
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed C–H acyloxylation of 2-phenylpyridine using oxygen as the oxidant†
Feifan Wang,
Zhiyang Lin,
Weisheng Yu,
Qingdong Hu,
Chao Shu and
Wu Zhang *
*
Key Laboratory of Functional Molecular Solids, Ministry of Education, Anhui Laboratory of Molecule-Based Materials, College of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000, China. E-mail: zhangwu@mail.ahnu.edu.cn
First published on 3rd May 2018
Abstract
An efficient copper-catalyzed direct o-acyloxylation of 2-phenylpyridine with carboxylic acids using oxygen as the oxidant has been developed. Various acids including aromatic acids, cinnamic acids and aliphatic acids are effective acyloxylation reagents and provide the desired products in moderate to excellent yields. The reaction proceeds well under an oxygen atmosphere, making this method potentially practical.
Introduction
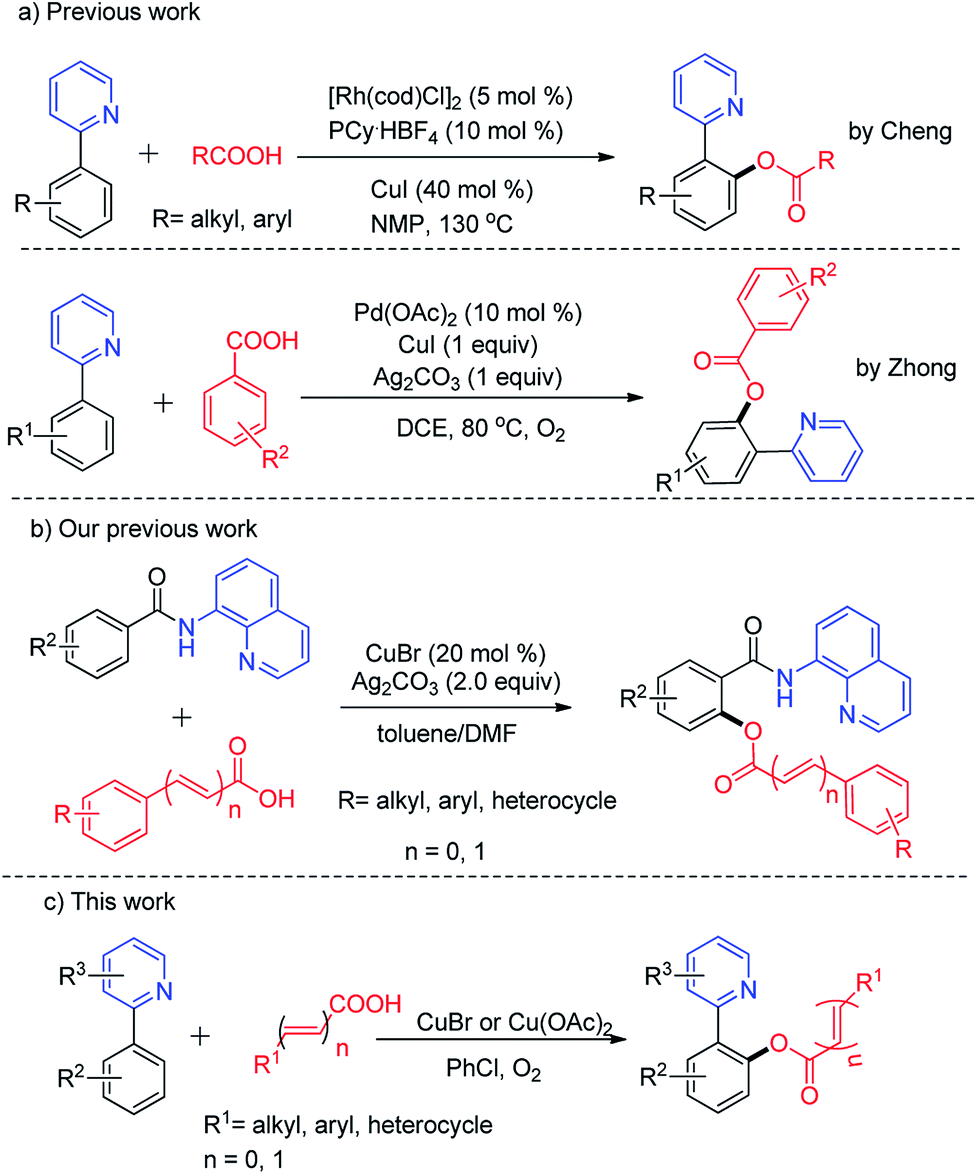
Transition-metal-catalyzed or -mediated C–H transformations have been widely used to synthesize many multifunctional molecules as a straightforward and atom economic method.1 The directing group assisted C–H bond activation and dehydrogenative cross-coupling are the most facile methods that have gained considerable attention over traditional cross-coupling methods.2,6a Copper complexes or salts have received increased attention as highly efficient catalysts to promote direct C–H bond functionalization for the construction of C–X, C–O, C–S and C–N bonds due to their abundance and low cost and toxicity over the past decades.3–5Accordingly, the importance of ester functionality in natural products, pharmaceuticals and functional materials has prompted chemists to investigate the development of regioselective C–O bond formation via C–H bond activation under transition metal catalysis. Recently, 2-phenylpyridine was elegantly identified as a versatile auxiliary in C–H transformations.6 And a number of o-benzoxylation protocols of 2-phenylpyridine have been developed with various acyloxyl sources such as benzyl alcohols or benzyl amines,7a anhydrides,7b sodium carboxylates,7c carboxylic acid,7d,e benzylic ethers,7f acyl chlorides,7g amides,7h aldehydes or methylarenes,7i aryl alkenes and alkynes.7j Very recently, Li and co-workers reported Rh(III)-catalyzed direct acyloxylation of sp2 C–H bonds with various carboxylic acids.8 Although various valuable acyloxylated products were obtained, the transformation still suffered from noble metal, and stoichiometric amount of silver oxidants. It would be ideal that this o-benzoxylation can be achieved under the oxidation of oxygen, which is often used as a clean terminal oxidant in copper-catalyzed reactions.9 Also, different oxidation states of copper (Cu(I), Cu(II), and Cu(III)) could be mutually converted with the assistance of O2.10
Continuing our research efforts in the development of novel copper-catalyzed C–H acyloxylation,11 we report now direct o-acyloxylation of 2-phenylpyridine with carboxylic acids including aromatic acids, cinnamic acids and aliphatic acids using oxygen as terminal oxidant with inexpensive copper catalysts, providing an operationally simple approach for the synthesis of benzoate (Scheme 1).
 | ||
| Scheme 1 Copper-catalyzed direct acyloxylation of C(sp2)–H bonds. | ||
Results and discussion
Initially, 3-methyl-2-phenylpyridine (1a, 1 equiv.) and benzoic acid (2a, 1.5 equiv.) were chosen as model substrates to establish the best reaction conditions. The catalytic activity of various copper sources, such as Cu(OAc)2·H2O, CuCl2·2H2O, CuBr2, CuSO4·5H2O, Cu(OAc)2, CuCl, CuBr, and CuI were investigated. Among them, CuBr was found to be superior and resulted in 88% yield (Table 1, entries 1–8). Decreasing the catalyst amount from 10 to 5 mol% resulted in the obvious decrease of the yield (Table 1, entry 9). In contrast, a little decrease of the yield was observed by increasing the catalyst loading to 15 mol% (Table 1, entry 10). The absence of copper catalyst fails to generate the desired products, which indicated that the copper catalyst played indispensable roles in this reaction (Table 1, entry 11). Then different solvents like toluene, benzene, DMSO, and DMF were screened, but they are not as effective as chlorobenzene (Table 1, entries 12–16).
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), solvent (2 mL) under oxygen for 24 h.b Isolated yield.c Under air.d Under Ar. | |||
| 1 | Cu(OAc)2·H2O (10) | PhCl | 85 |
| 2 | CuCl2·2H2O (10) | PhCl | 80 |
| 3 | CuBr2 (10) | PhCl | 53 |
| 4 | CuSO4·5H2O (10) | PhCl | 45 |
| 5 | Cu(OAc)2 (10) | PhCl | 87 |
| 6 | CuCl (10) | PhCl | 80 |
| 7 | CuBr (10) | PhCl | 88 |
| 8 | CuI (10) | PhCl | 48 |
| 9 | CuBr (5) | PhCl | 68 |
| 10 | CuBr (15) | PhCl | 83 |
| 11 | — | PhCl | NR |
| 12 | CuBr (10) | Toluene | 38 |
| 13 | CuBr (10) | DMF | 57 |
| 14 | CuBr (10) | DMSO | NR |
| 15 | CuBr (10) | 1,2-DCB | 73 |
| 16 | CuBr (10) | THF | 33 |
| 17 | CuBr (10) | PhCl | 70c |
| 18 | CuBr (10) | PhCl | —d |
This transformation also proceeded well under air, but the yield of 3aa was slightly lower (Table 1, entry 17). In contrast, the reaction did not occur under argon, indicating that oxygen was effective oxidant in the reaction.
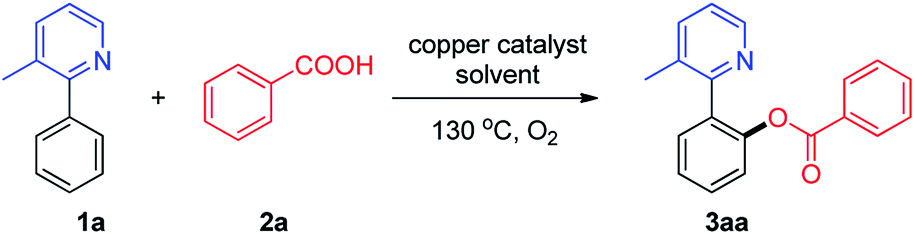
With the optimized conditions in hand, we investigated the substrate scope and generality of the copper-catalyzed C–H acyloxylation of 2-phenylpyridine with carboxylic acids. As shown in Scheme 2, a range of carboxylic acids were suitable for this reaction, including aromatic acids, cinnamic acids and aliphatic acids. In general, benzoic acid bearing electron-withdrawing groups exhibited higher reactivity than those bearing electron-donating groups. Although cinnamic acids and aliphatic acids exhibited lower reactivity than benzoic acids, moderate to good yields could be obtained and the result is superior to literature report using anhydrides as acyloxyl source.7b Gratifyingly, it was found that the o-acyloxylation of 3-methyl-2-phenylpyridine occurred with excellent levels of monosubstituted products, probably due to the steric hindrance. This protocol was compatible with a wide range of functional groups, such as alkyl, methoxy, chloro, bromo, aryl and trifluoromethyl groups.
 | ||
| Scheme 2 ortho-Acyloxylation reaction of 3-methyl-2-phenylpyridine with acids. aReaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), PhCl (2 mL), 130 °C, 24 h. bCu(OAc)2 (10 mol%). | ||
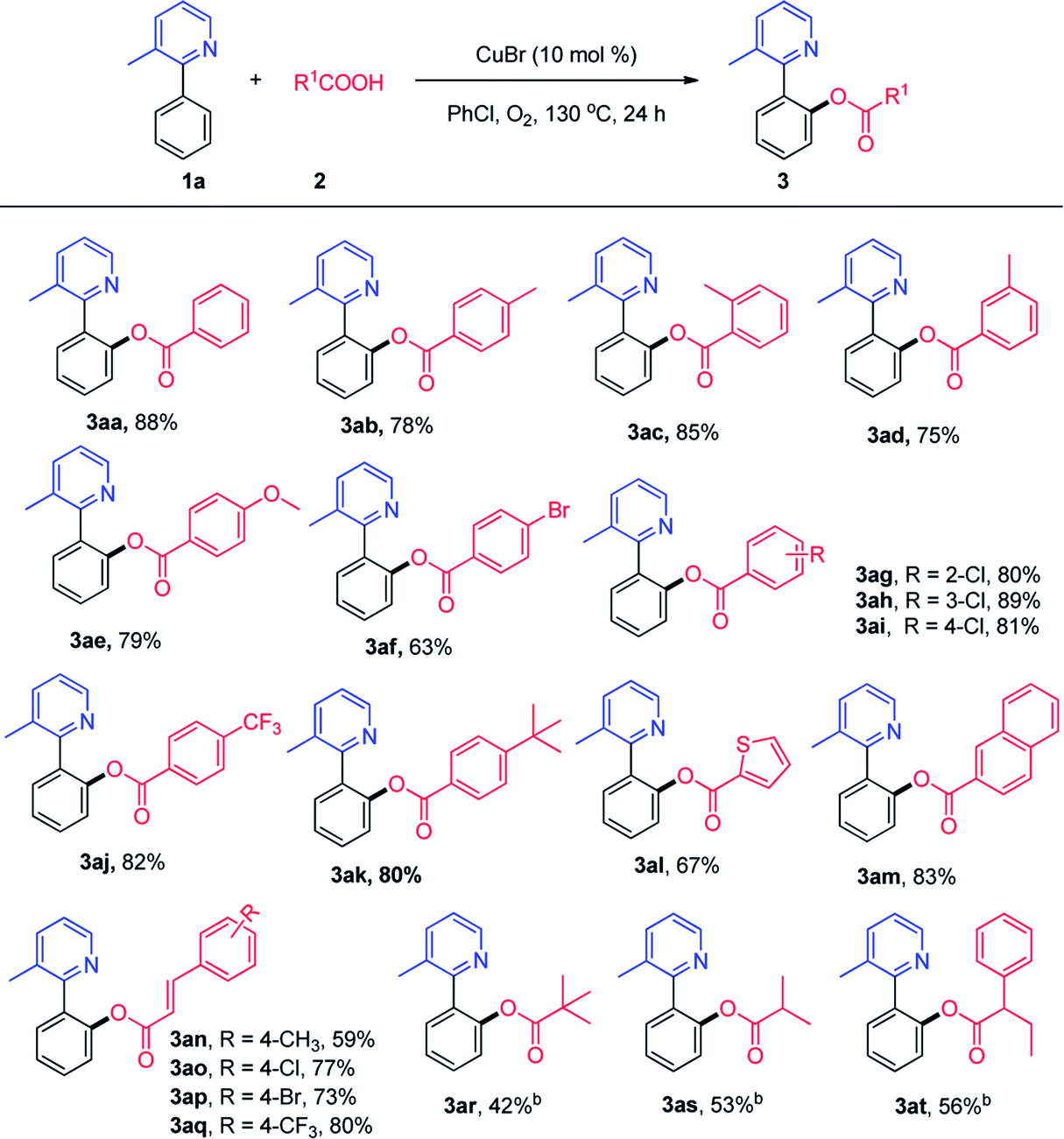
Then, we explored the viable substrate scope with variously decorated 2-arylpyridines (Scheme 3). 2-Phenylpyridine bearing a methyl group (3ca, 3da, and 3ea) in different positions reacted smoothly to afford the desired products in good yields. The methoxy and chloro groups at the ortho-position of the phenyl ring in 2-phenylpyridine did not exhibit a clear steric hindrance effect in this reaction (3ga and 3ha).
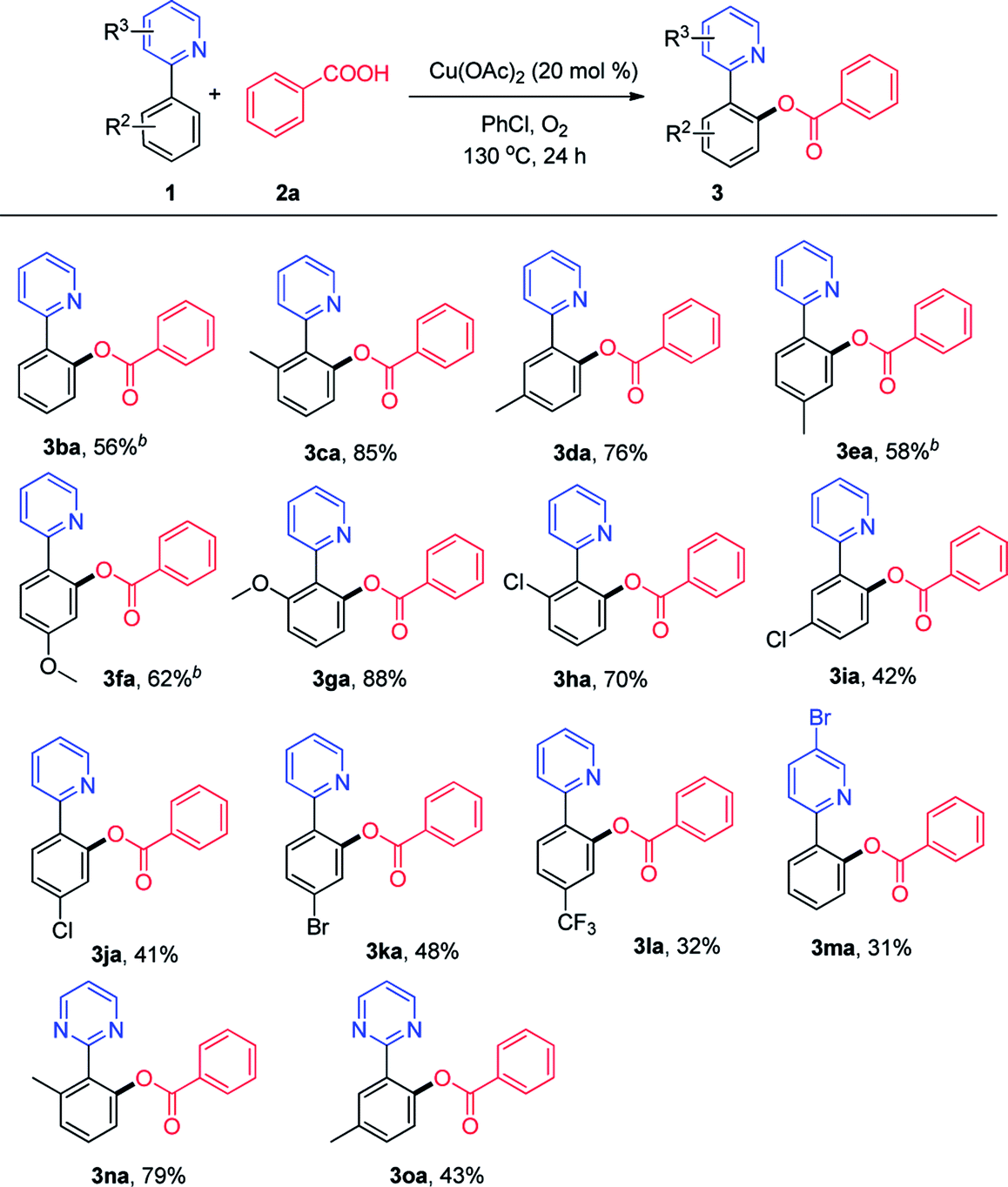 | ||
| Scheme 3 Scope of 2-phenylpyridines. aReaction conditions: 1 (0.1 mmol), 2a (0.3 mmol), Cu(OAc)2 (20 mmol%), PhCl (1 mL), 130 °C, 24 h. b11 h. | ||
Both electron-donating (OCH3, 3fa) and strong electron-withdrawing (CF3, 3la) functional groups on the phenyl ring of 2-phenylpyridine were well tolerated. We were pleased to find that the o-acyloxylation of 2-arylpyridines occurred with excellent levels of monoselectivity with methyl and chloro group at the meta-position (3da and 3ia). A good yield could be obtained using a halogen-bearing substrate, which can be used for further transformation.
In a gram-scale experiment, a satisfactory yield of 81% was obtained in the model reaction. The present acyloxylation provides an efficient and cost-effective approach for the synthesis of benzoates.
To gain insights on the reaction pathway of copper-catalyzed aerobic oxidative acyloxylation of 2-phenylpyridine with carboxylic acid, a series of experiments were carried out, as shown in Scheme 4. First, the intermolecular competition experiments between electronically differentiated substrates were conducted.12 In the intermolecular competition reaction of 2-phenylpyridine with p-toluylic acid and p-chlorobenzoic acid (2b and 2i) under the optimized conditions, the products 3ab and 3ai were obtained in 36% and 41% yield (Scheme 4a), indicating that the electron-deficient aromatic carboxylic acids reacted preferentially. Additionally, a scrambling test of 2-arylpyridines which bears an electron-donating and electron-withdrawing group (1e and 1j) with benzoic acid was conducted, and 3ea was isolated in 22% yield along with 7% of 3ja, indicating that the electron-rich arene proved to be inherently more reactive than their electron-deficient counterparts. Second, the desired product 3aa could be obtained in 74% or 82% yield when radical inhibitor 2,2,6,6-tetramethyl piperidine-N-oxide (TEMPO, 1 equiv.) or 2,6-di-tert-butyl-4-methylphenol (BHT, 1 equiv.) was added to the standard reaction, and ruling out the possibility of a radical pathway (Scheme 4-2). Third, the ortho-acyloxylation of the isotopically labeled substrates was investigated (Scheme 4-3).
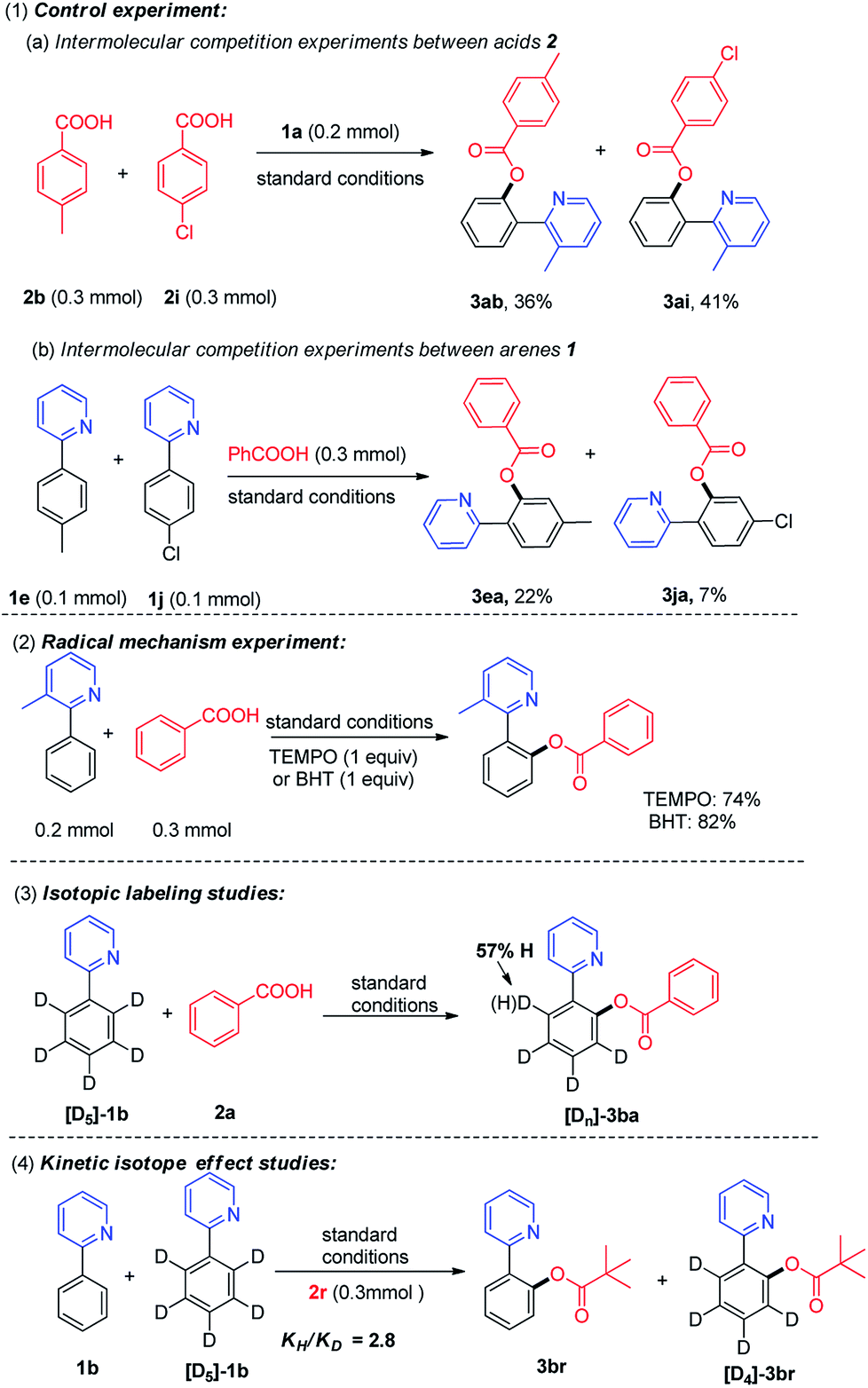 | ||
| Scheme 4 Intermolecular competition and KIE experiments. | ||
It was found that treating [D5]-1b with benzoic acid under the standard conditions afforded product [Dn]-3ba with a significant ortho-D/H exchange (see ESI†). The results confirmed that the initial ortho-CAr–H metalation is reversible. Finally, the intermolecular competition reaction between 1b and [D5]-1b with pivalic acid 2r in one vessel, established a kinetic isotopic effect (KIE) of KH/KD was 2.8. The results indicated that C–H bond cleavage in 2-phenylpyridine was involved in the rate-limiting step.
Subsequently, sodium benzoate was used rather than benzoic acid as acyloxyl sources. Trace of products was detected when sodium benzoate was used (Scheme S3, eqn (2)†). Based on our mechanistic studies and earlier precedent,7 a plausible mechanism for the copper-catalyzed ortho-acyloxylation of 2-phenylpyridine with benzoic acid is proposed in Scheme 5. Initially, Cu(I) was oxidized by oxygen to the Cu(II), and Cu(II) reacted with benzoic acids 2 affords cupric benzoate. Then, under the chelation assistance of the azo group, ortho-C–H bond electrophilic metalation of the phenyl ring of 2-arylpyridine with Cu(II) provides an active cyclometallated Cu(II) intermediate A. The fact that the 2-arylpyridine possessing electron-donating groups showed higher reactivity is consistent with the C–H bond electrophilic metalation regime. Subsequently, the intermediate A is oxidized to a Cu(III) intermediate B in the presence of Cu(II).13 Finally, reductive elimination from intermediate B delivered desired product 3b, accompanying with the regeneration of Cu(I) for the next cycle.
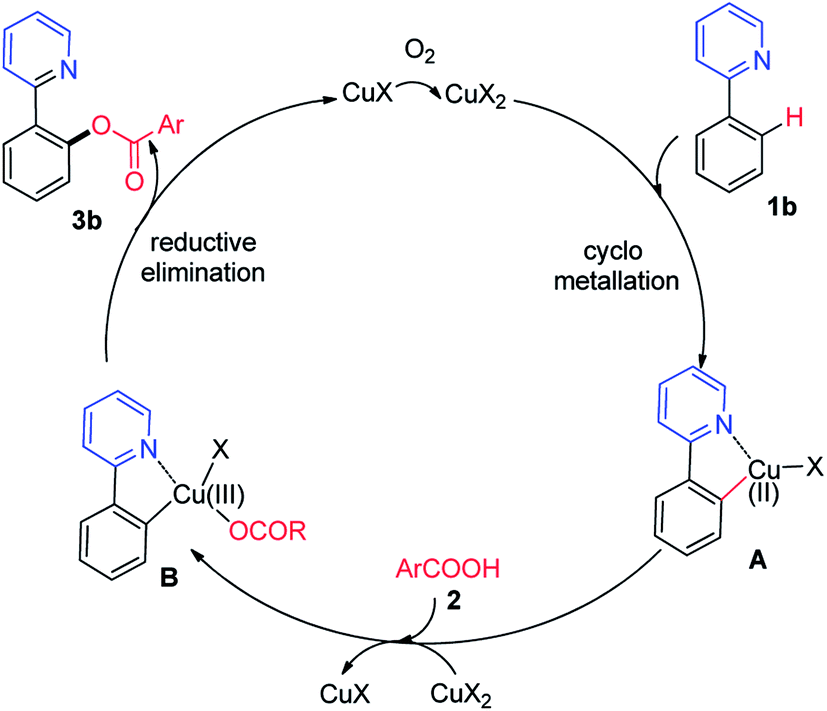 | ||
| Scheme 5 Plausible reaction mechanism. | ||
Conclusions
In summary, we first reported the copper-catalyzed C–H acyloxylation of 2-phenylpyridine with carboxylic acids under oxygen atmosphere. Various acids including aromatic acids, cinnamic acids and aliphatic acids are well compatible. This methodology has the advantages of simple operation, mild reaction condition with a broad substrate scope.Experimental
General information
All starting materials and reagents were commercially available and used directly without further purification. Most of 2-phenylpyridines were synthesized by the reactions between the corresponding 2-bromopyridine or 2-iodopyridine and phenylboronic acids according to the literature procedures.14 All known products gave satisfactory analytical data by NMR spectra, which corresponding to the reported literature values. Unknown compounds were confirmed by HRMS additionally. NMR spectra were determined at room temperature on Bruker Avance-300 or Bruker Avance-500 at 300 MHz or 500 MHz with tetramethylsilane (TMS) as an internal standard. Chemical shifts are given in δ relative to TMS, the coupling constants J are given in Hz. High-resolution mass spectra (HRMS) were recorded on Agilent 6200 LC/MS TOF using APCI or ESI in positive mode.Typical experimental procedure for the synthesis of 3
To a 10 mL reaction tube was added 3-methyl-2-phenylpyridine 1a (0.2 mmol), acid (0.3 mmol), CuBr (2.9 mg, 10 mol%), and chlorobenzene (2 mL) under oxygen atmosphere. The mixture was stirred at 130 °C for 24 h. The reaction mixture was then cooled to room temperature, diluted with ethyl acetate and quenched with saturated sodium chloride. The aqueous phase was extracted with ethyl acetate (3 × 10 mL). The organic layer was dried over Na2SO4. After concentration, the resulting residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 12/1) to afford the product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21272006), which provided financial support.Notes and references
- (a) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 24, 4087 Search PubMed; (b) S. Murru, C. S. Lott, B. McGough, D. M. Bernard and R. S. Srivastava, Org. Biomol. Chem., 2016, 14, 3681 RSC; (c) E. E. Galenko, V. A. Bodunov, A. V.Galenko, M. S. Novikov and A. F. Khlebnikov, J. Org. Chem., 2017, 82, 8568 CrossRef CAS PubMed; (d) R. Mandal and B. Sundararaju, Org. Lett., 2017, 19, 2544 CrossRef CAS PubMed; (e) S. Y. Ni, W. Z. Zhang, H. B. Mei, J. L. Han and Y. Pan, Org. Lett., 2017, 19, 2536 CrossRef CAS PubMed; (f) B. Wang, Y. J. Dai, W. Q. Tong and H. G. Gong, Org. Biomol. Chem., 2015, 13, 11418 RSC; (g) N. Ichiishi, A. J. Canty, B. F. Yates and M. S. Sanford, Org. Lett., 2013, 15, 5134 CrossRef CAS PubMed; (h) I. Nakamura, Y. Ishida and M. Terada, Org. Lett., 2014, 16, 2562 CrossRef CAS PubMed; (i) A. P. Thankachan, K. S. Sindhu, S. M. Ujwaldev and G. Anilkumar, Tetrahedron Lett., 2017, 58, 536 CrossRef CAS; (j) M. Zheng, F. Wu, K. Chen and S. F. Zhu, Org. Lett., 2016, 18, 3554 CrossRef CAS PubMed.
- (a) J. A. Gurak Jr, V. T. Tran, M. M. Sroda and K. M. Engle, Tetrahedron, 2017, 73, 3636 CrossRef; (b) R. Y. Zhu, M. E. Farmer, Y. Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed; (c) Y. Zheng, W. B. Song and L. J. Xuan, Org. Biomol. Chem., 2015, 13, 10834 RSC; (d) C. B. Yu, F. F. Li, J. Zhang and G. F. Zhong, Chem. Commun., 2017, 53, 533 RSC.
- (a) H. G. Wang, Y. Wang, C. L.Peng, J. C. Zhang and Q. Zhu, J. Am. Chem. Soc., 2010, 132, 13217 CrossRef CAS PubMed; (b) M. Kitahara, N. Umeda, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2011, 133, 2160 CrossRef CAS PubMed; (c) J. Gallardo-Donaire and R. Martin, J. Am. Chem. Soc., 2013, 135, 9350 CrossRef CAS PubMed; (d) S. Bhadra, C. Matheis, D. Katayev and L. J. Gooβen, Angew. Chem., Int. Ed., 2013, 52, 9279 CrossRef CAS PubMed; (e) G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932 CrossRef CAS PubMed; (f) C. Zhang, C. H. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (g) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (h) Y. Y. Weng, H. Zhou, C. Sun, Y. Y. Xie and W. K. Su, J. Org. Chem., 2017, 82, 9047 CrossRef CAS PubMed; (i) S. Seo and M. C. Willis, Org. Lett., 2017, 19, 4556 CrossRef CAS PubMed.
- (a) L. J. Yang, Z. Lu and S. S. Stahl, Chem. Commun., 2009, 42, 6460 RSC; (b) Y. Y. Li, M. C. Gao, B. X. Liu and B. Xu, Org. Chem. Front., 2017, 4, 445 RSC; (c) M. C. Gao, Y. Y. Li, Y. S. Gan and B. Xu, Angew. Chem., Int. Ed., 2015, 54, 8795 CrossRef CAS PubMed; (d) Y. Yamane, N. Miwa and T. Nishikata, ACS Catal., 2017, 7, 6872 CrossRef CAS; (e) M. M. Naik, V. P. Kamat and S. G. Tilve, Tetrahedron, 2017, 73, 5528 CrossRef CAS; (f) X. He, Y. Z. Xu, L. X. Kong, H. H. Wu, D. Z. Ji, Z. B. Wang, Y. G. Xu and Q. H. Zhu, Org. Chem. Front., 2017, 4, 1046 RSC; (g) J. Zhang, R. P. Jia and D. H. Wang, Tetrahedron Lett., 2016, 57, 3604 CrossRef CAS; (h) Y. Lin, M. Z. Cai, Z. Q. Fang and H. Zhao, Tetrahedron, 2016, 72, 3335 CrossRef CAS.
- X. X. Guo, D. W. Gu, Z. X. Wu and W. B. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed.
- (a) Z. K. Chen, B. J. Wang, J. T. Zhang, W. L. Yu, Z. X. Liu and Y. H. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (b) D. G. Zhou, F. Yang, X. Yang, C. X. Yan, P. P. Zhou and H. W. Jing, Org. Chem. Front., 2017, 4, 377 RSC; (c) R. Molteni, K. Edkins, M. Haehnel and A. Steffen, Organometallics, 2016, 35, 629 CrossRef CAS.
- (a) A. Banerjee, S. K. Rout, S. Guin and B. K. Patel, RSC Adv., 2014, 4, 55115 RSC; (b) W. H. Wang, F. Luo, S. H. Zhang and J. Cheng, J. Org. Chem., 2010, 75, 2415 CrossRef CAS PubMed; (c) L. P. Li, P. Yu, J. Cheng, F. Chen and C. D. Pan, Chem. Lett., 2012, 41, 600 CrossRef CAS; (d) Z. S. Ye, W. H. Wang, F. Luo, S. H. Zhang and J. Cheng, Org. Lett., 2009, 11, 3974 CrossRef CAS PubMed; (e) C.-J. Hu, X.-H. Zhang, Q.-P. Ding, T. Lv, S.-P. Ge and P. Zhong, Tetrahedron Lett., 2012, 53, 2465 CrossRef CAS; (f) N. Khatun, A. Banerjee, S. K. Santra, W. Ali and B. K. Patel, RSC Adv., 2015, 5, 36461 RSC; (g) W. H. Wang, C. D. Pan, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 3978 RSC; (h) S. L. Yedage and B. M. Bhanage, Synlett, 2015, 26, 2161–2169 CrossRef CAS; (i) Y. J. Bian, C. B. Xiang, Z. M. Chen and Z. Z. Huang, Synlett, 2011, 16, 2407 Search PubMed; (j) S. K. Rout, S. Guin, A. Gogoi, G. Majji and B. K. Patel, Org. Lett., 2014, 16, 1614 CrossRef CAS PubMed.
- C. J. Chen, Y. P. Pan, H. Q. Zhao, X. Xu, J. B. Xu, Z. Y. Zhang, S. Q. Xi, L. J. Xu and H. R. Li, Org. Chem. Front., 2018, 5, 415 RSC.
- (a) K. P. Bryliakov, Chem. Rev., 2017, 117, 11406 CrossRef CAS PubMed; (b) H. Yi, C. L. Bian, X. Hu, L. B. Niu and A. W. Lei, Chem. Commun., 2015, 51, 14046 RSC; (c) X. Wang, Y. B. Huang, Y. L. Xu, X. D. Tang, W. Q. Wu and H. F. Jiang, J. Org. Chem., 2017, 82, 2211 CrossRef CAS PubMed; (d) X. M. Li, H. Huang, C. G. Yu, Y. T. Zhang, H. Li and W. Wang, Org. Lett., 2016, 18, 5744 CrossRef CAS PubMed.
- (a) L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013 CrossRef CAS PubMed; (b) J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357 CrossRef CAS PubMed; (c) S. Chiba, L. Zhang and J.-Y. Lee, J. Am. Chem. Soc., 2010, 132, 7266 CrossRef CAS PubMed; (d) A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797 CrossRef CAS PubMed; (e) C. X. Wang, Y. D. Yang, D. K. Qin, Z. He and J. S. You, J. Org. Chem., 2015, 80, 8424 CrossRef CAS PubMed; (f) Q. Fu, D. Yi, Z. J. Zhang, W. Liang, S. Y. Chen, L. Yang, Q. Zhang, J. X. Ji and W. Wei, Org. Chem. Front., 2017, 4, 1385 RSC.
- F. F. Wang, Q. Y. Hu, C. Shu, Z. Y. Lin, D. W. Min, T. C. Shi and W. Zhang, Org. Lett., 2017, 19, 3636 CrossRef CAS PubMed.
- K. Raghuvanshi, D. Zell and L. Ackermann, Org. Lett., 2017, 19, 1278 CrossRef CAS PubMed.
- A. E. King, T. C. Brunold and S. S. Stahl, J. Am. Chem. Soc., 2009, 131, 5044 CrossRef CAS PubMed.
- X. F. Rao, C. Liu, J. S. Qiu and Z. L. Jin, Org. Biomol. Chem., 2012, 10, 7875 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for ESI and details of instruments and experimental procedures. See DOI: 10.1039/c8ra01974f |
| This journal is © The Royal Society of Chemistry 2018 |