
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvolution of cellular morphologies and crystalline structures in high-expansion isotactic polypropylene/cellulose nanofiber nanocomposite foams
Long Wang *a,
Yuta Hikimaa,
Masahiro Ohshima*a,
Takafumi Sekiguchib and
Hiroyuki Yanoc
*a,
Yuta Hikimaa,
Masahiro Ohshima*a,
Takafumi Sekiguchib and
Hiroyuki Yanoc
aDepartment of Chemical Engineering, Kyoto University, A4 Building, B1 Floor, Katsura Campus, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: wangl.kevin@cheme.kyoto-u.ac.jp; oshima@cheme.kyoto-u.ac.jp
bNew Business Development Division, SEIKO PMC Corp., Chiba 267-0056, Japan
cResearch Institute for Sustainable Humano-sphere, Kyoto University, Kyoto 611-0011, Japan
First published on 24th April 2018
Abstract
Herein, the development of cell morphology and crystalline microstructure of injection-molded isotactic polypropylene/cellulose nanofiber (PP/CNF) composite foams with 2–10-fold expansion ratios was investigated through scanning electron microscopy (SEM), wide-angle X-ray diffraction (WAXD), and small-angle X-ray scattering (SAXS). Compared with isotactic polypropylene (iPP) foams, the added CNF improved the cell morphology and resulted in a great reduction in cell size. Additionally, the PP lamella orientation and crystal type were notably altered during the core-back FIM process. As the expansion ratio increased, the original isotropic lamellae in the iPP foams were transformed into an oriented lamellar structure and then further transformed into a typical shish-kebab structure, while hybrid shish-kebab structures were simultaneously generated in the high-expansion PP/CNF nanocomposite foams. Accordingly, the highest content of β-crystals was observed in the low-expansion iPP foams. In contrast, the β-crystal content in PP/CNF composites decreased monotonously as the expansion ratio increased, which resulted from the combined effects of CNF's nucleating ability for α-crystals and the more dominant extensional flow effect assisted by the added CNF.
1. Introduction
Understanding the evolution of crystalline morphology and polymer structure during tensile deformation is crucial for manipulating the mechanical properties of polymeric products as well as for possible improvement of the materials. Upon tensile deformation, i.e., stretching deformation, the original lamellar microstructure of the semi-crystalline polymer can be transformed into a highly anisotropic fibrillar morphology.1–5 To explain the transformation of spherulite into a fibrous structure in the tensile deformation of semi-crystalline polymers, two prevailing but clearly different mechanisms have been proposed. One of the proposed mechanisms is that the deformation is achieved by breakage of the original lamellae into small crystal blocks and their subsequent arrangement into an oriented fibrillar structure.1–3 Stress-induced melting and crystallization is proposed as the other mechanism to account for the variations in crystalline morphology in the tensile deformation process.4,5 Moreover, recent studies based on “true stress–strain” experiments revealed that the aforementioned two mechanisms coexist during the tensile deformation of semi-crystalline polymer,6–8 as block slips within the crystalline lamellae occurred first during stretching deformation, and the stress-induced fragmentation and recrystallization of polymeric chains ensued under greater strain.As a widely investigated commercial polymer, isotactic polypropylene (iPP) is known to be a polymorphic material that has four modifications, namely, the monoclinic α-phase, trigonal β-phase, orthorhombic γ-phase, and smectic mesophase.9,10 The α-phase is thermodynamically stable and dominates under normal processing conditions. In contrast, the β-phase is thermodynamically metastable and can be obtained only under certain specific conditions such as crystallization in a temperature field,11 in a shear field,12–14 and in the presence of a β-crystal nucleating agent.15–17 Due to the differences in their supermolecular structures, iPP with α-phase crystals (α-iPP) exhibits a high modulus and high tensile strength but inferior toughness, while iPP with β-phase crystals (β-iPP) shows excellent toughness with relatively low strength.13,18 Because of the polymorphic nature of iPP, many studies have been carried out to investigate the variation in their morphology and structure during the deformation processes. These studies can be roughly divided into two types. One is studying the deformation-induced structural evolution of iPP with no β-crystal nucleating agent.12,13,19 The other is investigating the deformation behavior of iPP in the presence of a β-crystal nucleating agent.15–17,20
Microcellular foam injection molding (FIM), which uses a supercritical fluid such as supercritical carbon dioxide or nitrogen as a physical blowing agent, can manufacture lightweight plastic products with complex, three-dimensional geometries with low material usage and low energy consumption.21–23 In addition to being lightweight, due to the presence of cellular structures, such foam products usually reveal additional benefits, such as excellent energy absorption and good acoustic and thermal insulation in comparison with those of their solid counterparts.23–26 However, compared with those of batch or extrusion foams, injection-molded foams usually display a lower void fraction or foam expansion ratio, which is attributed to the fixed mold geometry in the regular FIM machines. To overcome this shortage, foam injection molding with core-back operation has been introduced, which can substantially increase the void fraction due to its expanded mold cavity volume during the injection molding process.22,27–30 Many studies have explored the foaming behavior and cell structures of various polymers prepared by FIM with core-back operation.22,27–33 It should be mentioned that core-back operation, which is similar to the regular drawing-induced deformation process, not only affects the evolution of cell structure but also influences the transition of crystal structure in the foam. However, to the best of the author's knowledge, no study has been conducted on the evolution of the polymer crystal structure during the core-back foaming process.
Owing to the weak melt strength of iPP, several approaches such as cross-linking,34 branching,28,32,35 blending,23 and nanoparticle addition22,24,33,36 have been investigated as ways to modify iPP to possess adequate melt strength and foaming processability. Cellulose nanofiber (CNF),37 which is derived from abundant and renewable resources, has highly promising applications as a cell nucleating agent and a reinforcing agent for plastic foams. The presence of internal hydrogen bonding provides CNF with superior mechanical properties such as high strength, an increased modulus, and a low coefficient of thermal expansion as well as a low density.38 In regard to microcellular foaming technology, however, only a few attempts have been made to fabricate polymeric foams containing CNF, and these efforts were carried out mainly through batch foaming.39–42 Additionally, the use of supercritical fluid can decrease the melt viscosity of the polymer during processing because of the plasticizing effect of the dissolved gas and thus can enhance the processability of polymer/CNF composites. The resultant low viscosity could reduce the shear stress exerted on the fiber during processing, contributing to reduced fiber breakage and the preservation of long fiber length and high aspect ratios.42 This behavior is especially helpful in the processing of composites containing natural fibers such as CNF, as it would offset the lack of thermal stability. Despite the great benefits, very little work has been conducted on the FIM of polymer foams mixed with CNF.
Recently, we have shown the feasibility of preparing PP/CNF composite foams with various contents of CNF using a core-back FIM technique.43 It was found that an optimum concentration of CNF existed, approximately 5 wt%, which displayed fine cellular structures and high mechanical performance. However, the relationships among the CNF content, expansion ratio, cellular morphology and crystalline microstructure in PP/CNF composite foams are yet to be determined. To investigate these relationships, injection-molded PP/CNF composite foams with 2–10-fold expansion ratios were prepared in this study. The maximum expansion ratio was much higher than that in our previous research (5-fold).20 The ensuing composite foams were characterized in terms of cell morphology, degree of orientation, crystal forms, crystallinity, and crystal size.
2. Experimental section
2.1. Materials
The iPP, grade F133A, used in this study was supplied by the Prime Polymer Corp., Tokyo, Japan. It has a melt flow rate (230 °C/2.16 kg) of 3.0 g/10 min and a weight-average molecular mass of 379 kg mol−1.To improve the dispersion of CNF in PP, the CNF was modified by alkenyl succinic anhydride (ASA) as described previously.43,44 In short, needle-leaf bleached kraft pulp, a derivative of cellulose, was modified using ASA in an N-methyl-2-pyrrolidone solvent. The hydrophobicity was controlled by adjusting the degree of substitution (DS). In this study, hydrophobic cellulose with a DS of 0.4 was used. Then, the PP/CNF composites were prepared using the following steps: mixing, kneading, drying, and melt extruding in a twin-screw extruder, followed by pelletizing for the FIM. During these processes, modified cellulose was defibrillated in an iPP matrix with shear force, which resulted in good dispersion in the PP resin.44 A 17 wt% PP/CNF masterbatch was pre-prepared by blending with pure iPP using a twin-screw extrusion process (KZW15-TW; Technovel Corp., Osaka, Japan), and the detailed preparation process can be found in our previous studies.43,44 Nitrogen (N2) purchased from Izumi Sangyo, Tokyo, Japan, with a purity of 99.9% was employed as the physical foaming agent.
2.2. Sample preparation
The foaming experiments were conducted with a 35 tonnage-clamping force injection mold machine (J35EL III-F, Japan Steel Work, Tokyo, Japan) equipped with a Trexel gas dosing system (SCF device SII TRJ-10-A-MPD, Trexel Inc., Showa Tansan, Japan). For the composites, the PP/CNF masterbatch was dry-blended with neat iPP, and a final concentration of 5 wt% was used.43 A mold with a rectangular cavity (70 mm × 50 mm) and an initial thickness of 1 mm was used.To prepare foams with different expansion ratios, FIM with core-back operation was applied. The polymer/N2 single phase mixture was injected and filled into the mold cavity, and the mold cavity was then quickly opened after a given dwelling time, which significantly expanded the cavity volume and reduced the pressure of the polymer injected into the mold. Foaming was induced by the rapid pressure decrease, and high-expansion foams could be prepared by increasing the expanded cavity volume. The manipulation of the core-back distance made it possible to prepare foam products with different expansion ratios. The details of core-back FIM operation have been described previously.26,32,43 The optimum dwelling time (core-back timing) was individually obtained for the neat iPP and PP/CNF composites for the studied processing conditions listed in Table 1.
| Processing variables | Values |
|---|---|
| Melt temperature (°C) | 190 (iPP), 210 (PP/CNF) |
| Mold temperature (°C) | 40 |
| Core-back rate (mm s−1) | 20 |
| Core-back distance (mm) | 1, 4, 6, 9 |
| Dwelling time (s) | 2.8 (iPP), 3.0 (PP/CNF) |
| Shot size (cm3) | 6.5 |
| Packing (dwelling) pressure (MPa) | 60 |
| Injection speed (mm s−1) | 70 |
| Injection pressure (MPa) | 180 |
| N2 content (wt%) | 0.2 |
2.3. Foam characterization
The microstructures of the foam samples were examined using a scanning electron microscope (Tiny-SEM Mighty-8, Technex Lab Co., Ltd., Tokyo, Japan). The specimens were cut from the middle of the foam samples and cryogenically fractured in liquid nitrogen, which were described in detail in our previous work.22 Prior to SEM observation, the fractured surfaces were coated with gold for 1 min using a VPS-020 Quick Coater (ULVAC KIKO, Ltd., Japan). Then, the cell density, N0, was calculated from the SEM images using the following expression:22,43
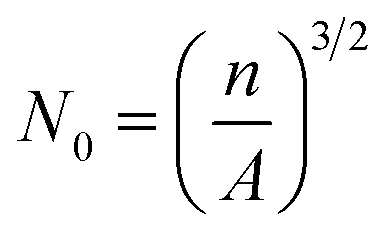 | (1) |
The open cell content (OCC) of the foam samples was measured using a gas pycnometer (AccuPycII, Shimadzu, Kyoto, Japan) under nitrogen atmosphere. The measured volume of the gas pycnometer excluded the specimen's open cell volume, and then the OCC was calculated by eqn (2).
 | (2) |
2.4. Two-dimensional wide-angle X-ray diffraction measurements
Two-dimensional wide-angle X-ray diffraction (2D WAXD) measurements were performed at beamline BL15U1 of the Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, China). The monochromated X-ray wavelength was 0.124 nm, and the distance from the specimen to the detector was 136 mm. An X-ray CCD detector (model SX165, Rayonix Co. Ltd., USA) with a resolution of 2048 × 2048 pixels (pixel size = 80 μm) was used for the detection of 2D WAXD images. It should be mentioned that the middle part of the injection-molded products was cut with a 1.0 mm thick piece for the 2D WAXD tests.33,47 The specimens were placed with the core-back direction perpendicular to the projection beams.Linear WAXD profiles were obtained from the circularly integrated intensities of the 2D WAXD patterns after background correction. The intensity profile was a function of the scattering vector, q, where q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ/λ, 2θ is the scattering angle, and λ is the wavelength of the incident beam. Then, the overall crystallinity Xc could be calculated by deconvoluting the peaks within the linear WAXD profiles as follows:45
sinθ/λ, 2θ is the scattering angle, and λ is the wavelength of the incident beam. Then, the overall crystallinity Xc could be calculated by deconvoluting the peaks within the linear WAXD profiles as follows:45
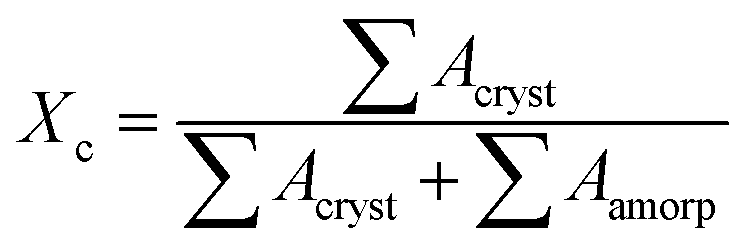 | (3) |
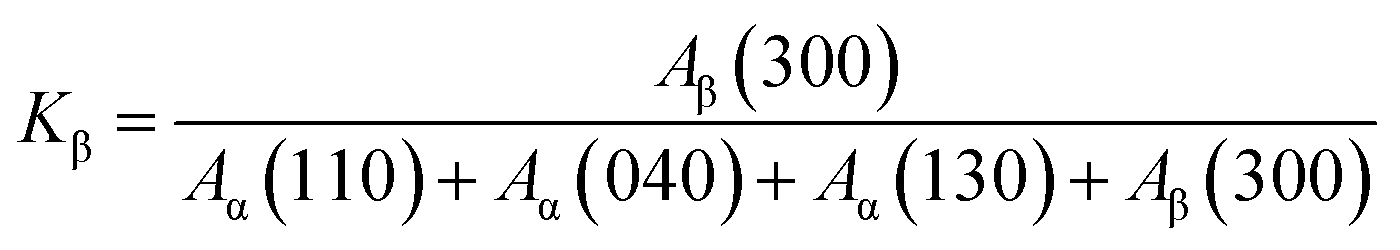 | (4) |
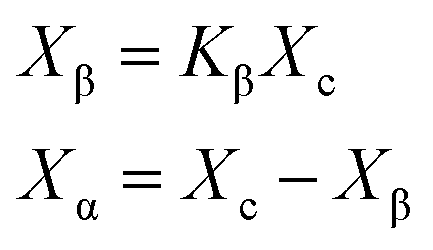 | (5) |
The azimuthal scanning profile was obtained by integrating the diffraction intensities azimuthally at a specific crystal reflection. Then, the orientation parameters, f, of the lamellar crystals in the FIM samples can be estimated by Herman's orientation function:45
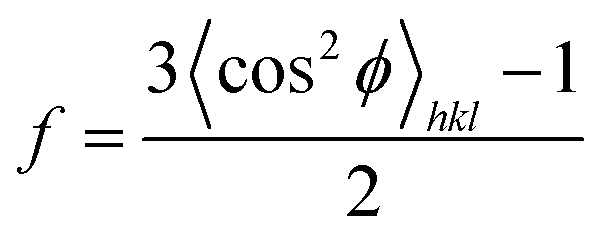 | (6) |
ϕ〉hkl can be defined as
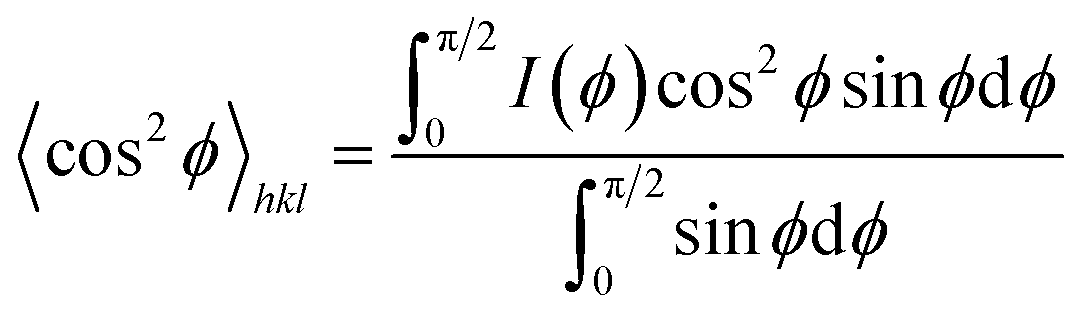 | (7) |
ϕ〉hkl is obtained using the Wilchinsky's method48 from the reflections of (110) and (040):
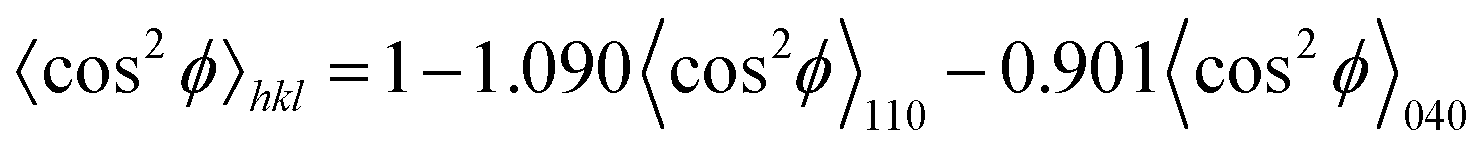 | (8) |
2.5. Small-angle X-ray scattering measurements
Small-angle X-ray scattering (SAXS) measurements were also carried out on beamline BL16B1 of the Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, China). An X-ray CCD detector (Model Mar165) with a resolution of 2048 × 2048 pixels was used to collect the SAXS images. The selected X-ray wavelength was 0.124 nm with a beam size of 80 × 80 μm2, and the sample-to-detector distance was kept at 2150 mm.3. Results and discussion
3.1. Cell morphology
Fig. 1 shows the overall cell morphology and the magnified microstructure of the core part of foam samples with a 2-fold expansion ratio (50% void fraction). The images illustrate the cell morphology of injection-molded foams in a cross-sectional area from a view parallel to the core-back direction. As expected, the FIM foams exhibited a cellular core layer and two solid (non-foamed) layers (approximately 350 μm in thickness). As shown in Fig. 1b, relatively large cells inevitably appeared in the core layer of the pure iPP foams due to its low melt elasticity and poor melt strength.22,32,33,43 The calculated average cell size and cell density of the 2-fold iPP foams were approximately 52 μm and 3.8 × 106 cells per cm3, respectively, which was consistent with the results of our previous work.32,43 Fig. 1c and d present SEM images of the PP/CNF composite foams, revealing that the addition of CNF greatly improved the cellular structures of PP. It was found that a higher cell density of approximately 4.6 × 108 cells per cm3 was achieved for the PP/CNF composite foams, reaching an increase of approximately 120 times compared with that of neat iPP. This substantial improvement in the foaming ability was certainly caused by the inclusion of CNF. In our previous work,33,43 the crystallization and rheological results showed that the added CNF worked not only as an effective crystal nucleating agent but also as a rheological modifier for PP, which would strongly affect the foaming behavior.49 Specifically, the formation of large numbers of smaller-sized crystals in the composites, because of the heterogeneous nucleating effect of CNF, would generate more bubble nucleation sites and greatly expedite the cell nucleation.50–52 Similarly, the CNF itself could also act as a cell nucleating agent and again enhance the bubble nucleation power. On the other hand, the presence of CNF in the PP matrix improved its melt strength and led to restricted cell growth and reduced cell coalescence. These effects were evident from the variation in cell sizes in the PP/CNF composite foams (Fig. 1d). Upon the addition of CNF, the average cell sizes were remarkably reduced from 52 μm in the neat iPP foams to 10.7 μm in the PP/CNF composite foams. | ||
| Fig. 1 SEM micrographs of (a) neat iPP and (c) PP/CNF composite foams. (b) and (d) are enlarged images of the inner regions of (a) and (c), respectively. The core-back direction is the horizontal. | ||
Fig. 2 illustrates the cellular structures of the iPP and PP/CNF composite foams prepared at various expansion ratios. It should be noted that the core-back distance, i.e., the distance by which the movable mold part was shifted, was changed from the original 1 mm to the final 10 mm to obtain the 10-fold foams, and foams with other expansion ratios were prepared correspondingly. SEM images of the core layer of the foams were taken from the view parallel to the core-back direction, and the optimum core-back timing was selected for each specimen. As shown in Fig. 2a, cells in the 5-fold expansion iPP foams became elliptical and deformed along the core-back direction, which was attributed to the extensional stress induced during the mold-opening operation.32,33,43 With increasing expansion ratio, the cells were further deformed and oriented along the core-back direction. The cells were greatly stretched in the 10-fold iPP foams. Additionally, as shown in Fig. 2c′, some tiny holes (bubbles) were generated in the deformed cell wall, indicating that stretching-induced secondary nucleation occurred during the core-back operation for high-expansion foams. This secondary nucleation was due to the rupture of the thinning cell wall during the extended stretching process.22,32,33 Interestingly this core-back operation could transform the closed cells of the low-expansion foams into open cells in the high-expansion foams.
 | ||
| Fig. 2 SEM images of (a) 5-fold iPP foams, (b) 7-fold iPP foams, (c) 10-fold iPP foams, (d) 5-fold PP/CNF composite foams, (e) 7-fold PP/CNF composite foams, (f) 10-fold PP/CNF composite foams. (c′) and (f′) are enlarged images of (c) and (f), respectively. The core-back direction is the horizontal. | ||
Compared with the iPP foams, a similar tendency of cell evolution with expansion ratio was observed in the PP/CNF composite foams, but the formation of much smaller cells accompanied the cell evolution. At the highest expansion ratio of 10-fold, the cells in the PP/CNF composites were severely deformed and regularly aligned in the core-back direction. Interestingly, in comparison with the neat iPP foams, large quantities of smaller cells were produced in the 10-fold PP/CNF composite foams, and a highly interconnected cellular structure was obtained, as shown in Fig. 2f′. To evaluate the degree of stretching-induced nucleation (small holes), the open cell content (OCC) was measured and is illustrated in Fig. 3. The OCC of the 2-fold injection-molded samples is almost zero and is not shown here. For the measurements, the solid (non-foamed) skin layer is removed and not considered in the OCC calculation. Generally, the OCC values of both the neat iPP and the PP/CNF composite foams were remarkably enhanced with the increase of the expansion ratio. This enhancement occurred because the cells were seriously deformed and stretched by the strong extensional force exerted by the mold-opening operation, and the resultant cell walls were apt to be thinner at the higher expansion ratios,32,33,43 which could easily cause secondary cell nucleation (stretching-induced cell nucleation) and initiate cell opening. Moreover, the added CNF markedly raised the OCC of the PP foams, and a very high OCC of approximately 94% was achieved in the 10-fold PP/CNF composite foams, which was due to the substantial enhancement of cell nucleation with the presence of CNF and the subsequent thinning of the cell wall. Therefore, these results showed that the added CNF acts not only as a cell nucleating agent but also as a cell opening agent, which might provide a viable method to produce large-scale open foams for potential use in areas such as acoustic insulation and oil spill cleanup.
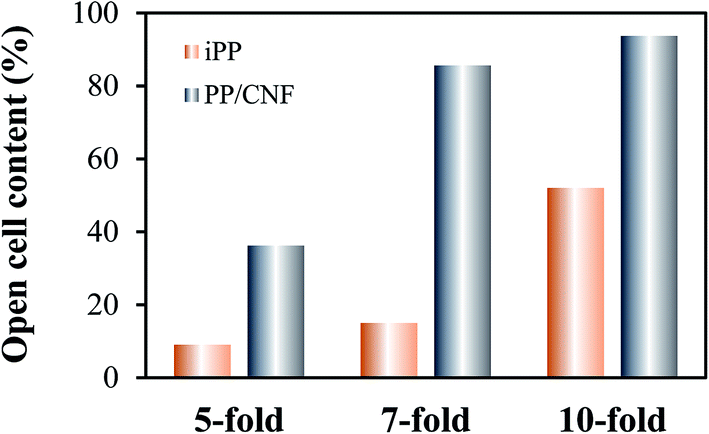 | ||
| Fig. 3 Open cell content of the neat iPP and PP/CNF composite foams as a function of the expansion ratio. | ||
3.2. Crystalline structure
Since the cell morphology was greatly changed during the core-back operation, it is important to study the crystalline evolution of the core-back FIM products. For this purpose, we first investigated the crystal structure and the molecular orientation of the solid (non-foamed) iPP and PP/CNF composites. Fig. 4 shows the 2D WAXD patterns of the FIM samples, viewed from the directions both parallel and perpendicular to the core-back direction. Typically, the 2D WAXD patterns of all specimens showed six diffraction reflections of α(110), β(300), α(040), α(130), α(131)/β(311), and α(041) from the inner to the outer circles, revealing that the injection-molded products contained α- and β-crystals.16,47 As shown in Fig. 4a and d, typical arclike diffractions were discerned in the skin layer of the solid iPP and the PP/CNF composites in the view parallel to the melt flow direction, viz., the direction perpendicular to the core-back direction, which indicated the existence of highly oriented lamellae in this layer.16,45 In contrast, in the core layer of these solid specimens (Fig. 4b and e), the diffraction patterns became isotropic diffraction circles, suggesting a random distribution of lamellae.14,53 Moreover, the 2D WAXD patterns in the core layer perpendicular to the flow direction, i.e., the direction parallel to the core-back direction, were also studied (Fig. 4c and f) and again showed isotropic diffraction circles, indicating that a random distribution of lamellae was generated in the inner layer of the non-foamed injection-molded samples.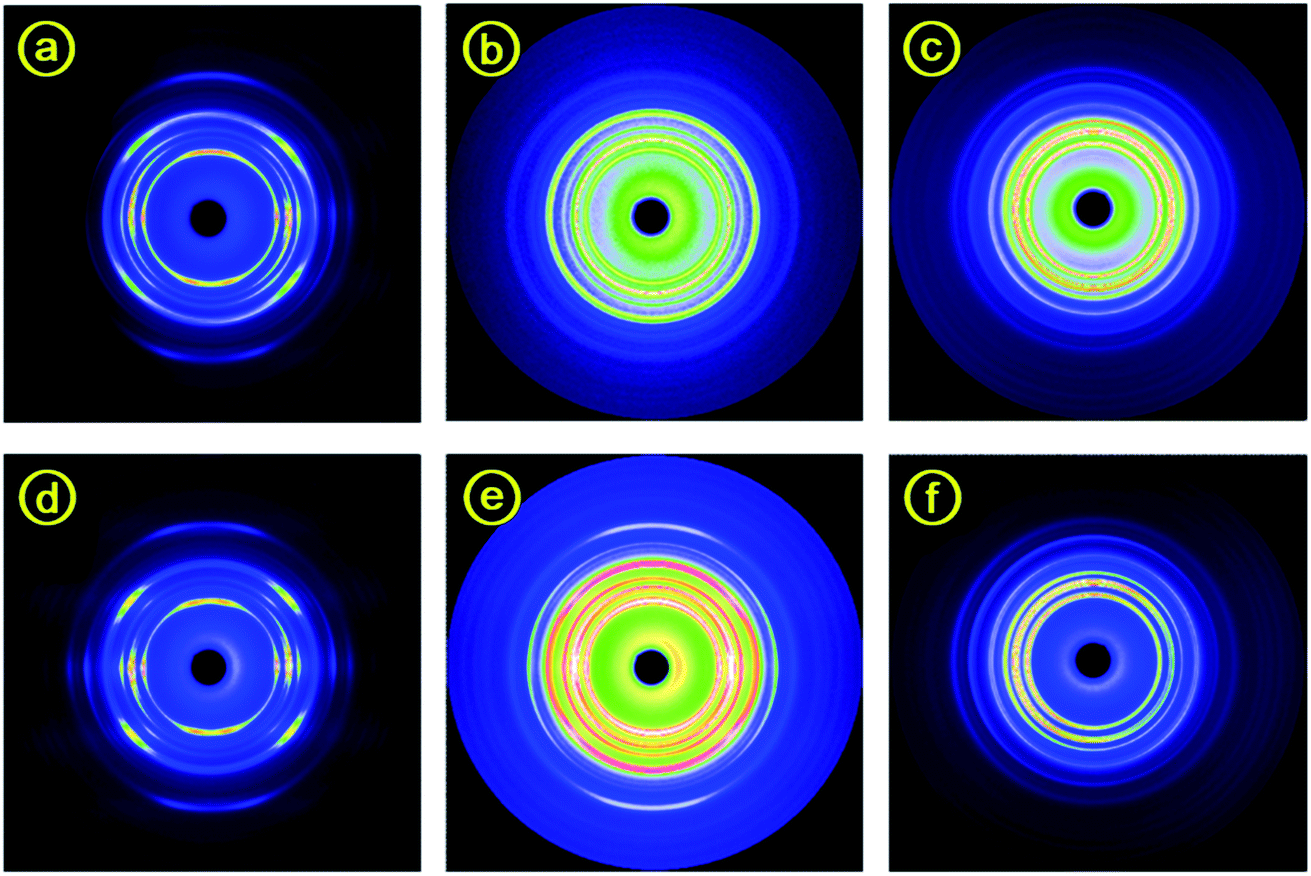 | ||
| Fig. 4 Typical selected 2D WAXD patterns for (a–c) solid (non-foamed) iPP, and (d–f) PP/CNF composites. (a and d) and (b and e) Show the view parallel to the flow melt direction from the skin and core layer, respectively. (c and f) Show the view perpendicular to the flow melt direction (parallel to the core-back direction) from the core layer. | ||
Fig. 5 shows representative 2D WAXD diffraction patterns at the core layer of injection-molded foams observed from a view parallel to the core-back direction. At a low 2-fold expansion ratio, as shown in Fig. 5a, mainly isotropic diffraction circles were observed for the neat iPP foams. Then, the diffraction circles of the iPP foams were transformed into arclike diffractions at a higher 5-fold expansion ratio, indicating that the lamellae were preferentially aligned in the core-back direction. Moreover, the arclike diffractions became sharper and narrower as the expansion ratio increased from 5- to 7- and 10-fold, which showed that a higher degree of orientation was achieved with high expansion. This result might be ascribed to the formation of a shish-kebab structure in the high-expansion foams.14,53 The evolution of the 2D WAXD patterns of the PP/CNF composites with the increase in the expansion ratio showed changes largely similar to those of the iPP foams.
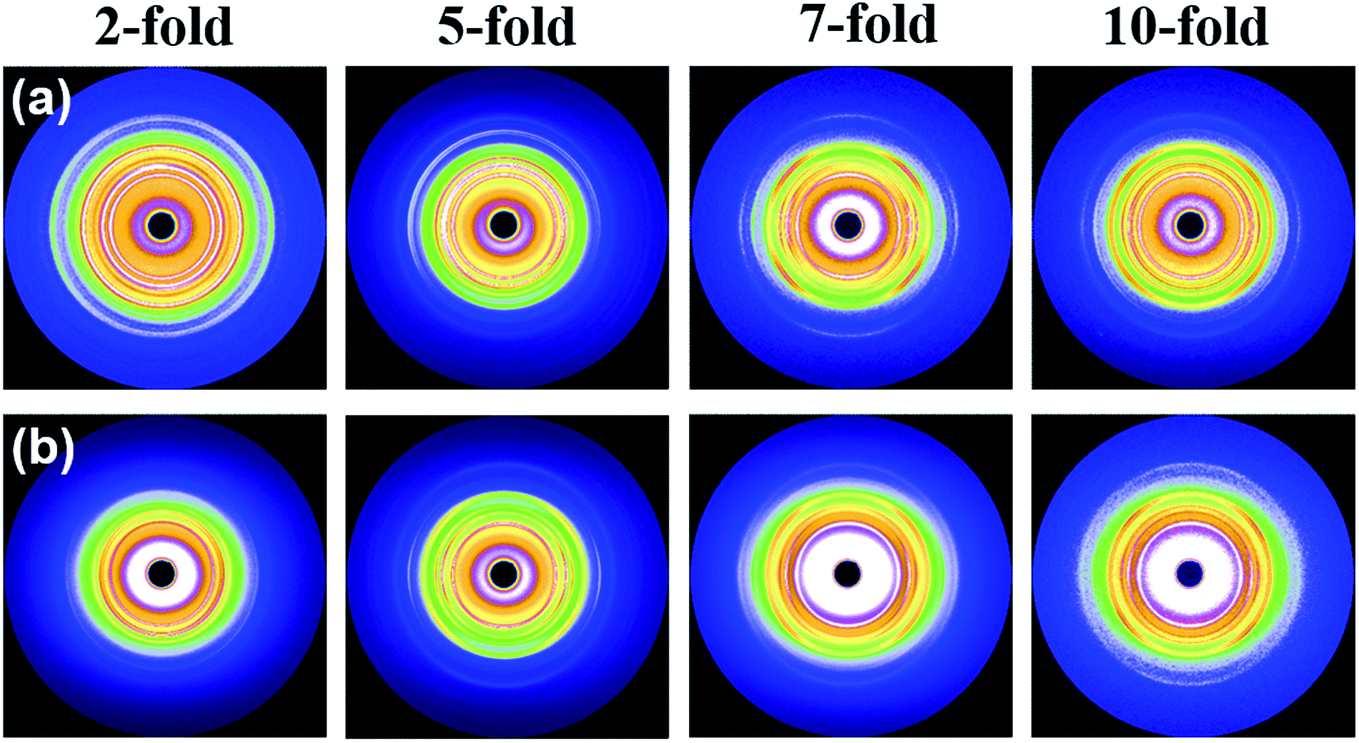 | ||
| Fig. 5 2D WAXD patterns of (a) neat iPP foams and (b) PP/CNF composite foams prepared at different expansion ratios. The core-back direction is the vertical. | ||
3.3. Oriented structure
The degree of orientation, f, of the neat iPP and that of the PP/CNF composite samples are shown in Fig. 6. For high-expansion foams (7- and 10-fold), the degree of orientation was very high, approximately 0.85, demonstrating that the orientation axes were preferentially parallel to the core-back direction.14,45,54 These highly oriented lamellar structures (shish-kebab structure) were mainly attributed to the intense extensional force applied through the shifting of the movable mold during the core-back operation. Under the extensional stress, the molecular chains tended to form stretched bundles, i.e., the precursors of the shish structure, which would promote the epitaxial growth of folded chain lamellae upon cooling and subsequently produce the shish-kebab structure.14,45,54 In contrast, at the low 2-fold expansion ratio, the core layer mainly consisted of isotropic crystals, which might be due to the weak extensional flow field as well as the rapid relaxation of the oriented nuclei. As Fig. 6 shows, a clear difference in the orientation parameter between iPP and the PP/CNF composite was observed at the middle expansion ratio of 5-fold, with values of 0.32 for the iPP and 0.81 for the PP/CNF composite foams. This phenomenon could be related to the added CNF, which contributes to the formation of oriented crystal structures. It should be noted that during the core-back operation, in addition to the macroscopic mechanical force, the cell growth of the foaming process would apply an extensional force to the PP molecule as well as the CNF.22,31–33 By assuming that the macroscopic mechanical force was largely equivalent when the expansion ratio was the same, the presence of CNF had additional benefits for the final oriented superstructure. On the one hand, the presence of CNF would promote the formation of extra oriented nuclei or crystals due to the anchoring effect of the CNF surface on the oriented nuclei or crystals as well as the stress amplification of extensional flow with the addition of CNF. The stress amplification effect is usually observed in polymer/filler composites.55–57 On the other hand, the added CNF benefited the preservation of more flow-induced oriented structures, ascribing the suppressing effect of CNF on the relaxation of the oriented molecular chains due to the enhanced molecular relaxation time.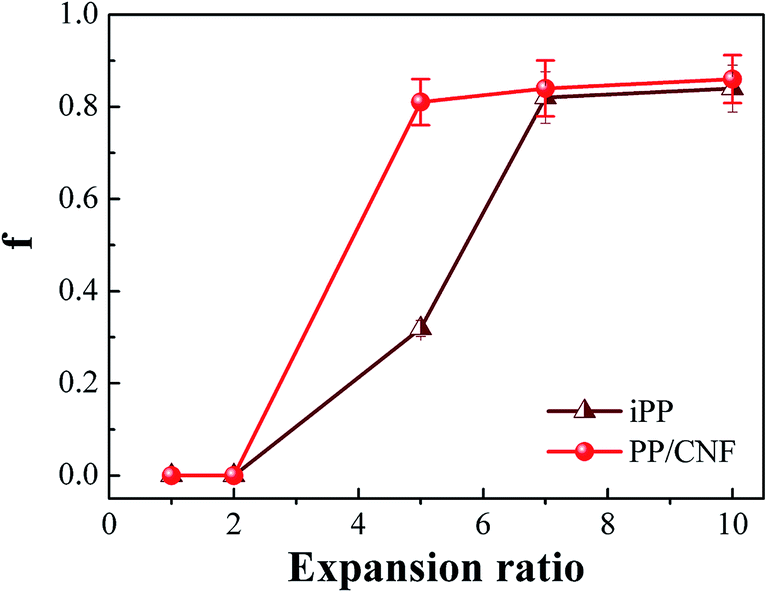 | ||
| Fig. 6 The orientation parameters of the neat iPP samples and PP/CNF composites. | ||
To further clarify the development of crystalline superstructure in the core-back FIM process, all the samples were characterized by SAXS measurements. Fig. 7 illustrates the typical SAXS patterns of the iPP and PP/CNF composite foams. The core-back direction was the vertical, which was considered to be the same as the fiber axis by assuring that the deformed scatters have a fiber symmetry.45,47,54 For the 7- and 10-fold foams, a weak equatorial streak and strong meridional maxima could be observed, which evidenced the existence of a shish-kebab structure with the shish aligned in the core-back direction and the kebab oriented perpendicularly to the core-back direction through epitaxial growth.14,45,54 Compared with the neat iPP, the PP/CNF composite foams showed a more intensive streak normal to the mold-opening direction, revealing a large-size or high-volume fraction of shish in the presence of CNF, which might be due to the role of CNF as a shish. In our recent paper, we revealed that the CNF was well-aligned along the cell wall in the core-back direction for 18-fold PP/CNF composite foams, and subsequent hybrid shish-kebab with CNF as a shish and classic shish-kebab were simultaneously obtained in the composite.33 Since similar 2D WAXD and SAXS diffraction patterns were obtained,33 it was reasonable to say that the classic shish-kebab and hybrid shish-kebab were also formed in the 7- and 10-fold PP/CNF composite foams. The 5-fold expansion foams showed again that more oriented structures were generated in the PP/CNF composites than in the iPP, which was consistent with the above 2D WAXD results. However, Fig. 7a and a′ show that no obvious scattering patterns were detected in the 2-fold foams of both iPP and PP/CNF composites, showing that the randomly distributed lamellae dominated in the low-expansion foams.45,54 Hence, the combined results of 2D WAXD and SAXS confirmed the existence of a shish-kebab structure in the core layers of high-expansion injection-molded foams, which was attributed to the strong stretching force applied during the core-back operation. The presence of CNF further promoted the formation of oriented lamellae and shish-kebab structure due to the more dominant extensional flow effect assisted by the added CNF.
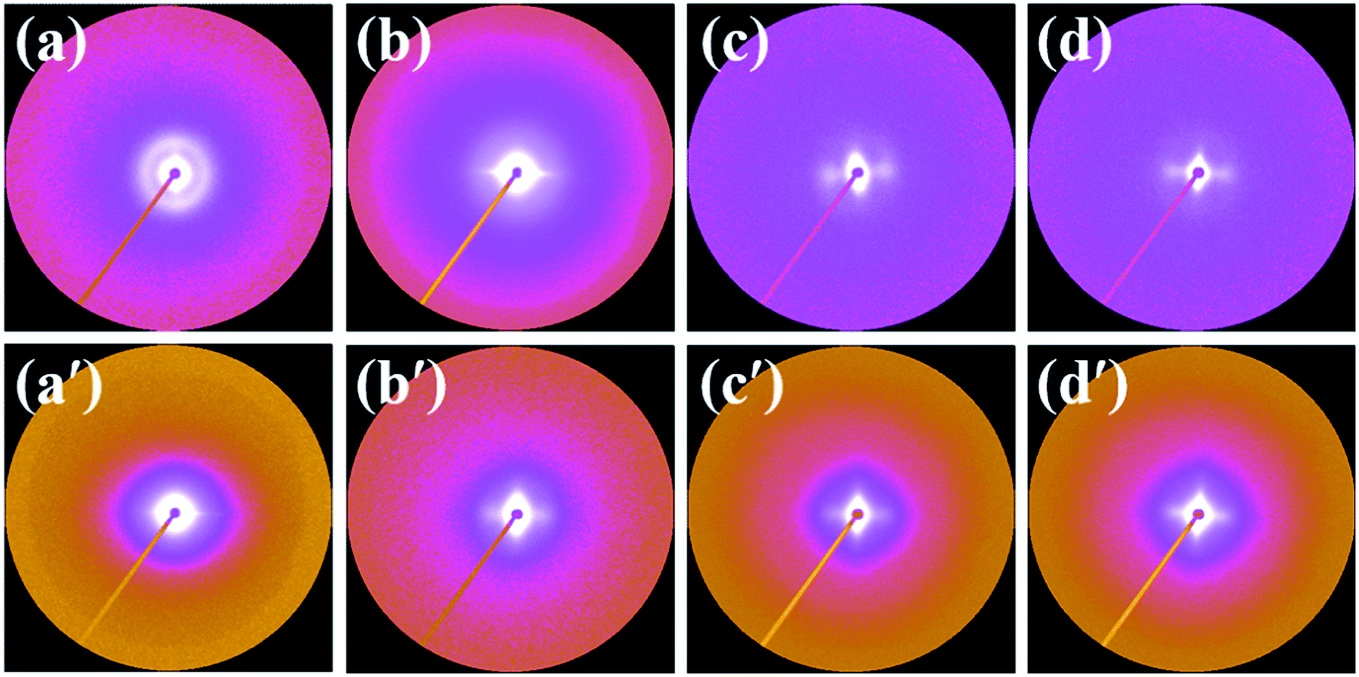 | ||
| Fig. 7 SAXS patterns of (a–d) neat iPP foams, and (a′–d′) PP/CNF composite foams prepared at expansion ratios of (a and a′) 2-fold, (b and b′) 5-fold, (c and c′) 7-fold, and (d and d′) 10-fold, respectively. The core-back direction is the vertical. | ||
3.4. Crystal forms and crystallinity
Fig. 8 shows the effect of the expansion ratio on the changes in the linear WAXD profiles of the injection-molded samples. The diffraction peaks detected at 2θ = 14.1°, 16°, 16.8°, 18.6°, 21.1°, and 21.8° were clearly observed for the solid (non-foamed) sample and corresponded to the α(110), β(300), α(040), α(130), α(131)/β(311), and α(041) reflections, respectively.14,47 The changes in the profiles as a function of the expansion ratio were also investigated. Compared with that of the solid (non-foamed) sample, the relative diffraction intensity of the β(300) plane in the iPP foams increased first at a 2-fold expansion ratio and then weakened gradually as the expansion ratio was increased further. Moreover, the presence of CNF seems to undermine the formation of β-crystals but contributes to the generation of α-crystals, and the relative diffraction intensity of the β(300) plane decreased as the expansion ratio increased.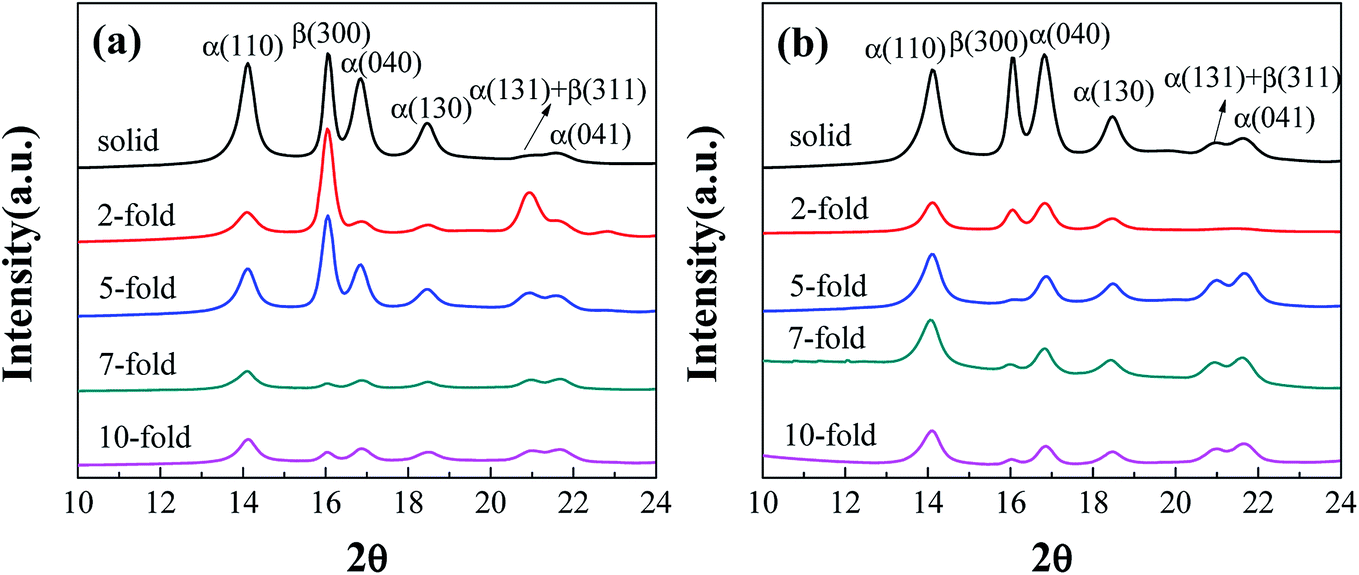 | ||
| Fig. 8 Linear WAXD intensity profiles of (a) neat iPP and (b) PP/CNF composite foams obtained from the 2D WAXD patterns. | ||
The relative content of β-crystal, Kβ, in the FIM samples is shown in Fig. 9, which revealed a dependency on the expansion ratio. For comparison, the non-foamed specimen was illustrated, which corresponded to a sample with an expansion ratio of 1. For the iPP samples, Kβ first increased from 0.24 to 0.66 as the expansion ratio increased from 1 (non-foamed) to 2-fold expansion foam. This might be due to the introduction of a relatively weak extensional flow, which would promote the α- to β-phase transformation. Although no obviously oriented lamellae were present in the 2-fold expansion foams, some oriented α-nuclei might form due to the applied extensional stress. The oriented α-nuclei would trigger the epitaxial growth of β-crystals, and thereby a high β-crystal content could be achieved.12,13,17 However, the Kβ of the iPP sample decreased with a further increase in the expansion ratio, revealing that with extensive stretching, the α-nuclei weakened the epitaxial growth of β-crystals, which was consistent with other studies regarding the crystal form changes of PP under high tensile force.15,16,47 Furthermore, the Kβ of the PP/CNF composites was lower than that of the iPP samples and decreased gradually with the increase in the expansion ratio, probably resulting from the combined suppression effects of the added CNF and of the high extensional force on the formation of β-crystals.
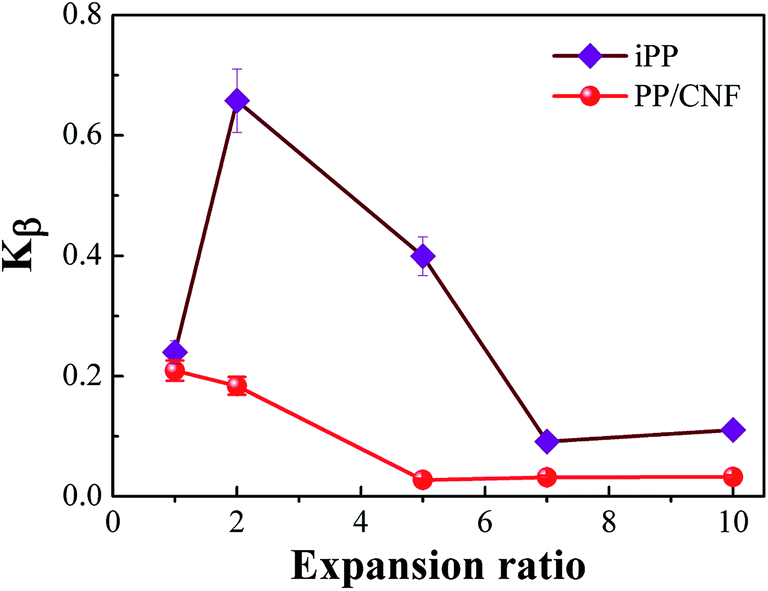 | ||
| Fig. 9 The relative amount of β-crystals of PP as a function of the expansion ratio for the neat iPP and PP/CNF composites. | ||
Fig. 10 depicts the total crystallinity and the crystallinity of α- and β-crystals of neat iPP and PP/CNF composites. As shown in Fig. 10a, it was demonstrated that the total crystallinity of the neat iPP increased slightly after foaming, which might be due to the expedited crystallization caused by the dissolved gas as well as the biaxial stretching force during foaming.31,42 For the foamed samples, the total crystallinity changed only a little and remained largely the same. Similarly, for the PP/CNF composites, the total crystallinity also increased marginally after foaming and then varied little across the various FIM foams. However, the changes to the expansion ratio and the addition of CNF greatly changed the distribution of α- and β-crystals in the prepared FIM samples.
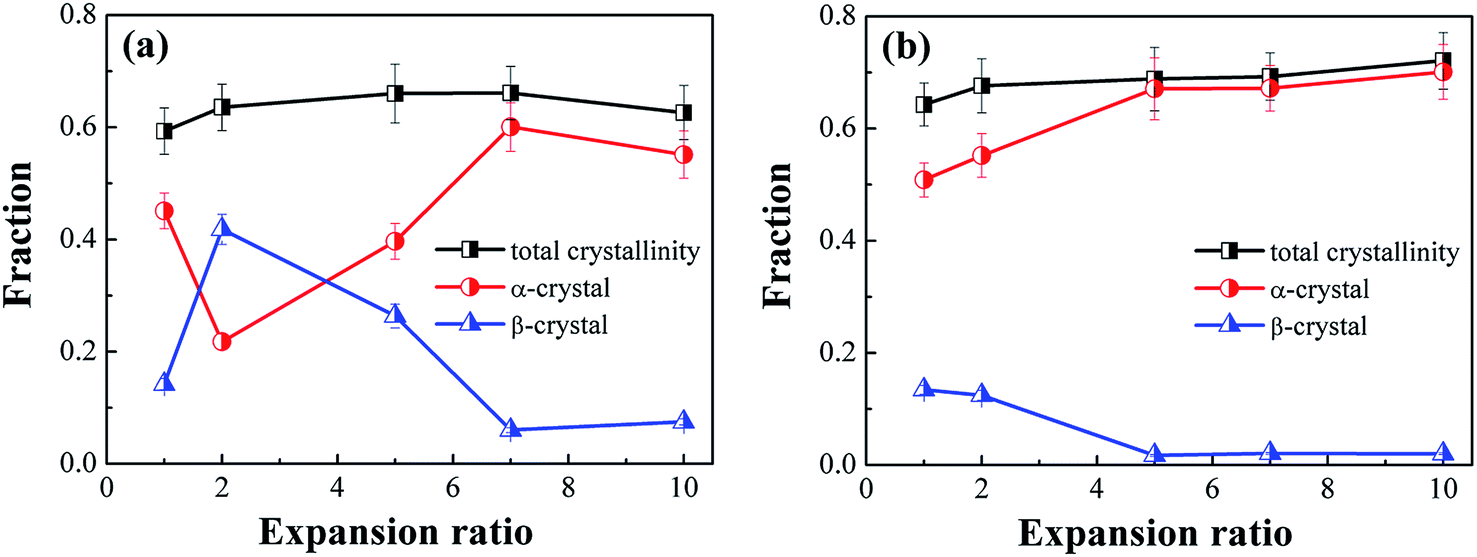 | ||
| Fig. 10 Evolution of α-crystal content, β-crystal content, and total crystallinity in the core layer of (a) neat iPP and (b) PP/CNF composites. | ||
For the non-foamed iPP, the degrees of crystallinity of the α- and β- crystals were 0.45 and 0.14, respectively, revealing that the α-phase crystals prevailed in the inner region of the solid injection-molded product. This result was consistent with the crystal form distribution in the regular injection molding process, as reported by other studies.14,58–60 After foaming, however, the 2-fold iPP foams demonstrated an adverse distribution of α- and β- crystals, displaying a high β-crystal content but a low content of α-crystals. This variation in the crystal forms was likely induced by the dissolved gas and/or additional extensional force exerted during foaming. To differentiate the effects of these two factors, injection-molded samples with dissolved gas but no mold-opening were prepared, which showed that the contents of α- and β-crystals were 0.51 and 0.11, respectively. Thus, it was reasonable to deduce that the imposition of extensional force rather than the dissolved gas had a dominant effect on the distribution of α- and β-crystals. Moreover, as the expansion ratio was further increased, the content of β-crystals in the iPP foams decreased and subsequently reached a low value at a high expansion ratio, manifesting the tendency toward the formation of α-crystals under the intensive stretching conditions. It is reported that under the flow field, the oriented α-nuclei are generated first, and afterward β-crystals grow on the surface of the α-nuclei.12,13,17,47 Under a weak extensional flow, the α-crystals are easily transformed into β-crystals, while a high extensional flow restricts the transformation to β-crystals and thereby results in α-crystals dominating the high-expansion iPP foams.12,13,15,17
However, the variation in β-crystals in PP/CNF composites was different from that in the iPP foams. The presence of CNF in the PP matrix leads to a lower content of β-crystals, and this difference was more obvious in the case with both CNF and a high extensional force, which produced a greatly decreased content of β-crystals in the high-expansion PP/CNF composite foams. This might be because CNF acts as an α-crystal nucleating agent for PP, which would be an adverse factor for the formation of β-crystals. Ljungberg et al. studied the crystallization behavior of iPP reinforced with various surface-modified cellulose whiskers and found that a low content of β-crystals was generated in PP with unmodified cellulose, while there were no β-crystals in the whiskers grafted with maleated PP.61 They noted that the more hydrophilic the cellulose surface becomes, the more favorable it is for the formation of β-crystals, which might have resulted from the crystal structure matching between the CNF surface and the PP crystals.61–63 Since a highly hydrophobic CNF was used here43,44 and since no β-crystals were found in our previous crystallization experiments on PP/CNF,43 these results indicated that the currently used hydrophobic CNF was beneficial for the formation of α-crystals rather than β-crystals. Additionally, the presence of CNF might have a stress amplification effect on the extensional flow, which would again benefit the formation of α-crystals, thus endowing PP/CNF composites with a lower content of β-crystals compared with that in their neat counterparts.55–57
3.5. Crystal size
The crystallite size Lhkl in the direction perpendicular to the (hkl) plane could be calculated by analyzing the pattern peaks of the WAXD profiles through the following expression:64|
Lhkl = K × λ/FWHM × cosθ
| (9) |
To examine the combined effect of CNF and core-back (stretching) operation on the lamellar arrangement, the crystal size L110 in the direction perpendicular to the α(110) plane was calculated and is shown in Fig. 11. This calculation revealed that the L110 of both neat iPP and PP/CNF composites tended to decrease with increasing expansion ratio. As is well known, the induced flow field increases the nucleation densities and promotes the growth rate of crystallites, leading to smaller crystal sizes.65–67 For the foams, a stronger extensional field was concurrent with the increased expansion ratio, which resulted in smaller crystal sizes for high expansion ratio foams. Moreover, at the same expansion ratio, CNF further reduced the crystal size, which was ascribed to the extra nucleating effect of CNF. Since there are more primary nuclei and nucleating sites in a constricted space, the growth of lamellae was prone to be inhibited in the PP/CNF composites and thereby produced smaller crystallites. Therefore, compared with the neat iPP samples, the PP/CNF composites always exhibited a smaller crystal size.
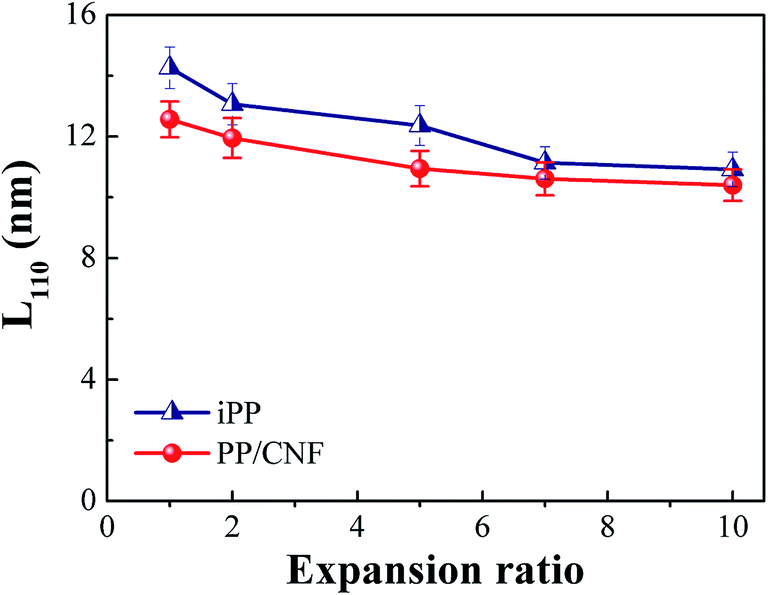 | ||
| Fig. 11 The crystal size L110 in the direction perpendicular to the α(110) plane as a function of the expansion ratio for the iPP and PP/CNF composites. | ||
3.6. Evolution mechanism of hierarchical structures
Based on the experimental results, a schematic diagram describing the effect of the expansion ratio on the crystal evolution in the inner region of the neat iPP and PP/CNF composites is depicted in Fig. 12. Before the start of core-back operation, the iPP and PP/CNF composites were already maintained in the cavity for 2.8 and 3.0 s, respectively, while cooling and crystallization occurred, which was caused by the cold mold (approximately 30 °C). The foaming temperature was monitored and recorded as 107 and 113 °C for the neat iPP and PP/CNF, respectively. From our previous fast scanning chip calorimetry (FSC) measurement,32,43 it is reasonable to say that the primary crystallization of PP had already commenced before the start of the core-back operation. However, the crystallization was far from completed, or else the high expansion ratio would not have been achieved because the melt would have been too stiff after full crystallization. Thus, some crystal nuclei or precursors were generated and remained in the polymer melt before the onset of the core-back manipulation.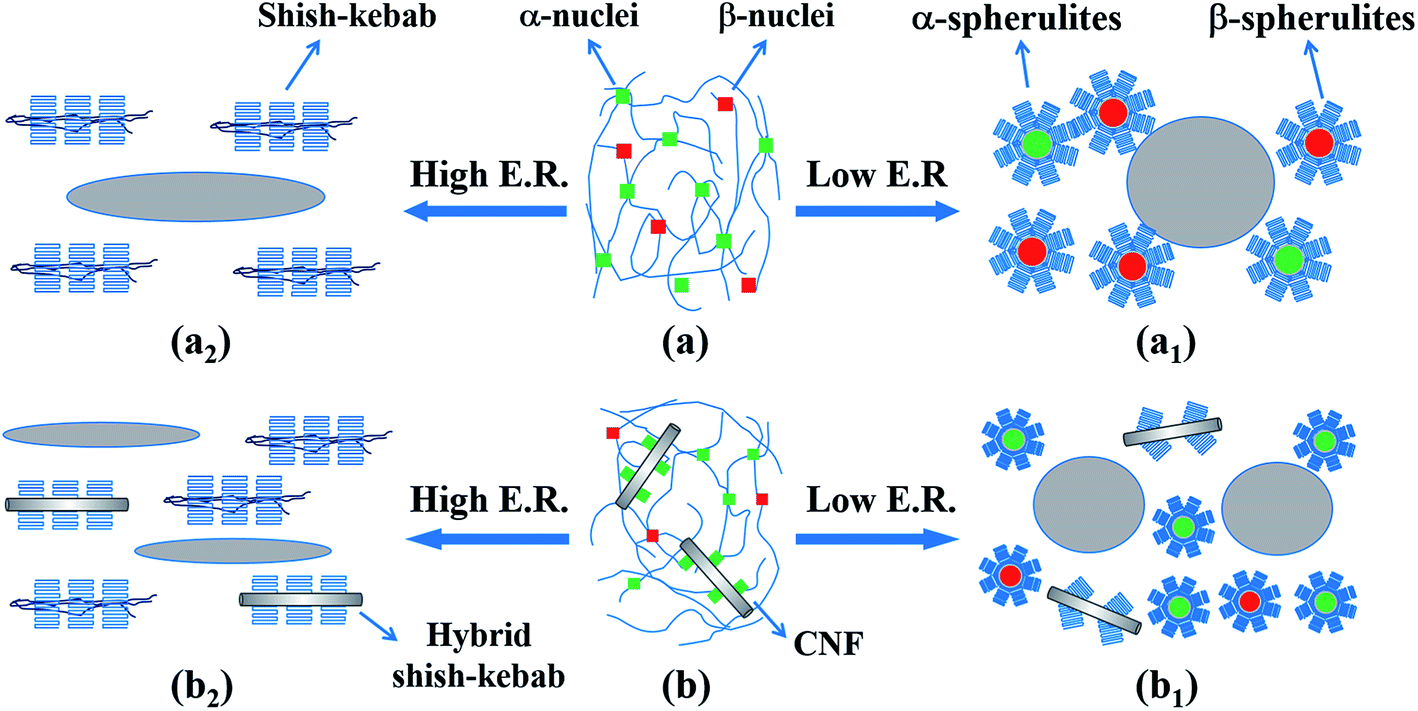 | ||
| Fig. 12 A schematic diagram showing the effect of expansion ratio (E. R.) on structural evolution in the inner region of (a) neat iPP and (b) PP/CNF composites. For clarity, the size of each indicator is not proportional to the real size. | ||
Generally, during the injection molding process, the molten polymer was subjected to a flow field, whereas the molecular chains were oriented along the melt flow direction during the melt filling stage.14,58–60 Even the final crystalline structure of the solid (non-foamed) specimen exhibited isotropic crystals, as some molecular chains in the core layer at relatively high temperature would be initially oriented along the flow direction. Thus, some oriented α-precursors were generated in the inner region and subsequently triggered the growth of β-nuclei.12,13,17,47 Fig. 12a and b illustrate the melt state immediately before the commencement of core-back operation for the neat iPP and PP/CNF composites, showing that α-nuclei dominated in the melt with smaller amounts of β-nuclei. For neat iPP foams at low expansion ratios, the relatively weak stretching force exerted by the mold-opening operation notably promoted the formation of β-crystals, and therefore β-crystals predominated in the prepared foams even though the main crystals were isotropic (Fig. 12a1). In addition, the high-expansion iPP foams were characterized by a shish-kebab structure due to the high extensional force (Fig. 12a2). This intensive stretching also reduced the content of β-crystals.12,15,16 At medium expansion ratios such as 5-fold, some preferentially oriented lamellae (kebab) were generated in the iPP foams with a medium content of β-crystals, which is not shown in the schematic for brevity. Regarding the PP/CNF composites, due to the nucleating ability of the hydrophobic CNF for α-crystals,61–63 α-crystals dominated even in the low-expansion PP/CNF composite foams. At high extensional forces, the CNF was also oriented and aligned along the core-back direction in addition to the oriented molecular chains, and subsequently hybrid shish-kebab and regular shish-kebab were simultaneously generated in the high-expansion PP/CNF composite foams. Since the crystal forms and crystalline structures strongly affect the mechanical and physical properties of the injection-molded foams, further studies on these factors will be conducted in the future.
4. Conclusion
Neat iPP and PP/CNF composite foams with expansion ratios as high as 10-fold were prepared using a core-back FIM process. It was found that the presence of CNF notably enhanced the foamability of PP, resulting in foams with much smaller cell sizes and higher cell densities. Moreover, the SEM results showed that the cellular structures were strongly dependent on the expansion ratio, and the cells were highly deformed and aligned along the core-back direction with increasing expansion ratio, which triggered highly fibrillated cellular structures at the maximum expansion of 10-fold. Furthermore, the evolution of the orientation, crystal form, crystallinity, and crystal size also depended heavily on the expansion ratio. The main crystalline morphology in the inner region of the FIM products transformed from the isotropic crystals of the low-expansion foams into the kebab structure of the medium-expansion foams and then to a shish-kebab structure for samples with a high expansion ratio, which was caused by the enhanced extensional force concurrent with the increased expansion ratio.Additionally, foaming endowed the 2-fold iPP foams with the highest content of β-crystals, which might be due to the relatively weak flow induced during the core-back operation and the resultant assistance in the transformation of α-crystals to β-crystals. In contrast, a high expansion ratio always weakened the generation of β-crystals and led to low β-crystal concentrations in high-expansion iPP foams, probably because a too high flow field undermined the formation of β-crystals. However, the β-crystal content in PP/CNF composites decreased monotonously as the expansion ratio increased, which was attributed to the combined suppression effects of the presence of CNF and a more dominant induced flow field. This study reveals that core-back operation strongly affects both the evolution of cell morphology and the crystalline structure of the injection-molded foams, which provides some tips for the application of injection-molded foams for structural components, especially when mechanical performance is a priority.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was conducted in associated with Grants-in-Aid for Scientific Research (B), number: 26289289 of the Japan Society for Promotion of Science (JSPS).References
- A. Peterlin and F. J. Baltá-Calleja, J. Appl. Phys., 1969, 40, 4238–4242 CrossRef CAS.
- A. Peterlin, J. Mater. Sci., 1971, 6, 490–508 CrossRef CAS.
- A. Peterlin, Colloid Polym. Sci., 1987, 265, 357–382 CAS.
- P. J. Flory and D. Y. Yoon, Nature, 1978, 272, 226–229 CrossRef CAS.
- W. Wu, G. D. Wignal and L. Mandelkern, Polymer, 1992, 33, 4137–4140 CrossRef CAS.
- Z. Jiang, Y. Tang, J. Rieger, H. F. Enderle, D. Lilge, S. V. Roth, R. Gehrke, Z. Wu, Z. Li and Y. Men, Polymer, 2009, 50, 4101–4111 CrossRef CAS.
- Y. Men, J. Rieger and G. Strobl, Phys. Rev. Lett., 2003, 91, 095502 CrossRef PubMed.
- R. Hiss, S. Hobeika, C. Lynn and G. Strobl, Macromolecules, 1999, 32, 4390–4403 CrossRef CAS.
- J. Varga, J. Macromol. Sci., Phys., 2002, 41, 1121–1171 CrossRef.
- B. Lotz, J. Wittmann and A. Lovinger, Polymer, 1996, 37, 4979–4992 CrossRef CAS.
- A. J. C. J. Lovinger and C. C. Grythe, J. Polym. Sci., Part B: Polym. Phys., 1977, 15, 641–656 CrossRef CAS.
- R. H. Somani, B. S. Hsiao, A. Nogales, H. Fruitwala, S. Srinivas and A. H. Tsou, Macromolecules, 2001, 34, 5902–5909 CrossRef CAS.
- J. Varga and J. Karger-Kocsis, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 657–670 CrossRef CAS.
- L. Wang and M. B. Yang, RSC Adv., 2014, 4, 25135–25147 RSC.
- Y. H. Chen, S. Yang, H. Q. Yang, G. J. Zhong, D. F. Fang, B. S. Hsiao and Z. M. Li, Polymer, 2016, 84, 254–266 CrossRef CAS.
- R. Y. Bao, Z. T. Ding, Z. Y. Liu, W. Yang, B. H. Xie and M. B. Yang, Polymer, 2013, 54, 1259–1268 CrossRef CAS.
- H. Huo, S. C. Jiang, L. J. An and J. C. Feng, Macromolecules, 2004, 37, 2478–2483 CrossRef CAS.
- S. C. Tjong, J. S. Shen and R. K. Y. Li, Polymer, 1996, 37, 2309–2316 CrossRef CAS.
- R. Bao, Z. Ding, G. Zhong, W. Yang, B. Xie and M. Yang, Colloid Polym. Sci., 2012, 290, 261–274 CAS.
- Z. Cai, Y. Zhang, J. Li, F. Xue, Y. Shang, X. He, J. Feng, Z. Wu and S. Jiang, Polymer, 2012, 53, 1593–1601 CrossRef CAS.
- S. Leduc and D. Rodrigue, Cell. Polym., 2005, 24, 313–327 CAS.
- R. Miyamoto, S. Yasuhara, H. Shikuma and M. Ohshima, Polym. Eng. Sci., 2014, 54, 2075–2085 CAS.
- X. Sun, H. Kharbas, J. Peng and L. S. Turng, Polymer, 2015, 56, 102–110 CrossRef CAS.
- J. Hou, G. Zhao, G. Wang, G. Dong and J. Xu, Mater. Des., 2017, 127, 115–125 CrossRef CAS.
- D. Jahani, A. Ameli, P. U. Jung, M. R. Barzegari, C. B. Park and H. Naguib, Mater. Des., 2014, 53, 20–28 CrossRef CAS.
- L. Wang, Y. Hikima, S. Ishihara and M. Ohshima, Polymer, 2017, 128, 119–127 CrossRef CAS.
- A. Ameli, D. Jahani, M. Nofar, P. U. Jung and C. B. Park, Compos. Sci. Technol., 2014, 90, 88–95 CrossRef CAS.
- A. N. J. Spörrer and V. Altstädt, J. Cell. Plast., 2007, 43, 313–330 CrossRef.
- J. Zhao, Q. Zhao, C. Wang, B. Guo, C. B. Park and G. Wang, Mater. Des., 2017, 131, 1–11 CrossRef CAS.
- J. Zhao, Q. Zhao, L. Wang, C. Wang, B. Guo, C. B. Park and G. Wang, Eur. Polym. J., 2018, 98, 1–10 CrossRef CAS.
- A. Ameli, M. Nofar, D. Jahani, G. Rizvi and C. B. Park, Chem. Eng. J., 2015, 262, 78–87 CrossRef CAS.
- L. Wang, S. Ishihara, M. Ando, A. Minato, Y. Hikima and M. Ohshima, Ind. Eng. Chem. Res., 2016, 55, 11970–11982 CrossRef CAS.
- L. Wang, S. Ishihara, Y. Hikima, M. Ohshima, T. Sekiguchi, A. Sato and H. Yano, ACS Appl. Mater. Interfaces, 2017, 9, 9250–9254 CAS.
- W. T. Zhai, H. Y. Wang, J. Yu, J. Y. Dong and J. S. He, Polym. Eng. Sci., 2008, 48, 1312–1321 CAS.
- S. J. Lee, L. Zhu and J. Maia, Polymer, 2015, 70, 173–182 CrossRef CAS.
- P. J. Gong, P. Buahom, M. P. Tran, M. Saniei, C. B. Park and P. Pötschke, Carbon, 2015, 93, 819–829 CrossRef CAS.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- J. Dlouhá, L. Suryanegara and H. Yano, Soft Matter, 2012, 8, 8704–8713 RSC.
- S. Y. Cho, H. H. Park, Y. S. Yun and H. J. Jin, Macromol. Res., 2013, 21, 529–533 CrossRef CAS.
- J. Dlouhá, L. Suryanegara and H. Yano, React. Funct. Polym., 2014, 85, 201–207 CrossRef.
- W. D. Ding, T. Kuboki, A. Wong, C. B. Park and M. Sain, RSC Adv., 2015, 5, 91544–91557 RSC.
- L. Wang, M. Ando, M. Kubota, S. Ishihara, Y. Hikima, M. Ohshima, T. Sekiguchi, A. Sato and H. Yano, Composites, Part A, 2017, 98, 166–173 CrossRef CAS.
- H. Yano, A. Sato, T. Yoshimura, Y. Igarashi, D. Kabusaki, F. Nakatsubo, et al., US pat. 9512304, December 6, 2016.
- R. H. Somani, L. Yang, B. S. Hsiao, T. Sun, N. V. Pogodina and A. Lustiger, Macromolecules, 2005, 38, 1244–1255 CrossRef CAS.
- A. Turner-Jones, J. M. Aizlewood and D. R. Beckett, Makromol. Chem., 1964, 75, 134–158 CrossRef CAS.
- Y. H. Chen, G. J. Zhong, Y. Wang, Z. M. Li and L. B. Li, Macromolecules, 2009, 42, 4343–4348 CrossRef CAS.
- Z. W. Wilchinsky, J. Appl. Phys., 1960, 31, 1969–1972 CrossRef CAS.
- K. Taki, D. Kitano and M. Ohshima, Ind. Eng. Chem. Res., 2011, 50, 3247–3252 CrossRef CAS.
- J. H. Han and C. D. Han, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 743–761 CrossRef CAS.
- S. N. Leung, C. B. Park and H. Li, Plast., Rubber Compos., 2006, 35, 93–100 CrossRef CAS.
- A. S. Wong, T. Y. Guo and C. B. Park, J. Supercrit. Fluids, 2013, 79, 142–151 CrossRef CAS.
- L. Wang and M. B. Yang, Polym. Int., 2014, 63, 1513–1522 CrossRef CAS.
- R. H. Somani, L. Yang, L. Zhu and B. S. Hsiao, Polymer, 2005, 46, 8587–8623 CrossRef CAS.
- Y. H. Chen, G. J. Zhong, J. Lei, Z. M. Li and B. S. Hsiao, Macromolecules, 2011, 44, 8080–8092 CrossRef CAS.
- J. Y. Wang, J. J. Yang, L. Deng, H. G. Fang, Y. Q. Zhang and Z. G. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 1364–1375 CAS.
- H. Tang, J. B. Chen, Y. Wang, J. Z. Xu, B. S. Hsiao, G. J. Zhong and Z. M. Li, Biomacromolecules, 2012, 13, 3858–3867 CrossRef CAS PubMed.
- M. R. Kantz, H. D. Newman and F. H. Stigale, J. Appl. Polym. Sci., 1972, 16, 1249–1260 CrossRef CAS.
- D. R. Fitchmun and Z. Mencik, J. Polym. Sci., Part B: Polym. Phys., 1973, 11, 951–971 CrossRef CAS.
- H. Dragaun, H. Hubeny and H. Muschik, J. Polym. Sci., Part B: Polym. Phys., 1977, 15, 1779–1789 CrossRef CAS.
- N. Ljungberg, J. Y. Cavaillé and L. Heux, Polymer, 2006, 47, 6285–6292 CrossRef CAS.
- D. T. Quillin, D. F. Caulfield and J. A. Koutsky, J. Appl. Polym. Sci., 1993, 50, 1187–1194 CrossRef CAS.
- D. T. Quillin, M. Yin, J. A. Koutsky and D. F. Caulfield, J. Appl. Polym. Sci., 1994, 52, 605–615 CrossRef CAS.
- L. E. Alexander, X-ray diffraction methods in polymer science, Wiley, New York, 1976 Search PubMed.
- G. Kumaraswamy, R. K. Verma, J. A. Kornfield, F. Yeh and B. S. Hsiao, Macromolecules, 2004, 37, 9005–9017 CrossRef CAS.
- I. Dukovski and M. Muthukumar, J. Chem. Phys., 2003, 118, 6648–6655 CrossRef CAS.
- P. W. Zhu, G. Edward and L. Nichols, J. Phys. D: Appl. Phys., 2009, 42, 245406 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |