaya, N. Yoshioka and H. Inoue, J. Inorg. Biochem., 2003, 94, 263–271 CrossRef CAS PubMed.
- N. O. Laschuk, I. I. Ebralidze, S. Quaranta, S. T. W. Kerr, J. G. Egan, S. Gillis, F. Gaspari, A. Latini and O. V. Zenkina, Mater. Des., 2016, 107, 18–25 CrossRef CAS.
- X. J. Wang, G. P. Li, T. Chen, M. X. Yang, Z. Zhang, T. Wu and H. Y. Chen, Nano Lett., 2008, 8, 2643–2647 CrossRef CAS PubMed.
- J. A. Anderson, C. D. Lorenz and A. Travesset, J. Comput. Phys., 2008, 227, 5342–5359 CrossRef.
- J. Glaser, T. D. Nguyen, J. A. Anderson, P. Lui, F. Spiga, J. A. Millan, D. C. Morse and S. C. Glotzer, Comput. Phys. Commun., 2015, 192, 97–107 CrossRef CAS.
- G. W. Slater, C. Holm, M. V. Chubynsky, H. W. de Haan, A. Dubé, K. Grass, O. A. Hickey, C. Kingsburry, D. Sean, T. N. Shendruk and L. Zhan, Electrophoresis, 2009, 30, 792–818 CrossRef CAS PubMed.
- J. D. Weeks, D. Chandler and H. C. Andersen, J. Chem. Phys., 1971, 54, 5237–5247 CrossRef CAS.
- I. M. Elminyawi, S. Gangopadhyay and C. M. Sorensen, J. Colloid Interface Sci., 1991, 144, 315–323 CrossRef CAS.
- I. Kryven, S. Lazzari and G. Storti, Macromol. Theory Simul., 2014, 23, 170–181 CrossRef CAS.
- M. V. Smoluchowski, Z. Phys. Chem., 1918, 92U, 129 Search PubMed.
- M. Y. Lin, H. M. Lindsay, D. A. Weitz, R. Klein, R. C. Ball and P. Meakin, J. Phys.: Condens. Matter, 1990, 2, 3093 CrossRef.
- M. Y. Lin, H. M. Lindsay, D. A. Weitz, R. C. Ball, R. Klein and P. Meakin, Nature, 1989, 339, 360 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed kinetics and calculation of rate constants of the formation dimers and trimers from primary single gold nanoparticles; additional experimental results including UV-vis and ATR-FTIR spectra; optical images of aqueous solutions of L–Au NPs with different concentrations of Fe ions; coarse grained molecular dynamics simulation details; multi-level simulation details; and video descriptions (PDF). Short videos generated from different points in the three types of simulations: Gold_beginning.mpg (MPG), Gold_end.mpg (MPG), Gold_Hemo_beginning.mpg (MPG), Gold_Hemo_end.mpg (MPG), Multiscale_1beginning.mpg (MPG), Multiscale_2middle.mpg (MPG), Multiscale_3end.mpg (MPG). See DOI: 10.1039/c8ra01996g |
| ‡ Questions regarding the simulations should be addressed to: E-mail: hendrick.dehaan@uoit.ca |
| This journal is © The Royal Society of Chemistry 2018 |