
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBicyclo[2.2.1]heptane containing N,N′-diarylsquaramide CXCR2 selective antagonists as anti-cancer metastasis agents†
Jin-Xin
Che
a,
Zhi-Long
Wang
b,
Xiao-Wu
Dong
 a,
You-Hong
Hu
b,
Xin
Xie
*bc and
Yong-Zhou
Hu
*a
a,
You-Hong
Hu
b,
Xin
Xie
*bc and
Yong-Zhou
Hu
*a
aZJU-ENS Joint Laboratory of Medicinal Chemistry, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, PR China. E-mail: huyz@zju.edu.cn
bState Key Laborarory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, PR China
cCAS Key Laboratory of Receptor Research, The National Center for Drug Screening, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, PR China. E-mail: xxie@mail.shcnc.ac.cn
First published on 21st March 2018
Abstract
CXCR1 and CXCR2 are CXC chemokine receptors (CXCRs), corresponding to cytokines of the CXC chemokine family. CXCR2 was found to be 77% homologous to CXCR1. Antagonism of the chemokine receptor CXCR2 has been proposed as a new strategy for the treatment of metastatic cancer. In order to find a CXCR2 selective antagonist, a bicyclo[2.2.1]heptane containing N,N′-diarylsquaramide (compound 2e) was identified by introducing a bridge ring system into the N,N′-diarylsquaramide skeleton, and it exhibited good CXCR2 antagonistic activity (CXCR2IC50 = 48 nM) and good selectivity (CXCR1IC50/CXCR2IC50 = 60.4). Furthermore, an in vitro biological assay of compound 2e also demonstrated its good anti-cancer metastatic effect against the pancreatic cancer cell line CFPAC1. In addition, compound 2e showed an extremely high stability in simulated intestinal fluid (SIF) and simulated gastric fluid (SGF), as well as in rat and human plasma, but not in rat and human liver microsomes. In vivo pharmacokinetic studies in rats indicated that 2e has an excellent PK profile (10 mg kg−1 po, Cmax = 2863 ng mL−1, t1/2 = 2.58 h). Moreover, molecular docking was further implemented to propose the preponderant configuration of compound 2e, providing important and useful guidelines for further development.
1. Introduction
CXCR1 and CXCR2 are CXC chemokine receptors (CXCRs) and G-protein coupled receptors, which correspond to cytokines of the CXC chemokine family. CXCR2 was found to be 77% homologous to CXCR1.1,2 The CXC chemokine receptor 1 (CXCR1) and CXC chemokine receptor 2 (CXCR2) can both be activated by chemokine CXCL8 (interleukin-8, IL-8) and granulocyte chemotactic protein-2. Furthermore, CXCR2 can also be activated by several other ELR+ CXC chemokines, including neutrophil-activating peptides and growth related oncogenes (GROα, β and γ).3 Several studies have demonstrated the intimate relationship between CXCR2 and different cancer types.4–9 Here, the blockade of CXCR2 represents an attractive strategy for the treatment of cancer metastasis.5,7,10–19 As shown in Fig. 1, CXCR1 or CXCR2 antagonists such as AZD5069, Danirixin, Reparixin, Ladarixin and SX-682 have entered clinical trials for different indications. Among these compounds, the CXCR2 selective antagonist AZD5069 is currently in phase II clinical trials for the treatment of metastatic head & neck cancer and metastatic pancreatic cancer, administered in combination with MEDI4736 (a PD-L1 inhibitor). In addition, SX-682 is also in phase I clinical trials for the treatment of metastatic melanoma, in combination with pembrolizumab. Therefore, developing novel CXCR2 selective antagonists may provide inspiration in future studies.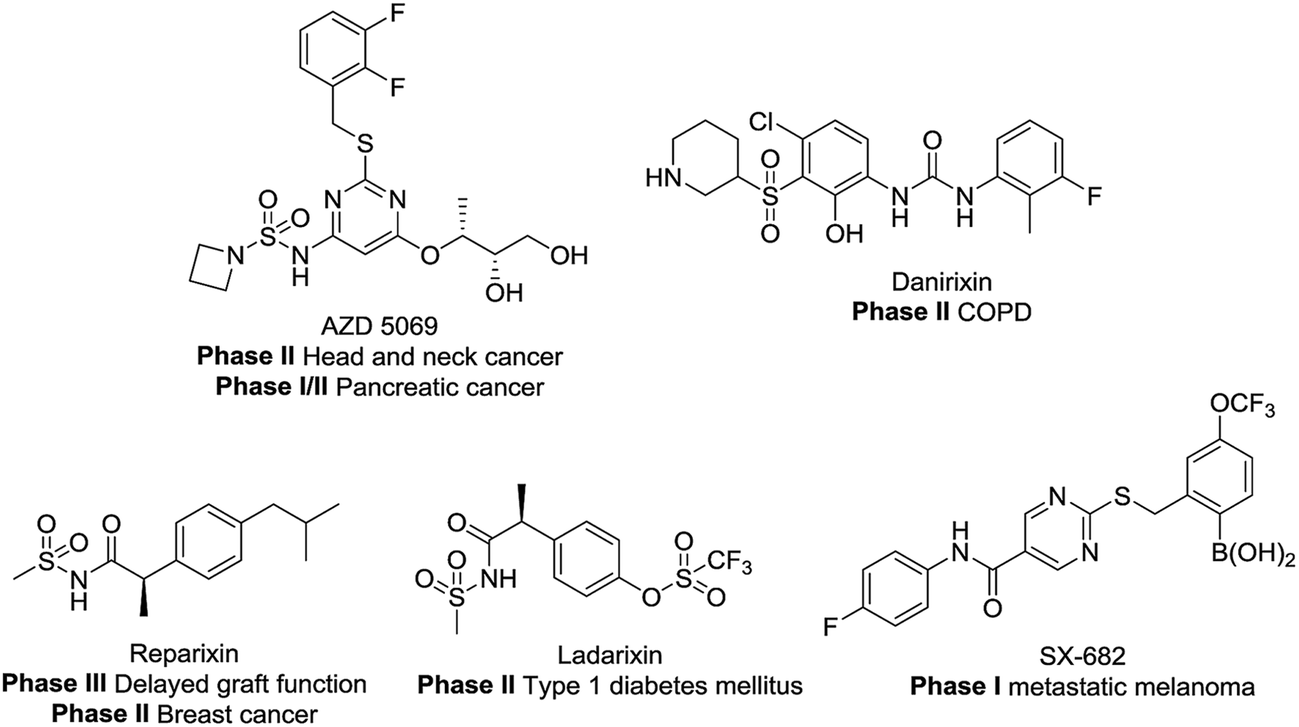 | ||
| Fig. 1 Structures of clinical compounds. | ||
Among numerous structural motifs in clinical compounds, polycyclic hydrocarbon scaffolds offer a variety of diverse features such as a unique three-dimensional shape and hydrophobic and pharmacokinetic properties.20–23 As shown in Fig. 2(A), the use of bicyclic motifs of polycyclic scaffolds, including bicyclo[2.2.1], bicyclo[2.2.2], bicyclo[4.4.1] and bicyclo[5.3.1], appears to be a widely used strategy in drug discovery.20 Furthermore, it is obvious that bridge ring systems play an important role in various pharmaceutical compounds.
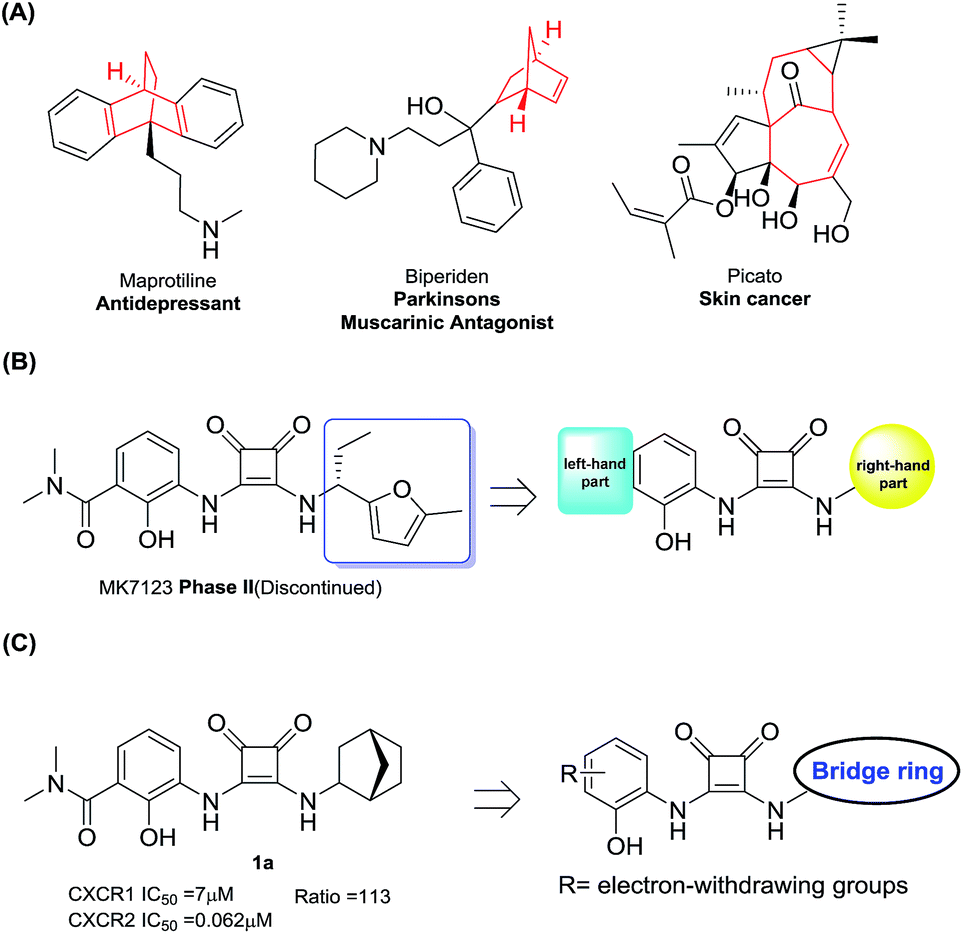 | ||
| Fig. 2 (A) Drugs containing bridge ring systems; (B) the right and left-hand parts of MK7123; (C) compound design rationale. | ||
The N,N′-diarylsquaramide CXCR1/2 antagonist MK7123 (SCH527123) has been discontinued in phase II clinical trials. The indications of MK7123 were chronic obstructive pulmonary disease (COPD), asthma and psoriasis. MK7123 exhibited 10-fold greater selectivity for CXCR1 over CXCR2.24 As shown in Fig. 2(B), due to the high tolerance and hydrophobic characteristics of the right-hand part of the compound,24–26 polycyclic hydrocarbon scaffolds were introduced to explore the CXCR2 selectivity and anti-cancer metastatic effect.
At first, a simple bicyclo[2.2.1] moiety was introduced to afford compound 1a, as shown in Fig. 2(C). Interestingly, compound 1a showed a 113-fold selectivity for CXCR2 over CXCR1 (CXCR1IC50 = 7 μM, CXCR2IC50 = 0.062 μM). In order to develop CXCR2 selective antagonists with much higher antagonistic potentials, promising pharmacokinetic profiles and good anti-cancer metastasis effects, other bridge ring systems were considered. Meanwhile, the introduction of different electron-withdrawing groups to the left-hand part of the structure was also evaluated.
2. Results and discussion
2.1 Chemistry
The synthesis of target compounds 1a–1f is illustrated in Scheme 1. The substituted aminophenol (compound 6) was derived from a commercially available starting material (3-nitrosalicylic acid 3). The acyl chloride containing compound 4 was obtained via an acylation reaction with thionyl chloride, and successive condensation in the presence of dimethylamine hydrochloride afforded the amide compound 5. Subsequently, the aniline compound 6 was acquired via reduction of the nitro-containing compound 5 in a H2 Pd/C system. After this, compound 6 underwent a nucleophilic substitution reaction with 3,4-diethoxycyclobut-3-ene-1,2-dione to provide the intermediate compound 7. Then the ethoxyl group was substituted with different bridge ring amines to provide the final products 1a–1f.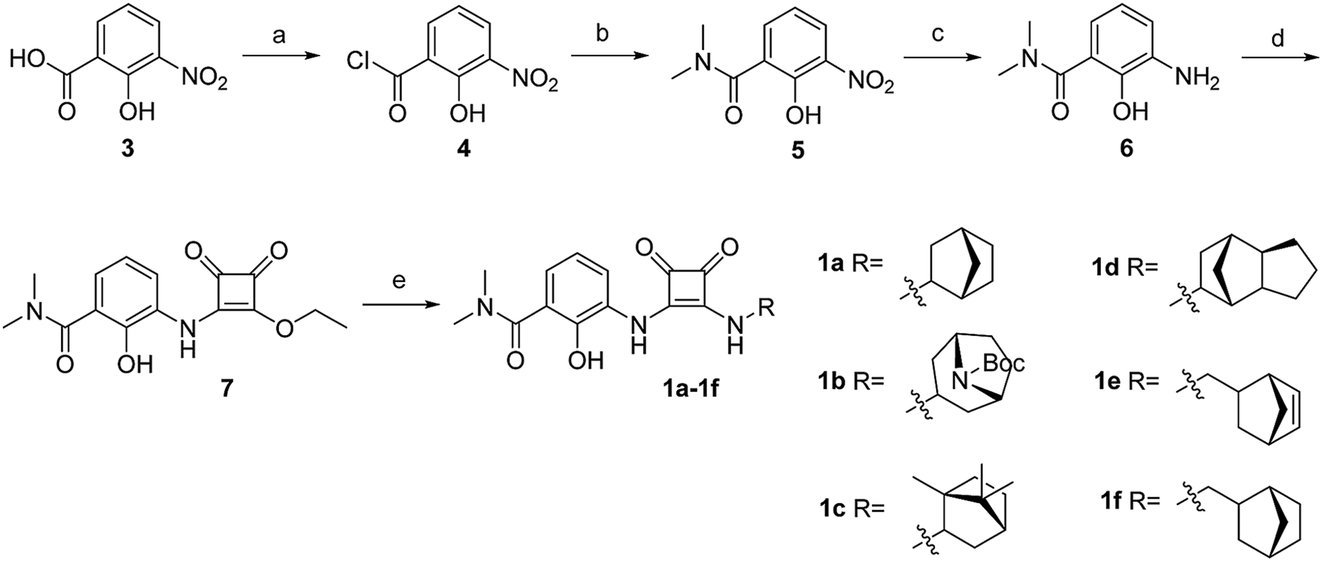 | ||
| Scheme 1 Synthetic route of compounds 1a–1f. (a) SOCl2, dichloromethane; (b) dimethylamine hydrochloride, TEA, dichloromethane; (c) Pd/C, H2, methanol; (d) 3,4-diethoxycyclobut-3-ene-1,2-dione, ethanol; (e) diverse bridge ring amines, ethanol. | ||
The synthesis of target compounds 2a–2h is illustrated in Scheme 2. Compounds 8a–8d with different electron-withdrawing groups were used as starting materials. Compounds 2a–2d were afforded via a 2-step nucleophilic substitution reaction with 3,4-dialkoxycyclobut-3-ene-1,2-dione and bridge-ring amines, respectively. The acyl chloride compound 4 was treated with different amines to provide amide compounds 10a–10c, and these nitryl containing intermediates were subsequently reduced to provide compounds 11a–11c. Compounds 11a–11c underwent a nucleophilic substitution reaction with 3,4-diethoxycyclobut-3-ene-1,2-dione to provide compounds 12a–12c. In the presence of bicyclo[2.2.1]heptan-2-amine, the target compounds 2e–2g were formed. The protecting group (–Boc) of compound 2g was easily removed using HCl saturated ethyl acetate to afford the target compound 2h.
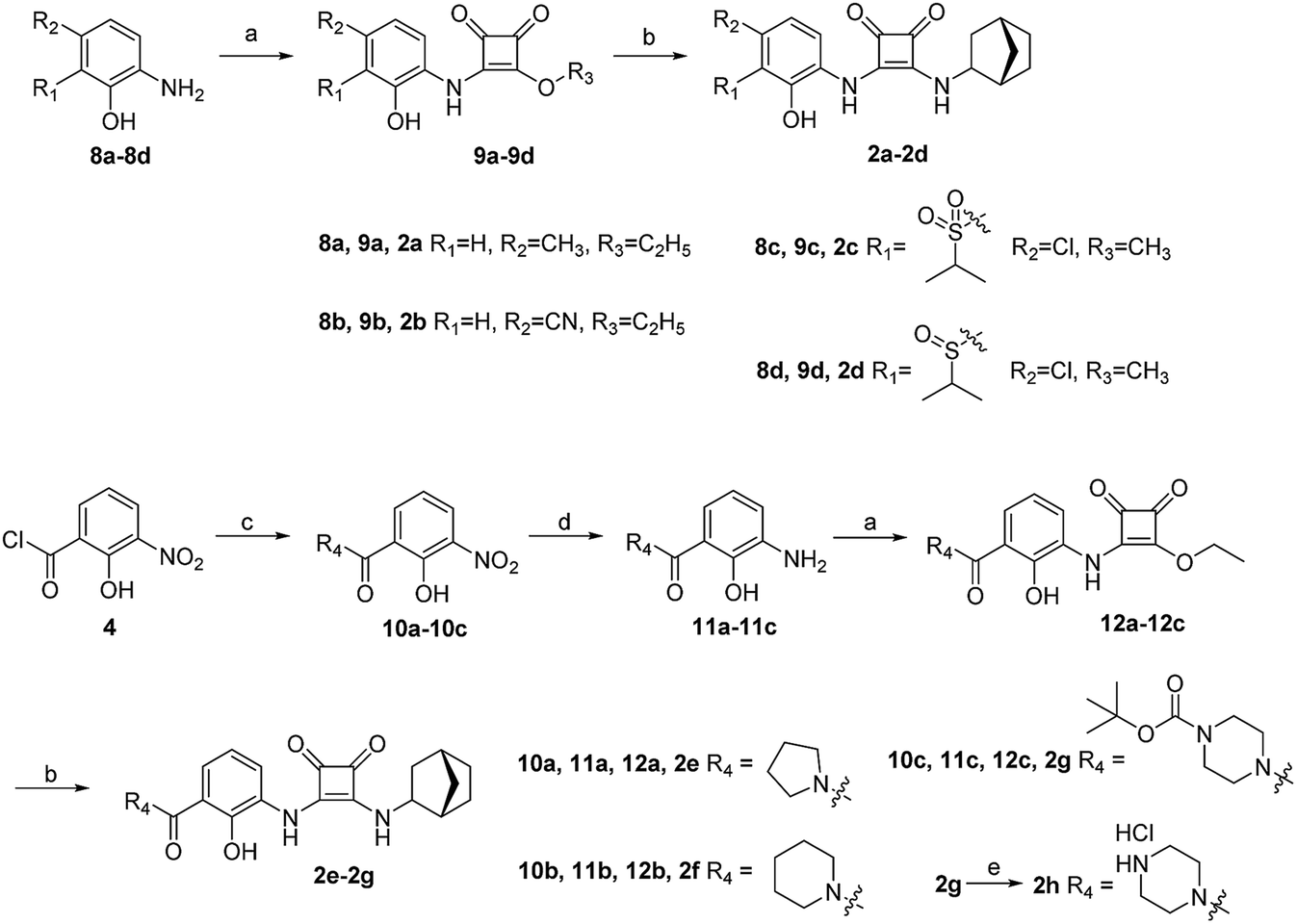 | ||
| Scheme 2 Synthetic route of compounds 2a–2h. (a) 3,4-diethoxycyclobut-3-ene-1,2-dione or 3,4-dimethoxycyclobut-3-ene-1,2-dione, ethanol; (b) (1S,4R)-bicyclo[2.2.1]heptan-2-amine, ethanol or methanol; (c) diverse amines, dichloromethane; (d) Pd/C, H2, methanol; (e) HCl saturated ethyl acetate. | ||
2.2 In vitro CXCR1 and CXCR2 antagonistic activity
As listed in Table 1, different bridge ring substituents were explored. Among these compounds, the bridge ring space size was found to be important for the corresponding CXCR2 affinities. When the polycyclic hydrocarbon scaffolds were enlarged (1b, CXCR2IC50 > 100 μM; 1c, CXCR2IC50 = 5.5 μM; 1d, CXCR2IC50 = 1 μM) or moved away from the parent nucleus (1e, CXCR2IC50 = 2.6 μM; 1f, CXCR2IC50 = 23.6 μM), the CXCR2 binding affinity of the corresponding analogues decreased dramatically.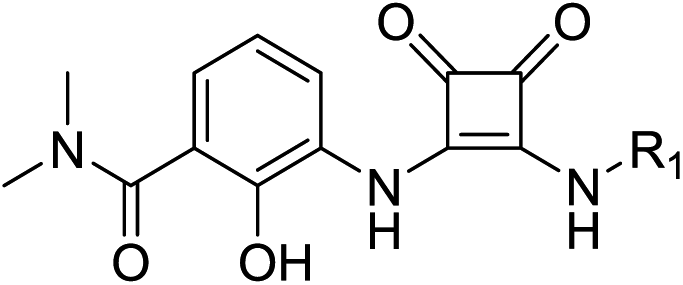
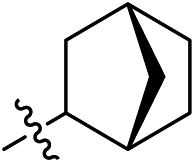
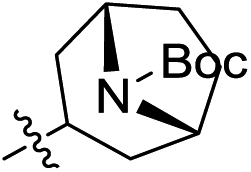
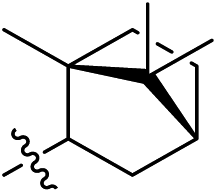
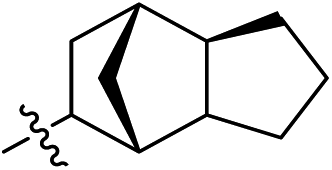
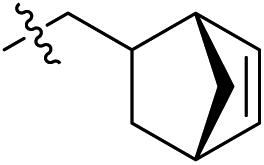
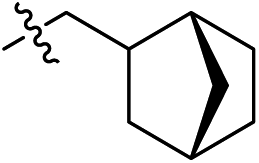
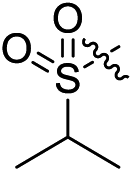
Subsequent structure–activity relationship (SAR) determinations resulted in changing the electron-withdrawing group on the left side of the structure. Throughout our assay, the introduction of an electron-donating methyl group (2a, CXCR2IC50 = 3.8 μM) or an electron-withdrawing cyano group (2b, CXCR2IC50 = 0.45 μM) at the R2 position resulted in loss of activity. Compared to compound 1a, after replacement of the R1 amide moiety with sulfone (2c, CXCR2IC50 = 0.12 μM) or sulfoxide (2d, CXCR2IC50 = 92 nM), compound 2c exhibited a similar selectivity but weaker binding affinity and compound 2d demonstrated a suitable CXCR2 antagonistic activity but with reduced selectivity. On the basis of this observation, subsequent modifications focused on the modification of the R1 amide moiety. Compound 2e showed an increased CXCR2 binding affinity (CXCR2IC50 = 48 nM), but a decreased selectivity ratio. When the amide along with the five-membered ring in 2e was enlarged to a six-membered ring (2f, CXCR2IC50 = 0.42 μM), the CXCR2 antagonistic affinity was found to be reduced approximately 10-fold. Comparing compound 2g (CXCR1IC50 = 5.6 μM and CXCR2IC50 = 0.42 μM) and 2h (CXCR1IC50 > 100 μM and CXCR2IC50 = 0.53 μM), the introduction of a hydrophilic center to the left-hand amide moiety (2h) led to a significant loss of CXCR1 antagonistic activity, resulting in an increased CXCR2 antagonistic selectivity (>188-fold) (Table 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | CXCR1 (μM) | CXCR2 (μM) | Ratioa | ||
| IC50 | SEM | IC50 | SEM | ||||
| a Ratio = CXCR1IC50/CXCR2IC50. | |||||||
| 2a | H | CH3 | 86.1 | 2.7 | 3.8 | 1.3 | 22.7 |
| 2b | H | CN | 42.7 | 1.5 | 0.45 | 0.14 | 94.9 |
| 2c |
|
Cl | 12.4 | 4.3 | 0.12 | 0.025 | 103.3 |
| 2d |
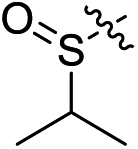
|
Cl | 6.5 | 3.0 | 0.092 | 0.019 | 70.7 |
| 2e |
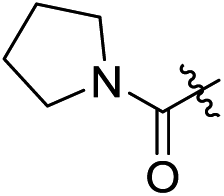
|
H | 2.9 | 0.3 | 0.048 | 0.0075 | 60.4 |
| 2f |
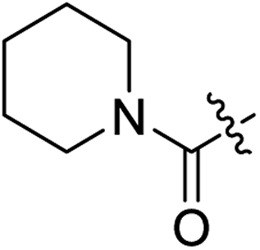
|
H | 10.3 | 5.6 | 0.59 | 0.061 | 17.5 |
| 2g |

|
H | 5.6 | 1.5 | 0.42 | 0.054 | 13.3 |
| 2h |
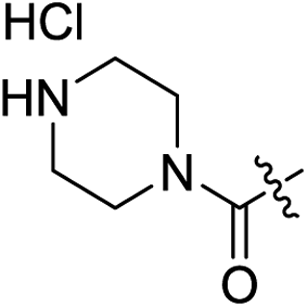
|
H | >100 | — | 0.53 | 0.17 | >188.7 |
2.3 Cell migration suppression by compound 2e
The highly metastatic CFPAC1 pancreatic cancer cell line was selected for the following in vitro biological evaluation. The migration inhibitory effect of compound 2e was studied based on a well-established wound healing assay and Transwell assay. At first, the cytotoxicity of compound 2e was evaluated to provide a suitable concentration range for the cell migration assays. As shown in Fig. 3(A), the result indicated that compound 2e displayed no inhibitory effect on the CFPAC1 cell line at different concentrations (6.25, 12.5, 25, 50, and 100 μM).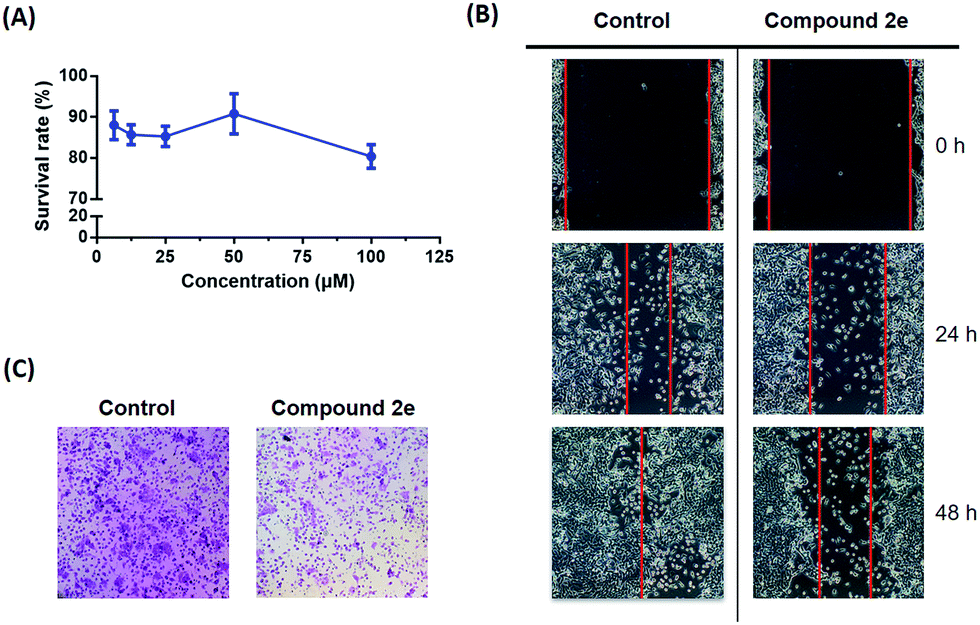 | ||
| Fig. 3 (A) The survival rate of compound 2e against the CFPAC1 cell line; (B) the images of CFPAC1 cell migration; (C) the images of the Transwell assay. | ||
Based on the cytotoxicity result, the cells were treated with 10 μM compound 2e. As a result, the wound area showed a time-dependent decrease in healing rate and the migration inhibitory rate was 20.2% after 48 h of treatment (Fig. 3(B)). To further address the anti-cancer metastatic effect of compound 2e, a subsequent Transwell assay was carried out. As shown in Fig. 3(C), 3 different visual fields were chosen randomly to take the count of the number of cells. The dosing group (727 ± 50 cells) traversed much less into the lower chamber of the Transwell than the control group (1221 ± 57 cells). Therefore, it is believed that compound 2e may modulate the migration capacity of CFPAC1 cells in vitro.
2.4 In vitro metabolism stability assays of compound 2e
In order to evaluate the druggability of compound 2e, the in vitro stability was tested. 2e was incubated with simulated intestinal fluid (SIF) and simulated gastric fluid (SGF), rat and human plasma, and rat and human liver microsomes. The relative concentration changes are shown in Table 3. Compound 2e exhibited a higher stability in SGF than in SIF after 45 min of treatment. In addition, 2e showed extreme stability in both rat and human plasma, as both stability rates were >99% after 45 min. However, in liver microsome assays, compound 2e was found to degrade at various levels. After 45 min of incubation, 84% of the compound was determined to remain in rat liver microsomes and 27% was found to remain in human liver microsomes. Therefore, improving the liver microsome stability of compound 2e remains a critical goal that is currently being addressed in our laboratory.| Compound | Remaining percentage (%) | |||||
|---|---|---|---|---|---|---|
| SIF and SGF stability | Plasma stability | Liver microsome stability | ||||
| SIF | SGF | Rat | Human | Rat | Human | |
| 2e | 92.2 ± 2.0 | >99 | >99 | >99 | 84.2 ± 3.4 | 27.4 ± 4.3 |
2.5 Oral pharmacokinetic profiles of compound 2e
Compound 2e was also tested in in vivo pharmacokinetic studies in rats (10 mg kg−1 PO). As shown in Table 4, the compound demonstrated a moderate clearance rate with a t1/2 of 2.58 h along with an excellent absorbance (Cmax of 2863 ng mL−1). The mean residence time (MRT) is 3.43 h and the area under the curve from 0 h to 24 h (AUC0–t) is 8820 h ng mL−1. This preliminary data suggests that this compound may constitute a reasonable starting point for further drug development.| PK parameters | Value |
|---|---|
| Dose | 10 mg kg−1 |
| K el | 0.285 ± 0.084 h−1 |
| t 1/2 | 2.58 ± 0.74 h |
| t max | 0.33 ± 0.14 h |
| C max | 2863 ± 94 ng mL−1 |
| AUC0–t | 8820 ± 3555 h ng mL−1 |
| AUC0–inf | 8940 ± 3455 h ng mL−1 |
| AUMC0–t | 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 509 ± 19743 h h ng mL−1 509 ± 19743 h h ng mL−1 |
| AUMC0–inf | 32103 ± 18610 h h ng mL−1 |
| MRTPO | 3.43 ± 0.73 h |
2.6 Molecular docking
In order to give a structural illustration for the experimental results above, molecular docking was performed to further explore the mechanism of the newly developed N,N′-diarylsquaramide derivatives and predict the preponderant configuration of compound 2e. Due to the absence of a crystal structure of CXCR2, the structure of CXCR1 was employed, which shares a 77% sequence similarity. The homologous modelling and docking were performed with the modules Align Sequence to Templates, Build Homology Models, Standard Dynamic Cascade and Flexible Docking (see Fig. S1†) implanted in Discovery Studio 2.5.According to the possible ligand binding site reported previously,27 the result of the docking study showed that the S configuration ((S)-2e, LibDockScore = 73.34) performed better than the R configuration ((R)-2e, LibDockScore = 52.10). As shown in Fig. 4(A), one of the ketone oxygens on the cyclobut-3-ene-1,2-dione part of (R)-2e is hydrogen-bonded to Arg80, and the bicyclo[2.2.1]heptane moiety of (R)-2e inserted into a hydrophobic pocket surrounded by Ser81, Ala249 and Val252. In contrast, (S)-2e exhibited a stronger binding mode. Interestingly, both of the ketone oxygens on the cyclobut-3-ene-1,2-dione part of (S)-2e are hydrogen-bonded to Arg80, and the amino groups on the cyclobut-3-ene-1,2-dione part of (S)-2e formed additional H-bonds with Tyr314 and Gly244. The bicyclo[2.2.1]heptane moiety of (S)-2e inserted into the same hydrophobic pocket surrounded by Ser81, Ala249 and Val252. Therefore, the antagonistic activity of (S)-2e may be better than that of (R)-2e, although this needs further biological verification.
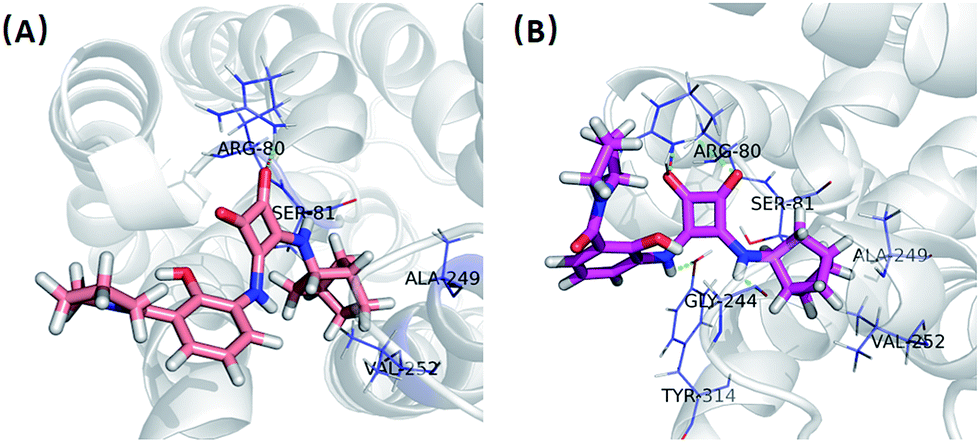 | ||
| Fig. 4 (A) Interaction mode of CXCR2 and (R)-2e; (B) interaction mode of CXCR2 and (S)-2e. | ||
3. Conclusions
In conclusion, a bridged ring system was applied and explored in the N,N′-diarylsquaramide skeleton. Upon optimization, compound 2e bearing bicyclo[2.2.1]heptane demonstrated good CXCR2 antagonistic activity and a suitable selectivity against CXCR1. In addition, good in vitro stability, reasonable oral pharmacokinetic profiles and a high anticancer metastatic effect were also demonstrated. Computational docking results illustrated the interaction mode of 2e, and (S)-2e may demonstrate better antagonistic activity. Further chiral resolution and in vivo biological tests of compound 2e are currently underway in our laboratory.4. Experimental section
4.1 CXCR1 and CXCR2 antagonistic activity assay
Human Embryonic Kidney 293 (HEK293) cells stably expressing Gα16 and CXCR1 or CXCR2 were seeded onto 96-well plates and incubated for 24 h. Cells were loaded with 2 μmol L−1 Fluo-4 AM in Hanks balanced salt solution (HBSS, containing KCl 5.4 mmol L−1, Na2HPO4 0.3 mmol L−1, KH2PO4 0.4 mmol L−1, NaHCO3 4.2 mmol L−1, CaCl2 1.3 mmol L−1, MgCl2 0.5 mmol L−1, Mg2SO4 0.6 mmol L−1, NaCl 137 mmol L−1, BSA 5 g L−1, glucose 5.6 mmol L−1, and sulfinpyrazone 250 μmol L−1, at pH 7.4) at 37 °C for 45 min. The excess dye was removed and 50 μL of the HBSS containing test compounds was added. After incubation at room temperature for 10 min, 25 μL HBSS containing IL-8 was dispensed into the well using a FlexStation II microplate reader (Molecular Devices, Sunnyvale, CA, USA) and the intracellular calcium change was recorded with an excitation wavelength of 485 nm and emission wavelength of 525 nm. The half maximal inhibitory concentrations (IC50) of compounds were determined using the “Analyze Data” “XY analyses” “Nonlinear regression (curve fit)” protocol implanted in GraphPad Prism software by constructing their dose–response curves, where the x axis is the log value of concentration and the y axis is the response value (for the curves of each compound, see Table S1†). Each experiment was repeated at least three times.4.2 Anti-proliferation assay
CFPAC1 cells were seeded in 96-well plates at a density of 4000 cells per well. After 24 h of adherence, cells were incubated with the medium alone or with medium containing test compounds for 72 h. Five different concentrations (6.25, 12.5, 25, 50, and 100 μM) of both antagonists were used. The cell viability was determined using MTT assays. The growth inhibition was calculated as % = [1 − (A/B)] × 100, where A and B were the absorbances of the treated and untreated cells, respectively.4.3 Cell migration assay
The cell migration assay was performed in a 24-well Transwell plate with an 8 μm polycarbonate sterile membrane (Corning Incorporated). The cells were plated in the upper chamber at 2 × 104 cells per insert in 200 μL of serum-free medium. Inserts were placed in wells containing 600 μL of medium supplemented with 10% FBS. After being cultured for 24 h, cells on the upper surface were detached with a cotton swab. Filters were fixed and cells in the lower filter were stained with 0.1% crystal violet for 15 minutes and photographed under a 100× microscope. 3 different visual fields were chosen randomly to take the count of the number of cells.4.4 Wound healing assay
CFPAC1 cells (5 × 105 cells per well) were seeded in a 6-well tissue culture plate and grown to 90% confluence. After the medium was removed, a gap with constant width was created in the center of the well by scratching the monolayer with a sterile micropipette tip. The cells were then rinsed with phosphate-buffered saline (PBS) thrice to remove cellular debris, and were subsequently exposed to 1‰ DMSO or 10 μM compound 2e. The wound closure was monitored and photographed at 0, 24, and 48 h using ImagePro software. The cell migration inhibitory rate was calculated as % = [1 − (0 h wound area − 24 h or 48 h wound area)/0 h wound area] × 100.4.5 Pharmacokinetic studies
This study was performed in strict accordance with the Laboratory Animal Management Regulations (State Scientific and Technological Commission Publication No. 8-27 Rev. 2017) and was approved by Zhejiang University Laboratory Animal Center (Hangzhou, China). SD rats (purchased from Zhaoyan (Suzhou, China) New Drug Research Center Co. Ltd) were administered compound 2e at doses of 10 mg kg−1 by oral gavage in the solvent (2% DMSO + 98% (0.5% MC)). Venous blood (100 μL) samples were collected at 0, 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h. Plasma was separated from whole blood by centrifugation and stored at −20 °C until analysis. Compound levels were determined using an Agilent 1200-API4000QTRAP LC/MS system. The Cmax, Tmax, t1/2 and AUC were evaluated using Analyst 1.5.1.4.6 SIF and SGF stability assays
SIF was prepared by dissolving 0.68 grams of pancreatin (Sigma) in 100 mL of 50 mM potassium phosphate buffer (pH 7.4). SGF was prepared by dissolving 3.2 g of pepsin (Sigma) in 1000 mL water containing 2.0 g of sodium chloride (Sinopharm Chemical Reagent Co., Ltd) and 7.0 mL of concentrated hydrochloric acid (Sinopharm Chemical Reagent Co., Ltd) (pH about 1.2). A 480 μL aliquot of the SIF buffer or SGF solution was added to 20 μL of a test article solution at 100 μg mL−1 to give a final solution. The mixture was incubated at 37 °C with gentle agitation. An aliquot of 100 μL of the reaction mixture was taken at 0, 15, and 45 minutes and quenched using 400 μL of methanol containing 1 μg mL−1 internal standard compound (from the in-house database). After quenching, the mixture was vortexed and centrifuged and 10 μL of the resulting solution was injected into a Shimadzu LCMS-2020 system. The percentage remaining of the test article at the incubation times of 15 and 45 minutes relative to that at 0 minutes was calculated using the peak area ratio of the test article versus the internal standard.4.7 Plasma stability assay
A 500 μL aliquot of rat plasma or human plasma (purchased from Shanghai Yuduo Biotechnology Company) was added to 5 μL of a test article solution at 100 μg mL−1 to give a final solution. The mixture was incubated at 37 °C with gentle agitation. An aliquot of 100 μL of the reaction mixture was taken at 0, 15, and 45 minutes and quenched using 400 μL of methanol containing 1 μg mL−1 internal standard compound (from the in-house database). After quenching, the mixture was vortexed and centrifuged and 10 μL of the resulting solution was injected into an API4000 + LC/MS system. The percentage remaining of the test article at the incubation times of 15 and 45 minutes relative to that at 0 minutes was calculated using the peak area ratio of the test article versus the internal standard.4.8 Liver microsome stability assay
1 mg mL−1 microsome solution (purchased from Ruide Research Institute for Liver Diseases (Shanghai) Co. Ltd) was mixed with 20 μL of 50 mM NADPH (Aladdin) solution to prepare a microsome-NADPH solution. 500 μL of the microsome-NADPH solution was pre-warmed at 37 °C for 5 minutes. 5 μL of a 100 μg mL−1 test article solution was then added to initiate the reaction. The incubation mixture was kept at 37 °C and 100 μL aliquots were taken at 0, 15, and 45 minutes. In each aliquot, the reaction was quenched using 400 μL of methanol containing 1 μg mL−1 internal standard compound (from the in-house database). After quenching, the mixtures were vortexed and centrifuged. The supernatant was transferred and 10 μL was injected into an API4000 + LC/MS system. The peak area ratio of a test article versus the internal standard was used in the calculation of the rate of disappearance of a test article.4.9 Chemistry
To a solution of compound 5 (105 mg, 0.5 mmol) in methanol, Pd/C (10 mg) was added and then heated under reflux for 6 h under a H2 atmosphere. The solution was filtered and the filtrate was concentrated to afford compound 6, which needed to be used directly for the next step. To a solution of compound 6 in ethanol, 3,4-diethoxycyclobut-3-ene-1,2-dione (128 mg, 0.75 mmol) was added and the solution was stirred for 12 h at room temperature. The solution was concentrated, the residue was dissolved in water and acidized to pH = 1–2 with 1 N HCl, then extracted with EtOAc (50 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, then filtered and concentrated. The residue was purified using silica gel chromatography to afford compound 7 (96 mg, 63% total yield) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.79 (s, 1H), 7.12 (m, 1H), 6.90 (t, J = 8.0 Hz, 1H), 4.88 (m, 2H), 3.20 (s, 6H), 1.53 (t, J = 7.0 Hz, 3H). ESI-MS: m/z = 303 [M–H]+.
:H2O = 1:4–1:1) to afford compound 1a–1e.
To a solution of compound 10a–10c in methanol, Pd/C was added, then heated under reflux for 6 h under a H2 atmosphere. The solution was filtered and the filtrate was concentrated to afford compounds 11a–11c, which needed to be used directly for the next step. To a solution of 11a–11c in ethanol, 3,4-diethoxycyclobut-3-ene-1,2-dione was added, and the solution was stirred for 12 h at room temperature. The solution was concentrated, the residue was dissolved in water and acidized to pH = 1–2 with 1 N HCl, then extracted with EtOAc (50 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, then filtered and concentrated. The residue was purified using silica gel chromatography to afford compounds 12a–12c.
:H2O = 1:4–1:1) to afford compounds 2a–2g.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Dr Bo Zhang (Hangzhou First People’s Hospital) for assistance with the cell migration assays. This work was supported by the National Natural Science Foundation of China (Grant No.: 81673294, 81472862 and 81425024).References
- W. E. Holmes, J. Lee, W. J. Kuang, G. C. Rice and W. I. Wood, Science, 1991, 253, 1278–1280 CrossRef CAS PubMed.
- P. M. Murphy and H. L. Tiffany, Science, 1991, 253, 1280–1283 CrossRef CAS PubMed.
- J. Vandercappellen, J. Van Damme and S. Struyf, Cancer Lett., 2008, 267, 226–244 CrossRef CAS.
- Y. Wang, Y. Qu, X. L. Niu, W. J. Sun, X. L. Zhang and L. Z. Li, Cytokine, 2011, 56, 365–375 CrossRef CAS.
- Y. S. Lee, I. Choi, Y. Ning, N. Y. Kim, V. Khatchadourian, D. Yang, H. K. Chung, D. Choi, M. J. LaBonte, R. D. Ladner, K. C. Nagulapalli Venkata, D. O. Rosenberg, N. A. Petasis, H. J. Lenz and Y. K. Hong, Br. J. Cancer, 2012, 106, 1833–1841 CrossRef CAS.
- J. K. Singh, G. Farnie, N. J. Bundred, B. M. Simoes, A. Shergill, G. Landberg, S. J. Howell and R. B. Clarke, Clin. Cancer Res., 2013, 19, 643–656 CrossRef CAS.
- C. W. Steele, S. A. Karim, J. D. Leach, P. Bailey, R. Upstill-Goddard, L. Rishi, M. Foth, S. Bryson, K. McDaid, Z. Wilson, C. Eberlein, J. B. Candido, M. Clarke, C. Nixon, J. Connelly, N. Jamieson, C. R. Carter, F. Balkwill, D. K. Chang, T. R. Evans, D. Strathdee, A. V. Biankin, R. J. Nibbs, S. T. Barry, O. J. Sansom and J. P. Morton, Cancer Cell, 2016, 29, 832–845 CrossRef CAS.
- W. L. Cheng, C. S. Wang, Y. H. Huang, M. M. Tsai, Y. Liang and K. H. Lin, Ann. Oncol., 2011, 22, 2267–2276 CrossRef PubMed.
- A. Li, X. J. Cheng, A. Moro, R. K. Singh, O. J. Hines and G. Eibl, Transl Oncol., 2011, 4, 20–28 CrossRef.
- K. M. Hertzer, G. W. Donald and O. J. Hines, Expert Opin. Ther. Targets, 2013, 17, 667–680 CrossRef CAS.
- B. Sharma, M. L. Varney, S. Saxena, L. Wu and R. K. Singh, Cancer Lett., 2016, 372, 192–200 CrossRef CAS.
- J. C. Acosta and J. Gil, Cancer Res., 2009, 69, 2167–2170 CrossRef CAS.
- S. Singh, M. Varney and R. K. Singh, Cancer Res., 2009, 69, 411–415 CrossRef CAS PubMed.
- G. Yang, D. G. Rosen, G. Liu, F. Yang, X. Guo, X. Xiao, F. Xue, I. Mercado-Uribe, J. Huang, S. H. Lin, G. B. Mills and J. Liu, Clin. Cancer Res., 2010, 16, 3875–3886 CrossRef CAS PubMed.
- B. Devapatla, A. Sharma and S. Woo, PLoS One, 2015, 10, e0139237 CrossRef.
- B. Otvos, D. J. Silver, E. E. Mulkearns-Hubert, A. G. Alvarado, S. M. Turaga, M. D. Sorensen, P. Rayman, W. A. Flavahan, J. S. Hale, K. Stoltz, M. Sinyuk, Q. Wu, A. Jarrar, S. H. Kim, P. L. Fox, I. Nakano, J. N. Rich, R. M. Ransohoff, J. Finke, B. W. Kristensen, M. A. Vogelbaum and J. D. Lathia, Stem Cells, 2016, 34, 2026–2039 CrossRef CAS.
- A. Purohit, M. Varney, S. Rachagani, M. M. Ouellette, S. K. Batra and R. K. Singh, Oncotarget, 2016, 7, 7280–7296 CrossRef.
- I. M. Stromnes and P. D. Greenberg, Cancer Cell, 2016, 29, 774–776 CrossRef CAS.
- X. Lu, J. W. Horner, E. Paul, X. Y. Shang, P. Troncoso, P. N. Deng, S. Jiang, Q. Chang, D. J. Spring, P. Sharma, J. A. Zebala, D. Y. Maeda, Y. A. Wang and R. A. DePinho, Nature, 2017, 543, 728–732 CrossRef CAS.
- T. P. Stockdale and C. M. Williams, Chem. Soc. Rev., 2015, 44, 7737–7763 RSC.
- T. H. Keller, A. Pichota and Z. Yin, Curr. Opin. Chem. Biol., 2006, 10, 357–361 CrossRef CAS PubMed.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS PubMed.
- M. P. Gleeson, A. Hersey, D. Montanari and J. Overington, Nat. Rev. Drug Discovery, 2011, 10, 197–208 CrossRef CAS PubMed.
- M. P. Dwyer, Y. N. Yu, J. P. Chao, C. Aki, J. H. Chao, P. Biju, V. Girijavallabhan, D. Rindgen, R. Bond, R. Mayer-Ezel, J. Jakway, R. W. Hipkin, J. Fossetta, W. Gonsiorek, H. Bian, X. D. Fan, C. Terminelli, J. Fine, D. Lundell, J. R. Merritt, L. L. Rokosz, B. Kaiser, G. Li, W. Wang, T. Stauffer, L. Ozgur, J. Baldwin and A. G. Taveras, J. Med. Chem., 2006, 49, 7603–7606 CrossRef CAS.
- J. R. Merritt, L. L. Rokosz, K. H. Nelson Jr, B. Kaiser, W. Wang, T. M. Stauffer, L. E. Ozgur, A. Schilling, G. Li, J. J. Baldwin, A. G. Taveras, M. P. Dwyer and J. Chao, Bioorg. Med. Chem. Lett., 2006, 16, 4107–4110 CrossRef CAS.
- J. Chao, A. G. Taveras, J. Chao, C. Aki, M. Dwyer, Y. Yu, B. Purakkattle, D. Rindgen, J. Jakway, W. Hipkin, J. Fosetta, X. Fan, D. Lundell, J. Fine, M. Minnicozzi, J. Phillips and J. R. Merritt, Bioorg. Med. Chem. Lett., 2007, 17, 3778–3783 CrossRef CAS.
- K. Salchow, M. E. Bond, S. C. Evans, N. J. Press, S. J. Charlton, P. A. Hunt and M. E. Bradley, Br. J. Pharmacol., 2010, 159, 1429–1439 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CXCR2 homologous modeling, CXCR1/2IC50 curve graphs, and NMR spectra of compounds 1a and 2e. See DOI: 10.1039/c8ra01806e |
| This journal is © The Royal Society of Chemistry 2018 |