
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Behaviour of the XH-*-π and YX-*-π interactions (X, Y = F, Cl, Br and I) in the coronene π-system, as elucidated by QTAIM dual functional analysis with QC calculations†
Satoko Hayashi *,
Yuji Sugibayashi and
Waro Nakanishi*
*,
Yuji Sugibayashi and
Waro Nakanishi*
Faculty of Systems Engineering, Wakayama University, 930 Sakaedani, Wakayama 640-8510, Japan. E-mail: hayashi3@sys.wakayama-u.ac.jp; nakanisi@sys.wakayama-u.ac.jp; Fax: +81 73 457 8253; Tel: +81 73 457 8252
First published on 3rd May 2018
Abstract
The dynamic and static nature of XH-*-π and YX-*-π in the coronene π-system (π(C24H12)) is elucidated by QTAIM dual functional analysis, where * emphasizes the presence of bond critical points (BCPs) in the interactions. The nature of the interactions is elucidated by analysing the plots of the total electron energy densities Hb(rc) versus Hb(rc) − Vb(rc)/2 [=(ħ2/8m)∇2ρb(rc)] for the interactions at BCPs, where Vb(rc) are the potential energy densities at the BCPs. The data for the perturbed structures around the fully optimized structures are employed for the plots in addition to those of the fully optimized structures. The plots are analysed using the polar coordinate of (R, θ) for the data of the fully optimized structures, while those containing the perturbed structures are analysed using (θp, κp), where θp corresponds to the tangent line of each plot and κp is the curvature. Whereas (R, θ) show the static nature, (θp, κp) represent the dynamic nature of the interactions. All interactions in X–H-*-π(C24H12) (X = F, Cl, Br and I) and Y–X-*-π(C24H12) (Y–X = F–F, Cl–Cl, Br–Br, I–I, F–Cl, F–Br and F–I) are classified by pure CS (closed shell) interactions and are characterized as having the vdW nature, except for X–H = F–H and Y–X = F–Cl, F–Br and F–I, which show the typical-HB nature without covalency. The structural features of the complexes are also discussed.
Introduction
Hydrogen bonds (HBs) and halogen bonds (XBs) are of current and continuous interest. HBs and XBs are fundamentally important for their ability to give rise to molecular association caused by the energy stabilization of the system.1–11 The direction-control through the formation of HBs plays a crucial role in all fields of chemical and biological sciences. The opening and closing of the duplex DNA structure in active proliferation at around room temperature is a typical example of the effect of HBs.12 HBs also play an important role in the very specific conformation of hormones with the HBs of the dimers controlling the characteristic biological properties.13 Conventional HBs of the shared proton interaction type4 are formed with atoms of the main group elements, which are usually not very strong in the neutral form (≤ approximately 40 kJ mol−1),1,5 albeit usually stronger than the van der Waals (vdW) interactions. Contributions from the charge transfer (CT) interaction become more important as the strength of HBs increases in addition to the vdW interactions, where attractive electrostatic interactions and the dispersion force mainly contribute to form the vdW adducts. Conversely, the attractive interactions, between the electrophilic σ*-orbitals of halogen or inter-halogen molecules with the non-bonding orbitals (n-orbitals), must be the driving force for the formation of typical XBs. The nature of XBs has been discussed based on the theoretical background of the molecular orbital description for the bonding and the σ-hole developed on the halogen atoms together with the stability based on the structural aspects.14 XBs are applicable to a wide variety of fields in chemical and biological sciences, such as crystal engineering, supramolecular soft matter and nanoparticles.π-orbitals also give rise to similar HBs and XBs with hydrogen halides and halogen or inter-halogen molecules, respectively. Similar to the case of n-orbitals, π-orbitals act as electron donors to form such adducts. The π-electron systems usually construct planar molecules. Benzene and coronene15 are the typical examples of the planar π-systems, together with graphene. Graphene shows unique physical properties. Graphene-based carbon allotropes, such as graphene, graphite, fullerenes16 and carbon nanotubes, have attracted considerable attention owing their many potential applications in nanotechnology, including nanoelectronics, energy storage and biosensing.17–19 Coronene, a typical planar molecule, is often employed as a model of graphene in the study of adsorption phenomena, even though it is suggested that coronene may, in certain cases, not be a good model of graphene due to the larger HOMO–LUMO gap in coronene.
We recently investigated the dynamic and static behaviour of the XH-*-π and/or YX-*-π interactions (π-HBs and/or π-XBs, respectively) (X, Y = F, Cl, Br and I) in the π-systems of benzene, π(C6H6),20,21 naphthalene, π(C10H8)22 and anthracene, π(C14H10).23 What is the behaviour of the π-HBs and π-XBs interactions in the coronene π-system, π(C24H12)? What are the differences and similarities in the interactions between π(C24H12) and π(C6H6), π(C10H8) and π(C14H10)? The nature of the interactions should be elucidated to obtain a better understanding of the chemistry arising from the interactions. The π-HB and π-XB interactions with the planer π(C24H12) system will supply an important starting point for the interactions with the bent π-systems, such as fullerenes and carbon nanotubes, and the circulene molecules, together with the non-covalent functionalization based on the interactions.24
Scheme 1 illustrates the structures of X–H⋯π(C24H12) (X = F, Cl, Br and I) and Y–X⋯π(C24H12) (Y–X = F–F, Cl–Cl, Br–Br, I–I, F–Cl, F–Br and F–I) to be elucidated in this work.25 The scope of the properties in the Y–X⋯π interactions have been demonstrated to be covered by those with Y–X = F–F, F–Cl, F–Br and F–I.10 The structural parameters are defined in Scheme 1 together with the types. The structures of the adducts will be called type ICor, if X–H or Y–X appears to interact with the coronene π-system through only a single site of X–H or Y–X. Namely, X–H or Y–X should be placed almost parallel to the normal line of the coronene molecular plane. Type ICor will be called type IACor, if X–H or Y–X interacts with a carbon atom in the central ring of coronene. On the other hand, the structure will be type IBCor, when X–H or Y–X is expected to interact with a carbon atom bearing no hydrogen atom in the outside ring of coronene, whereas it will be type ICCor when X–H or Y–X appears to interact with the midpoint between the adjacent carbon atoms bearing the hydrogen atoms of the outside ring of coronene. Type IDCor in Scheme 2 is discussed later.
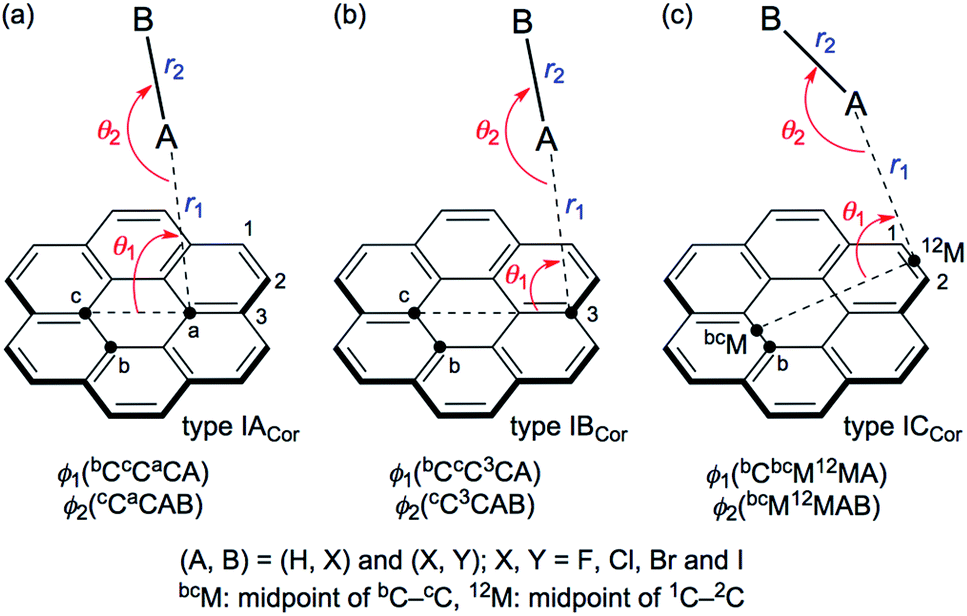 | ||
| Scheme 1 Structures of X–H⋯π(C24H12) and Y–X⋯π(C24H12) to be clarified with the definition of structural parameters and types, where A–B = X–H = F–H, Cl–H, Br–H and I–H and A–B = Y–X = F–F, Cl–Cl, Br–Br, I–I, F–Cl, F–Br and F–I. | ||
The QTAIM (quantum theory of atoms-in-molecules) approach, introduced by Bader,26,27 enables us to analyse the nature of chemical bonds and interactions.26–30 Interactions are defined by the corresponding bond paths (BPs), but we must be careful to use the correct terminology with this concept.31 The bond critical point (BCP) is an important concept in QTAIM and is a point along the BP at the interatomic surface where the charge density, ρ(r), reaches a minimum.32 This point is denoted by ρb(rc), as are the other QTAIM functions at BCPs, such as the Laplacians of ρb(rc) (∇2ρb(rc)), total electron energy densities Hb(rc), potential energy densities Vb(rc), kinetic energy densities Gb(rc) and kb(rc) (= Vb(rc)/Gb(rc)).33
In QTAIM, chemical bonds and interactions are classified by the signs of ∇2ρb(rc) and Hb(rc). Indeed, Hb(rc) − Vb(rc)/2 = 0 (∇2ρb(rc) = 0) corresponds to the borderline between the classical covalent bonds of shard shell (SS) interactions and the noncovalent closed shell (CS) interactions, but Hb(rc) = 0 appears to be buried in the noncovalent interactions of CS. (See eqn (S2) of the ESI† for the relation, (ħ2/8m)∇2ρb(rc) = Hb(rc) − Vb(rc)/2.) Therefore, it seems difficult to characterize the CS interactions, such as van der Waals (vdW) interactions,34,35 typical hydrogen bonds (t-HBs),2,3,36,37 interactions in molecular complexes formed through charge transfer (CT-MCs),38 trihalide ions (X3−)38 and interactions in trigonal bipyramidal adducts formed through CT (CT-TBPs).38 Then, we proposed employing the signs of the first derivatives of Hb(rc) − Vb(rc)/2 and Hb(rc) (d(Hb(rc) − Vb(rc)/2)/dr and dHb(rc)/dr, respectively) to characterize these interactions. The borderline between CT-MC and CT-TBP (containing X3−) is defined by d(Hb(rc) − Vb(rc)/2)/dr = 0, while that between vdW and t-HB is by dHb(rc)/dr = 0, as shown by the experimental results, with the presumption that the CS interactions are reasonably characterized as expected. The proposed definitions for the classification of interactions are summarized in Table S1 of the ESI,† together with those tentatively proposed,39 for convenience of discussion.
Recently, we proposed QTAIM dual functional analysis (QTAIM-DFA),40–43 according to QTAIM.26–29,44,45 QTAIM-DFA provides an excellent approach for evaluating, classifying and understanding weak to strong interactions in a unified form.40–43 In QTAIM-DFA, Hb(rc) are plotted versus Hb(rc) − Vb(rc)/2 [= (ħ2/8m) ∇2ρb(rc)]. In our treatment, data for perturbed structures around fully optimized structures are employed for the plots, in addition to those from the fully optimized structures.40–43 QTAIM-DFA can incorporate the classification of interactions by the signs of ∇2ρb(rc), Hb(rc), d(Hb(rc) − Vb(rc)/2)/dr and dHb(rc)/dr with the definitions, tentatively proposed.46 We have proposed the concept of “the dynamic nature of interactions” which originates from the data containing the perturbed structures.40a,41–43 Data from the fully optimized structures correspond to the static nature of interactions. QTAIM-DFA is applied to typical chemical bonds and interactions and rough criteria are established. The rough criteria can distinguish the chemical bonds and interactions in question from other types of interactions. QTAIM-DFA and these criteria are explained in the ESI using Schemes S1 and S2, Fig. S1 and eqn (S1)–(S6).† The basic concept of the QTAIM approach is also surveyed.
We consider QTAIM-DFA to be well-suited to elucidate the dynamic and static nature of the π-HBs and π-XBs interactions in π(C24H12), even though static behaviour of π-HBs in π(C24H12) has been discussed.47,48 In this study, we present the results of the investigations on the nature of the interactions. The interactions are classified and characterized based on the above criteria.
Methodological details in calculations
The structures were optimized using the Gaussian 09 programme package.49 The basis set system (BSS) from the Sapporo Basis Set Factory50 (BSS-S) was employed for the calculations. In the calculations with BSS-SA, the (7433211/743111/7411/2 + 1s1p) type was employed for I, the (743211/74111/721/2 + 1s1p) type for Br, the (63211/6111/31/2 + 1s1p) type for Cl and the (6211/311/21/2 + 1s1p) type for F with the (6211/311/21/2 + 1s1p) type for C and the (411/21/2 + 1s1p) type for H. BSS-SA was applied for the calculations at the M06-2X (M06-2X/BSS-SA) level of density functional theory (DFT). Optimized structures were confirmed by the frequency analysis. QTAIM functions were similarly calculated using the Gaussian 09 programme package49 with the same method of the optimizations and the data were analysed with the AIM2000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 51 and AIMAll52 programmes. The results obtained at the M06-2X/BSS-SA level of theory will be mainly discussed in the text.
51 and AIMAll52 programmes. The results obtained at the M06-2X/BSS-SA level of theory will be mainly discussed in the text.
For BSS-SB, the (743321/74321/742 + 1s1p) type was employed for I, the (74321/7421/72 + 1s1p) type for Br, the (6321/621/3 + 1s1p) type for Cl and the (621/31/2 + 1s1p) type for F with the (621/31/2 + 1s1p) type for C and the (31/3 + 1s1p) type for H. The calculations were also performed at the M06-2X/BSS-SB level of theory to search for the potential energy surface minima as the pre-optimizations, when necessary. M06-2X/BSS-SB is also employed to confirm the minima and BPs with BCPs around the interactions in question, if they are not obtained satisfactorily with M06-2X/BSS-SA.
The results obtained using M06-2X/BSS-SB are discussed in Tables S1 and S2 of the ESI† and/or the text, if necessary. M06-2X/BSS-SA was also applied to the benzene π-system for convenience of comparison. The calculations were similarly performed using MP2/6-311G(d,p)53,54 for the convenience of comparison. The results are collected in the ESI.†
Normal coordinates of internal vibrations (NIV) obtained by the frequency analysis were employed to generate the perturbed structures.41,42 This method is explained by eqn (1). A k-th perturbed structure (Skw) was generated by the addition of the normal coordinates of the k-th internal vibration (Nk) to the standard orientation of the fully optimized structure (So) in the matrix representation.55 The coefficient fkw in eqn (1) controls the difference in the structures between Skw and So: fkw are determined to satisfy eqn (1) for the interaction in question, where r and ro show the distances under investigation in the perturbed and fully optimized structures, respectively, and ao is the Bohr radius (0.52918 Å).56 Namely, the perturbed structures with NIV correspond to those with r being elongated or shortened by 0.05ao or 0.1ao, relative to ro. Nk of five digits are used to predict Skw. We refer to this method to generate the perturbed structures as NIV.
| Skw = So + fkw × Nk | (1) |
| r = ro + wao (w = (0), ±0.05 and ±0.1; ao = 0.52918 Å) | (2) |
| y = ao + a1x + a2x2 + a3x3 | (3) |
In the QTAIM-DFA treatment, Hb(rc) are plotted versus Hb(rc) − Vb(rc)/2 for five data points of w = 0, ±0.05 and ±0.1 in eqn (2). Each plot is analysed using a regression curve of the cubic function as shown in eqn (3), where (x, y) are (Hb(rc) − Vb(rc)/2, Hb(rc)) (Rc2 (square of correlation coefficient) > 0.99999 in usual).43
Results and discussion
Optimizations of X–H⋯π(C24H12) and Y–X⋯π(C24H12)
The structures were optimized for X–H⋯π(C24H12) and Y–X⋯π(C24H12). The optimizations were initially performed with M06-2X/BSS-SB, assuming the C1 symmetry. The X–H and Y–X components were placed in close proximity to types IACor, IBCor and ICCor together with type IDCor (see Schemes 1 and 2) in the optimization processes, but the systematic search was not performed. Each adduct finally converged to a structure with the C1 symmetry. The structures were optimized again with M06-2X/BSS-SA. The optimized structures are confirmed by all positive frequencies after the frequency analysis. Then, the C1 structures with all positive frequencies were further optimized, assuming the Cs symmetry in the cases where the C1 structures appeared to be very close to the Cs symmetry. The frequency analysis was also performed on the Cs structures. The IBCor and ICCor types were predicted for X–H⋯π(C24H12), while the IACor and ICCor types were used for Y–X⋯π(C24H12), when optimized with M06-2X/BSS-SA.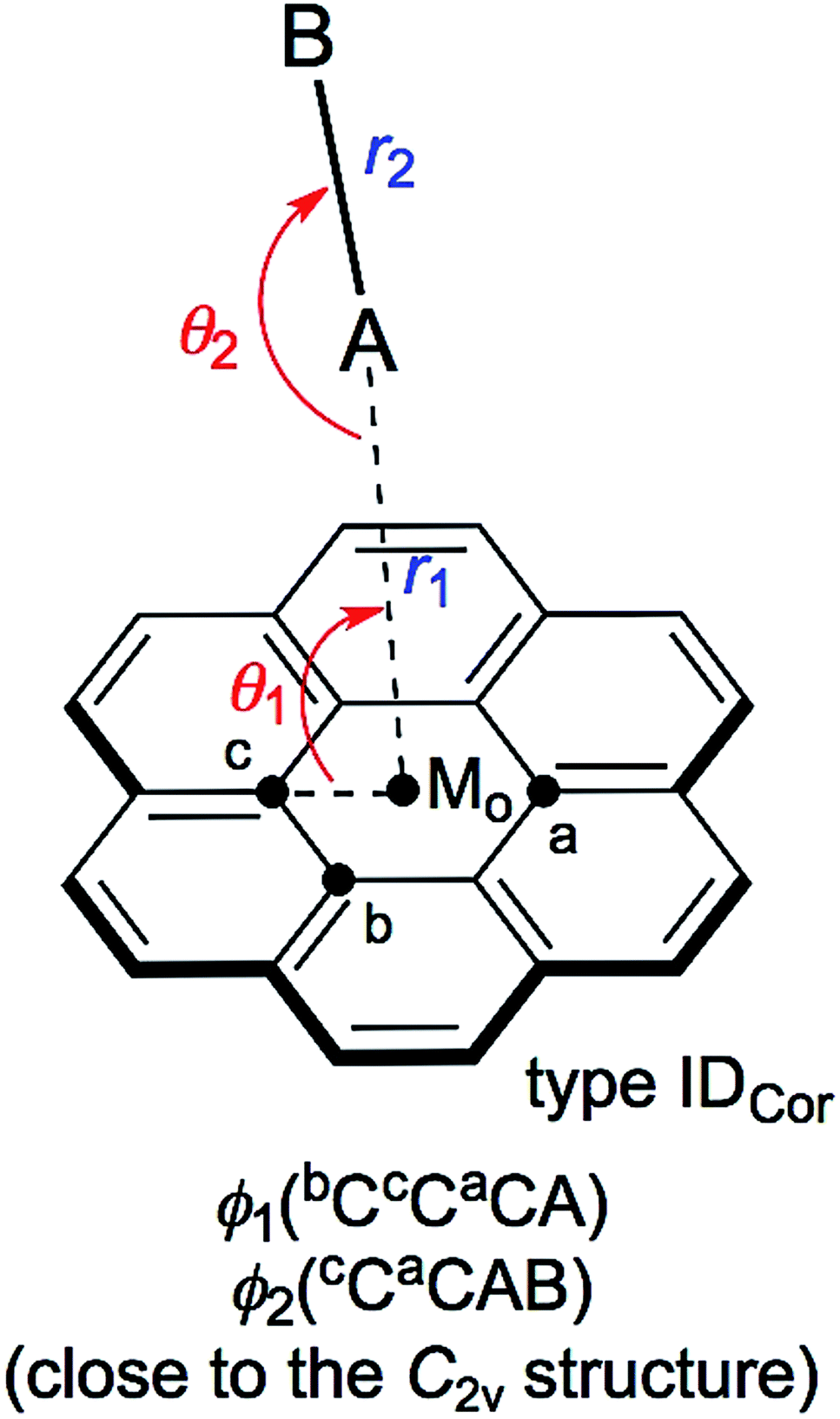 | ||
| Scheme 2 Structures of X–H⋯π(C24H12) and Y–X⋯π(C24H12) (A–B = X–H or Y–X: X, Y = F, Cl, Br and I). The structural parameters are defined, together with the types, where Mo is the centre point of C24H12. | ||
All positive frequencies were confirmed for all adducts, except for F–H⋯π(3C) (C1: IBCor), Cl–H⋯π(12M) (C1: ICCor), Cl–Cl⋯π(aC) (C1: IACor), Cl–Cl⋯π(12M) (C1: ICCor) and F–Cl⋯π(12M) (C1: ICCor). The motion of each imaginary frequency mainly corresponds to the angular displacements between π(C24H12) and X–H or Y–X. In the case of Cl–H⋯π(2C) (C1: IBCor), the calculation converged to Cl–H⋯π(12M) (C1: ICCor), which did not give positive frequencies only after the frequency analysis. Table 1 summarizes the structural parameters (r1, r2, θ1, θ2, ϕ1 and ϕ2) of X–H⋯π(C24H12) and Y–X⋯π(C24H12), defined in Scheme 1. The optimized structures are not shown in figures, but a number of them can be observed in Fig. 1 and 2. The magnitudes of the θ1, θ2, ϕ1 and ϕ2 values are close to 90°, 180°, 90° and 180° (or 0°), respectively, for the most cases.
| Y–X-*-π(C24H12), (symmetry: type) | r1, (Å) | r2, (Å) | θ1, (°) | θ2, (°) | ϕ1, (°) | ϕ2, (°) | ΔEESc,d, (kJ mol−1) | ΔEEntc,e, (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|
a See text for BSS-SA.b See Scheme 1 for the definition of the structural parameters.c ΔE = E(X–H⋯π(C24H12)/Y–X⋯π(C24H12)) − (E(X–H/Y–X) + E(C24H12)).d ΔEES represents ΔE on the energy surface.e ΔEEnt represents ΔE with the correction of the heat of enthalpy.f One imaginary frequency being predicted for each, of which motion mainly corresponds to the angular displacements between π(C24H12) and X–H or Y–X.g Br–H being placed above the midpoint between 2C and 3C, which is defined by type  . In this case, the r1 value is measured from 2C.h Close to the Cs symmetry, where Cl in F–Cl pointing to 12M, the midpoint between 1C and 2C. . In this case, the r1 value is measured from 2C.h Close to the Cs symmetry, where Cl in F–Cl pointing to 12M, the midpoint between 1C and 2C. |
||||||||
| F–H⋯π(3C) (C1: IBCor)f | 2.2609 | 0.9243 | 80.09 | 169.69 | −89.91 | 180.00 | −17.5 | −15.5 |
Br–H⋯π(2C) (C1:  )g )g |
2.6197 | 1.4243 | 78.28 | 166.98 | −114.35 | 52.29 | −16.8 | −10.0 |
| I–H⋯π(3C) (C1: IBCor) | 2.6427 | 1.6219 | 77.03 | 162.89 | −108.52 | −12.06 | −16.9 | −11.2 |
| F–H⋯π(12M) (Cs: ICCor) | 2.1815 | 0.9261 | 83.01 | 177.80 | −90.00 | 180.00 | −19.4 | −15.1 |
| Cl–H⋯π(12M) (C1: ICCor)f | 2.4502 | 1.2840 | 68.93 | 179.73 | −90.00 | 179.97 | −16.1 | −14.9 |
| Br–H⋯π(12M) (Cs: ICCor) | 2.5236 | 1.4244 | 68.68 | 167.96 | −90.00 | 0.00 | −17.2 | −10.3 |
| I–H⋯π(12M) (Cs: ICCor) | 2.5847 | 1.6222 | 68.98 | 164.38 | −90.00 | 0.00 | −17.3 | −12.2 |
| F–F⋯π(aC) (Cs: IACor) | 2.7873 | 1.3685 | 90.62 | 177.95 | −89.96 | 0.00 | −7.5 | −3.1 |
| Cl–Cl⋯π(aC) (C1: IACor)f | 3.0381 | 1.9950 | 90.95 | 178.16 | −89.95 | 0.03 | −16.0 | −13.5 |
| Br–Br⋯π(aC) (Cs: IACor) | 3.1293 | 2.2912 | 90.95 | 177.03 | −89.95 | 0.00 | −20.1 | −14.1 |
| I–I⋯π(aC) (Cs: IACor) | 3.3116 | 2.6768 | 89.61 | 178.88 | −89.92 | 180.00 | −23.5 | −20.2 |
| F–Cl⋯π(aC) (Cs: IACor) | 2.9409 | 1.6257 | 90.82 | 177.56 | −90.07 | 0.00 | −20.4 | −16.5 |
| F–Br⋯π(aC) (Cs: IACor) | 3.0096 | 1.7632 | 92.21 | 174.94 | −90.11 | 0.00 | −27.4 | −22.8 |
| F–I⋯π(aC) (Cs: IACor) | 3.1554 | 1.9216 | 93.14 | 178.55 | −90.00 | 0.00 | −35.6 | −32.0 |
| F–F⋯π(12M) (Cs: ICCor) | 2.9010 | 1.3677 | 72.64 | 162.12 | −90.00 | 0.00 | −7.0 | −3.5 |
| Cl–Cl⋯π(12M) (C1: ICCor)f | 3.0107 | 1.9990 | 87.28 | 178.12 | −90.00 | 0.00 | −14.3 | −13.2 |
| Br–Br⋯π(12M) (C1: ICCor) | 3.0801 | 2.2975 | 90.46 | 176.98 | −88.52 | 65.27 | −17.8 | −13.3 |
| I–I⋯π(12M) (Cs: ICCor) | 3.3370 | 2.6788 | 86.08 | 179.26 | −90.00 | 180.00 | −20.6 | −16.5 |
| F–Cl⋯π(12M) (C1: ICCor)f,h | 2.8523 | 1.6323 | 91.82 | 178.00 | −90.02 | −0.09 | −21.7 | −19.3 |
| F–Br⋯π(12M) (Cs: ICCor) | 2.8616 | 1.7699 | 98.68 | 178.24 | −90.00 | 180.00 | −30.0 | −25.3 |
| F–I⋯π(12M) (Cs: ICCor) | 3.0277 | 1.9269 | 94.21 | 179.61 | −90.00 | 0.00 | −36.5 | −32.8 |
 | ||
Fig. 1 Molecular graphs for F–H-*-π(C24H12) (C1: IBCor) (a), Br–H-*-π(C24H12) (C1:  ) (b), I–H-*-π(C24H12) (C1: IBCor) (c), F–H-*-π(C24H12) (Cs: ICCor) (d), Cl–H-*-π(C24H12) (C1: ICCor) (e), Br–H-*-π(C24H12) (Cs: ICCor) (f) and I–H-*-π(C24H12) (Cs: ICCor) (g), calculated with M06-2X/BSS-SA. BCPs are denoted by red dots, RCPs by yellow dots, CCPs by green dots and BPs by pink lines. Carbon atoms are in black and hydrogen atoms are in grey, with fluorine, chlorine, bromine and iodine atoms in dark yellow, green, dark purple and purple, respectively. The contour plot of ρ(r) is also drawn for each on the plane containing the H-*-3C(C24H12) moiety for type IBCor with the H-*-2C(C24H12) moiety for type ) (b), I–H-*-π(C24H12) (C1: IBCor) (c), F–H-*-π(C24H12) (Cs: ICCor) (d), Cl–H-*-π(C24H12) (C1: ICCor) (e), Br–H-*-π(C24H12) (Cs: ICCor) (f) and I–H-*-π(C24H12) (Cs: ICCor) (g), calculated with M06-2X/BSS-SA. BCPs are denoted by red dots, RCPs by yellow dots, CCPs by green dots and BPs by pink lines. Carbon atoms are in black and hydrogen atoms are in grey, with fluorine, chlorine, bromine and iodine atoms in dark yellow, green, dark purple and purple, respectively. The contour plot of ρ(r) is also drawn for each on the plane containing the H-*-3C(C24H12) moiety for type IBCor with the H-*-2C(C24H12) moiety for type  or on the plane of the H-*-12M(C24H12) moiety for type ICCor, where the contour plot is drawn on each plane. or on the plane of the H-*-12M(C24H12) moiety for type ICCor, where the contour plot is drawn on each plane. | ||
 | ||
| Fig. 2 Molecular graphs for F–F-*-π(C24H12) (Cs: IACor) (a), F–Cl-*-π(C24H12) (Cs: IACor) (b), F–Br-*-π(C24H12) (Cs: IACor) (c), F–I-*-π(C24H12) (Cs: IACor) (d), F–F-*-π(C24H12) (Cs: ICCor) (e), F–Cl-*-π(C24H12) (C1: ICCor) (f), F–Br-*-π(C24H12) (Cs: ICCor) (g) and F–I-*-π(C24H12) (Cs: ICCor) (h), calculated with M06-2X/BSS-SA. BCPs are denoted by red dots, RCPs by yellow dots and BPs by pink lines. Carbon atoms are in black and hydrogen atoms are in grey, with fluorine, chlorine, bromine and iodine atoms in dark yellow, green, dark purple and purple, respectively. The contour plot of ρ(r) is also drawn for each on the plane containing the X-*-3C(C24H12) moiety for type IBCor or on the plane of X-*-12M(C24H12) moiety for type ICCor, where the contour plot is drawn on each plane. | ||
However, significant deviations are observed in some cases. The (ϕ1, ϕ2) values of (−114.4°, 52.3°) for Br–H⋯π(2C) (C1:  ) are the typical example, taken from the intermediate structure between Br–H⋯π(2C) (C1: IBCor) and typical Br–H⋯π(12M) (Cs: ICCor). The lack of convergence of Cl–H⋯π(C24H12) to the IBCor type with all positive frequencies is related to the formation of Br–H⋯π(2C) (C1:
) are the typical example, taken from the intermediate structure between Br–H⋯π(2C) (C1: IBCor) and typical Br–H⋯π(12M) (Cs: ICCor). The lack of convergence of Cl–H⋯π(C24H12) to the IBCor type with all positive frequencies is related to the formation of Br–H⋯π(2C) (C1:  ).
).
This is a very gentle potential energy surface around the inter-conversion between Cl–H⋯π(C24H12) (ICCor) and the related structure. Similarly, for the cases discussed above, all positive frequencies only were not predicted for F–H⋯π(3C) (C1: IBCor) and Cl–H⋯π(12M) (C1: ICCor) in X–H⋯π(C24H12). This is also owing to the very gentle potential energy surface around the motions of the imaginary frequencies for Cl–Cl⋯π(aC) (C1: IACor), Cl–Cl⋯π(12M) (C1: ICCor) and F–Cl⋯π(12M) (C1: ICCor) in Y–X⋯π(C24H12). Nevertheless, with the exception of Cl–H⋯π(12M) (C1: ICCor), positive frequencies only are predicted for these cases when the calculations are performed with M06-2X/BSS-SB. The results are collected in Table S1 of the ESI.†
The energy differences between X–H⋯π(C24H12) and Y–X⋯π(C24H12) and the components, ΔE (= E(X–H⋯π(C24H12)/Y–X⋯π(C24H12)) − (E(X–H/Y–X) + E(C24H12))) (ΔEES and ΔEEnt), are also given in Table 1. ΔEES and ΔEEnt represent ΔE on the energy surface and ΔE with the collections by the enthalpy for the formation of the adducts at 25 °C, respectively. The plot of ΔEEnt versus ΔEES gave a (very) good correlation (y = 0.992x + 3.85: Rc2 = 0.955 (n (number of data points) = 21)) even though the data for Cl–H⋯π(12M) (C1: ICCor), Br–H⋯π(12M) (Cs: ICCor), Br–H⋯π(2C) (C1:  ), Br–Br⋯π(aC) (Cs: IACor) and Cl–Cl⋯π(12M) (C1: ICCor) appear to deviate somewhat from the correlation (Fig. S2 of the ESI†). A much better correlation was obtained if the data for the five species are omitted from the correlation (y = 1.000x + 3.90: Rc2 = 0.986 (n = 16)). Therefore, ΔEES can be used for the discussion of ΔE.
), Br–Br⋯π(aC) (Cs: IACor) and Cl–Cl⋯π(12M) (C1: ICCor) appear to deviate somewhat from the correlation (Fig. S2 of the ESI†). A much better correlation was obtained if the data for the five species are omitted from the correlation (y = 1.000x + 3.90: Rc2 = 0.986 (n = 16)). Therefore, ΔEES can be used for the discussion of ΔE.
After the elucidation of the structural feature of X–H⋯π(C24H12) and Y–X⋯π(C24H12), molecular graphs, contour plots, negative Laplacians and trajectory plots are examined next.
Molecular graphs, contour plots, negative Laplacians and trajectory plots for X–H-*-π(C24H12) and Y–X-*-π(C24H12)
Fig. 1 illustrates the molecular graphs for F–H-*-π(C24H12) (C1: IBCor), Br–H-*-π(C24H12) (C1: ), I–H-*-π(C24H12) (C1: IBCor), F–H-*-π(C24H12) (Cs: ICCor), Cl–H-*-π(C24H12) (C1: ICCor), Br–H-*-π(C24H12) (Cs: ICCor) and I–H-*-π(C24H12) (Cs: ICCor), calculated with M06-2X/BSS-SA. Each molecular graph contains the contour plot of ρ(r) drawn on the plane containing the H-*-3C moiety for F–H-*-π(C24H12) (IBCor) and I–H-*-π(C24H12) (IBCor) with the H-*-2C moiety for Br–H-*-π(C24H12) (
), I–H-*-π(C24H12) (C1: IBCor), F–H-*-π(C24H12) (Cs: ICCor), Cl–H-*-π(C24H12) (C1: ICCor), Br–H-*-π(C24H12) (Cs: ICCor) and I–H-*-π(C24H12) (Cs: ICCor), calculated with M06-2X/BSS-SA. Each molecular graph contains the contour plot of ρ(r) drawn on the plane containing the H-*-3C moiety for F–H-*-π(C24H12) (IBCor) and I–H-*-π(C24H12) (IBCor) with the H-*-2C moiety for Br–H-*-π(C24H12) ( ) or on the plane of H-*-12M moiety for X–H-*-π(C24H12) (ICCor), albeit partially. Fig. 2 shows the molecular graphs for the IACor and ICCor types of F–X-*-π(C24H12) (X = F, Cl, Br and I), calculated with M06-2X/BSS-SA. The contour plot of ρ(r) is drawn for each adduct partially, similar to Fig. 1.
) or on the plane of H-*-12M moiety for X–H-*-π(C24H12) (ICCor), albeit partially. Fig. 2 shows the molecular graphs for the IACor and ICCor types of F–X-*-π(C24H12) (X = F, Cl, Br and I), calculated with M06-2X/BSS-SA. The contour plot of ρ(r) is drawn for each adduct partially, similar to Fig. 1.
In Fig. 1, all expected BCPs are clearly observed, including those for the XH-*-π and YX-*-π interactions in question, together with ring critical points (RCPs) and cage critical points (CCPs), if such exist. The structural feature is visualized well by the molecular graphs. The BPs for H-*-π and X-*-π in question seem linear for most of X–H-*-π(C24H12) and Y–X-*-π(C24H12), although some seem somewhat bending. BCPs are well located at the (three-dimensional) saddle points of ρ(r). Negative Laplacians and trajectory plots are drawn for X–H-*-π(C24H12), similar to Fig. 1 and are shown in Fig. S3 and S4 of the ESI,† respectively. Negative Laplacians and trajectory plots are also drawn for Y–X-*-π(C24H12), similar to Fig. 2, and are shown in Fig. S5 and S6 of the ESI,† respectively. The behaviour of the BCPs is well-visualized through ∇2ρ(r) as shown in Fig. S3 and S5 of the ESI.† All BCPs in X–H-*-π(C24H12) and Y–X-*-π(C24H12) are placed in the blue areas of the negative Laplacians; therefore, the interactions corresponding to the BCPs should be classified by the CS interactions. The space around the species around the interactions in question is well divided into atoms, as demonstrated in Fig. S4 and S6 of the ESI.†
Survey of X–H-*-π(C24H12) and Y–X-*-π(C24H12) interactions, evaluated with M06-2X/BSS-SA
How can the X–H-*-π(C24H12) and Y–X-*-π(C24H12) interactions be described? The interactions can be defined by the corresponding BPs, although we must be careful to use the correct terminology with this concept.31 As shown in Fig. 1 and 2, BPs for the adducts appear to be straight, with the exception of X–H-*-π(C24H12) (C1: IBCor) (X = Br and I) and Y–X-*-π(12M) (C1: ICCor) (Y–X = F–Cl and Br–Br). The lengths of BPs (rBP) and the straight-line distances (RSL) evaluated with M06-2X/BSS-SA, are collected in Table S3 of the ESI† together with the ΔrBP (= rBP − RSL) values. The ΔrBP value are 0.68 Å for F–Cl-*-π(12M) (C1: ICCor), 0.41 Å for Br–Br-*-π(12M) (C1: ICCor), 0.35 Å for Br–H-*-π(2C) (C1: IBCor) and 0.17 Å for I–H-*-π(3C) (C1: IBCor). However, the ΔrBP values are smaller than 0.064 Å for X–H-*-π(C24H12) and smaller than 0.015 Å for Y–X-*-π(C24H12) (Cs: IACor) (X, Y = F, Cl, Br and I), as shown in Table S3.† Therefore, the H-*-π and X-*-π interactions in the coronene π-system can be approximated as straight lines, except for the four species, although ΔrBP = 0.064 Å for F–H-*-π(3C) (C1: IBCor). The plot of rBP versus RSL for the adducts gave an excellent correlation (y = 0.966x + 0.1079; Rc2 = 0.999 (n = 16)), if the data of the four species are neglected from the correlation (not shown in the figure).QTAIM functions are evaluated for the H-*-π and X-*- π interactions at BCPs in X–H-*-π(C24H12) and Y–X-*-π(C24H12) (X, Y = F, Cl, Br and I) using the M06-2X functional. The obtained values are presented in Table 2. Fig. 3 shows the plot of Hb(rc) versus Hb(rc) − Vb(rc)/2 for the data in Table 2 and those from the perturbed structures around the fully optimized structures. All data in Fig. 3 appear in the region of Hb(rc) − Vb(rc)/2 > 0 and Hb(rc) > 0, and therefore, all interactions in question are classified by the pure CS interactions.
| Y–X-*-π(C24H12), (symmetry: type) | ρb(rc), (eao−3) | c∇2ρb(rc)c, (au) | Hb(rc), (au) | kb(rc)d | R, (au) | θ, (°) | Freq, (cm−1) | kf, (mDyne Å−1) | θp, (°) | κp, (au−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| a See text for BSS-SA and BSS-SB.b Data are given at BCP, which is shown by X-*-π.c c∇2ρb(rc) = Hb(rc) − Vb(rc)/2, where c = ħ2/8m.d kb(rc) = Vb(rc)/Gb(rc).e One imaginary frequency being predicted for each.f BCP (and BP) being not detected.g Perturbed structures for Cl–H-*-π(2C) (C1: IACor) are generated employing w = −0.1, −0.05, (0), 0.01 and 0.015 in eqn (2); therefore, some intervals in the plot are shorter than others. | ||||||||||
| M06-2X/BSS-SA | ||||||||||
| F–H-*-π(3C) (C1: IBCo)e | 0.0146 | 0.0053 | 0.0005 | −0.948 | 0.0053 | 84.4 | 84.4 | 0.024 | 113.0 | 172.1 |
Br–H-*-π(2C) (C1:  ) ) |
0.0090 | 0.0037 | 0.0012 | −0.801 | 0.0039 | 71.6 | 60.4 | 0.025 | 82.4 | 162.5 |
| I–H-*-π(3C) (C1: IBCor) | 0.0089 | 0.0035 | 0.0012 | −0.785 | 0.0037 | 70.5 | 49.1 | 0.017 | 83.7 | 156.9 |
| F–H-*-π(12M) (Cs: ICCor) | 0.0172 | 0.0057 | 0.0001 | −0.992 | 0.0057 | 89.1 | 134.4 | 0.059 | 129.1 | 242.5 |
| Cl–H-*-π(12M) (C1: ICCor)e,f | Non | Non | Non | Non | Non | Non | Non | Non | Non | Non |
| Br–H-*-π(12M) (Cs: ICCor) | 0.0099 | 0.0038 | 0.0012 | −0.814 | 0.0039 | 72.6 | 60.4 | 0.027 | 86.4 | 181.1 |
| I–H-*-π(12M) (Cs: ICCor) | 0.0093 | 0.0035 | 0.0012 | −0.798 | 0.0037 | 71.4 | 53.5 | 0.020 | 83.7 | 171.6 |
| F–F-*-π(aC) (Cs: IACor) | 0.0097 | 0.0056 | 0.0023 | −0.735 | 0.0061 | 67.3 | 72.5 | 0.035 | 71.7 | 32.9 |
| Cl–Cl-*-π(aC) (C1: IACor)e | 0.0113 | 0.0050 | 0.0015 | −0.828 | 0.0052 | 73.6 | 74.5 | 0.036 | 87.2 | 96.5 |
| Br–Br-*-π(aC) (Cs: IACor) | 0.0115 | 0.0047 | 0.0012 | −0.847 | 0.0048 | 75.1 | 59.2 | 0.024 | 87.9 | 121.0 |
| I–I-*-π(aC) (Cs: IACor) | 0.0109 | 0.0040 | 0.0011 | −0.846 | 0.0042 | 75.1 | 44.9 | 0.013 | 89.4 | 144.6 |
| F–Cl-*-π(aC) (Cs: IACor) | 0.0131 | 0.0058 | 0.0014 | −0.858 | 0.0060 | 76.0 | 79.5 | 0.037 | 92.0 | 108.4 |
| F–Br-*-π(aC) (Cs: IACor) | 0.0137 | 0.0056 | 0.0012 | −0.880 | 0.0057 | 77.9 | 67.4 | 0.033 | 94.5 | 135.3 |
| F–I-*-π(aC) (Cs: IACor) | 0.0134 | 0.0049 | 0.0009 | −0.904 | 0.0050 | 80.1 | 66.6 | 0.027 | 104.1 | 227.6 |
| F–F-*-π(12M) (Cs: ICCor) | 0.0081 | 0.0046 | 0.0020 | −0.716 | 0.0050 | 66.2 | 61.2 | 0.026 | 68.1 | 24.5 |
| Cl–Cl-*-π(12M) (C1: ICCor)e | 0.0124 | 0.0053 | 0.0017 | −0.814 | 0.0055 | 72.6 | 170.6 | 0.070 | 86.4 | 98.1 |
| Br–Br-*-π(12M) (C1: ICCor) | 0.0132 | 0.0051 | 0.0014 | −0.843 | 0.0053 | 74.8 | 54.1 | 0.032 | 89.3 | 178.7 |
| I–I-*-π(12M) (Cs: ICCor) | 0.0108 | 0.0039 | 0.0012 | −0.821 | 0.0040 | 73.1 | 48.1 | 0.012 | 87.1 | 107.5 |
| F–Cl-*-π(12M) (C1: ICCor)e | 0.0163 | 0.0067 | 0.0016 | −0.867 | 0.0069 | 76.8 | 73.8 | 0.023 | 95.8 | 92.0 |
| F–Br-*-π(12M) (Cs: ICCor) | 0.0189 | 0.0070 | 0.0011 | −0.918 | 0.0071 | 81.4 | 108.7 | 0.042 | 109.6 | 220.8 |
| F–I-*-π(12M) (Cs: ICCor) | 0.0177 | 0.0059 | 0.0005 | −0.951 | 0.0059 | 84.7 | 106.8 | 0.039 | 124.8 | 375.2 |
|
||||||||||
| M06-2X/BSS-SB | ||||||||||
| F–H-*-π(3C) (Cs: IBCor) | 0.0142 | 0.0053 | 0.0014 | −0.852 | 0.0055 | 75.6 | 115.5 | 0.051 | 78.7 | 68.4 |
| Cl–H-*-π(2C) (C1: IACo)g | 0.0118 | 0.0041 | 0.0009 | −0.870 | 0.0042 | 77.0 | 310.7 | 0.231 | 75.7 | 535 |
| Br–H-*-π(2C) (C1: IBCor) | 0.0092 | 0.0033 | 0.0008 | −0.871 | 0.0034 | 77.1 | 59.2 | 0.025 | 82.0 | 12.2 |
| I–H-*-π(3C) (C1: IBCor) | 0.0093 | 0.0033 | 0.0009 | −0.847 | 0.0034 | 75.2 | 49.5 | 0.019 | 84.7 | 81.0 |
| Cl–H-*-π(12M) (C1: ICCor) | 0.0108 | 0.0036 | 0.0007 | −0.895 | 0.0037 | 79.2 | 24.6 | 0.003 | 81.9 | 149.9 |
 | ||
| Fig. 3 QTAIM-DFA plots of Hb(rc) versus Hb(rc) − Vb(rc)/2 for X–H-*-π(C24H12) and Y–X-*-π(C24H12) (X, Y = F, Cl, Br and I). Marks and colours are shown in the figure, where circle and square marks correspond to the data evaluated with M06-2X/BSS-SA. | ||
Nature of X–H-*-π(C24H12) and Y–X-*-π(C24H12) interactions, evaluated with M06-2X/BSS-SA
The plots of Hb(rc) versus Hb(rc) − Vb(rc)/2 in Fig. 3 are analysed according to eqn (S3)–(S6) of the ESI,† which provide the QTAIM-DFA parameters of (R, θ) and (θp, κp). Table 2 collects the frequencies, correlated to NIV employed to generate the perturbed structures and the force constants, kf. The nature of the interactions in question is classified and characterized based on the QTAIM-DFA parameters, employing the standard values (criteria) as the reference. Table 3 summarizes the predicted nature of H-*-π in X–H-*-π(C24H12) and X-*-π in Y–X-*-π(C24H12), employing the θ and θp values evaluated with M06-2X/BSS-SA.| Y–X-*-π(C24H12), (symmetry: type) | θ, (°) | θp, (°) | Predicted nature | Y–X-*-π(C24H12), (symmetry: type) | θ, (°) | θp, (°) | Predicted nature |
|---|---|---|---|---|---|---|---|
| a See text for BSS-SA.b One imaginary frequency being predicted for each.c Classified by the pure closed shell (CS) interactions and characterized as the typical hydrogen bonds (t-HB) with no covalency.d Very close to the regular CS (r-CS) interactions and characterized as t-HB with covalency (t-HBwc).e Predicted to be the vdW interactions appeared in the p-CS region. | |||||||
| X–H-*-π(C24H12) | |||||||
| F–H-*-π(3C) (C1: IBCor)b | 84.4 | 113.0 | p-CS/t-HBncc | F–H-*-π(12M) (Cs: ICCor) | 89.1 | 129.1 | p-CS/t-HBncc,d |
| Br–H-*-π(2C) (C1: IBCor) | 71.6 | 82.4 | p-CS/vdWe | Br–H-*-π(12M) (Cs: ICCor) | 72.6 | 86.4 | p-CS/vdWe |
| I–H-*-π(3C) (C1: IBCor) | 70.5 | 83.7 | p-CS/vdWe | I–H-*-π(12M) (Cs: ICCor) | 71.4 | 83.7 | p-CS/vdWe |
|
|||||||
| Y–X-*-π(C24H12) | |||||||
| F–F-*-π(aC) (Cs: IACor) | 67.3 | 71.7 | p-CS/vdWe | F–F-*-π(12M) (Cs: ICCor) | 66.2 | 68.1 | p-CS/vdWe |
| Cl–Cl-*-π(aC) (C1: IACor)b | 73.6 | 87.2 | p-CS/vdWe | Cl–Cl-*-π(12M) (C1: ICCor)b | 72.6 | 86.4 | p-CS/vdWe |
| Br–Br-*-π(aC) (Cs: IACor) | 75.1 | 87.9 | p-CS/vdWe | Br–Br-*-π(12M) (C1: ICCor) | 74.8 | 89.3 | p-CS/vdWe |
| I–I-*-π(aC) (Cs: IACor) | 75.1 | 89.4 | p-CS/vdWe | I–I-*-π(12M) (Cs: ICCor) | 73.1 | 87.1 | p-CS/vdWe |
| F–Cl-*-π(aC) (Cs: IACor) | 76.0 | 92.0 | p-CS/t-HBncc | F–Cl-*-π(12M) (C1: ICCor)b | 76.8 | 95.8 | p-CS/t-HBncc |
| F–Br-*-π(aC) (Cs: IACor) | 77.9 | 94.5 | p-CS/t-HBncc | F–Br-*-π(12M) (Cs: ICCor) | 81.4 | 109.6 | p-CS/t-HBncc |
| F–I-*-π(aC) (Cs: IACor) | 80.1 | 104.1 | p-CS/t-HBncc | F–I-*-π(12M) (Cs: ICCor) | 84.7 | 124.8 | p-CS/t-HBncc |
As summarized in Table 3, the θ and θp values in X–H-*-π(C24H12) decrease in the order of X–H- = F–H- > Br–H- > I–H-, even though θp for X–H- = I–H- appears to be somewhat larger than that for the case of X–H- = Br–H-. The results show that θ and θp in X–H-*-π(C24H12) are controlled by the electronegativity of X. Namely, the values will be larger if the polarity of the Xδ−–Hδ+ type becomes larger. Conversely, θ and θp in Y–X-*-π(C24H12) become larger in the order of Y–X- = F–F- < Cl–Cl- < Br–Br- < I–I- < F–Cl- < F–Br- < F–I-. These results would be the reflection of two factors. The first is the softness of X. The θ and θp values become larger with increasing softness of X. The second factor is the polarity of Yδ−–Xδ+. The θ and θp values increase with increasing polarity, resulting in the larger extension of σ*(X–Y) at the X side. This is very interesting because the θ and θp values are larger for Y–X- = F–Cl-, relative to the case of Y–X- = I–I-. The predicted nature is discussed next.
It would be instructive to review the criteria before the detailed discussion of the nature for H-*-π and X-*-π. The criteria specify that θ < 180° (Hb(rc) − Vb(rc)/2 > 0) for the CS interactions and θ > 180° (Hb(rc) − Vb(rc)/2 < 0) for the SS interactions. The CS interactions for θ < 180° are sub-divided into the pure CS interactions for 45° < θ < 90° (Hb(rc) > 0) and the regular CS interactions for 90° < θ < 180° (Hb(rc) < 0). The θp value plays an important role in characterizing the interactions. In the pure CS region of 45° < θ < 90°, the character of interactions will be the vdW type for 45° < θp < 90° and the typical-HB type (t-HB) with no covalency (t-HBnc) for 90° < θp < 125°, where θp = 125° is tentatively given, corresponding to θ = 90°. The regular CS (90° < θ < 180°) and SS (180° < θ) interactions are not discussed here, since the interactions in this region are not detected in this work.
The θ values are less than 90° for all X–H-*-π(C24H12) and Y–X-*-π(C24H12) interactions examined in this work. Therefore, the H-*-π and X-*-π interactions are all classified by the pure CS interactions. On the other hand, the θp values are less than 90° for all interactions with the exception of F–H-*-π(C24H12) of the IBCor and ICCor types and F–X-*-π(C24H12) (X = Cl, Br and I) of the IACor and ICCor types. The interactions in X–H-*-π(C24H12) and Y–X-*-π(C24H12) are all characterized as the vdW nature for those with θp < 90°. The interactions with θp > 90° are characterized to have the nature of typical hydrogen bonds with no covalency (t-HBnc). However, the nature of the H-*-π interactions in F–H-*-π(12M) (Cs: ICCor) should be examined carefully. The θp value is 129.1°, which is larger than 125°. The results suggest that the H-*-π interaction should be characterized as t-HB with covalency (t-HBwc). However, the θ value of 89.1° is less than 90°, therefore, the interaction must have no covalency. In this case, the θ value should have the priority to the θp value in the prediction of the nature of the interaction, since θp is only given tentatively corresponding to θ = 90°. Therefore, the H-*-π interaction in F–H-*-π(12M) (Cs: ICCor) would be better characterized as t-HBnc. However, the interaction appears to be close to the borderline area between t-HBnc and t-HBwc, since θ = 89.1° is close to 90°, while θp = 129.1° > 125°.
The X–H-*-π(C24H12) and Y–X-*-π(C24H12) interactions (X, Y = F, Cl, Br and I) were also analysed for the IDCor type with M06-2X/BSS-SA (see Scheme 2). The results of this analysis are discussed next.
Nature of X–H-*-π(C24H12) and Y–X-*-π(C24H12) interactions of the IDCor type, evaluated with M06-2X/BSS-SA
Indeed, the IDCor type is not optimized for X–H-*-π(C24H12) with M06-2X/BSS-SA, even though they are optimized when calculated at the MP2 level. The nature of the Y–X-*-π(C24H12) interactions around the main axis of π(C24H12) is also very interesting. Therefore, X–H-*-π(C24H12) and Y–X-*-π(C24H12) are optimized assuming the C2v symmetry. The structural parameters are presented in Table S4 of the ESI,† and are defined in Scheme 2. Table S5 of the ESI† presents the QTAIM-DFA parameters of (R, θ) and (θp, κp) evaluated with M06-2X/BSS-SA, together with the frequencies correlated to NIV employed to generate the perturbed structures and the force constants kf.The nature of the interactions in question is classified and characterized based on the QTAIM-DFA parameters, employing the standard values as the reference. Table 4 summarizes the predicted nature of the H-*-π and X-*-π interactions in X–H-*-π(C24H12) and Y–X-*-π(C24H12) of the C2v symmetry, respectively, employing the θ and θp values evaluated with M06-2X/BSS-SA. As summarized in Table 4, the θ and θp values in X–H-*-π(C24H12) (C2v: IDCor) decrease in the order of X–H- = F–H- > Cl–H- > I–H- > Br–H-. On the other hand, the θp values in Y–X-*-π(C24H12) increase in the order of F–F- < Cl–Cl- < I–I- and F–Cl- < Br–Br- < F–Br- < F–I-. The θ and θp are smaller than 90° for all interactions in X–H-*-π(C24H12) (C2v: IDCor) and Y–X-*-π(C24H12) (C2v: IDCor) (see Table 4). Therefore, the H-*-π and X-*-π interactions are all classified by the pure CS interactions and are characterized to be of the vdW nature (p-CS/vdW).
| Y–X-*-π(C24H12), (symmetry: type) | θ, (°) | θp, (°) | Predicted nature | Y–X-*-π(C24H12), (symmetry: type) | θ, (°) | θp, (°) | Predicted nature |
|---|---|---|---|---|---|---|---|
| a See text for BSS-SA.b Classified by the p-CS interactions and characterized as the vdW interactions. | |||||||
| F–H-*-π(Mo) (C2v: IDCor) | 70.4 | 79.5 | p-CS/vdWb | Cl–H-*-π(Mo) (C2v: IDCor) | 69.7 | 75.5 | p-CS/vdWb |
| Br–H-*-π(Mo) (C2v: IDCor) | 69.5 | 76.1 | p-CS/vdWb | I–H-*-π(Mo) (C2v: IDCor) | 69.6 | 76.5 | p-CS/vdWb |
| F–F-*-π(Mo) (C2v: IDCor) | 65.9 | 66.6 | p-CS/vdWb | ||||
| Cl–Cl-*-π(Mo) (C2v: IDCor) | 66.7 | 74.4 | p-CS/vdWb | F–Cl-*-π(Mo) (C2v: IDCor) | 67.4 | 75.1 | p-CS/vdWb |
| Br–Br-*-π(Mo) (C2v: IDCor) | 69.0 | 76.0 | p-CS/vdWb | F–Br-*-π(Mo) (C2v: IDCor) | 69.9 | 76.1 | p-CS/vdWb |
| I–I-*-π(Mo) (C2v: IDCor) | 69.1 | 75.1 | p-CS/vdWb | F–I-*-π(Mo) (C2v: IDCor) | 70.6 | 77.3 | p-CS/vdWb |
Nature of X–H-*-π(C24H12) and Y–X-*-π(C24H12) versus that of X–H-*-π(C6H6) and Y–X-*-π(C6H6)
The Y–X-*-π(C6H6) and X–H-*-π(C6H6) interactions (X, Y = F, Cl, Br and I) are similarly evaluated with M06-2X/BSS-SA. The results are presented in Table S6 and S7 of the ESI.† Fig. 4 shows the plots of θ and θp for Y–X-*-π(C24H12) versus those of Y–X-*-π(C6H6) for convenience of comparison. As shown in Fig. 4, the θ and θp values for Y–X-*-π(aC: C24H12) (IACor) appear to be somewhat smaller than those for Y–X-*-π(C6H6) (Cs: IBBzn), respectively, if those of the same Y–X are compared, whereas the values for Y–X-*-π(12M: C24H12) (ICCor) are predicted to be larger than those for Y–X-*-π(C6H6) (Cs: IBBzn), respectively. Conversely, the θ and θp values for Y–X-*-π(Mo: C24H12) (C2v: IDCor) are very close to those for Y–X-*-π(C6H6) (C2v: IDBzn), respectively, if those of the same Y–X are compared.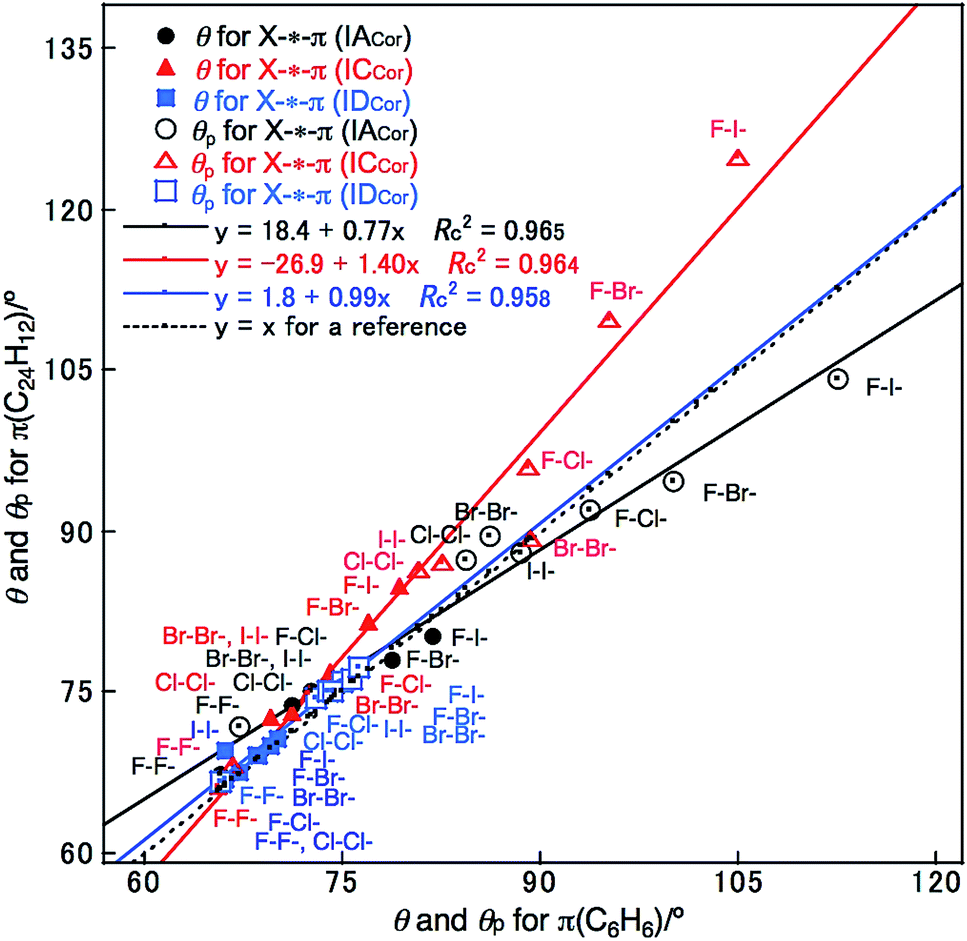 | ||
| Fig. 4 Plots of θ and θp for Y–X-*-π(C24H12) (X, Y = F, Cl, Br and I) versus those for Y–X-*-π(C6H6), respectively, evaluated with M06-2X/BSS-SA. | ||
What is the reason for the predicted results shown in Fig. 4? The charge developed on the C and H atoms of benzene and coronene is examined as the possible origin of these results. Scheme 3 shows the charge evaluated based on the natural population analysis (Qn) with MP2/6-311G(d,p).57 The outside CC–H bonds in coronene are predicted to be substantially positively charged relative to the case of benzene, and the inside AC6 atoms are almost neutral, resulting in the negative charge accumulated on the BC atoms (see, Scheme 3). The results show that the θ and θp values for Y–X-*-π(C24H12) would be larger than those for Y–X-*-π(C6H6), respectively, if Qn for the former or around the interaction is smaller than for the latter. For the small range of the interactions in the adducts, the electron–electron repulsion may play a more important role in the strength of the X-*-π interaction rather than the attractive interaction such as the CT interaction.
 | ||
| Scheme 3 Natural charges (Qn) on the C and H atoms in benzene and coronene evaluated with MP2/6-311G(d,p). | ||
The θ and θp values for X–H-*-π(C24H12) are similarly plotted versus those for X–H-*-π(C6H6), as shown in Fig. S9 of the ESI.† In this case, the θ and θp values increase in the order of π(Mo: C24H12) (IDCor) < π(C6H6) ≤ π(3C: C24H12) (IBCor) < π(12M: C24H12) (ICCor), if those of the same X–H are compared. These results appear to be in close agreement to those for Y–X-*-π(C24H12) with Y–X-*-π(C6H6) (see, Fig. S9 of the ESI†), even though small differences between the two cases are observed.
The H-*-π and X-*-π interactions in the bent π-systems are also of highly interest. An investigation of such interactions is currently in progress.
Conclusions
QTAIM-DFA was applied to the X–H-*-π(C24H12) (X = F, Cl, Br and I) and Y–X-*-π(C24H12) (Y–X = F–F, Cl–Cl, Br–Br, I–I, F–Cl, F–Br and F–I) interactions, which must be of fundamental importance. The structures were optimized mainly at the M06-2X/BSS-SA level of theory. Four types of structures were optimized for X–H⋯π(C24H12) and Y–X⋯π (C24H12) (types IACor, IBCor, ICCor and IDCor) (see, Schemes 1 and 2). The IBCor and ICCor types were predicted for X–H⋯π(C24H12), while the IACor and ICCor types were for Y–X⋯π(C24H12), if optimized with M06-2X/BSS-SA. All BCPs expected are clearly observed in the molecular graphs drawn on the optimized structures.QTAIM-DFA parameters of (R, θ) and (θp, κp) are calculated for H-*-π in X–H-*-π(C24H12) and X-*-π in Y–X-*-π(C24H12) by analysing the plots of Hb(rc) versus Hb(rc) − Vb(rc)/2 at BCPs. The θ values are smaller than 90° for all X–H-*-π(C24H12) and Y–X-*-π(C24H12) interactions, and are therefore classified as the pure CS interactions. The θp values are larger than 90° for F–H-*-π(C24H12) of the IBCor and ICCor types and F–X-*-π(C24H12) (X = Cl, Br and I) of the IACor and ICCor types; therefore, they have the t-HBnc nature. The H-*-π interaction in F–H-*-π(C24H12) (Cs: type ICCor) appear to be present close to the borderline area between t-HBnc and t-HBwc, since θ = 89.1°, which is close to 90°, while θp = 129.1° > 125°. The H-*-π and X-*-π interactions other than above have the vdW nature due to θp < 90°. The θ and θp values are smaller than 90° for all interactions in question in X–H-*-π(C24H12) (C2v: IDCor) and Y–X-*-π(C24H12) (C2v: IDCor). Therefore, the H-*-π and X-*-π interactions around the main axis of π(C24H12) in the adducts are all predicted to have the nature of p-CS/vdW. The θ and θp values for Y–X-*-π(Mo: C24H12) (C2v: IDCor) are very close to the corresponding values for Y–X-*-π(C6H6) (C2v: IDBzn), respectively. Conversely, the θ and θp values for Y–X-*-π(aC: C24H12) (IACor) appear to be somewhat smaller than the corresponding values for Y–X-*-π(C6H6) (Cs: IBBzn), respectively, whereas the values for Y–X-*-π(12M: C24H12) (ICCor) are predicted to be larger than those for Y–X-*-π(C6H6) (Cs: IBBzn), respectively.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research (No. 17K05785) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, NY, 1960 Search PubMed.
- S. Scheiner, Hydrogen Bonding, A Theoretical Perspective, Oxford University Press, Oxford, U.K., 1997 Search PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology (IUCr Monographs on Crystallography), Oxford University Press, Oxford, 1999 Search PubMed.
- G. Gilli and P. Gilli, The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory (IUCr Monographs on Crystallography), Oxford University Press, Oxford, 2009 Search PubMed.
- S. Hayashi, K. Matsuiwa, M. Kitamoto and W. Nakanishi, J. Phys. Chem. A, 2013, 117, 1804–1816 CrossRef CAS PubMed.
- M. Meot-Ner (Mautner), Chem. Rev., 2005, 105, 213–284 CrossRef.
- G. Buemi, Intramolecular Hydrogen Bonds. Methodologies and Strategies for Their Strength Evaluation. in Hydrogen Bonding – New Insights, Vol. 3, Challenges and Advances in Computational Chemistry and Physics, ed. S. J. Grabowski, Springer, New York, 2006, ch. 2 Search PubMed.
- Halogen Bonding: Fundamentals and Applications, ed. P. Metrangolo and G. Resnati, Series: Structure and Bonding, vol. 126, Springer, New York, 2008 Search PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- (a) A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed; (b) M. Alberti, A. Aguilar, J. M. Lucas and F. Pirani, J. Phys. Chem. A, 2010, 114, 11964–11970 CrossRef CAS PubMed.
- See references cited ref. 5.
- C. Otto, G. A. Thomas, K. Rippe, T. M. Jovin and W. L. Peticolas, Biochemistry, 1991, 30, 3062–3069 CrossRef CAS PubMed.
- The temperature effect on the low frequency Raman spectra of hormones was investigated, which should be closely related to the specific conformation, together with the intra- and inter-molecular interactions employing QTAIM. See, V. A. Minaeva, B. F. Minaev, G. V. Baryshnikov, N. V. Surovtsev, O. P. Cherkasova, L. I. Tkachenko, N. N. Karaush and E. V. Stromylo, Opt. Spectrosc., 2015, 118, 214–223 CrossRef CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- (a) S. Yoshimoto, E. Tsutsumi, O. Fujii, R. Narita and K. Itaya, Chem. Commun., 2005, 1188–1190 RSC; (b) T. Kato and T. Yamaba, Chem. Phys. Lett., 2005, 403, 113–118 CrossRef CAS; (c) M. D. Watson, F. Jäckel, N. Severin, J. P. Rabe and K. Müllen, J. Am. Chem. Soc., 2004, 126, 1402–1407 CrossRef CAS PubMed; (d) X. Chi, C. Besnard, V. K. Thorsmølle, V. Y. Butko, A. J. Taylor, T. Siegrist and A. P. Ramirez, Chem. Mater., 2004, 16, 5751–5755 CrossRef CAS.
- M. Bühl and A. Hirsch, Chem. Rev., 2001, 101, 1153–1183 CrossRef.
- Carbon Nanomaterials: Synthesis, Structure, Properties and Applications, ed. R. B. Mathur, B. P. Singh and S. Pande, CRC Press, Taylor & Francis, Boca Raton, FL, USA, 2017 Search PubMed.
- C. Cheng, S. Li, A. Thomas, N. A. Kotov and R. Haag, Chem. Rev., 2017, 117, 1826–1914 CrossRef CAS PubMed.
- (a) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed; (b) M. D. Watson, A. Fechtenkotter and K. Müllen, Chem. Rev., 2001, 101, 1267–1300 CrossRef CAS PubMed; (c) H. Seyler, B. Purushothaman, D. J. Jones, A. B. Holmes and W. W. H. Wong, Pure Appl. Chem., 2012, 84, 1047–1067 CrossRef CAS; (d) X. Feng, W. Pisula and K. Müllen, Pure Appl. Chem., 2009, 81, 2203–2224 CrossRef CAS; (e) J.-L. Wang, J. Yan, Z.-M. Tang, Q. Xiao, Y. Ma and J. Pei, J. Am. Chem. Soc., 2008, 130, 9952–9962 CrossRef CAS PubMed; (f) W. Pisula, X. Feng and K. Müllen, Adv. Mater., 2010, 22, 3634–3649 CrossRef CAS PubMed; (g) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed; (h) L. Chen, Y. Hernandez, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS PubMed; (i) J. Wei, B. Han, Q. Guo, X. Shi, W. Wang and N. Wei, Angew. Chem., Int. Ed., 2010, 49, 8209–8213 CrossRef CAS PubMed; (j) J. Li, M. Kastler, W. Pisula, J. W. F. Robertson, D. Wasserfallen, A. C. Grimsdale, J. Wu and K. Müllen, Adv. Funct. Mater., 2007, 17, 2528–2533 CrossRef CAS; (k) W. Jin, T. Fukushima, A. Kosaka, M. Niki, N. Ishii and T. Aida, J. Am. Chem. Soc., 2005, 127, 8284–8285 CrossRef CAS PubMed; (l) W. Jin, Y. Yamamoto, T. Fukushima, N. Ishii, J. Kim, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 9434–9440 CrossRef CAS PubMed; (m) K. Müllen and J. P. Rabe, Acc. Chem. Res., 2008, 41, 511–520 CrossRef PubMed.
- Y. Sugibayashi, S. Hayashi and W. Nakanishi, Phys. Chem. Chem. Phys., 2015, 17, 28879–28891 RSC . The Y–X-*-π(C6H6) interactions are evaluated somewhat stronger than the X–H-*-π(C6H6) interactions (X, Y = F, Cl, Br and I).
- Y. Sugibayashi, S. Hayashi and W. Nakanishi, ChemPhysChem, 2016, 17, 2579–2589 CrossRef CAS PubMed.
- S. Hayashi, Y. Sugibayashi and W. Nakanishi, RSC Adv., 2016, 6, 49651–49660 RSC.
- S. Hayashi, Y. Sugibayashi and W. Nakanishi, RSC Adv., 2017, 7, 31858–31865 RSC.
- (a) D. Umadevi, S. Panigrahi and G. N. Sastry, Acc. Chem. Res., 2014, 47, 2574–2581 CrossRef CAS PubMed; (b) N. N. Karaush, G. V. Baryshnikov, V. A. Minaeva and B. F. Minaev, New J. Chem., 2015, 39, 7815–7821 RSC; (c) N. N. Karaush, G. V. Baryshnikov and B. F. Minaev, RSC Adv., 2015, 5, 24299–24305 RSC . See also the references cited therein.
- The stacked structures for the interaction between halogen molecules and coronene were reported, in addition to the T-shaped (edge-to-face) structures. The X–H-*-π(C24H12) and Y–X-*-π(C24H12) interactions of the 3c–4e type are discussed in this paper, while the nature of the interactions in the stacked structures will be discussed elsewhere. For the stacked structures, see D. Y. Kim, J. M. L. Madridejos, M. Ha, J.-H. Kim, D. C. M. Yang, C. Baig and K. S. Kim, Chem. Commun., 2017, 53, 6140–6143 RSC.
- (a) Atoms in Molecules. A Quantum Theory, ed. R. F. W. Bader, Oxford University Press, Oxford, UK, 1990 Search PubMed; (b) C. F. Matta and R. J. Boyd, An Introduction to the Quantum Theory of Atoms in Molecules In The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, ed. C. F. Matta and R. J. Boyd, Wiley-VCH, Weinheim, Germany, 2007, ch. 1 Search PubMed.
- (a) R. F. W. Bader, T. S. Slee, D. Cremer and E. Kraka, J. Am. Chem. Soc., 1983, 105, 5061–5068 CrossRef CAS; (b) R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS; (c) R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS; (d) F. Biegler-König, R. F. W. Bader and T. H. Tang, J. Comput. Chem., 1982, 3, 317–328 CrossRef; (e) R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS; (f) T. H. Tang, R. F. W. Bader and P. MacDougall, Inorg. Chem., 1985, 24, 2047–2053 CrossRef CAS; (g) F. Biegler-König, J. Schönbohm and D. Bayles, J. Comput. Chem., 2001, 22, 545–559 CrossRef; (h) F. Biegler-König and J. Schönbohm, J. Comput. Chem., 2002, 23, 1489–1494 CrossRef PubMed.
- J. Molina and J. A. Dobado, Theor. Chem. Acc., 2001, 105, 328–337 CrossRef CAS.
- J. A. Dobado, H. Martinez-Garcia, J. Molina and M. R. Sundberg, J. Am. Chem. Soc., 2000, 122, 1144–1149 CrossRef CAS.
- Some specific interactions are tried to analyze employing QTAIM. See, I. S. Bushmarinov, M. Y. Antipin, V. R. Akhmetova, G. R. Nadyrgulova and K. A. Lyssenko, J. Phys. Chem. A, 2008, 112, 5017–5023 CrossRef CAS PubMed , for an example.
- Interactions would be easily imaged by means of QTAIM if they can be defined as the corresponding bond paths (BPs), especially for experimental chemists. However, it is demonstrated that the detection of the BPs between two atoms in a molecule emerging from natural alignment of the gradient vector held of the one-electron density of a molecule is neither necessary nor a sufficient condition for the presence of a chemical bond between those atoms.58 In this connection, it is pointed out that the terms line paths (LPs) and line critical points (LCPs) should be used in place of BPs and BCPs, respectively.58b Consequently, the dynamic and static nature in this work should be regarded as the investigation performed at LCPs on LPs corresponding to the XH…π and YX…π interactions in the corannulene π-system. Nevertheless, the interactions expected between π(C24H14) and XH or YX are clearly detected by BPs with BCPs which is another reason to use BPs and BCPs in this work.
- Critical points (CPs) are characterized by the rank (ω) and the signature (σ). The CPs of the species in the three-dimensional space are classified by ω = 3, which generally corresponds to all species. On the other hand, σ is defined by the simple algebraic sum of the signs of ∂2ρb(rc)/∂ri2 (ri = x, y and z for i = 1, 2 and 3, respectively), where the + and – signs of ∂2ρb(rc)/∂ri2 are counted as +1 and –1, respectively. Therefore, σ = –3, –1, 1 and 3 correspond to attractors (nuclei), bond critical points (BCPs), ring critical points (RCPs) and cage critical points (CCPs), respectively. Namely, BCP is characterized by (ω, σ) = (3, –1).26.
- The borderline between the pure-CS and regular-CS interactions is described by kb(rc) (= Vb(rc)/Gb(rc)) = –1 and that between the regular-CS and SS interactions by kb(rc) = –2. For the characterization of interactions by means of kb(rc), see (a) R. Bianchi, G. Gervasio and D. Marabello, Inorg. Chem., 2000, 39, 2360–2366 CrossRef CAS PubMed; (b) E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 12, 5529–5542 CrossRef.
- Molecular Interactions. From van der Waals to Strongly Bound Complexes, ed S. Scheiner, Wiley, New York, 1997. For example, Search PubMed (a) A. van der Avoird, P. E. S. Wormer and R. Moszynski, Theory and Computation of Vibration, Rotation and Tunneling Motions of Van der Waals Complexes and their Spectra, ch. 4; (b) J. E. Del Bene and L. Shavitt, The Quest for Reliability in Calculated Properties of Hydrogen-bonded Complexes, ch. 5; (c) T. A. Ford, Ab Initio Predictions of the Vibrational Spectra of Some Molecular Complexes: Comparison with Experiment, ch. 6. See also other chapters.
- For vdW interactions, see (a) C. E. H. Dessent and K. Müller-Dethlefs, Chem. Rev., 2000, 100, 3999–4021 CrossRef CAS PubMed; (b) P. E. S. Wormer and A. van der Avoird, Chem. Rev., 2000, 100, 4109–4144 CrossRef CAS PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- (a) M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC; (b) E. Espinosa, M. Souhassou, H. Lachekar and C. Lecomte, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 563–572 CrossRef; (c) E. Espinosa, C. Lecomte and E. Molins, Chem. Phys. Lett., 1999, 300, 745–748 CrossRef CAS; (d) E. Espinosa, I. Alkorta, I. Rozas, J. Elguero and E. Molins, Chem. Phys. Lett., 2001, 336, 457–461 CrossRef CAS; (e) C. Gatti and L. Bertini, Acta Crystallogr., Sect. A: Found. Crystallogr., 2004, 60, 438–449 CrossRef PubMed.
- (a) Chemistry of Hypervalent Compounds, ed K.-y. Akiba, Wiley-VCH, New York, 1999 Search PubMed; (b) W. Nakanishi, Hypervalent Chalcogen Compounds in Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. A. Devillanova, Royal Society of Chemistry, Cambridge, 2006, ch. 10.3, pp. 644–668 Search PubMed.
- The border between t-HB and CT-MC is tentatively defined as θp = 90° − tan−1 ([dHb(rc)/dr]/[d(Hb(rc) − Vb(rc)/2)/dr] = 90° − tan−1 (dHb(rc)/d(Hb(rc) − V = (rc)/2) = 150°, where θ = 115° corresponds to θp = 150°. The border between Cov-w and Cov-s is similarly tentatively defined as R = [(Hb(rc) − Vb(rc)/2)2 + (Hb(rc))2]1/2 = 0.15 au.
- (a) W. Nakanishi, S. Hayashi and K. Narahara, J. Phys. Chem. A, 2009, 113, 10050–10057 CrossRef CAS PubMed; (b) W. Nakanishi, S. Hayashi and K. Narahara, J. Phys. Chem. A, 2008, 112, 13593–13599 CrossRef CAS PubMed.
- W. Nakanishi and S. Hayashi, Curr. Org. Chem., 2010, 14, 181–197 CrossRef CAS.
- W. Nakanishi and S. Hayashi, J. Phys. Chem. A, 2010, 114, 7423–7430 CrossRef CAS PubMed.
- W. Nakanishi, S. Hayashi, K. Matsuiwa and M. Kitamoto, Bull. Chem. Soc. Jpn., 2012, 85, 1293–1305 CrossRef CAS.
- See also, D. Cremer and E. Kraka, Angew. Chem., Int. Ed., 1984, 23, 627–628 CrossRef.
- QTAIM-DFA is successfully applied to analyse weak to strong interactions in the gas phase. It could also be applied to the interactions in crystals and that in larger systems containing bioactive materials. The methodological improvement is inevitable to generate perturbed structures suitable for the systems.
- The concept of a dynamic molecular graph was recently proposed by Cortés-Guzmán and co-workers through the investigation of the Born–Oppenheimer molecular dynamics (BOMD), which was exemplified by [Fe{C-(CH2)3}(CO)3]. The investigation illustrates the change in the behavior of the molecular graph.59 The concept of the dynamic molecular graph would be closely related to that of the dynamic nature of interactions predicted by employing the perturbed structures generated with the normal coordinate of internal vibrations (NIV). In this treatment, the selected vibration for NIV must contain the motion of the interaction in question most effectively among all the zero-point internal vibrations. A structural catastrophe is confirmed not to occur for the perturbed and fully optimized structures for the elucidation of the dynamic nature of the interaction in question with NIV.54.
- (a) J. Granatier, P. Lazar, M. Otyepka and P. Hobza, J. Chem. Theory Comput., 2011, 7, 3743–3755 CrossRef CAS PubMed; (b) P. Lazar, F. Karlický, P. Jurečka, M. Kocman, E. Otyepková, K. Šafářova and M. Otyepka, J. Am. Chem. Soc., 2013, 135, 6372–6377 CrossRef CAS PubMed; (c) M. A. Hussain, D. Vijay and G. N. Sastry, J. Comput. Chem., 2016, 37, 366–377 CrossRef CAS PubMed.
- (a) J. Kysilka, M. Rubes, L. Grajciar, P. Nachtigall and O. Bludsky, J. Phys. Chem. A, 2011, 115, 11387–11393 CrossRef CAS PubMed; (b) Y. Wang, H. Qian, K. Morokuma and S. Irle, J. Phys. Chem. A, 2012, 116, 7154–7160 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Noro, M. Sekiya and T. Koga, Theor. Chem. Acc., 2012, 131, 1124 CrossRef.
- The AIM2000 program (Version 2.0) is employed to analyze and visualize atoms-in-molecules: F. Biegler-König, J. Comput. Chem., 2000, 21, 1040–1048 CrossRef.
- T. A. Keith, AIMAll (Version 17.11.14), TK Gristmill Software, Overland Park KS, USA, 2017, aim.tkgristmill.com Search PubMed.
- (a) C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef; (b) J. Gauss, J. Chem. Phys., 1993, 99, 3629–3643 CrossRef CAS; (c) J. Gauss, Ber. Bunsen-Ges. Phys. Chem., 1995, 99, 1001–1008 CrossRef CAS.
- The basis set of the (5211111111/411111111/31111) type was employed for I in the calculations of I–H⋯π(C24H12) and Y–I⋯π(C24H12) (Y = F and I), which was obtained from EMSL Basis Set Exchange Library. The basis set is called 6–311G(d) for I. (See also ref. 60 and 61).
- For the m × n matrix representation, m corresponds to the number of atoms and n (=3) to the x, y and z components of the space.
- The values of w = (0), ±0.1 and ±0.2 in r = ro + wao were employed for the perturbed structures in POM (partial optimization method) in ref. 40b and 41 because the bond orders become 2/3 and 3/2 times larger at w = +0.2 and –0.2, respectively, relative to the original values at w = 0. However, it seems better to employ the perturbed structures closer to the fully optimized one, which will reduce errors in the QTAIM functions at the perturbed structures generated by using NIV and/or POM (see ref. 40–43). Therefore, w = (0), ±0.05 and ±0.1 for r = ro + wao were used for the analysis herein.
- The reliable Qn values could not be obtained if evaluated with M062X/BSS-SA.
- (a) R. F. W. Bader, J. Phys. Chem. A, 2009, 113, 10391–10396 CrossRef CAS PubMed; (b) C. Foroutan-Nejad, S. Shahbazian and R. Marek, Chem.–Eur. J., 2012, 18, 4982–4993 CrossRef PubMed; (c) M. Garcıa-Revilla, E. Francisco, P. L. A. Popelier and A. M. Pendás, ChemPhysChem, 2013, 14, 1211–1218 CrossRef PubMed; (d) Z. A. Keyvani, S. Shahbazian and M. Zahedi, Chem.–Eur. J., 2016, 22, 5003–5009 CrossRef CAS PubMed.
- F. Cortés-Guzmán, T. Rocha-Rinza, J. M. Guevara-Vela, G. Cuevas and R. M. Gómez, Chem.–Eur. J., 2014, 20, 5665–5672 CrossRef PubMed.
- D. Feller, J. Comput. Chem., 1996, 17, 1571–1586 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: QTAIM-DFA approach, computational data, and the fully optimized structures given by Cartesian coordinates, together with total energies of the coronene π-system. See DOI: 10.1039/c8ra01862f |
| This journal is © The Royal Society of Chemistry 2018 |