
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of a cation-exchange reaction of CuS nanoparticles and fluorescent copper nanoparticles in a DNA biosensor†
Weiling Song,
Nan Zhang,
Zhenzhu Luan,
Xiaoru Zhang * and
Peng He*
* and
Peng He*
Key Laboratory of Sensor Analysis of Tumor Marker, Ministry of Education, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: zhangxr7407@126.com
First published on 23rd April 2018
Abstract
A novel detection method based on the cation-exchange reaction of CuS nanoparticles (CuS NPs) combined with poly T-templated fluorescent Cu nanoparticles (Cu NPs) was developed. First, CuS NPs-magnetic bead conjugates were prepared through the hybridization of DNA. Competition with target DNA resulted in the release of CuS NPs, and exonuclease III catalysis could lead to recycling of the target DNA. Then, the CuS NPs released into the supernatant were subjected to a cation-exchange reaction after the addition of AgNO3. The obtained Cu2+ could form fluorescent Cu NPs using poly T DNA as a template. The fluorescence intensity of the Cu NPs could be used to determine the concentration of the target DNA. To further increase the detection sensitivity, two types of DNA decorated magnetic beads were used. After Exo III digestion for two cycle processes, more CuS NPs entered the supernatant. Hence, a stronger fluorescence intensity was found after the cation-exchange reaction and the formation of fluorescent Cu NPs. The developed method is convenient and low cost with good sensitivity and selectivity.
Recently, inorganic nanoparticles have attracted much attention in the field of biosensing owing to their ideal optical and electronic properties, high stability and easy separability.1 Among all these applications, the cation-exchange reaction is a very interesting one. Since its discovery in 2004,2 many studies on the synthesis of nanomaterials using this versatile technique have emerged.3 In the field of biosensors, Zhong et al. used CdSe nanocrystals and ZnS nanocrystal clusters as markers. After a cation-exchange reaction between these nanoparticles and Ag+ ions, thousands of divalent cations were released into the solution, which could turn on the fluorescence of fluorogenic dyes.4 CdSe nanocrystals or ZnS nanocrystal clusters were used to produce Cd2+ or Zn2+ and the later one is more biocompatible. Tang et al. developed a signal-on photoelectrochemical immunoassay based on silver nanolabels-assisted ion-exchange reaction with CdTe quantum dots.5 Tan et al. combined the catalytic DNAzyme with a cation-exchange reaction for an ultrasensitive, multiplex, fluorescent immunosorbent assay.6 In our previous study, a cation-exchange reaction of nontoxic CuS NPs was used to obtain a large concentration of Cu2+, which can be used to enhance the chemiluminescence of a luminol–H2O2 system.7 Later, we developed a colorimetric assay for single-nucleotide polymorphisms and DNA methyltransferase based on a cation-exchange reaction of CuS NPs and the click chemistry of azide- and alkyne-functionalized gold nanoparticles.8 Compared to the acid dissolution of CuS NPs using a strong acid, e.g., HNO3,9 the reaction conditions for the cation-exchange of CuS NPs are mild and more suitable for real biological sample detection. However, the operation process is complicated, especially for the click reaction of functionalized gold nanoparticles. Hence, to make this strategy more practical, it is necessary to develop a more convenient and economical method to measure the Cu2+ released by the cation-exchange reaction.
Recently, DNA-templated fluorescent nanoclusters or nanoparticles, including Ag,10 Au,11 Pt12 and Cu,13 have attracted much attention than traditional organic dyes and quantum dots because of their strong fluorescence, high photostability and low toxicity. Among them, poly T-templated fluorescent copper nanoparticles (Cu NPs) can be synthesized in a few minutes.14 Hence, fluorescent probe Cu NPs are more suitable for in situ syntheses and applications in biosensors.15 Based on the above consideration, here, we attempted to make use of the advantage of the cation-exchange reaction of CuS NPs and DNA-templated fluorescent Cu NPs together. The CuS NPs were used to label biomolecules. After the recognition and purification steps, quantitative amount of released Cu2+ caused by the cation-exchange reaction can be used to produce fluorescent Cu NPs. Based on the fluorescent intensity of the Cu NPs, the target could be easily quantified. To further enhance the detection sensitivity, an exonuclease III-assisted cascade signal amplification was introduced. After the cycling cleavage process, a simple and low-cost strategy for the sensitive detection of DNA was developed, and this method can be used for the early diagnosis of other bio-active molecules.
As shown in Fig. 1A, biotin modified DNA1 could be immobilized on streptavidin-modified magnetic beads (MBs) through the interaction between biotin and streptavidin. Then, CuS NP-labelled DNA2 was added to form the complex MB-DNA1/DNA2-CuS. In the absence of the target DNA, MB-DNA1/DNA2-CuS complex with two protruding 3′ ends was stable against digestion by Exo III.16 Therefore, no free CuS NPs existed in the supernatant. When the target DNA was added, longer DNA duplex could form on the CuS NPs with a blunt 3′-terminus on the DNA2 and release MB-DNA1. Thus, Exo III could catalyze the step-wise removal of mononucleotides from the 3′-terminus of the DNA2, releasing the target DNA and CuS NPs labelled mononucleotides. The released target DNA could participate in the next cycle of the hybridization and cleavage process (“cycle I”). As a result, many CuS NPs entered the supernatant after the magnetic separation. When addition of AgNO3 solution to the supernatant, a cation-exchange reaction was initiated, and the resultant Cu2+ can react with poly T via reduction by SA to obtain fluorescent Cu NPs. In this way, when more target DNAs are present, more cupric ions are obtained due to the cation-exchange reaction, and more fluorescent Cu NPs are produced in situ, resulting in a higher fluorescent intensity. Hence the fluorescent intensity of the fluorescent Cu NPs can be used to determine the concentration of the target DNA.
 | ||
| Fig. 1 Experimental schematic diagram of (A) for cycle I; (B) for cycle I + cycle II process. | ||
The as-prepared CuS NPs and Cu NPs were characterized by transmission electron microscopy (TEM). The images in Fig. 2A and B showed that the nanocrystals were nearly monodispersed and were highly dispersed and spherical in shape. The average size of the CuS NPs is approximately 10 nm. While the average size for the Cu NPs templated by poly T40 is approximately 5 nm. This confirmed the formation of CuS NPs and Cu NPs. As shown in Fig. 2C, the maxima fluorescence excitation and emission of the Cu NPs were at 340 and 625 nm, respectively. Thus, the Stokes shift was as long as 285 nm, making it an ideal fluorescent probe due to the low background signal. Compared to the blank, the fluorescent Cu NPs exhibited a bright orange-red color under UV light (see the inset in Fig. 2C). The UV-vis absorption spectrum of the Cu NPs exhibited a peak at 340 nm (Fig. 2D), which is the same as the fluorescence excitation spectrum. The conditions for the preparation of the fluorescent Cu NPs were optimized in Fig. S1 of ESI.†
 | ||
| Fig. 2 (A) TEM image of CuS NPs; (B) TEM image of fluorescent Cu NPs; (C) excitation and emission of fluorescent copper nanoparticles. Inset from left to right: photographs of the blank and fluorescent Cu NPs under UV light; (D) absorption spectra of fluorescent Cu NPs. | ||
For sensing purposes, the influence of Ag+ ions on the fluorescent intensity should be eliminated. As shown in Fig. 3A, when 40 μM Cu2+ was added to the poly T-SA system, an obvious fluorescence signal was obtained. However, when different concentrations of Ag+ (from 4.0 × 10−2 M to 4.0 × 10−8 M) were added to the poly T-SA system, the fluorescence signal was very low and can be ignored. Ag+ had almost no effect on the Cu2+-poly T-SA systems over a wide concentration range. Thus, we confirmed that the fluorescence signal was only caused by the formation of Cu NPs.
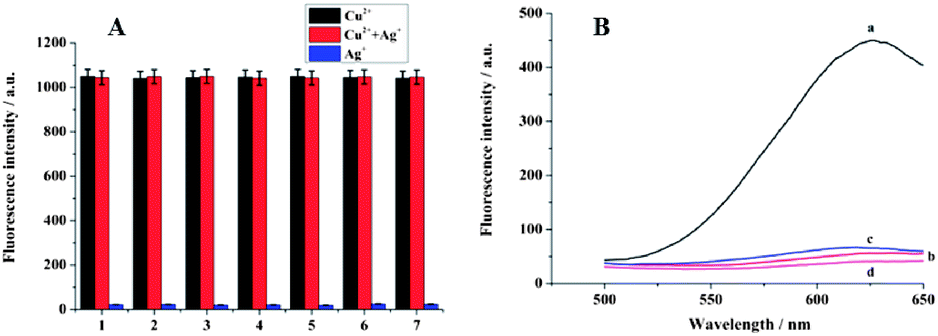 | ||
| Fig. 3 The feasibility of the cation-exchange reaction of CuS NPs and the formation of fluorescent Cu NPs. (A) The influence of Ag+ on the fluorescence of the system. The concentration of Ag+ was (1) 4.0 × 10−2 M; (2) 4.0 × 10−3 M; (3) 4.0 × 10−4 M; (4) 4.0 × 10−5 M; (5) 4.0 × 10−6 M; (6) 4.0 × 10−7 M; (7) 4.0 × 10−8 M. The concentration of Cu2+ was 40 μM. (B) Fluorescence signals obtained in a series of control experiments: (a) in the presence of 28 units Exo III and 1.0 × 10−6 M target DNA; (b) addition of 2.5 × 10−5 M GSH to solution (a); (c) in the presence of 1.0 × 10−6 M target DNA but without Exo III; (d) in the absence of Exo III and target DNA. | ||
To test the feasibility of this assay, a series of control experiments were designed, as shown in Fig. 3B. In the absence of the target DNA and Exo III, the fluorescent intensity was negligible (curve d), because no CuS NPs were introduced into the supernatant. When 1.0 × 10−6 M target DNA was added without Exo III, only a small fluorescence signal was obtained (curve c), since the recycling cleavage process was not initiated. However, the fluorescent intensity increased substantially for the cycle I process when 1.0 × 10−6 M target DNA and 28 units of Exo III (the optimization of the detection conditions was shown in Fig. S2 of ESI†) were added, which indicated that a large number of Cu2+ ions and fluorescent Cu NPs were produce. Furthermore, when 2.5 × 10−5 M glutathione was added to the above mixture, a sharp decrease in the fluorescence signal was observed (curve b) due to the formation of non-fluorescent coordination complexes through a Cu–S bond,17 which demonstrated the formation of fluorescent Cu NPs from another point of view.
Inductively coupled plasma-mass spectrometry (ICP-MS) was used to detect the Cu2+ produced by the cation-exchange reaction. From the results for the Cu2+ concentration applied in the cycle I route shown in Table 1, we can see that the content of cupric ions is related to the concentration of the target DNA. When more target DNAs were added, a higher concentration of cupric ions was measured, which confirmed our design principle.
| CDNA = 0 | CDNA = 1.0 × 10−9 M | CDNA = 1.0 × 10−8 M | |
|---|---|---|---|
| Cu2+ concentration applied cycle I route (M) | <0.000 | 7.19 × 10−6 | 2.78 × 10−5 |
| Cu2+ concentration applied cycle I + cycle II route (M) | <0.000 | 3.31 × 10−5 | 1.22 × 10−4 |
In the above investigation, we confirmed that cation-exchange reaction and in situ formation of fluorescent Cu NPs could be combined and used for the fluorescence detection method (see Fig. S3 of the ESI† for the detection of target DNA through cycle I route). To further enhance the sensitivity of our biosensor, a strategy consisting of two amplification steps was designed. As shown in Fig. 1B, the complexes MB-DNA1/DNA2-CuS and MB-DNA3/DNA4-CuS with two protruding 3′ ends each were prepared first. When target DNA was introduced, in addition to “cycle I”, the released MB-DNA1 can compete to bind with MB-DNA3/DNA4-CuS complex, forming MB-DNA1/DNA4-CuS complex. This complex with a blunt 3′-terminus on DNA4 can be further digested by Exo III too, forming cycle II. After the Exo III digestion for cycle I and cycle II processes, substantial number of CuS NPs entered the supernatant. Hence, after the cation-exchange reaction, more Cu2+ can be produced. As a result, more fluorescent Cu NPs and stronger fluorescent intensity can be observed after the two cycle processes.
The feasibility of this experiment was identical to that in Fig. 3B, except for the further amplification with cycle I + cycle II. As shown in Fig. 4A, when 1.0 × 10−6 M target DNA and 50 units Exo III were added (see the optimization of the detection conditions in Fig. S4 of the ESI†), a much greater enhancement in the fluorescent intensity (curve a) was observed compared to that in (curve a) of Fig. 3B, for the same amount of target DNA. At the same time, ICP-MS was also used to test the role of cycle II. Compared to the first and second lines in Table 1, we can see that after two cycles, more cupric ions were detected for the same concentration of the target DNA, which confirmed the amplification effect of cycle II.
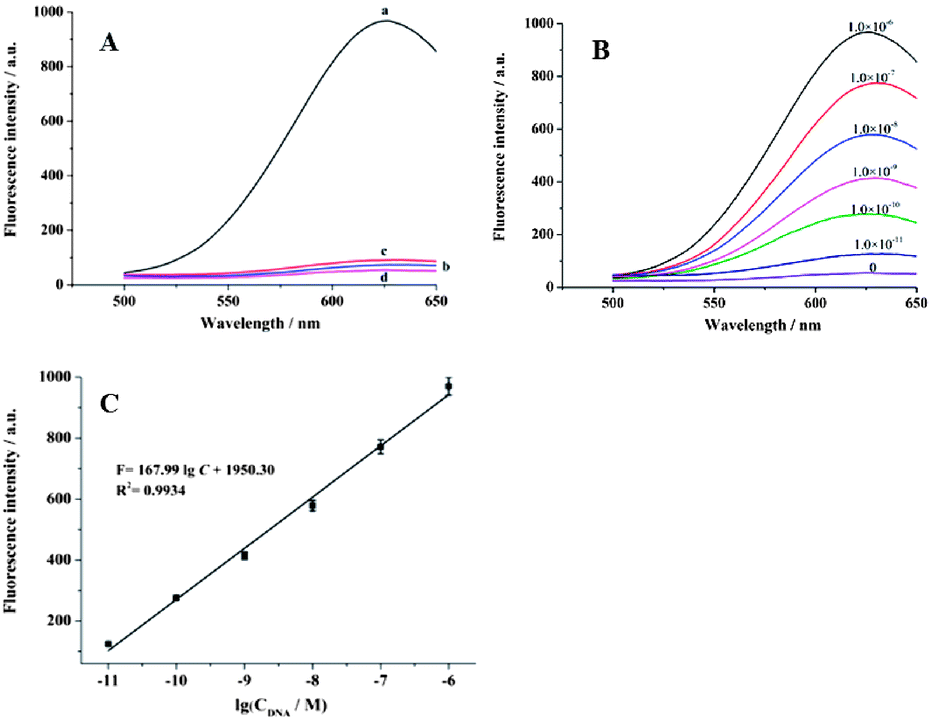 | ||
| Fig. 4 (A) Fluorescence signal obtained by a series of control experiments: (a) in the presence of 50 units Exo III and 1.0 × 10−6 M target DNA; (b) addition of 2.5 × 10−5 M GSH to the solution (a); (c) in the presence of 1.0 × 10−6 M target DNA without Exo III; (d) in the absence of Exo III and target DNA; (B) the relationship between the DNA concentration and the fluorescent intensity; (C) the linear relationship between target DNA concentration and fluorescence intensity. | ||
To achieve the best performance, the reaction temperature, reaction time for cycle I + cycle II and the amount of Exo III were optimized using 1.0 × 10−8 M target DNA. From the results shown in Fig. S4 of the ESI,† we can see that for the cycle I + cycle II process, the reaction can be performed at 37 °C for 90 min in presence of 50 units Exo III.
Under optimum detection conditions, different concentrations of the target DNA were introduced. As shown in Fig. 4B, the fluorescent intensity increased as the target DNA concentration increased in the range from 1.0 × 10 −11 M to 1.0 × 10 −6 M. The linear regression equation was F = 167.99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logC + 1950.30 (F represents FL intensity; C is the concentration of target DNA) with a coefficient of R2 = 0.9934 (Fig. 4C). A detection limit of 6.0 × 10−12 M was estimated using 3σ. For this cycle I + cycle II process, the sensitivity is approximately 100-fold higher than that of cycle I process (Fig. S3 in the ESI†). The advantage of our method lies in its no-toxicity, cheap and convenient usage. The reaction time for the cation-exchange reaction and the formation of fluorescent CuNPs is only several minutes. The slowest process is the amplification process for cycle I + cycle II, which takes about 90 min to complete Exo III-stimulated cascade recycling process. In this work, we confirmed the feasibility of combining the cation-exchange reaction with the in situ formation of fluorescent Cu NPs and demonstrated that this method can be used in a DNA biosensor. This strategy can be applied to other fluorescent biosensors after fine design.
logC + 1950.30 (F represents FL intensity; C is the concentration of target DNA) with a coefficient of R2 = 0.9934 (Fig. 4C). A detection limit of 6.0 × 10−12 M was estimated using 3σ. For this cycle I + cycle II process, the sensitivity is approximately 100-fold higher than that of cycle I process (Fig. S3 in the ESI†). The advantage of our method lies in its no-toxicity, cheap and convenient usage. The reaction time for the cation-exchange reaction and the formation of fluorescent CuNPs is only several minutes. The slowest process is the amplification process for cycle I + cycle II, which takes about 90 min to complete Exo III-stimulated cascade recycling process. In this work, we confirmed the feasibility of combining the cation-exchange reaction with the in situ formation of fluorescent Cu NPs and demonstrated that this method can be used in a DNA biosensor. This strategy can be applied to other fluorescent biosensors after fine design.
Control experiments were performed to determine the selectivity of the present sensor by testing the fluorescence response to complementary target DNA, single-base mismatched DNA (1 MT), two-base mismatched DNA (2 MT) and random DNA, respectively. As shown in Fig. 5A, the fluorescence intensity of 1.0 × 10−8 single-base mismatched DNA was approximately 33.6% to that of the target DNA with the same concentration. Even higher fluorescent signal was observed for 1 MT with concentration of 1.0 × 10−6 M. As for 2 MT, fluorescence signals were much lower for both 1.0 × 10−8 M and 1.0 × 10−6 M 2 MT indicating much better selectivity for two-base mismatched DNA. No significant difference in fluorescence signals was observed between the random DNA and the blank group. This result suggested the high selectivity of the developed method for the discrimination of a perfect complementary target from base mismatched sequences.
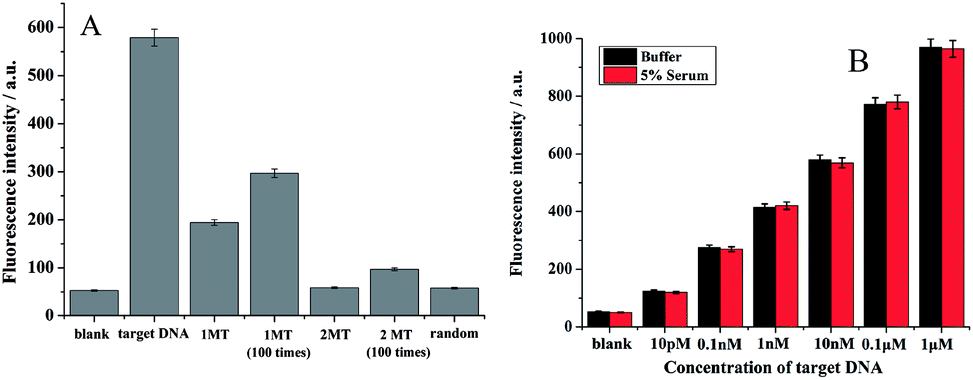 | ||
| Fig. 5 (A) The fluorescence responses for the blank and four DNA sequences including random DNA, single-base mismatched (1 MT) and complementary target DNA. The concentrations of various DNA sequences were 10 nM. Error bars represent the standard deviation of three repetitive measurements; (B) fluorescence intensity of different concentrations of target DNA in buffer and 5% serum. | ||
A practical clinical analysis in complex system was investigated using 5% diluted human serum samples spiked with different concentrations of target DNA. Blank experiment was carried out in buffer or diluted human serum without any oligonucleotides. From the results shown in Fig. 5B, we can see that the fluorescence responses increased with the increase in the concentration of target DNA in the diluted human serum, and the fluorescence signals in diluted human serum were identical to those obtained in the buffer solutions. These results suggested that the developed method had good potential for practical sensing in biological samples.
Meanwhile, we further compared fluorescent assay with quantitative real-time polymerase chain reaction (RT-qPCR) for target DNA dissolved in MCF-7 cell lysate. From Fig. S6 in ESI† we can see that the results obtained by the developed method shows an acceptable agreement with those obtained by RT-qPCR.
Conclusions
A novel detection method based on the cation-exchange reaction of CuS NPs with DNA-templated fluorescent Cu NPs was developed. This method has the merit of mild detection conditions, rapid response for cation-exchange reaction and the in situ formation of Cu NPs. Compared with our previously developed cation-exchange-based chemiluminescence amplification and Cu(I)-based click chemistry of functionalized AuNPs method, this strategy is much simpler and less expensive, only requiring a single DNA template to form fluorescent Cu NPs. After the detailed design, a variety of DNA circulating amplification processes can be introduced, and the sensitivity can be amplified accordingly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by the National Natural Science Foundation of China (No. 21775080, 21705086, 21505082), Key Research and Development project of Shandong Province (No. 2017GSF221004).Notes and references
- (a) J. Zhou, Y. Yang and C. Zhang, Chem. Rev., 2015, 115, 11669 CrossRef CAS PubMed; (b) E. Han, L. Ding and H. X. Ju, Anal. Chem., 2011, 83, 7006 CrossRef CAS PubMed.
- D. H. Son, S. M. Hughes, Y. Yin and A. P. Alivisatos, Science, 2004, 306, 1009 CrossRef CAS PubMed.
- (a) L. D. Trizio and L. Manna, Chem. Rev., 2016, 116, 10852 CrossRef PubMed; (b) B. D. Mao, C. H. Chuang, F. Lu, L. X. Sang, J. Zhu and C. Burda, J. Phys. Chem. C, 2013, 117, 648 CrossRef CAS; (c) B. J. Beberwyck, Y. Surendranath and A. P. Alivisatos, J. Phys. Chem. C, 2013, 117, 19759 CrossRef CAS; (d) L. W. Mi, Y. F. Chen, Z. Zheng, H. W. Hou, W. H. Chen and S. Z. Cui, RSC Adv., 2014, 4, 19257 RSC.
- (a) J. Li, S. Schachermeyer, Y. Wang, Y. Yin and W. Zhong, Anal. Chem., 2009, 81, 9723 CrossRef CAS PubMed; (b) J. Li, T. Zhang, J. Ge, Y. Yin and W. Zhong, Angew. Chem., Int. Ed., 2009, 48, 1588 CrossRef CAS PubMed; (c) J. Yao, K. Flack, L. Ding and W. Zhong, Analyst, 2013, 138, 3121 RSC; (d) J. Yao, X. Han, S. Zeng and W. Zhong, Anal. Chem., 2012, 84, 1645 CrossRef CAS PubMed; (e) J. Yao, S. Schachermeyer, Y. Yin and W. Zhong, Anal. Chem., 2011, 83, 402 CrossRef CAS PubMed.
- Y. Lin, Q. Zhou, D. Tang, R. Niessner, H. Yang and D. Knopp, Anal. Chem., 2016, 88, 7858 CrossRef CAS PubMed.
- R. Hu, T. Liu, X. B. Zhang, Y. H. Yang, T. Chen, C. C. Wu, Y. Liu, G. Z. Zhu, S. Y. Huan, T. Fu and W. H. Tan, Anal. Chem., 2015, 87, 7746 CrossRef CAS PubMed.
- X. R. Zhang, H. X. Liu, R. J. Li, N. B. Zhang, Y. Xiong and S. Y. Niu, Chem. Commun., 2015, 51, 6952 RSC.
- X. R. Zhang, Y. Zhang, F. T. Chen, Y. Li and S. S. Zhang, Chem. Commun., 2016, 52, 13261 RSC.
- J. Wang, G. Liu and A. Merkoi, J. Am. Chem. Soc., 2003, 125, 3214 CrossRef CAS PubMed.
- (a) A. A. Zinchenko, K. Yoshikawa and D. Baigl, Adv. Mater., 2005, 17, 2820 CrossRef CAS; (b) G. Y. Lan, W. Y. Chen and H. T. Chang, RSC Adv., 2011, 1, 802 RSC.
- L. A. Stearns, R. Chhabra, J. Sharma, Y. Liu, W. T. Petuskey, H. Yan and J. C. Chaput, Angew. Chem., 2009, 121, 8646 CrossRef.
- R. Seidel, L. C. Ciacchi, M. Weigel, W. Pompe and M. Mertig, J. Phys. Chem. B, 2004, 108, 10801 CrossRef CAS.
- (a) C. F. Monson and A. T. Woolley, Nano Lett., 2003, 3, 359 CrossRef CAS; (b) T. P. Qing, Z. H. Qing, Z. G. Mao, X. X. He, F. Z. Xu, L. Wen, D. G. He, H. Shi and K. M. Wang, RSC Adv., 2014, 4, 61092 RSC; (c) Z. H. Qing, X. X. He, D. G. He, K. Wang, F. Z. Xu, T. P. Qing and X. Yang, Angew. Chem., Int. Ed., 2013, 52, 9719 CrossRef CAS PubMed.
- (a) Z. H. Qing, X. X. He, T. P. Qing, K. Wang, H. Shi, D. G. He, Z. Zou, L. Yan, F. Z. Xu, X. S. Ye and Z. Mao, Anal. Chem., 2013, 85, 12138 CrossRef CAS PubMed; (b) J. P. Cao, W. Wang, B. Bo, X. X. Mao, K. M. Wang and X. L. Zhu, Biosens. Bioelectron., 2017, 90, 534 CrossRef CAS PubMed.
- (a) Z. Z. Dong, L. Zhang, M. Qiao, J. Ge, A. L. Liu and Z. H. Li, Talanta, 2016, 146, 253 CrossRef CAS PubMed; (b) L. Zhang, Q. Y. Cai, J. Li, J. Ge, J. Y. Wang, Z. Z. Dong and Z. H. Li, Biosens. Bioelectron., 2015, 69, 77 CrossRef CAS PubMed; (c) J. Ge, L. Zhang, Z. Z. Dong, Q. Y. Caia and Z. H. Li, Anal. Methods, 2016, 8, 2831 RSC; (d) J. Ge, Z. Z. Dong, L. Zhang, Q. Y. Cai, D. M. Bai and Z. H. Li, RSC Adv., 2016, 6, 91077 RSC.
- (a) X. Wang, T. Hou, T. Lu and F. Li, Anal. Chem., 2014, 86, 9626 CrossRef CAS PubMed; (b) S. Bi, X. Jia, J. Ye and Y. Dong, Biosens. Bioelectron., 2015, 71, 427 CrossRef CAS PubMed.
- Y. Hu, Y. Wu, T. Chen, X. Chu and R. Yu, Anal. Methods, 2013, 5, 3577 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01799a |
| This journal is © The Royal Society of Chemistry 2018 |