
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel (benzimidazolyl)isoquinolinols and evaluation as adenosine A1 receptor tools†
Sameek Singha,
Samantha L. Cooperb,
Jacqueline R. Glennb,
Jessica Beresfordb,
Lydia R. Percivalb,
Joel D. A. Tyndall a,
Stephen J. Hillb,
Laura E. Kilpatrickb and
Andrea J. Vernall*a
a,
Stephen J. Hillb,
Laura E. Kilpatrickb and
Andrea J. Vernall*a
aSchool of Pharmacy, University of Otago, PO Box 56, Dunedin 9054, New Zealand. E-mail: andrea.vernall@otago.ac.nz; Tel: +64 3 479 4518
bPhysiology, Pharmacology and Neuroscience Division, School of Life Sciences, University of Nottingham, Nottingham, NG7 2UH, UK
First published on 3rd May 2018
Abstract
G protein-coupled receptors (GPCRs) constitute the largest family of transmembrane receptors in eukaryotes. The adenosine A1 receptor (A1AR) is a class A GPCR that is of interest as a therapeutic target particularly in the treatment of cardiovascular disease and neuropathic pain. Increased knowledge of the role A1AR plays in mediating these pathophysiological processes will help realise the therapeutic potential of this receptor. There is a lack of enabling tools such as selective fluorescent probes to study A1AR, therefore we designed a series of (benzimidazolyl)isoquinolinols conjugated to a fluorescent dye (31–35, 42–43). An improved procedure for the synthesis of isoquinolinols from tetrahydroisoquinolinols via oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and atmospheric oxygen is reported. This synthetic method offers advantages over previous metal-based methods for the preparation of isoquinolinols and isoquinolines, which are important scaffolds found in many biologically active compounds and natural products. We report the first synthesis of the (benzimidazolyl)isoquinolinol compound class, however the fluorescent conjugates were not successful as A1AR fluorescent ligands.
Introduction
Adenosine receptors (ARs) are class A G protein-coupled receptors (GPCRs), classified in four distinct subtypes – A1, A2A, A2B and A3. Adenosine A1 receptor (A1AR) is expressed throughout the human body with predominant distribution in brain, heart, kidney and adipose tissue and is an attractive therapeutic target.1 To this end, A1AR ligands are of interest and are being developed to treat various pathological conditions such as atrial fibrillation, angina pectoris, congestive heart failure, diabetes, neuropathic pain and renal disorders.2 Fluorescent ligands have emerged as useful and powerful tools to study real-time and live cell dynamic processes of GPCRs.3 Fluorescent ligands have been developed for ARs,4 including some with high AR subtype selectivity for A3AR.5 There is one report of fluorescent dansyl-linked N6 NECA derivatives with subtype selectivity for rat A1AR,6 however the short excitation wavelength of the dansyl fluorophore is not ideal for many applications. When the same research group conjugated the lead pharmacophore-linker to longer excitation wavelength fluorophores A3AR selective probes ensued.6 Therefore there remains a need to develop fluorescent ligands with high subtype selectivity for A1AR.One approach to the design of a fluorescent ligand is to identify an existing pharmacophore with the requisite selectivity and affinity for the target receptor, and then identify a location that a linker and fluorophore can be attached.5 With this design in mind, it is then necessary to synthesise and test a range of pharmacophore, pharmacophore-linker and fluorescent ligands as modifications have the potential to alter the affinity and efficacy of the parent pharmacophore. In an effort to develop a selective A1AR fluorescent ligand we chose to avoid a xanthine amine congener (XAC)-based pharmacophore because of subtype selectivity concerns as a range of XAC-fluorescent conjugates have been reported as either non AR subtype selective or A3AR selective.7 Cosimelli et al. recently reported a series of (benzimidazolyl)isoquinolines as selective antagonists for human A1AR (hA1AR), among them ligands 1–3 (Fig. 1),8 which were used as the inspiration for the fluorescent ligand design reported herein.
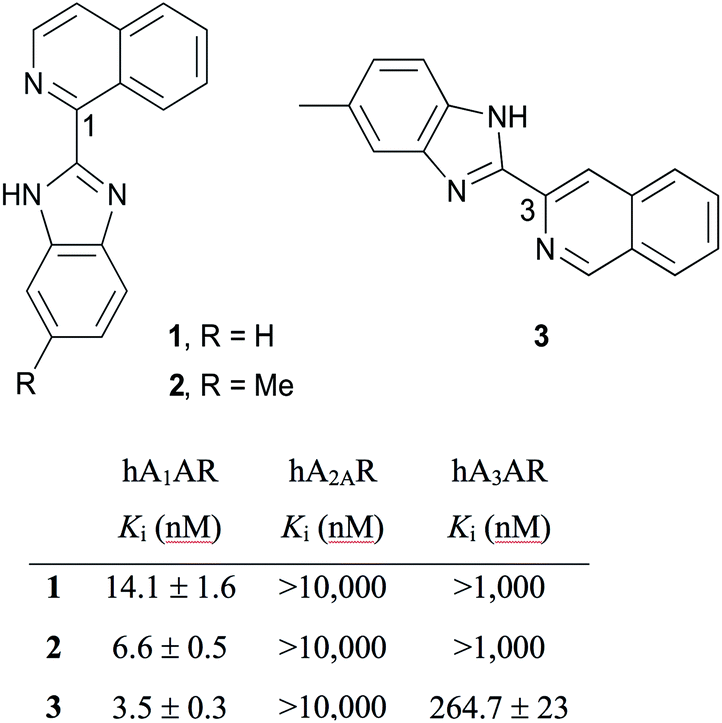 | ||
| Fig. 1 A1AR subtype selective (benzimidazolyl)isoquinolines with previously reported AR affinities.8 | ||
Structure–activity relationships (SAR) from the Cosimelli et al. study indicated small, lipophilic benzimidazole substituents such as methyl and ethylthio groups in some positions improved A1AR affinity and in others abolished it.8 This sensitivity to slight positional change, along with potential buried, lipophilic interactions these groups could be making with the A1AR to enhance affinity led us to discount linker substitution from the benzimidazole ring. Since 1,3-substituted isoquinoline analogues such as VUF5455 (ref. 9) and LUF6096 (ref. 10) have been reported as allosteric ligands for A3AR, albeit with quite different substituents, we chose to instead explore the previously unexplored isoquinoline C5–8 positions for linker attachment. A1AR ligands such as 1–3 lack a reactive functional group for linker and subsequently fluorophore attachment in these positions, therefore we designed a series of (benzimidazolyl)isoquinolinols. To the best of our knowledge, there are no previous literature reports on the synthesis of (benzimidazolyl)isoquinolinols. The hydroxyl group offers accessible chemistry for linker attachment, e.g. via an ether bond that remains unionised at physiological pH. Succinimidyl-6-[2-(p-{(E)-2-[4,4-difluoro-5-(2-thienyl)-3a,4a-diaza-4-bora-s-indacen-3-yl]-ethenyl}phenoxy)acetylamino]hexanoate (BODIPY630/650-SE) was chosen as the fluorophore for coupling because of its success in developing fluorophores for studying GPCRs in previous studies4,5,11 and its suitable spectroscopic properties as an acceptor in bioluminescence resonance energy transfer (BRET) assays.12
We envisaged using the Pictet–Spengler reaction13 in our synthesis to form a hydroxytetrahydroisoquinoline carboxylic acid followed by aromatisation to give a hydroxyisoquinoline carboxylic acid. This required an aromatisation reaction tolerant of functional groups such as a phenol, amine and carboxylic acid. Although isoquinoline is an important pharmacophore in various natural products, metal co-ordination ligands and marketed drugs such as papaverine, ripasudil, quinisocaine and moxaverine, a general method for the construction of isoquinolines with broad functional group tolerance, including the synthesis of isoquinolinols, is lacking. Aromatisation of tetrahydroisoquinoline to isoquinoline is usually carried out using heavy metals such as palladium with harsh conditions with poor functional group tolerance.14–21 Protection/deprotection of functional groups is another common strategy employed for successful aromatisation to prepare functionalised isoquinolines.22,23 Use of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) has been reported for the aromatisation of tetrahydroisoquinoline22,24,25 and dihydroisoquinoline26,27 to isoquinolines at high temperature. In this study, we explored the use of DDQ to aromatise tetrahydroisoquinols to isoquinolinols under mild conditions as an alternate to heavy metal-based or harsh reaction conditions.
Results and discussion
The synthesis of 1-substituted 6-hydroxyl tetrahydroisoquinoline 5 began with attempts to carry out a Pictet–Spengler reaction of 3-(2-aminoethyl)phenol hydrobromide 4 with glyoxylic acid monohydrate following the procedure Li et al.28 (no spectroscopic data provided for 5, ref. 29), however these were unsuccessful. Pleasingly, following the synthesis of 5 reported by Maillard et al.29 tetrahydroisoquinoline 5 was obtained in good yield, the point of difference between the Li et al.28 and Maillard et al.29 procedures being addition of triethylamine to the amine hydrobromide salt prior to addition of glyoxylic acid monohydrate (Scheme 1). The neutralisation presumably meant the imine could form more readily and hence the Pictet–Spengler reaction could proceed. Attempts to aromatise carboxylic acid 5 using DDQ and isolate the hydroxyisoquinoline carboxylic acid (using optimised conditions described below) were problematic, in part due to difficulty analysing and purifying the polar carboxylic acid. Therefore 5 was esterified to give methyl ester 6 (compound 6 was previously reported by Ma et al.30 but with no spectroscopic data). Synthesis of the 3-substituted tetrahydroisoquinolinol series began with Pictet–Spengler condensation of (+/−)-m-tyrosine 8 with formaldehyde to give 9 according to a literature procedure reporting the synthesis of 9,31 followed by esterification to give 10. The synthesis of methyl ester 10 from 9 has previously been reported18,25,32–34 however none of these reports include spectroscopic data for 10. The first attempt to synthesise the 7-hydroxyl tetrahydroisoquinoline series via condensation of tyramine with glyoxylic acid monohydrate according to the procedure for 5 (except without trimethylamine as the amine was not a salt) was not successful. Failure of this reaction was attributed to the lack of an activating group in tyramine, compared to the presence of an activating para hydroxyl relative to the point of condensation for 4 and 8. Instead, Fmoc-protected tyramine 12 was reacted with glyoxylic acid monohydrate to give a carboxylic acid according to the synthesis of this carboxylic acid reported by Maillard et al.29 which was esterified to give 13 in low yield over 2 steps. NMR spectra of 13 showed a mixture of isomers that were confirmed as rotamers rather than regioisomers via variable temperature NMR experiments.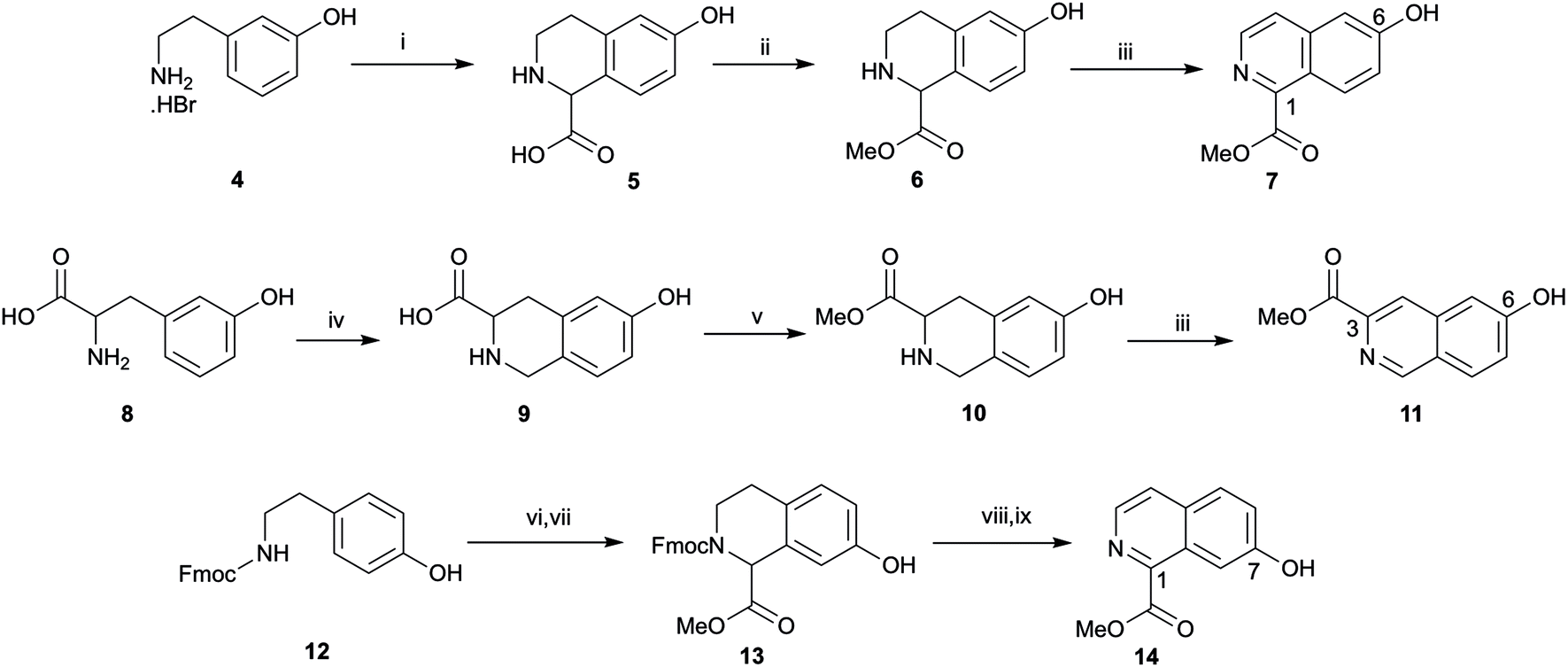 | ||
| Scheme 1 Synthesis of isoquinolines. Reagents and conditions: (i) Et3N, glyoxylic acid monohydrate, EtOH, 71%; (ii) SOCl2, MeOH, 65 °C, quantitative; (iii) DDQ, THF, 1,4 dioxane, 45 °C, 49–52%; (iv) HCl, H2O, formaldehyde (37 wt% in H2O) 100 °C, 48%; (v) H2SO4, MeOH, 96%; (vi) glyoxylic acid monohydrate, H2SO4, CH3CO2H; (vii) SOCl2, MeOH, 18% over two steps; (viii) DMSO, MeOH, 60 °C; (ix) DDQ, THF, 1,4 dioxane, 45 °C, 61% over two steps. | ||
There is one report of the aromatisation of an unprotected hydroxytetrahydroisoquinoline (10) with DDQ in patent literature (conversion of 10 to 11, no spectroscopic data for 11 provided25) however in our hands all initial attempts to aromatise 6 or 10 using DDQ with varying temperatures and solvents and with either oxygen excluded or in a closed reaction vessel failed. When 6 and DDQ were dissolved in THF and dioxane and heated to 100 °C under a N2 atmosphere the partially oxidised methyl 6-hydroxy-3,4-dihydroisoquinoline-1-carboxylate was isolated as the major product. However, at moderate temperature and with vigorous stirring in a flask open to the atmosphere, 6 and DDQ were reacted to give the desired product 7 in moderate yield. To our knowledge, isoquinoline 7 has only been reported once before in the literature in a Japanese patent35 but no spectroscopic data was provided. The procedure to convert 6 to 7 was also used to successfully aromatise 10 to 11. Since the aromatised product did not form without vigorous stirring open to the atmosphere it is likely that DDQ mediates formation of a dihydroisoquinoline, which is then aromatised by oxygen as the active oxidant. Oxygen has previously been reported as an oxidant in aromatisation reactions36,37 and indeed Dong et al.38 used a base in dimethyl sulfoxide (DMSO) along with oxygen from air for conversion of N-tosyltetrahydroisoquinolines into isoquinolines. Dong et al.38 also isolated an imine under an argon atmosphere instead of the isoquinoline product, analogous to the partially oxidised imine intermediate (methyl 6-hydroxy-3,4-dihydroisoquinoline-1-carboxylate) isolated in our study with exclusion of air. Formation of an iminium ion intermediate (3,4-dihydroisoquinoline) has also been reported for DDQ-based cross-dehydrogenative coupling reactions of tetrahydroisoquinolines.39
Diethylamine was initially used for Fmoc deprotection of 13 however subsequent aromatisation with DDQ provided poor yield of 14, most likely due to residual diethylamine. High temperature NMR spectra in DMSO-d6, carried out to study rotamers of 13 revealed cleavage of Fmoc, and indeed there is precedent in the literature that Fmoc cleavage can be conducted in neat DMSO at high temperature.40 Thus, Fmoc cleavage of 13 in DMSO, followed by DDQ and air mediated aromatisation afforded 14 in comparable yield to 7 and 11. The moderate yield of DDQ-air mediated aromatisation is unclear but may be due to low solubility in organic solvent of these compounds and susceptibility to methyl ester hydrolysis during basic work up.
Hydrolysis of 7 and 14 under basic conditions provided 15 and 16, which were condensed with o-phenylenediamine or 2,4-diaminotoluene in hot polyphosphoric acid (PPA) to afford 1-(benzimidazolyl)isoquinolinols 17–19 in low yield (Scheme 2). This low yield was likely due to a number of factors, including poor double dehydration yield and a difficult isolation of a product with low organic solvent solubility from the viscous reaction mixture. NMR spectra of 17–19 showed a mixture of conformers, which was elucidated to be tautomers using variable solvent and high temperature NMR experiments and high performance liquid chromatography (HPLC) (discussed in ESI†). The phenolic group of 17–19 was reacted with tert-butyl bromoacetate to afford corresponding tert-butyl esters 20–22, which were reacted with trifluoroacetic acid to give carboxylic acids 23–25. Carboxylic acid 24 was coupled to three different amino linkers – a C8 alkyl, short polyethylene glycol (PEG) or L-Ala-L-Ala short peptide to give 27, 28 or 29 respectively. Carboxylic acid 23 underwent amide coupling to give PEG-linked 26 while the 7-substituted carboxylic acid 25 was converted to PEG-linked 30. These linkers were selected to explore variations in the physicochemical properties of the resulting fluorescent probes, in particular in light of lipophilicity and non-specific membrane binding. Boc deprotection of 26–30 provided amines that were reacted with BODIPY630/650-SE to give fluorescent conjugates 31–35. 3-(Benzimidazolyl)isoquinolinol-based fluorescent conjugates 42 and 43 were synthesised using analogous methodology (Scheme 3). Absorption and emission spectra of fluorescent compounds 31–35 and 42–43 showed excitation maxima at 624 nm and emission maxima at either 641 nm or 642 nm (detailed in ESI†).
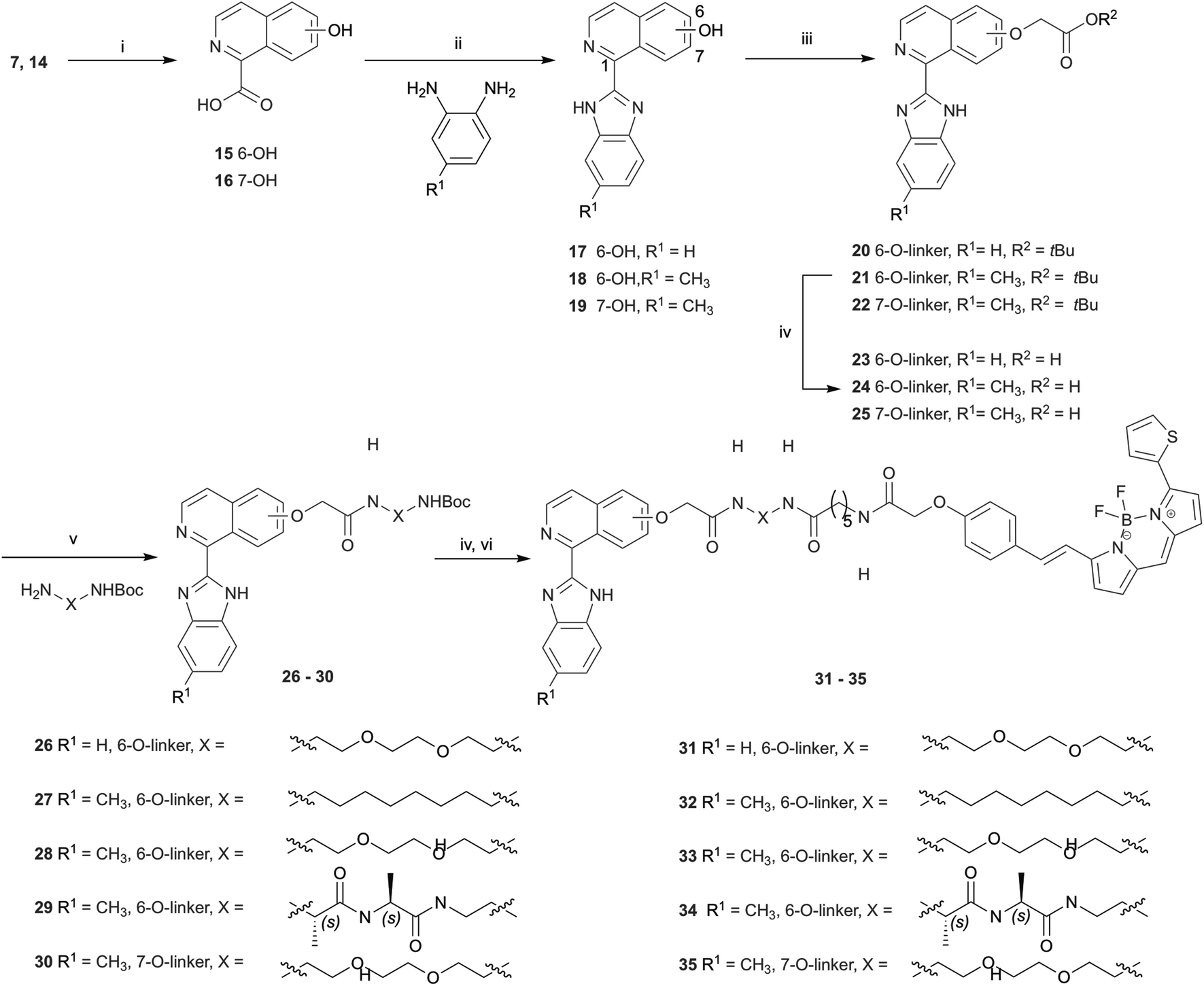 | ||
| Scheme 2 Synthesis of 1-(1H-benzimidazol-2-yl)isoquinolinols. Reagents and conditions: (i) LiOH, THF, H2O, 70–96%; (ii) PPA, 250 °C, 21–38%; (iii) tert-butyl bromoacetate, K2CO3, THF, 60 °C, 66–96%; (iv) TFA, DCM, 60% – quantitative; (v) HATU, DIPEA, DMF, 29–69%; (vi) BODIPY630/650-SE, DIPEA, DMF, 33–84%. | ||
 | ||
| Scheme 3 Synthesis of 3-(1H-benzimidazol-2-yl)isoquinolinols. Reagents and conditions: (i) LiOH, THF, H2O, 90%; (ii) PPA, 250 °C, 26%; (iii) tert-butyl bromoacetate, K2CO3, THF, 60 °C, 94%; (iv) TFA, DCM, quantitative; (v) HATU, DIPEA, DMF, 37–39%; (vi) BODIPY630/650-SE, DIPEA, DMF, 73–86%. | ||
Pharmacology
Fluorescent ligands 31–35 and 42–43 were analysed for their ability to bind to A1AR according to a previously reported bioluminescence resonance energy transfer (NanoBRET) saturation binding assay using the novel luciferase NanoLuc (NLuc; Promega Corporation, USA).12 HEK293 cell lines stably transfected with N-terminal NLuc labelled A1AR were treated with increasing concentrations of the (benzimidazolyl)isoquinolinol fluorescent ligands, in the presence and absence of 1 μM DPCPX (a high affinity, non-fluorescent, selective A1AR antagonist) to determine levels of non-specific binding. Unfortunately, all ligands 31–35 and 42–43 did not show any indication of binding to A1AR, for example data shown for 31 and 33 (Fig. 2a and b) (other compound data in ESI Fig. S16†). Fluorescent ligands 31–34 were then analysed for their ability to bind to NLuc-labelled A3AR in analogous NanoBRET experiments, using A3AR selective MRS1220 to establish non-specific binding. Ligands 32 and 34 did not show any specific binding to NLuc-A3AR (data not shown), while 31 and to a lesser extent 33 (Fig. 2c and d) showed a small degree of binding to NLuc-A3AR. However, the specific binding window was small and the standard error's of the calculated Kd values for specific binding to NLuc-A3AR were large (31 Kd = 162 ± 65.5 nM, n = 5; 33 Kd = 534 ± 254 nM, n = 4).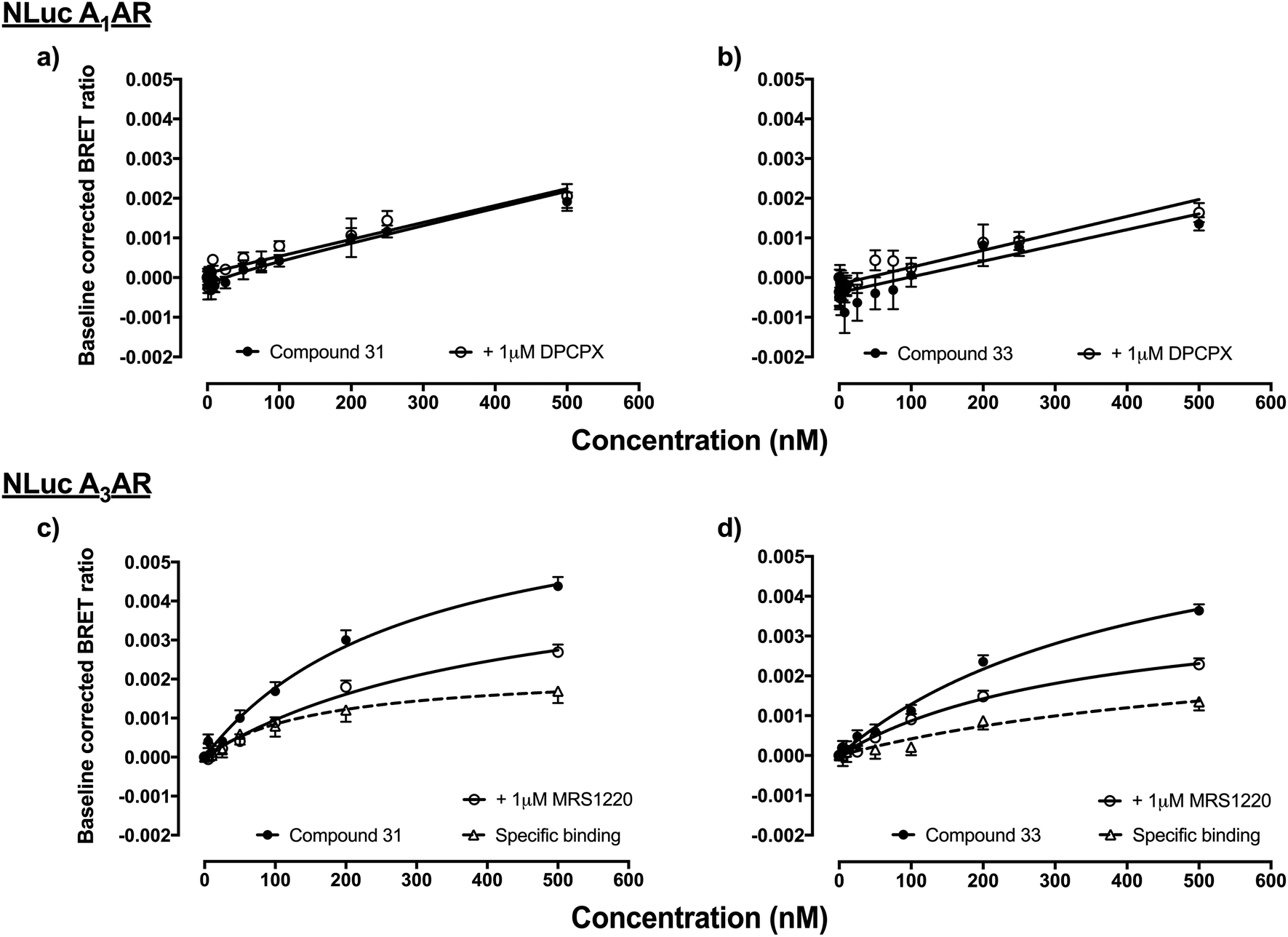 | ||
| Fig. 2 HEK293 cells stably transfected with N-terminally NLuc-tagged A1AR or A3AR were treated with increasing concentrations of fluorescent ligand and the BRET ratio measured after direct addition of the NLuc substrate furimazine (10 μM).12 Non-specific binding was assessed in the absence and presence of 1 μM DPCPX (NLuc-A1AR) or 1 μM MRS1220 (NLuc-A3AR). Pooled data of raw BRET ratio's were baseline corrected (minus vehicle + furimazine BRET ratios) so that data are expressed as fold increase in BRET ratios over basal. (a) NLuc-A1AR and 31, (b) NLuc-A1AR and 33, (c) NLuc-A3AR and 31 and (d) NLuc-A3AR and 33. Data represents four–seven independent experiments (in triplicate). | ||
This NanoBRET assay is an excellent way to assess new fluorescent ligands41 since it measures the interaction of the fluorescent ligand and receptor directly without relying on the ability of the test ligand to compete with another tracer such as a radioligand. In this study the level of fluorescent ligand non-specific/membrane-binding was defined by measuring BRET in the presence, for A3AR, of a competing high concentration of unlabelled MRS 1220. Therefore this measure of specificity is defined by the ability of MRS 1220 (a[1,2,4]triazolo[1,5-c]quinazolinyl pharmacophore) to prevent binding of the fluorescent compounds, which implies each would be interacting with the same or an overlapping region of the ligand binding pocket of the receptor. It is clear from Fig. 2c and d, however, that the non-specific binding component (obtained in the presence of MRS 1220) is not linear and is better fit with a saturable binding curve. It therefore remains a possibility (along with non-specific membrane interactions) that 31 and 33 may be interacting with an additional region of A3AR distinct from the orthosteric binding site of MRS 1220.
A possible explanation for the lack of binding of fluorescent ligands 31–35 and 42–43 to NLuc A1AR, is steric occlusion of binding due to the presence of the N-terminal NanoLuc tag. To investigate this, NanoBRET was used to quantify the specific binding of CA200645, a known fluorescent non selective AR ligand at NLuc A1AR. Saturable binding was observed, with minimal non-specific binding and a large observation window (Fig. 3A; Kd = 110 nM + 18.46 n = 5) consistent with previous NanoBRET observations of CA200645 binding to NLuc A1AR or NLuc A3AR.12 NanoBRET assay plates were then washed and imaged using a IX Ultra confocal plate reader (MetaXpress, Molecular Devices) with fluorescence intensity quantified per well using a modified multiple wavelength cell scoring algorithm.42 Saturable binding of CA200645 was observed at NLuc A1AR (Kd = 189.3 nM ± 69.6; n = 4, data not shown), with a Kd value consistent with that measured by NanoBRET (Fig. 3A). The binding of CA200645 to untagged A1AR stably expressed in HEK293, was also quantified using this fluorescent imaging-based technique with the resultant estimated binding affinity comparable to that seen for NLuc tagged A1AR (144.8 nM ± 63.6; n = 3; data not shown). From these results it can be concluded that the NanoLuc tag does not occlude binding of CA200645 to the receptor, therefore it seems unlikely this tag is the reason for the poor binding of fluorescent ligands 31–35 and 42–43.
 | ||
| Fig. 3 HEK293 cells stably expressing N-terminal NLuc tagged A1AR were treated with increasing concentrations of CA200645 (5–500 nM; 1 h at 37 °C; A). Non specific binding was defined using 1 μM DPCPX, an A1AR selective antagonist. The NanoLuc substrate furimazine was added (10 μM), with luminescence and fluorescence emissions recorded using a Pherastar FS. Data was pooled from independent experiments (n = 5) and baseline corrected (minus vehicle + furimazine BRET ratios) so that data are expressed as fold increase in BRET ratios over basal and where appropriate fit using one site saturation binding (mean ± SEM). For competition experiments (B), NLuc A1AR were co-incubated with a fixed concentration of CA200645 and increasing concentrations of unlabelled ligand (1 h at 37 °C). Total CA200645 binding and vehicle are shown by the black and white bars respectively. Data were pooled from five independent experiments and are expressed as mean ± SEM. | ||
In light of the poor binding of fluorescent ligands 31–35 and 42–43 to A1AR as measured using NanoBRET competition binding experiments, a subset of (benzimidazolyl)isoquinolinols described in Schemes 2 and 3 (1, 17–19, 28, 30, 37 and 40) and literature reference compound 1 were tested using NanoBRET competition assays. HEK293 NLuc A1AR cells were treated with a fixed concentration of CA200645 (25 nM) and increasing concentrations of unlabelled ligand. The A1AR selective antagonist DPCPX was used as a positive control. No displacement of CA200645 binding was observed with benzimidazolyl)isoquinolinols 1, 17–19, 28, 30, 37 and 40 until ligand concentrations were in excess of 10−6 M (Fig. 3B; representative (benzimidazolyl)isoquinolinols shown). With the exception of DPCPX, it was not possible to calculate a pKi value for all compounds studied (DPCPX pKi; 8.82 ± 0.16). To our surprise, no displacement of CA200645 was observed when using 1, which was previously reported as a high affinity and selective A1AR ligand (hA1AR Ki = 14.1 ± 1.6 nM, Fig. 1) in a [3H]DPCPX binding assay in membranes obtained from CHO cells stably expressing hA1AR.8 Interestingly both 1-(1H-1,3-benzodiazol-2-yl)isoquinolin-6-ol 17 and 3-(6-methyl-1H-1,3-benzodiazol-2-yl)isoquinolin-6-ol 37 both showed better (although still limited) binding to NLuc A1AR than 1 (Fig. 3B).
Experimental
Full details of the synthesis of 1, 4–43, including all methodology, chemical synthesis, pharmacology, equipment and compound characterisation can be found in the ESI,† along with a comprehensive tautomer study of 18. All compounds tested in the BRET assay were shown by analytical HPLC to possess >95% purity. As a representative examples of a DDQ aromatisation reaction, conversion of 6 to 7 was as follows: to a solution of 6 (0.17 g, 0.83 mmol) in 1,4-dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (10 mL 1:1, v/v) at 45 °C was added DDQ (0.38 g, 1.67 mmol) and the reaction was stirred vigorously at 45 °C for 5 h with the mouth of the flask open to the atmosphere to allow mixing of air. 1,4-Dioxane (10 mL) was added and the reaction mixture filtered, the filtrate was diluted with EtOAc and washed three times with sat aq. NaHCO3 solution. The organic washings were combined, washed once with water, brine solution, then dried over MgSO4, concentrated under reduced pressure and purified by silica gel flash column chromatography (30–50% EtOAc/hexane) to provide 7 (84 mg, 0.413 mmol, yield 49%) as an off-white solid. 1H NMR (400 MHz, MeOD-d4) δ 4.04 (s, 3H, OCH3), 7.16 (d, J = 2.5 Hz, 1H, ArH), 7.28 (dd, 1H, J = 2.5, 9.3 Hz, ArH), 7.73 (d, 1H, J = 5.7 Hz, ArH), 8.30 (d, 1H, J = 5.7 Hz, ArH), 8.52 (d, 1H, J = 9.3 Hz, ArH). 13C NMR (101 MHz, MeOD-d4) δ 53.24, 108.72, 122.70, 122.89, 124.25, 129.49, 141.07, 141.83, 149.28, 161.18, 167.61. HRMS calculated for C11H10NO3 (M + H)+, 204.0655; found, 204.0647.
:THF (10 mL 1:1, v/v) at 45 °C was added DDQ (0.38 g, 1.67 mmol) and the reaction was stirred vigorously at 45 °C for 5 h with the mouth of the flask open to the atmosphere to allow mixing of air. 1,4-Dioxane (10 mL) was added and the reaction mixture filtered, the filtrate was diluted with EtOAc and washed three times with sat aq. NaHCO3 solution. The organic washings were combined, washed once with water, brine solution, then dried over MgSO4, concentrated under reduced pressure and purified by silica gel flash column chromatography (30–50% EtOAc/hexane) to provide 7 (84 mg, 0.413 mmol, yield 49%) as an off-white solid. 1H NMR (400 MHz, MeOD-d4) δ 4.04 (s, 3H, OCH3), 7.16 (d, J = 2.5 Hz, 1H, ArH), 7.28 (dd, 1H, J = 2.5, 9.3 Hz, ArH), 7.73 (d, 1H, J = 5.7 Hz, ArH), 8.30 (d, 1H, J = 5.7 Hz, ArH), 8.52 (d, 1H, J = 9.3 Hz, ArH). 13C NMR (101 MHz, MeOD-d4) δ 53.24, 108.72, 122.70, 122.89, 124.25, 129.49, 141.07, 141.83, 149.28, 161.18, 167.61. HRMS calculated for C11H10NO3 (M + H)+, 204.0655; found, 204.0647.
Conclusions
Our overall goal was to design new A1AR selective fluorescent ligands, however it can be concluded that a (benzimidazolyl)isoquinolinol scaffold linked via the C5 or C6 isoquinoline position to a fluorophore is not suitable for this purpose. Although exploration of other regions of the (benzimidazolyl)isoquinoline scaffold (such as from the benzimidazole) for linker attachment are possible, in light of the poor A1AR binding observed for 1, the selection of a different A1AR selective pharmacophore may be optimal. Although the end biological aim of A1AR fluorescent ligands was not achieved, a method for aromatisation of tetrahydroisoquinolinols to give isoquinolinols and conformational (tautomer) studies of (benzimidazolyl)isoquinolinols are reported. Since there are only two reports25,43 for non-metal-mediated aromatisation of unprotected tetrahydroisoquinolinol to isoquinolinol, for which experimental details are unclear and no yields are provided, the method described herein using DDQ and atmospheric oxygen in mild conditions will prove useful for the preparation of biologically important isoquinolines and isoquinolinols.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a University of Otago Research Grant and the School of Pharmacy, University of Otago and the University of Nottingham. S. S. is supported by a Doctoral Scholarship from the School of Pharmacy, University of Otago. The authors thank Dr Leigh Stoddart of the Physiology, Pharmacology and Neuroscience group at the University of Nottingham for making the NLuc A3AR construct and Promega Corporation for making the NLuc A1AR construct.References
- J.-F. Chen, H. K. Eltzschig and B. B. Fredholm, Nat. Rev. Drug Discovery, 2013, 12, 265–286 CrossRef CAS PubMed.
- C. E. Müller and K. A. Jacobson, Biochim. Biophys. Acta, 2011, 1808, 1290–1308 CrossRef PubMed.
- L. A. Stoddart, C. W. White, K. Nguyen, S. J. Hill and K. D. Pfleger, Br. J. Pharmacol., 2016, 173, 3028–3037 CrossRef CAS PubMed.
- E. Kozma, P. S. Jayasekara, L. Squarcialupi, S. Paoletta, S. Moro, S. Federico, G. Spalluto and K. A. Jacobson, Bioorg. Med. Chem. Lett., 2013, 23, 26–36 CrossRef CAS PubMed.
- A. J. Vernall, S. J. Hill and B. Kellam, Br. J. Pharmacol., 2014, 171, 1073–1084 CrossRef CAS PubMed.
- M. Macchia, F. Salvetti, S. Barontini, F. Calvani, M. Gesi, M. Hamdan, A. Lucacchini, A. Pellegrini, P. Soldam and C. Martini, Bioorg. Med. Chem. Lett., 1998, 8, 3223–3228 CrossRef CAS PubMed.
- A. J. Vernall, L. A. Stoddart, S. J. Briddon, H. W. Ng, C. A. Laughton, S. W. Doughty, S. J. Hill and B. Kellam, Org. Biomol. Chem., 2013, 11, 5673–5682 CAS.
- B. Cosimelli, S. Taliani, G. Greco, E. Novellino, A. Sala, E. Severi, F. Da Settimo, C. La Motta, I. Pugliesi, L. Antonioli, M. Fornai, R. Colucci, C. Blandizzi, S. Daniele, M. L. Trincavelli and C. Martini, ChemMedChem, 2011, 6, 1909–1918 CrossRef CAS PubMed.
- Z.-G. Gao, J. E. Van Muijlwijk-Koezen, A. Chen, C. E. Müller, A. P. Ijzerman and K. A. Jacobson, Mol. Pharmacol., 2001, 60, 1057–1063 CrossRef CAS PubMed.
- L. H. Heitman, A. Goblyos, A. M. Zweemer, R. Bakker, T. Mulder-Krieger, J. P. van Veldhoven, H. de Vries, J. Brussee and A. P. IJzerman, J. Med. Chem., 2009, 52, 926–931 CrossRef CAS PubMed.
- Z. Ma, L. Du and M. Li, J. Med. Chem., 2014, 57, 8187–8203 CrossRef CAS PubMed.
- L. A. Stoddart, E. K. M. Johnstone, A. J. Wheal, J. Goulding, M. B. Robers, T. Machleidt, K. V. Wood, S. J. Hill and K. D. G. Pfleger, Nat. Methods, 2015, 12, 661–663 CrossRef CAS PubMed.
- E. D. Cox and J. M. Cook, Chem. Rev., 1995, 95, 1797–1842 CrossRef CAS.
- H.-P. Buchstaller, L. Burgdorf, D. Finsinger, C. Amendt, M. Grell, C. Sirrenberg, F. Zenkeand Preparation of isoquinoline derivatives as kinase inhibitors, WO2005082858A2, Merck Patent Gmbh, 2005.
- H.-P. Buchstaller, L. Burgdorf, D. Finsinger, F. Stieber, C. Sirrenberg, C. Amendt, M. Grell, F. Zenke and M. Krier, Bioorg. Med. Chem. Lett., 2011, 21, 2264–2269 CrossRef CAS PubMed.
- K. Iwasa, M. Moriyasu, Y. Tachibana, H.-S. Kim, Y. Wataya, W. Wiegrebe, K. F. Bastow, L. M. Cosentino, M. Kozuka and K.-H. Lee, Bioorg. Med. Chem., 2001, 9, 2871–2884 CrossRef CAS PubMed.
- F. Navas and P. K. Spearing, Preparation of isoxazole compounds as therapeutic farnesoid X receptor agonists, US pat., 20080096921A1, Smithkline Beecham Corporation, 2008.
- E. F. Calderwood, M. Duffey, A. E. Gould, P. D. Greenspan, B. Kulkarni, M. J. Lamarche, R. S. Rowland, M. Tregay, and T. J. Vos, Bicyclic compounds with kinase inhibitory activity, WO2007067444A1, Millennium Pharmaceuticals, Inc., 2007.
- S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS PubMed.
- W. Yao, Y. Zhang, X. Jia and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 1390–1394 CrossRef CAS PubMed.
- R. Chen, J. Qi, Z. Mao and S. Cui, Org. Biomol. Chem., 2016, 14, 6201–6204 CAS.
- A. M. M. Mjalli, D. Jones, D. R. Gohimmukkula, G. Huang, J. Zhu, M. Rao, R. C. Andrews, and T. Ren, Benzazole derivatives, compositions, and methods of use as b-secretase inhibitors, WO2006099379A2, Transtech Pharma, Inc., 2006.
- T. R. Burke Jr, P. L. Russ and V. E. Marquez, Heterocycles, 1992, 34, 757–764 CrossRef.
- A. Jirgensons, E. Loza, M. Charlton, P. W. Finn, D. P. L. Ribas, and A. Saint-Leger, Novel N-acyl-arylsulfonamide derivatives as aminoacyl-tRNA synthetase inhibitors, WO2016129983A1, Latvian Institute Of Organic Synthesis, 2016.
- A. Abeywardane, S. R. Brunette, M. J. Burke, T. M. Kirrane Jr, C. C. Man, D. R. Marshall, A. K. Padyana, H. Razavi, R. Sibley, K. L. L. Smith, R. J. Snow, R. J. Sorcek, H. Takahashi, S. J. Taylor, M. R. Turner, E. R. R. Young, Q. Zhang, Y. Zhang, and R. M. Zindell, Pyrazole derivatives which inhibit leukotriene production, WO2014014874A1, Boehringer Ingelheim International Gmbh, 2014.
- V. K. Tiwari, N. Kamal and M. Kapur, Org. Lett., 2017, 19, 262–265 CrossRef CAS PubMed.
- A. G. Schultz, W. G. McMahon, R. R. Staib and R. K. Kullnig, Tetrahedron Lett., 1987, 28, 4929–4932 CrossRef CAS.
- J. J. Li and J. A. Tino, Tetrahydroisoquinoline analogs useful as growth hormone secretagogues, WO2001085695A1, Bristol-Myers Squibb Co., 2001.
- M. C. Maillard, F. A. Brookfield, S. M. Courtney, F. M. Eustache, M. J. Gemkow, R. K. Handel, L. C. Johnson, P. D. Johnson, M. A. Kerry, F. Krieger, M. Meniconi, I. Muñoz-Sanjuán, J. J. Palfrey, H. Park, S. Schaertl, M. G. Taylor, D. Weddell and C. Dominguez, Biorg. Med. Chem., 2011, 19, 5833–5851 CrossRef CAS PubMed.
- D. Ma, W. Wu, G. Yang, J. Li, J. Li and Q. Ye, Bioorg. Med. Chem. Lett., 2004, 14, 47–50 CrossRef CAS PubMed.
- P. L. Ornstein, M. B. Arnold, N. K. Augenstein and J. W. Paschal, J. Org. Chem., 1991, 56, 4388–4392 CrossRef CAS.
- A. M. M. Mjalli, D. Jones, D. R. Gohimmukkula, G. Huang, J. Zhu, M. Rao, R. C. Andrews, and T. Ren, Benzazole derivatives, compositions, and methods of use as b-secretase inhibitors, WO2006099379A2, Transtech Pharma, Inc., 2006.
- S. Seiwert, L. Beigelman, B. Buckman, V. Serebryany, and A. D. Stoycheva, Preparation of proline tripeptides and analogs as inhibitors of hepatitis C virus replication for treating hepatitis C infection and liver fibrosis, WO2010045266A1, InterMune, Inc., USA, 2010.
- D. S. Karanewsky, J. Fotsing, C. Tachdjian, and M. Arellano, Identification of human T2R receptors that respond to bitter compounds that elicit the bitter taste in compositions, and the use thereof in assays to identify compounds that inhibit (block) bitter taste in compositions and use thereof, WO2011106114A1, Senomyx, Inc., USA, 2011.
- S. Ishiguro, N. Kawaguchi, M. Nakakoshi, S. Shimada, M. Seya, M. Nomoto, M. Okue, and H. Tomizuka, Preparation of isoquinoline derivatives as protease inhibitors, JP07304744A, Snow Brand Milk Prod Co Ltd, Japan, 1995.
- S. Chen, Y. Li, P. Ni, B. Yang, H. Huang and G.-J. Deng, J. Org. Chem., 2017, 82, 2935–2942 CrossRef CAS PubMed.
- T. Shamim, M. Gupta and S. Paul, J. Mol. Catal. A: Chem., 2009, 302, 15–19 CrossRef CAS.
- J. Dong, X.-X. Shi, J.-J. Yan, J. Xing, Q. Zhang and S. Xiao, Eur. J. Org. Chem., 2010,(36), 6987–6992 CrossRef CAS.
- A. S.-K. Tsang, P. Jensen, J. M. Hook, A. S. K. Hashmi and M. H. Todd, Pure Appl. Chem., 2011, 83, 655–665 CrossRef CAS.
- S. Höck, R. Marti, R. Riedl and M. Simeunovic, CHIMIA International Journal for Chemistry, 2010, 64, 200–202 CrossRef.
- L. A. Stoddart, L. E. Kilpatrick and S. J. Hill, Trends Pharmacol. Sci., 2018, 39, 136–147 CrossRef CAS PubMed.
- M. A. Arruda, L. A. Stoddart, K. Gherbi, S. J. Briddon, B. Kellam and S. J. Hill, Front. Pharmacol., 2017, 8, 908 CrossRef PubMed.
- B. Franck, G. Blaschke and G. Schlingloff, Angew. Chem., 1963, 75, 957–965 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See \DOI: 10.1039/c7ra13148h |
| This journal is © The Royal Society of Chemistry 2018 |