
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular radical cyclization approach to access highly substituted indolines and 2,3-dihydrobenzofurans under visible-light†
Clarice A. D. Caiuby a,
Akbar Alia,
Vinicius T. Santanab,
Francisco W. de S. Lucasa,
Marilia S. Santosa,
Arlene G. Corrêaa,
Otaciro R. Nascimentob,
Hao Jianga and
Márcio W. Paixão*a
a,
Akbar Alia,
Vinicius T. Santanab,
Francisco W. de S. Lucasa,
Marilia S. Santosa,
Arlene G. Corrêaa,
Otaciro R. Nascimentob,
Hao Jianga and
Márcio W. Paixão*a
aCenter of Excellence for Research in Sustainable Chemistry (CERSusChem), Department of Chemistry, Federal University of São Carlos – UFSCar, Rodovia Washington Luís, km 235 – SP-310, São Carlos, São Paulo, Brazil 13565-905. E-mail: mwpaixao@ufscar.br
bGroup of Molecular Biophysics “Sérgio Mascarenhas”, São Carlos Institute of Physics – IFSC/USP, University of São Paulo, Avenida Trabalhador São Carlense, 400, São Carlos, SP 13560-970, Brazil
First published on 5th April 2018
Abstract
The combination of visible-light and tris(trimethylsilyl)silane promoting intramolecular reductive cyclization protocol for the synthesis of functionalized indolines and 2,3-dihydrobenzofurans has been developed. The transformations occur in the absence of transition metal and additional photocatalyst. In addition, quantum yield (Φ) was determined and electron paramagnetic resonance spectroscopy was performed to better understand the reaction pathway.
Introduction
Oxygen- and nitrogen-containing heterocyclic scaffolds are important components of a variety of important natural products and prevalent structural motifs in biologically active molecules, agrochemicals and designed materials.1In particular, indolines and 2,3-dihydrobenzofurans have attracted considerable interest from both synthetic and medicinal chemistry due to the presence of these privileged scaffolds in pharmaceutical2 and naturally occurring alkaloids3 (Fig. 1). Under this scenario, highly decorated architectures containing those systems exhibit noteworthy anti-HIV inhibitor,4 antihypertensive5 and anti-inflammatory6 activities.
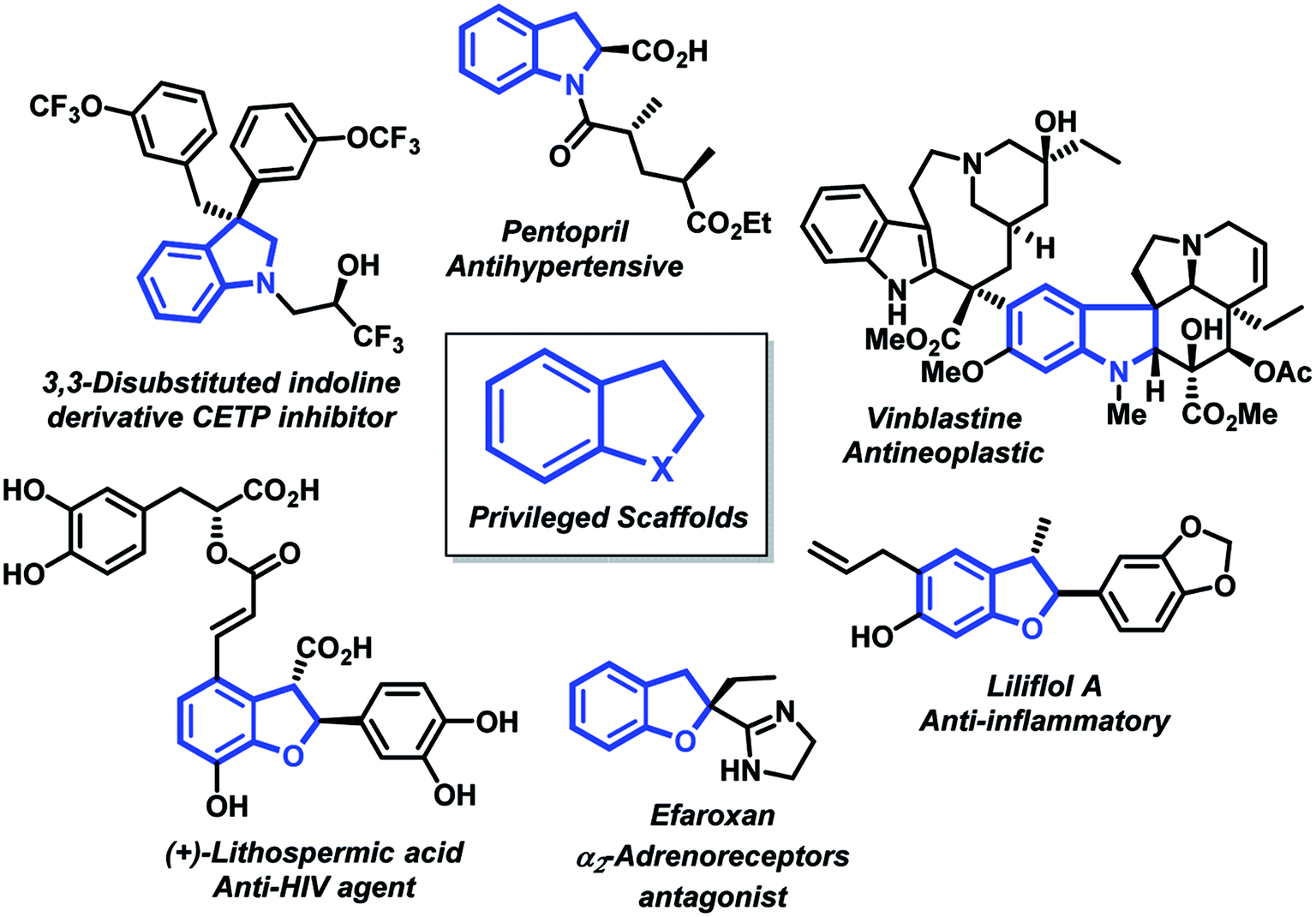 | ||
| Fig. 1 Selected examples of biologically active indolines and 2,3-dihydrobenzofurans. | ||
As a consequence of their high biological potentials, significant effort has been devoted to the development of efficient protocols for the synthesis of these heterocyclic frameworks.7 Towards this end, some traditional methods involves oxidation processes,8 Lewis or Bronsted acids9 and transition-metal-catalyzed reactions – for example, the intramolecular hydro functionalization of alkenes, aryl functionalization, the heteroannulation of 1,3-dienes and transition-metal-catalyzed dicarbofunctionalization of unactivated olefins via tandem cross-coupling/cyclization process to afford a variety of oxygen and nitrogen heterocycles.10
Aside from these approaches, free-radical mediated reactions have been developed to access these unique heterocyclic cores.11 However, a large number of the seminal single-electron-transfer strategies requires stoichiometric quantities of hazardous reagents (e.g. tributyltin hydride or AIBN) as radical generators – in which implicates environmental concerns.12
Nevertheless, the field of synthetic organic photochemistry is rapidly growing in importance as it represents an efficient and environment-friendly alternative to the previous described methods.13 The photon energy allows to access reactive intermediates that cannot be accessed by other strategies employing a clean energy source under mild conditions. Under this concept, either UV or visible light mediated approaches have been described to access indolines and dihydrobenzofurans (Scheme 1).
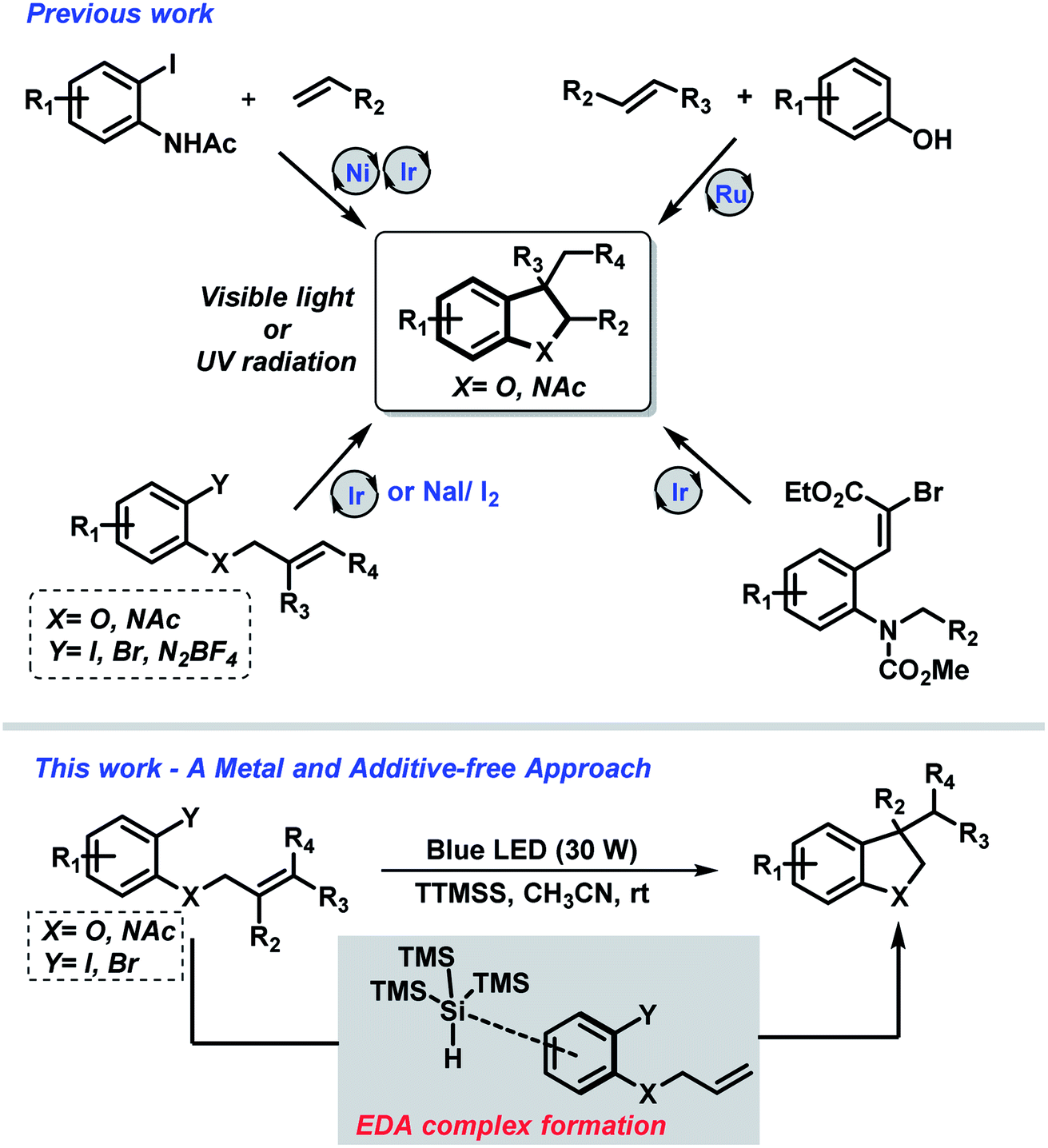 | ||
| Scheme 1 Photo-promoted strategies for synthesis of indolines and 2,3-dihydrobenzofurans. | ||
Very recently, Li and co-workers introduced an elegant metal-free carboiodination protocol for the syntheses of highly decorated indolines and 2,3-dihydrobenzofurans under UV radiation.14
On the other hand, the use of visible-light as a source of energy for promoting chemical transformation has emerged as an interesting topic in organic synthesis.15 In this sense, different research groups have demonstrated the syntheses of nitrogen- and oxygen-containing heterocyclic scaffolds using Cu(I),16 Ir(III)17 and Ru(II)18 based complexes under visible-light irradiation. Moreover, Jamison and coworkers have described the combination of nickel- and photoredox-catalysis (Ni/Ru) for the regioselective synthesis of 3-substituted indolines from alkenes and 2-iodoacetanilides.19
Although the advances of metal promoted photoredox strategies in the activation of small molecules, these approaches also revealed some drawbacks such as catalysts cost, removal from the reaction medium and the need of an excess of additive. In this context, electron donor–acceptor (EDA) complex have been explored as an alternative.20
Nevertheless, in spite of the considerable advances that have been made in this field, the design of general and mild photochemical strategies that improves efficiency, lower cost and decrease waste are highly desirable.21 In this regard, our research group has recently described a metal-free photochemical protocol for the synthesis of N-heterocyclic compounds.22 Encouraged by this work, which took advantage of the EDA complex formation – herein, we report the extension of this approach towards the synthesis of highly substituted indolines and 2,3-dihydrobenzofurans.
Results and discussion
We began our investigation performing a screening of different solvents and the starting material (see ESI Tables S2 and S4†). The N-allyl-N-2-haloanilines were evaluated, considering the halogen atom and nature of the substituent on the nitrogen. The N-allyl-N-(2-iodophenyl)acetamide 1a was chosen as model substrate, employing reaction conditions similar to our previously reported synthesis of the privileged indole and oxindole scaffolds (Table 1, for complete details see ESI†). Pleasing, the reaction of 1a in the presence of 1 equiv. of tris(trimethylsilyl)silane (TTMSS), acetonitrile as solvent under a compact fluorescent lamp (CFL) irradiation for 24 hours provided 47% of the targeted indoline 2a (Table 1, entry 1). The addition of ethanol (5 equiv.) as hydrogen source did not change the reaction yield (Table 1, entry 2). We also evaluated the amount of TTMSS and found that with 2 equiv. proved beneficial to the yield of the desired product (Table 1, entry 3).
|
|||||
|---|---|---|---|---|---|
| Entry | Silane | hν | Temp | Time (h) | Yieldb (%) |
| a Unless otherwise specified reactions were performed using 1a (0.20 mmol), silane (2 equiv.), MeCN (4 mL).b Yield of isolated products.c Reaction vessel placed between two 12 W CFL lamp.d 1 equiv. of TTMSS was used.e Reaction was performed with 1 equiv. of TTMSS and 5 equiv. of EtOH.f Irradiated with a white light-emitting diode (LEDs) strip.g UV radiation (40 W).h Irradiated with a blue LEDs strip under argon atmosphere and degasified solvent. | |||||
| 1c,d | TTMSS | CFL | rt | 24 | 47 |
| 2c,e | TTMSS | CFL | rt | 24 | 50 |
| 3c | TTMSS | CFL | rt | 24 | 70 |
| 4f | TTMSS | LEDs | rt | 24 | 92 |
| 5g | TTMSS | UV | rt | 0.5 | 65 |
| 6f | Et3SiH | LEDs | rt | 48 | — |
| 7f | PhSiH3 | LEDs | rt | 48 | — |
| 8 | TTMSS | Darkness | rt | 72 | 70 |
| 9 | TTMSS | Darkness | 50 °C | 5 | 72 |
| 10h | TTMSS | LEDs | rt | 5 | 89 |
| 11 | — | Darkness | 50 °C | 48 | — |
| 12f | — | LEDs | rt | 48 | — |

To our delight, using white-light LEDs as the source of visible irradiation showed a significant effect on chemical efficiency – thus providing indoline 2a in 90% yield (Table 1, entry 4). Under UV-irradiation (254 nm) for 30 min at room temperature, the model reaction provides product 2a in 65% along with byproducts. These findings were attributed to high energy of the irradiation which can decrease the reaction selectivity (Table 1, entry 5). Moreover, the influence of the organosilane was also investigated. Meanwhile, when Et3SiH and PhSiH3 were used instead of TTMSS, the reaction did not occur, and the starting material were completely recovered (Table 1, entries 6 and 7).
It is worth mentioning that the TTMSS-based radical can also be generated under aerobic conditions,23 therefore, control experiments were conducted with light absence to evaluate this parallel pathway for the tandem reductive/cyclization process. Conducting the reaction at room temperature and in the darkness delivered the indoline ring in 70% after a long reaction time (72 hours, Table 1, entry 8).
When the reaction temperature was increased to 50 °C, similar range of chemical efficiency could be observed in a short reaction period (Table 1, entry 9). Although the generation of the TTMSS-based radical under thermal conditions was feasible, it showed to be less effective than the photochemical process. It is well reported that molecular oxygen – even under air atmosphere conditions – can spontaneously react with TTMSS at room temperature to generate a silyl-centered radical.24 Having that in mind, we then evaluated the influence of oxygen in the radical generation of the described methodology. When the reaction was carried out under degasified solvent, argon atmosphere and blue LEDs irradiation, the desired indoline 1a could be obtained in 89% chemical yield (Table 1, entry 10). Furthermore, performing the reaction under air atmosphere, low temperature and darkness, there was no product formation and the starting material could be completely recovered (see ESI† for details). These results provided strong evidences that support the essential role of the visible-light in the radical generation event of this protocol. Next, complementary experiments showed that the combination of TTMSS and visible light were essential for this transformation (Table 1, entries 11 and 12).
We then sought to determined the wavelength required for an efficient silane-mediated atom transfer process of 1a (Table 2).
|
||||
|---|---|---|---|---|
| Entry | λb (nm) | Solvent | Time (h) | Yieldc (%) |
| a Unless otherwise specified reactions were performed using 1a (0.20 mmol), TTMSS (2 equiv., 0.40 mmol) and specified solvent (4 mL).b Irradiated with a LEDs strip.c Yield of isolated products. | ||||
| 1 | 632 | MeCN | 12 | 46 |
| 2 | 520 | MeCN | 7 | 85 |
| 3 | 450 | MeCN | 5 | 98 |
| 4 | 450 | Acetone | 24 | 93 |
The use of a red-light LEDs (632 nm) resulted in a low yield after 12 hours (Table 2, entry 1) – meanwhile, under a green-light LEDs (520 nm) irradiation, the reaction was completed after 7 hours with an increment on the chemical yield (Table 2, entry 2). To our delight, the highest yield was achieved employing blue-light LEDs irradiation (Table 2 – entry 3), therefore, under the model reaction conditions the desired product was obtained in 98% yield after 5 h.
With the optimized reaction conditions in hand, we examined the generality of the reaction protocol (Table 3). In general, the N-allyl-2-iodophenyl-acetamides bearing electron-withdrawing groups on the aromatic ring (2b–2e) reacted efficiently to afford the corresponding indoline products in moderate to excellent yields. To our delight, substrates bearing additional halogen e.g. Cl – functionality that allow subsequent functionalization by transition metal mediated cross-coupling reactions25 – underwent the reaction efficiently, affording the corresponding product in good yield (2f = 78% y). Subsequently, we observed that the substitution pattern on the aromatic ring of the substrate did not affect the chemical efficiency (compared 2f vs. 2g), however a substantial difference in the reaction times were necessary. The protocol was also compatible with substrates bearing electron-donating groups, thus rendering products 2j and 2k in 60% and 83% respectively. As an important demonstration of the reactivity, N-allyl-2-bromophenyl-acetamide derivatives where also effective under our reaction conditions, providing the cyclization products 2i and 2l, albeit in diminished yields.

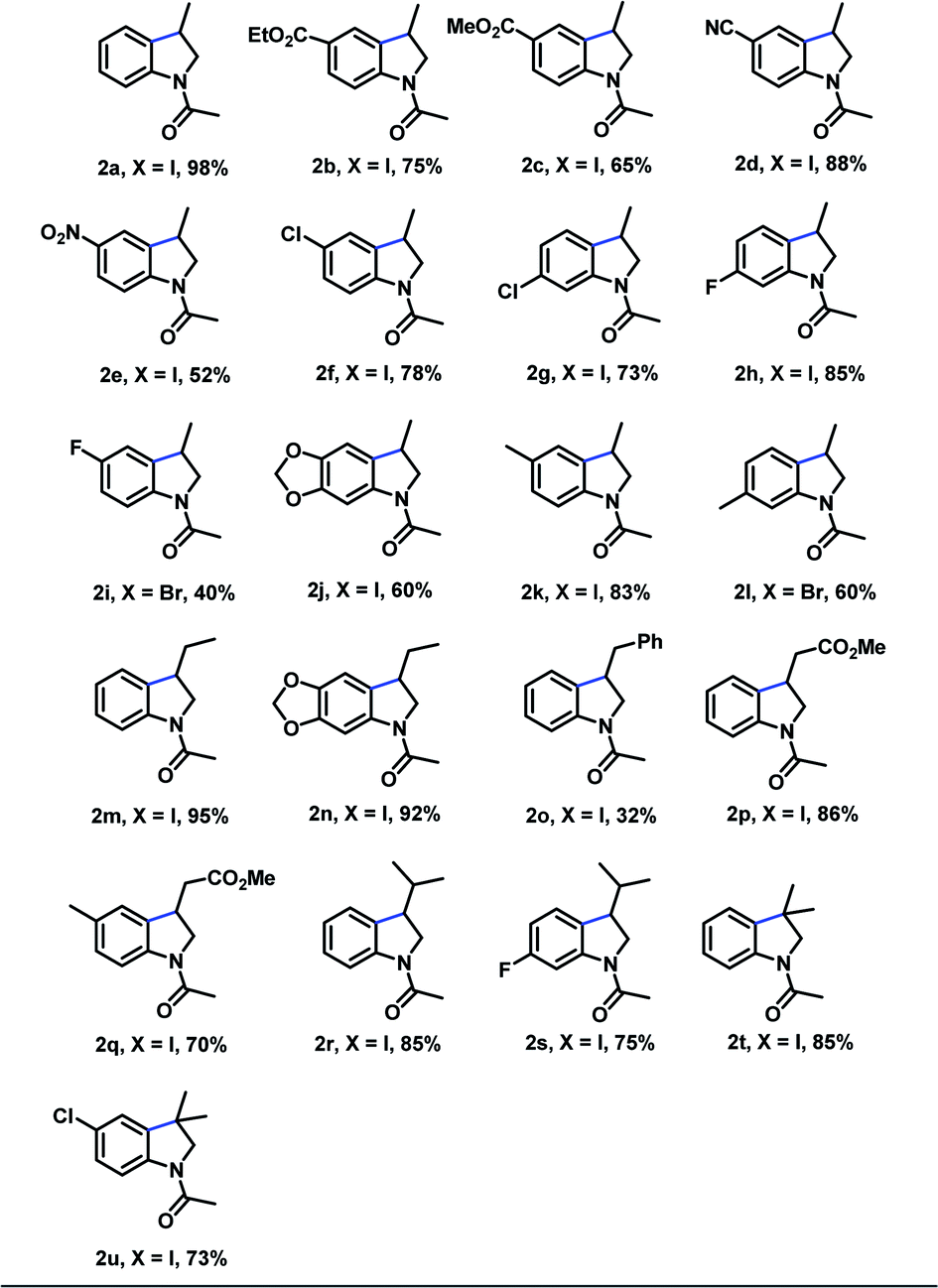
To further assess the functional-group tolerance of the metal-free photo-initiated synthesis of indolines, we explored the reactivity of different substrates having different substituents on the olefinic portion. Notably, a series of β-mono substituted olefins bearing different functional group, such as methyl (2m and 2n), aryl (2o) and methyl ester (2p and 2q) are compatible with this reaction conditions, and all the reaction produced desired products in moderate to good yields. Substrates having two substituents on the terminal carbon of the olefin provided the products 2r and 2s in good yields. Finally, α-disubstituted substrates were converted into the desired indoline products 2t and 2u in good yields.
Based on the encouraging results for the synthesis of the indoline framework, we envisioned to extended the TM-free visible-light-induced photochemical methodology to the synthesis of 2,3-dihydrobenzofurans. To this end, a brief optimization was further performed, especially regarding the wavelength range of the light source. Among those screened, white LEDs provided optimum yield of the 2,3-dihydrobenzofuran (for details see the ESI†). Subjecting the substrate (1-(allyloxy)-2-iodobenzene) to a slightly modified version of the optimized conditions – however, in the absence of both TTMSS and visible light, no conversion to the desired cyclization product were observed, with the starting material being fully recovered.
At this point, our attention turned toward the preparation of some examples of oxygen-heterocycle in the optimized conditions (Table 4). For substrate bearing terminal alkene, high conversion was observed. With respect to the alkene component elaboration, the reaction was also tolerant to alkyl-substitution partners (4b and 4c).

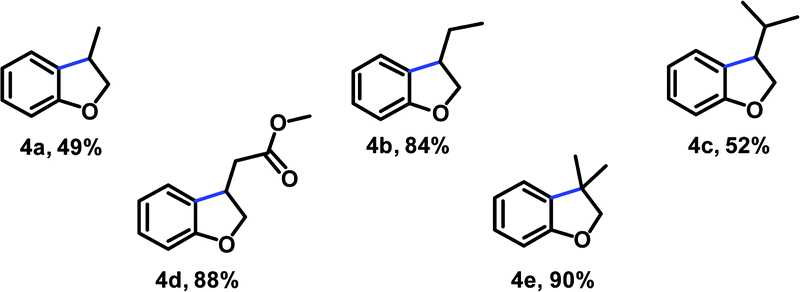
The double bond was also amenable further structural variations, therefore, 4d having a terminal electron withdrawing group undergoes sequential reduction/cyclization in high yield. Finally, the potential of this methodology was further demonstrated in the synthesis of dihydrobenzofuran 4e which contains a quaternary center.
In order to gain further insight into the mechanism of this reaction, a series of control experiments were also pursued. Therefore, we started by performing UV-visible spectroscopic studies to investigate the behavior of all the components of the reaction under visible light. Individual solutions of 1a, TTMSS, indoline 2a and reaction mixture were prepared and the absorbances were measured (Fig. 2). The solutions were prepared in duplicate, one of which was kept under light irradiation and the other in the light absence (see ESI† for further details). The solid lines represent all measurement performed in the darkness, whereas dashed lines correspond to the spectra of the solutions exposed to the irradiation. Firstly, the individual evaluation of compounds 1a (red lines), TTMSS (black lines) and 2a (blue lines) did not showed a significant change in the absorbance spectra after exposure to the blue LEDs irradiation (Fig. 2).
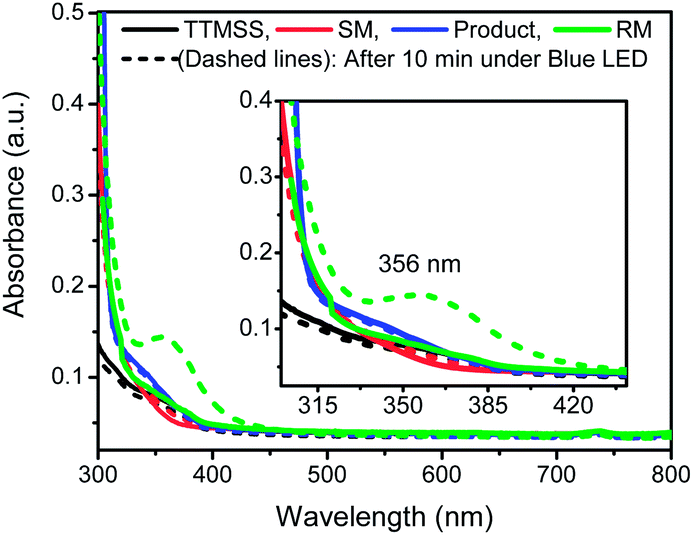 | ||
| Fig. 2 Absorbance spectra for TTMSS solutions (0.02 mol L−1, black lines), 1a (0.01 mol L−1, red lines), 2a (0.01 mol L−1, blue lines) and the reaction mixture of TTMSS (0.02 mol L−1) + 1a (0.01 mol L−1) (RM, green lines). SM: starting material, RM: reaction mixture. | ||
However, a new absorption band (λ = 356 nm) was detected for the reaction mixture composed by TTMSS and 1a (RM, green lines) upon the radiation incidence – which suggests the formation of an intermediate that absorbs nearly the visible light region. This species would be generated through electron donor–acceptor association between 1a and TTMSS (EDA complex), and therefore acts as a promoter of the transformation.
Complementarily, fraction of light absorbed experiments revealed the reaction mixture absorbs almost 100% of the light fraction at 450 nm, supporting the experimental data in which the reaction is promoted efficiently by blue LEDs light. These findings added to lower irradiance of the green and red LEDs (85% and 3% respectively) corroborate the lower yield and reactivity observed for the reaction under these light sources (for details see ESI†).
Trapping experiments were performed and afforded strong evidence that the reaction proceeds via a single electron pathway. When the reaction was accomplished in the presence of a radical scavenger – 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) – there was a complete inhibition of the desired transformation. Furthermore, MS analysis of the reaction mixture showed the presence of the starting material 1a (which was fully recovered) along with the formation of the TTMSS adduct with TEMPO, implying the formation of silyl-centered radical under visible-light irradiation.
Additionally, spin trapping experiments were also performed. In this study, compound 1a was used as model substrate and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as spin trap.26 Thus, carrying out the reaction either in the darkness or in the absence of TTMSS no signal was observed. On the other hand, subjecting substrate 1a to a blue LEDs irradiation, in the presence of TTMSS and DMPO, resulted in the EPR spectrum (red line) presented in Fig. 3. In the same figure, the simulated spectrum (black line) using the EasySpin package27 is presented.
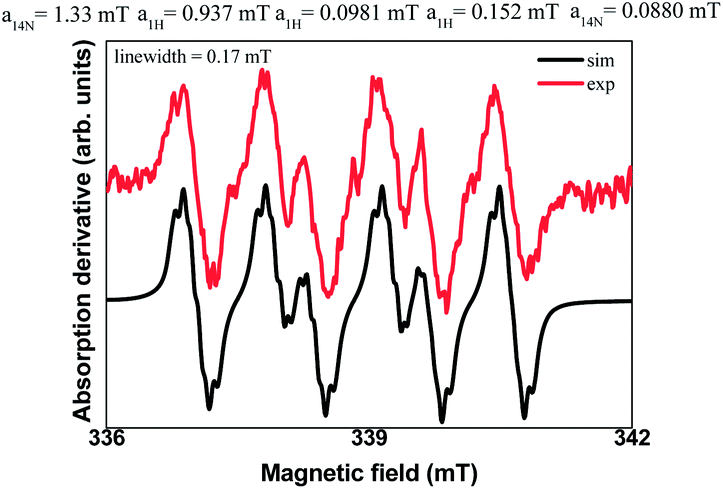 | ||
| Fig. 3 EPR spectra of the reaction mixture with DMPO under blue LEDs radiation (30 W). | ||
The hyperfine splitting observed was first simulated considering a spin-1/2 (NO˙ radical) coupled to the closest N and H nuclei of the DMPO molecule, resulting in the hyperfine coupling constants a14N = 1.33 mT, a1H = 0.94 mT with a 0.4 mT linewidth. However, considering the possible products formed between the DMPO and the intermediary radicals of the reaction, several steps were taken into account in order to improve the simulation.
The best simulation was obtained considering two additional hydrogen atoms that are closer to the bond of the trapped radical with the DMPO (position 3) and a more distant nitrogen (both nitrogen- and hydrogens atoms provided from 2a), that must also be in the trapped radical. The hyperfine coupling constants that best fit the data are a1H = 0.0981 mT, a1H = 0.152 mT, and a14N = 0.088 mT with a linewidth of 0.17 mT. The radical species captured by the DMPO may be the alkyl radical intermediate, leading to the formation of the adduct 5 (Scheme 2). The nuclei used in the simulation are highlighted in the adduct 5. Moreover, no EPR signal was detected after irradiation of a TTMSS solution in acetonitrile.
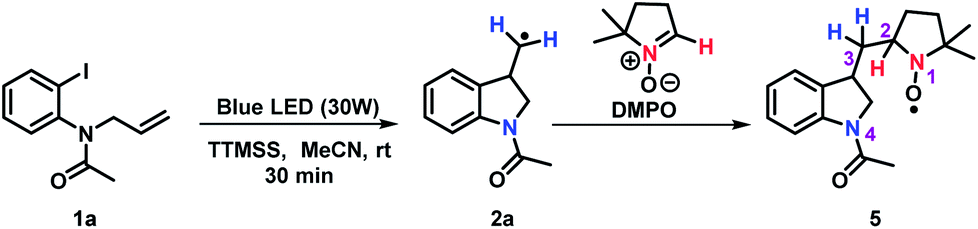 | ||
| Scheme 2 Reaction between 1a and the trapping reagent DMPO. | ||
Likewise, quantum yield experiments were performed to provide additional insights about radical chain propagation. The quantum yield of the specified photo reaction between 1a and TTMSS to provide 2a, was obtained using the photodecomposition of potassium trioxalatoferrate(III) trihydrate as the actinometer. The quantum yield average for the reaction was Φ = 28.37, which suggests that a single photon triggers 28 radical steps, indicating that transformation can occur through radical-chain process.28 This fact corroborates the hypothesis that TTMSS has an important role as a radical initiator.
In light of these experimental results and previous work,22 a plausible radical/SET pathway based on excited EDA complexes is outlined in Fig. 4. Upon light irradiation, the photochemical transformation would occur via a reactive excited complex (1a – TTMSS)*. The generation of such complex, can be rationalized through two distinct mechanisms – (a) the formation of an electron donor–acceptor association (EDA complex) which is irradiated at the wavelength of its CT band, or (b) local irradiation of individual bands of either components, leads to excited form, which can combine with its nonexcited counterpart, thus leading to the corresponding exciplex. Sequentially, over either an electron-transfer or energy-transfer events the homolytic cleavage of the Si–H bond generates the silicon-centered radical (I). Then, a dehalogenation reaction of the substrate 1a occurs through the abstraction of the iodine by the silicon radical, forming an aryl radical (II). The intermediate II passes through a 5-exo-trig cyclisation, to generate the alkyl radical III, followed by abstraction of hydrogen to form the final product 2a.
 | ||
| Fig. 4 Mechanistic proposal for photo-promoted indolines synthesis. | ||
Conclusions
In summary, we have described a mild and metal-free visible light mediated protocol for the synthesis of highly substituted indolines that proceeds via formation of an EDA complex between N-allyl-2-haloanilines and the organosilane reagent, TTMSS. Several functional groups were well tolerated affording the nitrogen heterocycles mostly in good to excellent yields. The methodology has also been expanded to oxygen-containing substrates for the synthesis of 2,3-dihydrobenzofurans. Mechanistic insights were obtained through UV-visible spectroscopy, quantum yield, and EPR analysis indicating that the association substrate-TTMSS is essential to start the photo-transformation, which proceeds through a radical chain propagation. The features of this methodology such as environmentally friendly radical generation and highly valued structures reveal the potential of this chemistry to access a variety of complex scaffolds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. A. D. Caiuby and A. Ali acknowledge CNPq for their Master and PhD fellowships, respectively. FAPESP is also acknowledged for the postdoctoral fellowship to F. W. de S. Lucas and M. S. Santos. M. W. Paixão and A. G. Corrêa gratefully acknowledge financial support from CNPq (INCT – Catálise) and FAPESP (13/07600-3, 14/50249-8 and 15/17141-1). The authors also acknowledge GSK for the financial support.Notes and references
- (a) X. X. Guo, D. W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed; (b) B. Eftekhari-Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958–3043 CrossRef CAS PubMed; (c) N. R. Candeias, L. C. Branco, P. M. P. Gois, C. A. M. Afonso and A. F. Trindade, Chem. Rev., 2009, 109, 2703–2802 CrossRef CAS PubMed; (d) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051–3060 RSC.
- (a) J. E. Wilson, R. Kurukulasuriya, M. Reibarkh, M. Reiter, A. Zwicker, K. Zhao, F. Zhang, R. Anand, V. J. Colandrea, A. M. Cumiskey, A. Crespo, R. A. Duffy, B. A. Murphy, K. Mitra, D. G. Johns, J. L. Duffy and P. Vachal, ACS Med. Chem. Lett., 2016, 7, 261–265 CrossRef CAS PubMed; (b) P. Mayer, P. Brunel and T. Imbert, Bioorg. Med. Chem. Lett., 1999, 9, 3023–3026 CrossRef.
- D. Zhang, H. Song and Y. Qin, Acc. Chem. Res., 2011, 44, 447–457 CrossRef CAS PubMed.
- I. S. Abd-Elazem, H. S. Chen, R. B. Bates and R. C. C. Huang, Antiviral Res., 2002, 55, 91–106 CrossRef CAS PubMed.
- A. Rakhit, M. E. Hurley, V. Tipnis, J. Coleman, A. Rommel and H. R. Brunner, J. Clin. Pharmacol., 1986, 26, 156–164 CrossRef CAS PubMed.
- E. D. Coy, L. E. Cuca and M. Sefkow, Bioorg. Med. Chem. Lett., 2009, 19, 6922–6925 CrossRef CAS PubMed.
- L. Pouységu, A. V. Avellan and S. Quideau, J. Org. Chem., 2002, 67, 3425–3436 CrossRef.
- (a) S. J. Gharpure and Y. G. Shelke, Org. Lett., 2017, 19, 5406–5409 CrossRef CAS PubMed; (b) M. Rueping, C. Brinkmann, A. P. Antonchick and I. Atoresei, Org. Lett., 2010, 12, 4604–4607 CrossRef CAS PubMed.
- (a) For Pd-catalyzed reactions see: E. D. Coy, L. Jovanovic and M. Sefkow, Org. Lett., 2010, 12, 1976–1979 CrossRef PubMed; (b) A. R. O. Venning, P. T. Bohan and E. J. Alexanian, J. Am. Chem. Soc., 2015, 137, 3731–3734 CrossRef CAS PubMed; (c) For Cu-catalyzed reaction see: J. Huang, Y. Chen, J. Chan, M. L. Ronk, R. D. Larsen and M. M. Faul, Synlett, 2011, 1419–1422 CrossRef CAS; (d) For Ru-catalyzed reaction see: M. K. Manna, S. K. Bhunia and R. Jana, Chem. Commun., 2017, 53, 6906–6909 RSC; (e) For Rh-catalyzed reaction see: T. A. Davis, C. Wang and T. Rovis, Synlett, 2015, 26, 1520–1524 CrossRef CAS PubMed.
- (a) D. Liu, G. Zhao and L. Xiang, Eur. J. Org. Chem., 2010, 3975–3984 CrossRef CAS; (b) A. M. Hyde and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 177–180 CrossRef CAS PubMed; (c) M. D. Ganton and M. A. Kerr, Org. Lett., 2005, 7, 4777–4779 CrossRef CAS PubMed; (d) M. S. Pinaud, P. Diaz, J. Martinez and F. Lamaty, Tetrahedron Lett., 2007, 63, 3340–3349 CrossRef; (e) J. lnanaga, O. Ujikawa and M. Yamaguchi, Tetrahedron Lett., 1991, 32, 1737–1740 Search PubMed; (f) R. W. Charnley, B. W. Puffer and P. H. Dussault, J. Am. Chem. Soc., 2014, 136, 5821–5823 CrossRef PubMed; (g) S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2011, 133, 1778–1780 CrossRef CAS PubMed; (h) C. Chen, J. Hu, J. Su and X. Tong, Tetrahedron Lett., 2014, 55, 3229–3231 CrossRef CAS; (i) T. T. Schempp, B. E. Daniels, S. T. Staben and C. E. Stivala, Org. Lett., 2017, 19, 3616–3619 CrossRef CAS PubMed.
- For selected reviews see: (a) D. P. Curran, C. H. T. Chen, S. J. Geib and A. J. B. Lapierre, Tetrahedron, 2004, 60, 4413–4424 CrossRef CAS; (b) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMedFor a recent example see: (c) M. Hartmann and A. Studer, Angew. Chem., Int. Ed., 2014, 53, 8180–8183 CrossRef CAS PubMed.
- (a) J. N. Johnston, M. A. Plotkin, R. Viswanathan and E. N. Prabhakaran, Org. Lett., 2001, 3, 1009–1011 CAS; (b) H. Fuwa and M. Sasaki, Org. Lett., 2007, 9, 3347–3350 CrossRef CAS PubMed; (c) Y. Ueno, X. Chino and M. Okawara, Tetrahedron Lett., 1982, 23, 2575–2576 CrossRef CAS.
- (a) D. Ravelli, D. Dondi, M. Fagnonia and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC; (b) J. R. Chen, X. Q. Hu, L. Q. Lu and W. J. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed; (c) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10266 CrossRef CAS PubMed; (e) T. Peglow, S. Blechert and E. Steckhan, Chem. Commun., 1999, 433–434 RSC; (f) H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303–12306 CrossRef CAS PubMed; (g) W. Guo, L.-Q. Lu, Y. Wang, Y.-N. Wang, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 53, 2265–2269 CrossRef PubMed; (h) X.-D. Xia, Y.-L. Ren, J.-R. Chen, X.-L. Yu, L.-Q. Lu, Y.-Q. Zou, J. Wan and W.-J. Xiao, Chem.–Asian J., 2015, 10, 124–128 CrossRef CAS PubMed; (i) X.-D. Xia, L.-Q. Lu, W.-Q. Liu, D.-Z. Chen, Y.-H. Zheng, L.-Z. Wu and W.-J. Xiao, Chem.–Eur. J., 2016, 22, 8432–8437 CrossRef CAS PubMed.
- X. Yang, W. Liu, L. Li, W. Wei and C. J. Li, Chem.–Eur. J., 2016, 22, 15252–15256 CrossRef CAS PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) T. P. Yoon, M. A. Ischay and J. Du, Nature, 2010, 2, 529–532 Search PubMed; (d) E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCuske and D. W. C. MacMillan, Science, 2017, 355, 380–385 CrossRef CAS PubMed; (e) D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef PubMed.
- B. Michelet, C. Deldaele, S. Kajouj, C. Moucheron and G. Evano, Org. Lett., 2017, 19, 3576–3579 CrossRef CAS PubMed.
- (a) H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303–12306 CrossRef CAS PubMed; (b) S. K. Pagire and O. Reiser, Green Chem., 2017, 19, 1721–1725 RSC.
- T. R. Blum, Y. Zhu, S. A. Nordeen and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 11056–11059 CrossRef CAS PubMed.
- S. Z. Tasker and T. F. Jamison, J. Am. Chem. Soc., 2015, 137, 9531–9534 CrossRef CAS PubMed.
- For a review on photoreactions via EDA complexes see: (a) C. Lima, T. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CASFor selected exemples see: (b) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (c) E. Arceo, A. Bahamonde, G. Bergonzini and P. Melchiorre, Chem. Sci., 2014, 5, 2438–2442 RSC; (d) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS PubMed; (e) Y.-Y. Liu, X.-Y. Yu, J.-R. Chen, M.-M. Qiao, X. Qi, D.-Q. Shi and W. Xiao, Angew. Chem., Int. Ed., 2017, 56, 9527–9531 CrossRef CAS PubMed; (f) R. Wang, W. Guan, Z.-B. Han, F. Liang, T. Suga, X. Bi and H. Nishide, Org. Lett., 2017, 19, 2358–2361 CrossRef CAS PubMed; (g) V. Quint, F. Morlet-Savary, J.-F. Lohier, J. Laleveé, A.-C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436–7441 CrossRef CAS PubMed.
- H. Jiang, J. R. Bak, F. J. López-Delgado and K. A. Jørgensen, Green Chem., 2013, 15, 3355–3359 RSC.
- G. P. Silva, A. Ali, R. C. Silva, H. Jiang and M. Paixão, Chem. Commun., 2015, 51, 15110–15113 RSC.
- (a) C. Chatgilialoglu, Acc. Chem. Res., 1992, 188, 188–194 CrossRef; (b) C. Chatgilialoglu, Chem.–Eur. J., 2008, 14, 2310–2320 CrossRef CAS PubMed.
- A. B. Zaborovskiy, D. S. Lutsyk, R. E. Prystansky, V. I. Kopylets, V. I. Timokhin and C. Chatgilialoglu, J. Organomet. Chem., 2004, 689, 2912–2919 CrossRef CAS.
- (a) P. Ruiz-Castillo, D. G. Blackmond and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 3085–3092 CrossRef CAS PubMed; (b) R. A. Green and J. F. Hartwig, Angew. Chem., Int. Ed., 2015, 54, 3768–3772 CrossRef CAS PubMed; (c) L. Li, S. Zhao, A. J. Pangu, M. Diane and M. R. Biscoe, J. Am. Chem. Soc., 2014, 136, 14027–14030 CrossRef CAS PubMed.
- E. G. Janzen, Acc. Chem. Res., 1971, 4, 31–40 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, additional experiments, characterisation of products, and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c8ra01787e |
| This journal is © The Royal Society of Chemistry 2018 |