DOI:
10.1039/C8RA01696H
(Paper)
RSC Adv., 2018,
8, 14879-14887
Efficient activation of persulfate by Fe3O4@β-cyclodextrin nanocomposite for removal of bisphenol A†
Received
26th February 2018
, Accepted 9th April 2018
First published on 19th April 2018
Abstract
Experimental studies were conducted to investigate the degradation of bisphenol A (BPA) by using persulfate (PS) as the oxidant and Fe3O4@β-cyclodextrin (β-CD) nanocomposite as a heterogeneous activator. The catalytic activity was evaluated in consideration of the effect of various parameters, such as pH value, PS concentration and Fe3O4@β-CD load. The results showed that 100% removal of BPA was gained at pH 3.0 with 5 mM PS, 1.0 g L−1 Fe3O4@β-CD, and 0.1 mM BPA in 120 min. Further, the catalytic activity of Fe3O4@β-CD nanocomposite was observed as much higher when compared with Fe3O4 nanoparticles alone. The sulfate and hydroxyl radicals referred to in the Fe3O4@β-CD/PS system were determined as the reactive species responsible for the degradation of BPA by radical quenching and electron spin resonance (ESR) tests. In addition, the catalyst also possessed with accepted reusability and stability. On the basis of the results of the effect of chloride ions (Cl−), β-CD was found to play a crucial role in reducing interference from Cl− ions, and lead to achieve higher removal efficiency for BPA in Fe3O4@β-CD/PS system. A possible mechanistic process of BPA degradation was proposed according to the identified intermediates by gas chromatography-mass spectroscopy (GC-MS).
1. Introduction
Bisphenol A (BPA), a known endocrine disrupter, has been extensively used as important industrial intermediate and monomer in the manufacture of epoxy resins, polyacrylates, polyesters, polycarbonate plastics and other chemical products.1 Because of its relatively high solubility, low biodegradability, and low volatility, BPA can be easily detected in food, paper, surface waters, wastewater treatment plants and even drinking waters.2,3 Moreover, BPA can disrupt the functions of hormones in living organisms due to their estrogenic activity and poses toxicological risks to humans and animals even at a low level.3,4 Thus, many approaches, such as adsorption,5,6 biological treatment,7,8 electrochemical method,9 sonolysis,10 ozonation,11 photocatalytic degradation,4,12 and chemical oxidation13 have been proposed for the elimination of BPA. Of the above various remediation approaches, advanced oxidation processes (AOPs) are considered as one of the most efficient and rapid approaches in terms of completely eliminating the target detrimental pollutants.14,15
Among various AOPs, Fenton method has been extensively applied to remove refractory organics in waste water, because this method can generate selective and highly reactive oxidizing species to remove a wide variety of contaminants using hydrogen peroxide (H2O2).14 Fe2+/H2O2 is the most extensively investigated system for the decomposition of organic contaminants as reported in the literature.16 However, the system has certain shortcomings associated with the homogeneous Fenton method, such as the difficulty to separate and recover dissolved iron ions, the formation of iron sludge residues, ineffective utilization of formed hydroxyl radicals (˙OH).14 To overcome these drawbacks, recent investigations are moving towards the utilization of Fenton-like system based on heterogeneous catalysts. The fabrication of iron-based heterogeneous catalysts (i.e., Fe3O4) with low Fe dissolution have been increasingly studied in recent years to replace the homogeneous Fe2+/H2O2 system.17
To date, increasing attention has been paid to the sulfate radicals (SO4˙−)-based advanced oxidation processes (SR-AOPs) due to its high mineralization efficiency of organic chemicals at trace levels and also as an alternative for AOPs. In contrast to ˙OH generated from H2O2, SO4˙− is a stronger oxidant for its high standard redox potential (2.5–3.1 V) and has a longer lifetime.18 Furthermore, SO4˙− has been reported to be more effective than ˙OH in degrading persistent organic pollutants due to its higher selective and lower scavenging effect of S2O82− on SO4˙−.18,19 SO4˙− can be generated through activation of PS (persulfate) or PMS (peroxymonosulfate) with heating,20 UV-light irradiation,12,21 activated carbon,22,23 transition metal ions (i.e., Fe2+, Co2+ and Ag+).24 Among the above transition metal ions, Co2+ has been reported to be very active in terms of activating PMS to generate SO4˙−.25 Meanwhile, Fe2+ has been considered as an alternative technology to activate PS/PMS to produce SO4˙− due to the environmentally friendly nature.26 It was reported that the coupling of transition metal ions with PS to be the good combination for the formation of SO4˙− and exhibited high degradation efficiency in a wide pH range.25,27 However, the adverse effects of leaching ions on ecology and human health need to be considered in developing metal oxide (i.e., Fe3O4) based heterogeneous PS activation catalytic systems. Yan et al.28 and Tan et al.29 have studied the degradation of sulfamonomethoxine and acetaminophen using Fe3O4 magnetic nanoparticles, respectively. They also reported that the activation of persulfate by Fe3O4 magnetic nanoparticles has great potential applications for the degradation of organic contaminants. Recently, to improve the catalytic activity of heterogeneous catalysts based on Fe3O4 magnetic nanoparticles, polymers were used to modify the surface of pure Fe3O4 to enhance the degradation of pollutants.30 Zhou et al.31 studied the combination of glutathione with Fe3O4 can be used as heterogeneous catalyst to enhance Fenton-like activity of degrading 2,4-dichlorophenol at neutral pH. Sun et al.32 also reported that the Fe3O4 coated by ascorbic acid formed a new heterogeneous activator of persulfate to improve the degradation of 2,4-dichlorophenol. Our previous studies have reported Fe3O4@β-CD could form a ternary complex (Fe2+-β-CD-pollutant) on the surface of catalysts when H2O2 was activated by Fe3O4@β-CD nanocomposite, promoting the ˙OH radicals to directly attack contaminants; The catalytic activity of Fe3O4@β-CD was also demonstrated as higher degradation efficiency than those of Fe3O4 alone.33
In the current study, we focused on the development of high performance Fe3O4@β-CD nanocomposite to destruct BPA from contaminant environment and the investigation of its role as a heterogeneous catalyst for activating PS. The influence of different factors (i.e., pH value, PS concentration and catalyst load and Cl− scavenger) on the BPA degradation was studied and the catalyst reusability was also tested. The involved radical in the degradation reactions were identified and the possible degradation pathway of BPA was also examined.
2. Experimental
2.1 Materials
The materials used in this paper were described in the Text S1 of the ESI.†
2.2 Preparation and characterization of catalysts
The Fe3O4@β-CD nanocomposite was fabricated using a simple and one-step coprecipitation method.33 Briefly, Fe2(SO4)3 (0.01 mol) and FeSO4·7H2O (0.01 mol) were dissolved in 25 mL 0.5 mol L−1 H2SO4 aqueous solution and heated to 80 ± 5 °C. After 0.5 h of vigorous stirring under a N2 stream, then 250 mL of 4.0 mol L−1 NaOH solution (including 12.8 g β-CD) was added drop-wise into the heated Fe2+/Fe3+ solution at 80 °C under continuously mechanical stirring. After 2 h reaction, the generated precipitate was collected by magnetic separation and then washed with deionized water and absolute ethanol for several times. Finally, the precipitate was dried under vacuum at 60 °C for 8 h and then stored in a desiccator for further experiments. Characterization of the prepared catalysts was described in ESI Text S2.†
2.3 Catalytic degradation experiment
All experiments were performed in a 50 mL conical flask placed on a constant temperature rotary shaker with the rotate speed of 150 rpm in the dark at room temperature (25 ± 1 °C). Typically, an appropriate amount of catalyst was dispersed into 10 mL solution of 0.1 mM BPA. Then, a known concentration of PS in the form of oxidant was added into the pH-adjusted solution to initiate the degradation reaction. The final amount of catalyst and PS concentration were 0.5–2.0 g L−1 and 1–10 mM, respectively. Samples were taken at predetermined time intervals using a 5 mL syringe and filtered immediately through a 0.22 μm filter. Before filtrating, excess ethanol was immediately added to quench the reaction. The catalyst gathered from the reaction solution was washed with 2 mL methanol for five times. The washing liquid was collected and mixed together for further analysis. To test the reusability and stability of the catalyst, the catalyst was gathered with a magnet, washed with deionized water, dried under vacuum, and reused for the next reaction under similar experimental conditions. Each degradation test was carried out at least in duplicate, and all the results were expressed as a mean experimental value.
2.4 Sample analysis
The concentration of BPA was determined with a high performance liquid chromatograph (HPLC; ELITE P1201) equipped with a C18 reversed-phase column (4.6 mm × 250 mm) and a UV detector at wavelength of 278 nm. The mobile phase used for BPA was a mixture of methanol and water (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, v/v) at a constant flow rate of 1.0 mL min−1 with a constant column temperature of 30 °C. The aromatic degradation intermediates of BPA were determined by GC-MS, and the detailed parameters were found in ESI Text S3.† ESR studies were performed by using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a spin-trapping agent. The solution containing 40 mM DMPO, 1.0 g L−1 catalyst was prepared, and then 5 mM PS was added into the solution to initiate the reaction. After 2 min reaction, samples were taken and analyzed on a JES-X320 spectrometer at ambient temperature. The ESR instrument was conducted at the center field of 327.0 mT with a modulation frequency of 100 kHz, and power of 1 mW with scan time of 30 s. The degradation of BPA was analyzed by using a total organic carbon (TOC) Analyzer (TOC-L, Shimadzu).
:30, v/v) at a constant flow rate of 1.0 mL min−1 with a constant column temperature of 30 °C. The aromatic degradation intermediates of BPA were determined by GC-MS, and the detailed parameters were found in ESI Text S3.† ESR studies were performed by using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a spin-trapping agent. The solution containing 40 mM DMPO, 1.0 g L−1 catalyst was prepared, and then 5 mM PS was added into the solution to initiate the reaction. After 2 min reaction, samples were taken and analyzed on a JES-X320 spectrometer at ambient temperature. The ESR instrument was conducted at the center field of 327.0 mT with a modulation frequency of 100 kHz, and power of 1 mW with scan time of 30 s. The degradation of BPA was analyzed by using a total organic carbon (TOC) Analyzer (TOC-L, Shimadzu).
3. Results and discussion
3.1 Characterization of the catalysts
The characterizations of catalysts were provided in Fig. S1−S5.† Furthermore, the physical and chemical properties of these catalysts studied in this study were listed in Table S1 in ESI.† These results obviously showed that the catalysts were successfully fabricated by the simple one-step coprecipitation method.
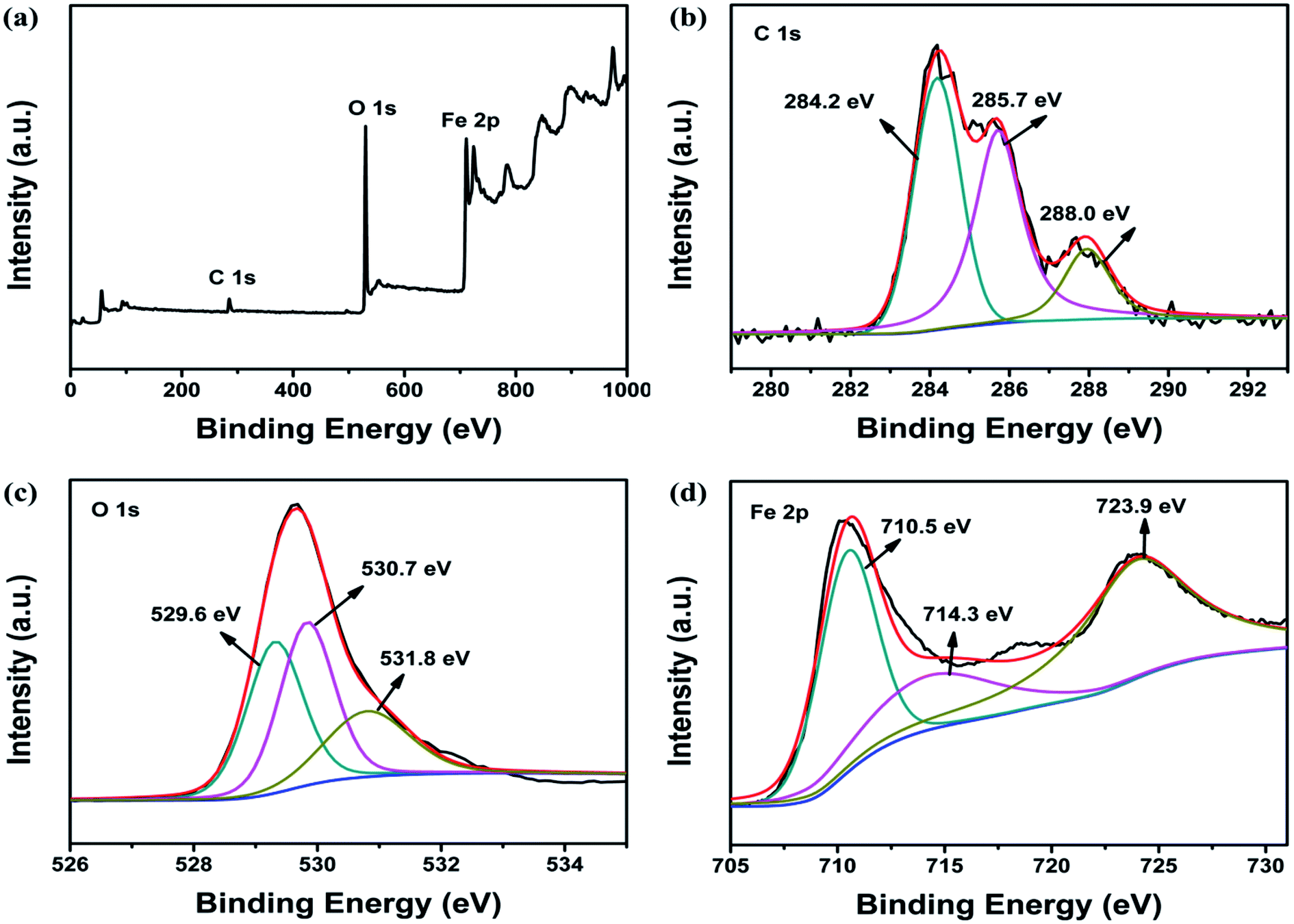
To better characterize the chemical composition of the as-synthesized nanocomposite, the XPS spectrum of Fe3O4@β-CD was recorded. The wide survey scan of XPS spectrum as shown in Fig. 1a indicated the presence of C 1s, O 1s, and Fe 2p. The peak for the C 1s spectrum in Fig. 1b could be curve-fitted into three peak constitutions with binding energy at 284.2, 285.7 and 288.0 eV, which were connected with the carbon atoms of C–C, C–O and C–O–C/C–O–Fe, respectively.34 In Fig. 1c, the O 1s spectrum was integrated into three curves with peak position around 530 eV. The lower binding energy peak at 529.6 eV was associated with the Fe–O bands from Fe3O4, while the one at 530.7 eV was ascribed to C–O of β-CD. And the peak at 531.8 eV should be arisen from Fe–O–C between Fe3O4 and β-CD.35 As shown in Fig. 1d, the binding energy peaks at 710.5 and 723.9 eV in the high resolution Fe 2p spectrum were indicative of the occurrence of Fe 2p3/2 and Fe 2p1/2 peaks of Fe3O4, respectively. Meanwhile, the peak positioned at 710.5 and 714.3 eV could be indexed to the Fe2+ and Fe3+, respectively.35,36 Therefore, the analysis results of XPS further confirm the successful fabrication of Fe3O4@β-CD.
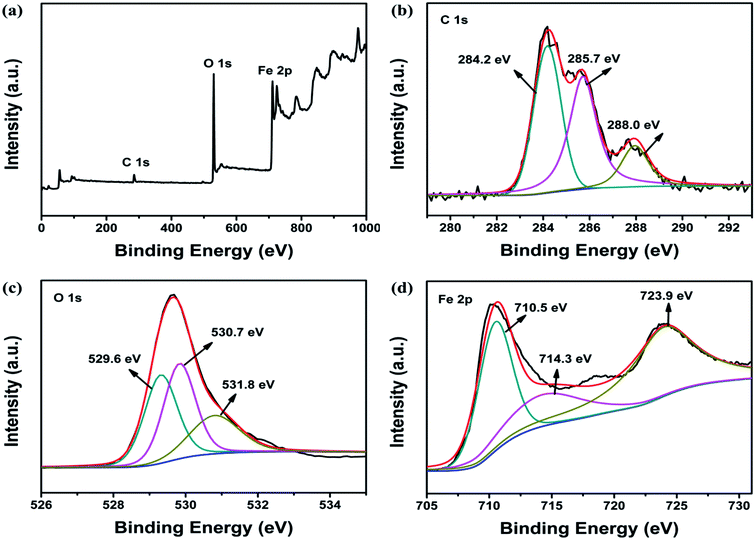 |
| | Fig. 1 XPS spectra of Fe3O4@β-CD: (a) survey spectrum, (b) C 1s, (c) O 1s, (d) Fe 2p. | |
3.2 BPA degradation
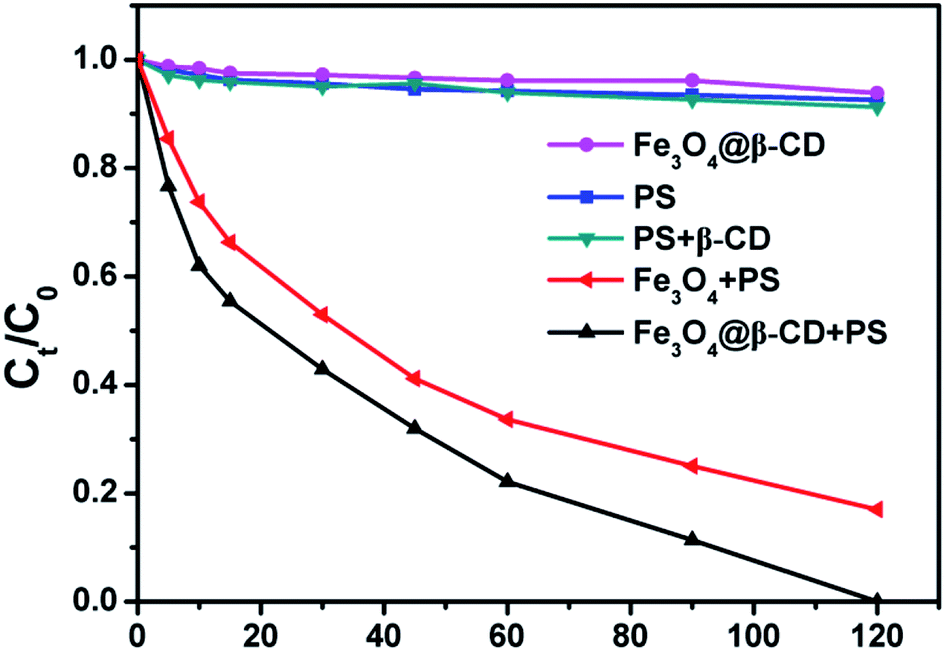
Batch experiments were conducted to compare the removal efficiencies of BPA in various systems at pH 3.0 with initial BPA concentration of 0.1 mM. As can be observed from Fig. 2, BPA was not effectively removed by PS, PS/β-CD, or Fe3O4@β-CD nanocomposite alone, whereas BPA was efficiently degraded when Fe3O4@β-CD or Fe3O4 used in the presence of PS. It can also be observed that only a small amount of BPA being diminished by the PS/β-CD, or Fe3O4@β-CD nanocomposite alone. This may be due to PS decomposition and the surface adsorption of the catalyst. In the presence of only PS, about 7% removal efficiency of BPA was observed, which can be attributed to SO4˙− from the PS decomposition. These results also indicated that the removal of BPA was insignificant by PS, PS/β-CD, or Fe3O4@β-CD nanocomposite alone when compared to the rapid removal of BPA by heterogeneous degradation. The removal and degradation percentages of BPA were 100% and 83% in the Fe3O4@β-CD/PS and Fe3O4/PS system in 120 min, respectively. The results revealed that the dissociation of PS was mainly activated by Fe3O4 nanoparticles. Furthermore, the degradation of BPA followed the pseudo-first-order reaction in kinetics, which may be described with pseudo-first-order rate degradation: ln(C0/Ct) = kobst, where kobs is the apparent rate constant (min−1), and C0, Ct is the concentration of BPA at initial and given time t, respectively. As shown in Fig. S6 of the ESI,† the kobs values of BPA degradation were evaluated as 0.0252 min−1 (R2 = 0.984) for Fe3O4@β-CD nanocomposite and 0.0161 min−1 (R2 = 0.978) for Fe3O4 nanoparticles. The results indicated that the removal of BPA using Fe3O4@β-CD nanocomposite as the heterogeneous catalyst was notably higher than that for Fe3O4 nanoparticles alone. The superior catalytic activity of Fe3O4@β-CD nanocomposite could be ascribed to the synergy effect of β-CD in the Fe3O4@β-CD nanocomposite. In particular, PS was activated by the Fe3O4@β-CD nanocomposite to generate SO4˙− and ˙OH. Meanwhile, the organic pollutant was trapped in the β-CD cavity to form the construction of a ternary complex (iron-β-CD-pollutant),33,37 which caused the generated SO4˙− and ˙OH to increase the possibility of attacking organic pollutant and accelerated degradation process. The ternary complex was also demonstrated by (500 MHz) 1H NMR (Fig. S7†). Therefore, Fe3O4@β-CD coupled with PS can be used as an effective catalytic system for the degradation of BPA.
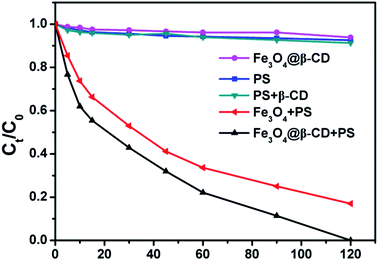 |
| | Fig. 2 Degradation of BPA in different systems. Conditions: [BPA] = 0.1 mM, [PS] = 5 mM, [Fe3O4] = 1.0 g L−1, [Fe3O4@β-CD] = 1.0 g L−1, [β-CD] = 20 mg L−1, pH 3.0 and T = 25 °C. | |
3.3 Effects of pH, PS concentration and catalyst load on BPA degradation
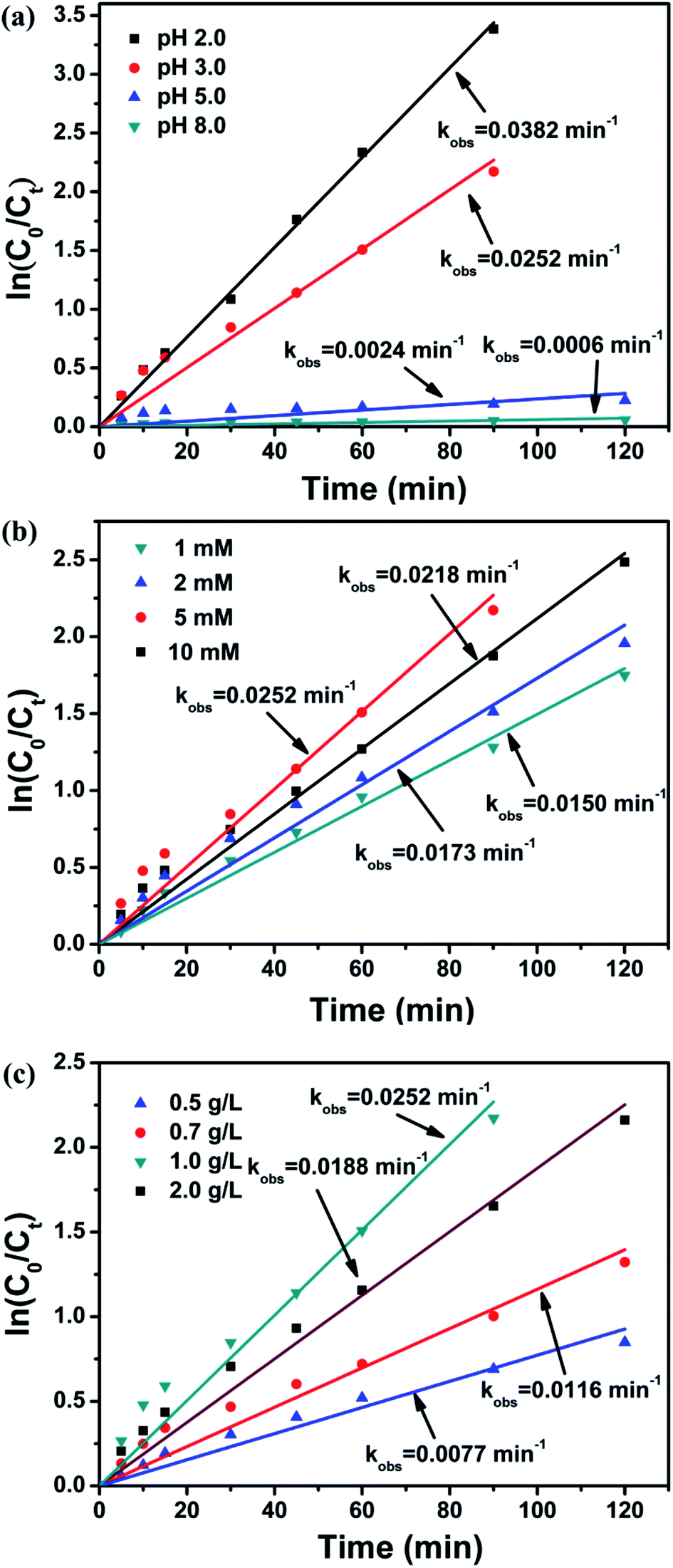
It is well known that the behavior of heterogeneous AOPs is strongly associated with the solution pH for contaminants degradation.33,38,39 Recently, it has been also reported that the degradation processes that focused on iron oxide activated PS and Fe3O4/PMS system were influenced by pH.28,29 Therefore, the degradation of BPA was studied at different solution pH. As observed in Fig. 3a, the kobs value decreased obviously from 0.0382 to 0.0006 min−1 as pH increased from 2.0 to 8.0, which demonstrated that acidic pH condition favored the degradation of BPA in the Fe3O4@β-CD/PS system. However, the more obvious removal efficiency of BPA at pH 2.0 would be owed to the higher concentration of Fe dissolved from Fe3O4@β-CD nanocomposite, which motivated the formation of SO4˙− through homogenous AOPs. In brief, the dissolved Fe2+ reacted with PS can generate more SO4˙−. Then, SO4˙− reacts with Fe2+ at diffusion control rate and an excess amount of Fe2+ in solution can undergo homogenous reaction and thereby further facilitate the reaction.40
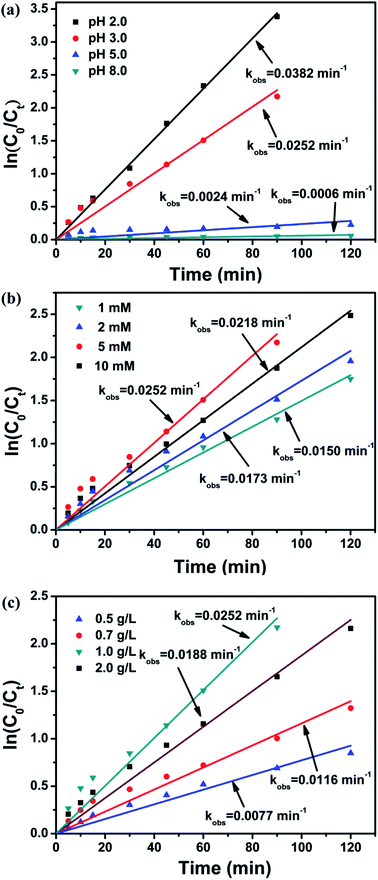 |
| | Fig. 3 Factorial effects of heterogeneous reaction for BPA degradation: (a) pH value (b) PS concentration (c) Fe3O4@β-CD load. Conditions: [BPA] = 0.1 mM, [PS] = 5 mM, [Fe3O4@β-CD] = 1.0 g L−1, pH 3.0 and T = 25 °C. | |
The pseudo-first-order plots for BPA degradation as a function of PS concentration (Fig. 3b) were observed. It is obvious that the kobs value increased significantly from 0.0150 to 0.0252 min−1 as the PS concentration increased from 1 to 5 mM. Moreover, the availability of PS concentration is the vital factor to control the generation of radicals at a low PS concentration. However, with a higher PS concentration (10 mM), the kobs value decreased to 0.0218 min−1, which may be related to the scavenging effect of SO4˙− by PS at the fixed catalyst load. In case of excessive PS concentration, there is a competitive reaction between BPA and PS for the consumption of SO4˙−, which limits the degradation of BPA. The SO4˙− can react with PS to generate S2O8˙−, as expressed by the following reaction.19,41
| | |
SO4˙− + S2O82− → SO42− + S2O8˙−
| (1) |
Further, the SO4˙−/S2O82− reaction with a rate constant of 6.1 × 105 mol L−1 s−1 (reaction (1))41 competes with the SO4˙−/BPA reaction.
The kinetics of BPA degradation was also investigated at different catalyst load (Fig. 3c). The kobs value increased rapidly from 0.0077 to 0.0252 min−1 with an increasing load of Fe3O4@β-CD from 0.5 to 1.0 g L−1, and then slightly decreased to 0.0188 min−1 with further increased Fe3O4@β-CD load up to 2.0 g L−1. The enhancement of reaction rate may be ascribed to the increasing number of active sites for the formation of SO4˙− as the Fe3O4@β-CD load increased. Another possible reason is that high concentration of Fe dissolved from increasing Fe3O4@β-CD nanocomposite will accelerate the generation of SO4˙− and removal of BPA. And the slight decrease of BPA removal may be considered as the agglomeration of nanoparticles and the scavenging of SO4˙− by excess Fe2+ through the following reaction.24,37
| | |
SO4˙− + Fe2+ → Fe3+ + SO42−
| (2) |
3.4 Reusability and stability of Fe3O4@β-CD
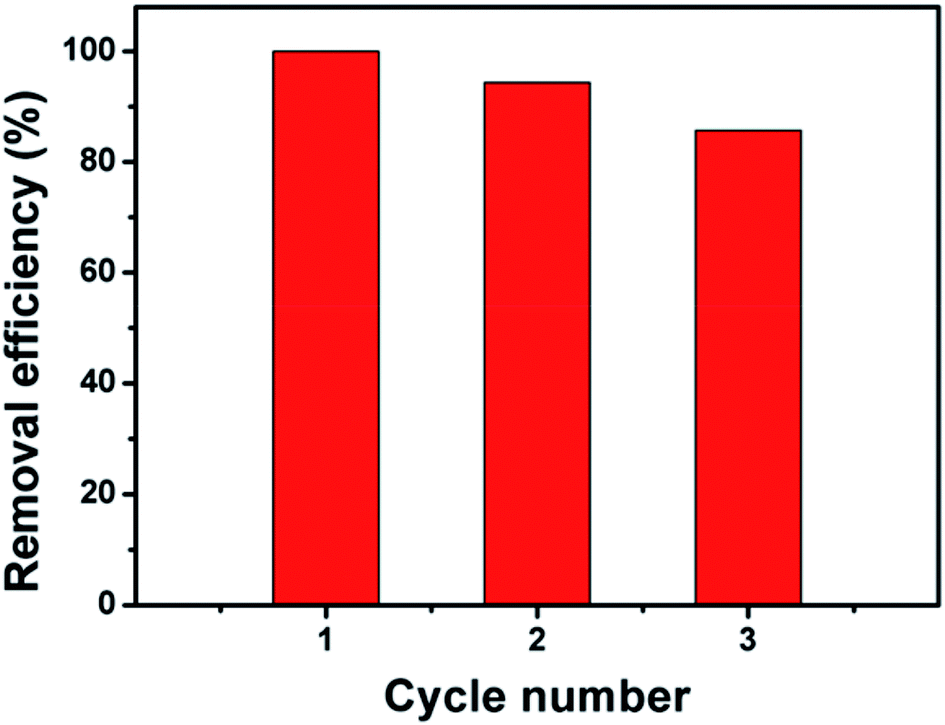
In order to evaluate the reusability and stability of Fe3O4@β-CD nanocomposite, three batches of BPA degradation were carried out under the similar experimental conditions. As shown in Fig. 4, it is obvious that the removal efficiency of BPA was steadily declined with increase of the cycle number, indicating that the reaction activity of catalyst reduced gradually during three successive runs after 120 min of reaction. The decreased activity might be due to the loss of active catalytic sites caused by the leaching iron ions from the catalyst. In order to ascertain the change in surface area, the BET properties of the Fe3O4@β-CD after 120 min reaction were also studied (Table S1†). After three runs, the BET surface area was increased slightly from 104.19 to 119.80 m2 g−1, which may be caused by the leaching of iron ions. As displayed in Fig. S8,† the iron concentrations leaching into the reaction solutions arrived to 0.53 mg L−1 at pH 3 when 0.1 mM BPA was completely degraded. However, the XRD pattern of the reused Fe3O4@β-CD nanocomposite before reaction and reused was observed with similar diffraction peaks (Fig. S1, ESI†). The mass spectra of pure β-CD, Fe3O4@β-CD and reused Fe3O4@β-CD were shown in Fig. S9,† indicating that the most abundant ion was determined as [β-CD] + at m/z 1157 (identified as β-CD) and the β-CD remained untouched during the reaction. The results confirm that the Fe3O4@β-CD nanocomposite still remained in quasi-spherical and almost uniform (Fig. S5, ESI†).
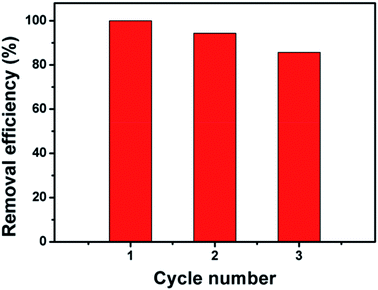 |
| | Fig. 4 Reusability of Fe3O4@β-CD for BPA degradation. Conditions: [BPA] = 0.1 mM, [PS] = 5 mM, [Fe3O4@β-CD] = 1.0 g L−1, pH 3.0 and T = 25 °C. | |
To further estimate the influence of iron ions leaching into the reaction solutions undergo a homogeneous Fenton reaction, iron salt based on the maximal concentration of leaching Fe ions was used to conduct contrast experiments under similar experimental condition. Fig. S10† showed that the degradation efficiency of BPA was after 120 min of reaction, indicating the homogenous reaction only provided a small percentage contribution of degrading BPA.
Thus, Fe3O4@β-CD still had a high stability and a good reusability after the recycling tests, suggesting the Fe3O4@β-CD as an efficient heterogeneous catalyst can be used for a longer reaction time.
3.5 Activation mechanism of PS on Fe3O4@β-CD
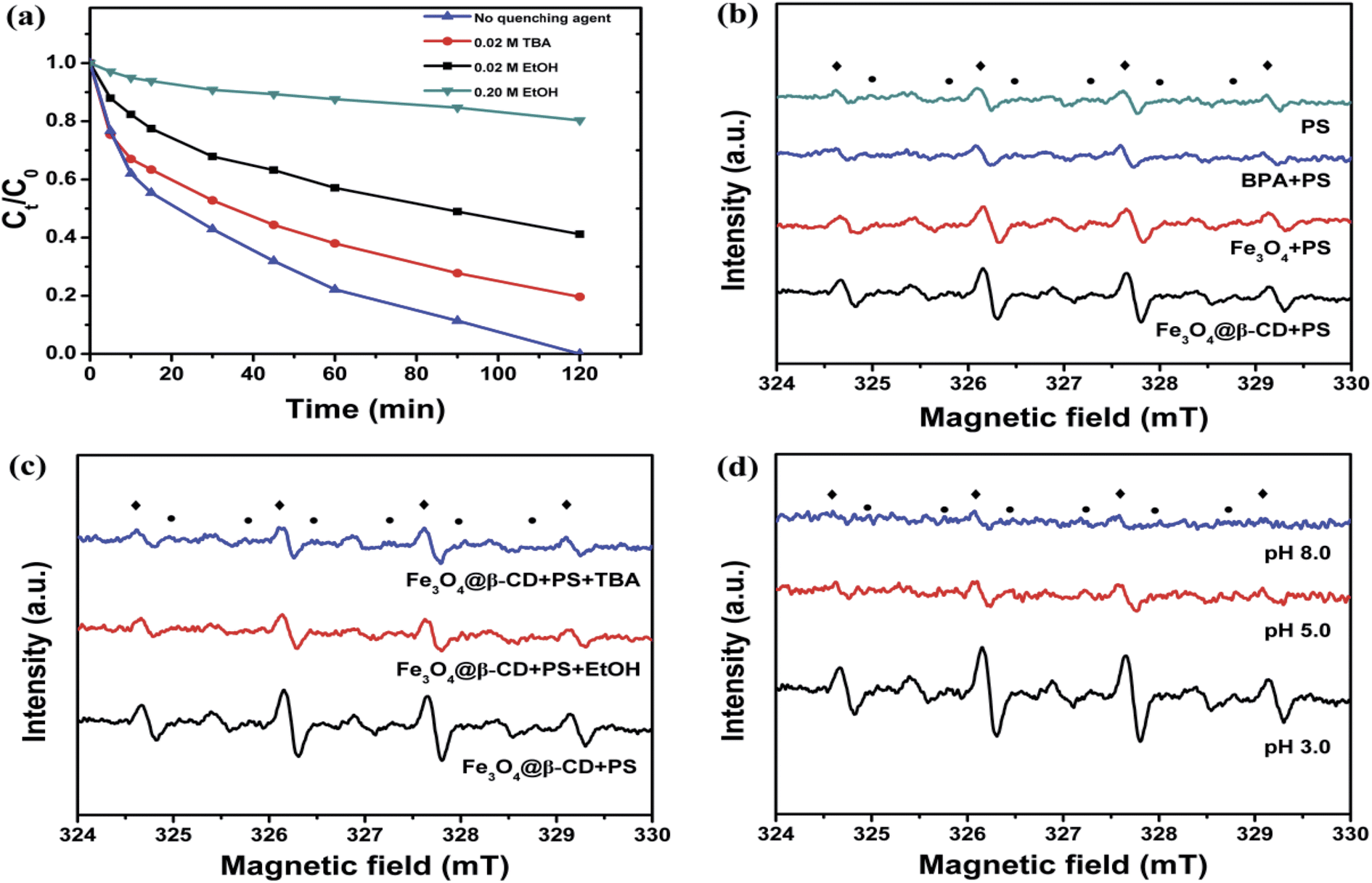
It is reported in the literature that two different reactive radicals (i.e., SO4˙− and ˙OH) could be generated for the decomposition of organic contaminants when PS is activated by metal oxide at room temperatures.39,42 To identify the major reactive radical species generated from the activation of PS, two alcohols (EtOH and TBA) were spiked into reaction solution as radical quenchers. In general, EtOH (containing α-hydrogen) is an effective quencher for both SO4˙− and ˙OH due to the high reaction rate constants with SO4˙− (k = (1.6–7.7) × 107 mol L−1 s−1) and ˙OH (k = (1.2–2.8) × 109 mol L−1 s−1), respectively.43 However, TBA (without α-hydrogen) is chosen as another effective quencher for ˙OH but not for SO4˙− because the reaction rate constant with ˙OH (k = (3.8–7.6) × 108 mol L−1 s−1) is much higher than that with SO4˙− (k = (4–9.1) × 105 mol L−1 s−1).43 Based on the above properties, the contribution of SO4˙− and ˙OH on the degradation of BPA could be differentiated by the quenching tests with EtOH and TBA. As shown in Fig. 5a, when no radical quencher was added, 100% of BPA was removed in 120 min. However, the addition of 0.02 M EtOH and TBA led to decreasing removal of BPA to 58.8% and 80.4% in 120 min, respectively. More addition of 0.2 M EtOH further decreased the BPA removal to 20.7%. The much more obviously decrease of BPA removal caused by EtOH than TBA indicated that the mainly reactive radical species generated during the activation of PS by Fe3O4@β-CD were sulfate radicals.
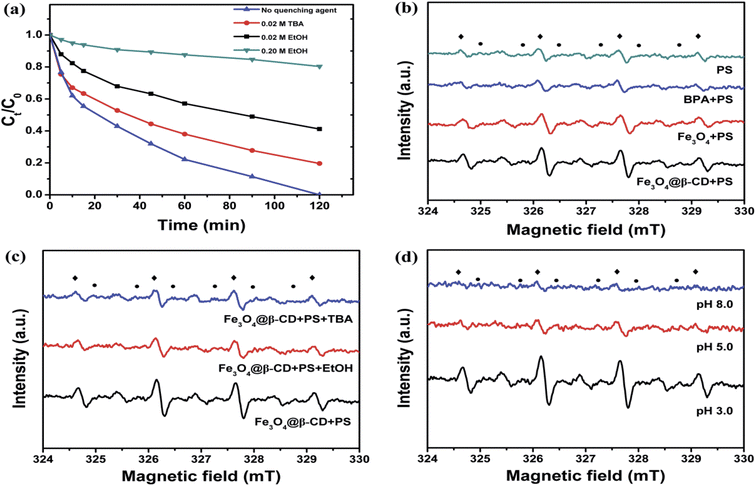 |
| | Fig. 5 (a) Removal of BPA without/with radical quencher of EtOH (0.02 M and 0.2 M) and TBA (0.02 M). (b) ESR spectra of various systems. (c) ESR spectra of Fe3O4@β-CD/PS system with/without EtOH and TBA (0.02 M). (d) The influence of pH value on the ESR spectra. Conditions: [PS] = 5 mM, [Fe3O4@β-CD] = 1.0 g L−1, [DMPO] = 40 mM (◆: DMPO-˙OH; ●: DMPO-SO4˙−), T = 25 °C. | |
ESR/DMPO studies were performed to further confirm the generation of involved radical species (SO4˙−, ˙OH) in the Fe3O4@β-CD and PS catalytic system as shown in Fig. 5b–d. The fourfold characteristic peak with an intensity ratio of 1:2:2:1 in accordance with those of DMPO-˙OH adducts and the pattern of typical DMPO-SO4˙− adducts were observed in the ESR spectra of Fe3O4@β-CD/PS system. It can also be observed from Fig. 5b that both Fe3O4@β-CD/PS and Fe3O4/PS systems could have the ability to generate SO4˙− and ˙OH radicals. Also, the observations suggested that the generation of radicals was independent of the presence of β-CD. Furthermore, when the PS was added into BPA solution, the relative intensity of DMPO-SO4˙− and DMPO-˙OH signals decreased, which suggested that these radicals were prior to reacting with BPA. The addition of 0.02 M EtOH and TBA decreased the intensity of the signals of DMPO-SO4˙− and DMPO-˙OH adducts in the Fe3O4@β-CD/PS/EtOH or TBA when compared with that of Fe3O4@β-CD/PS system as shown in Fig. 5c. This suggests that the addition of 0.02 M EtOH and TBA scavenged only a small part of generated radicals, which was in agreement with the experimental results as presented in Fig. 5a. Finally, the influence of pH value on the relative intensity of DMPO-SO4˙− and DMPO-˙OH signals were studied, and Fig. 5d showed that the DMPO-SO4˙− and DMPO-˙OH signals reduced as the pH increased from 3.0 to 8.0. This may be due to the fact that the dissolved iron from catalyst surface was lowered, which leads to a decreased number of radicals generated at much higher pH. From the above results, it can be confirmed that both sulfate and hydroxyl radicals were involved for the destruction of BPA. The reaction process can proceed as following reactions.44
| | |
SO4˙− + BPA → SO42− + BPA˙+
| (3) |
| | |
BPA˙+ + H2O → (OH) BPA˙ + H+
| (4) |
| | |
˙OH + BPA → (OH) BPA˙
| (5) |
| | |
(OH) BPA˙ → intermediates → CO2+ H2O
| (6) |
3.6 Influence of Cl− scavenger
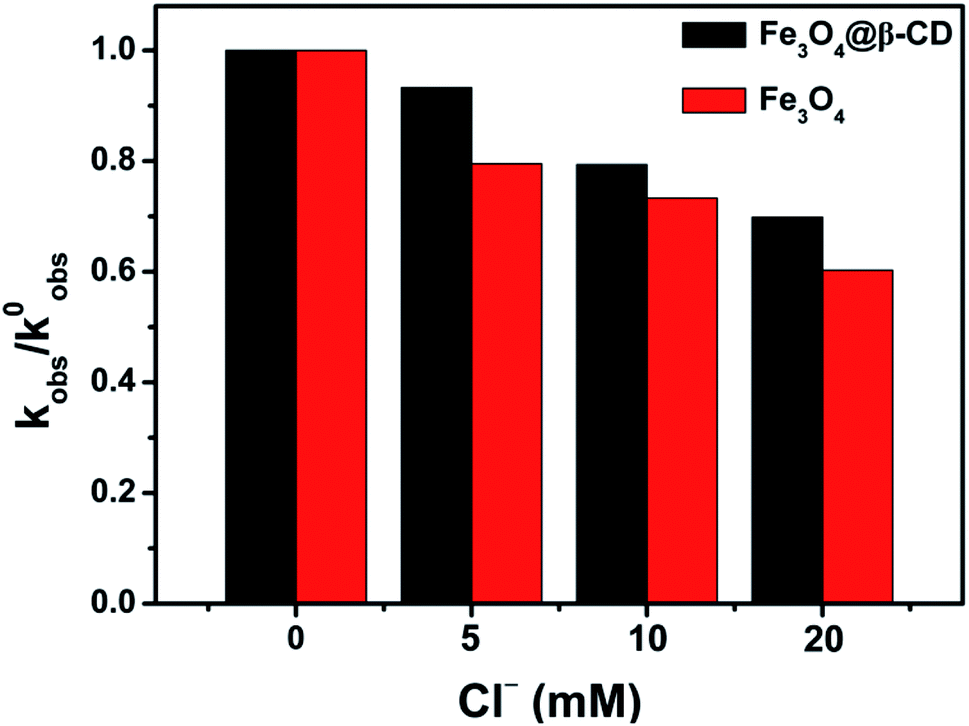
It has been previously studied that the existence of the chloride ion could serious inhibit the AOPs effectiveness during wastewater treatment because it is a radical scavenger.45,46 It is thermodynamically spontaneous for PS to oxidize Cl− to produce less reactive chlorine species (i.e., Cl2, Cl˙ and Cl2˙), which can rapidly combine with another chloride and reduce the oxidation efficiency of sulfate radicals.46,47 Therefore, further experiments were conducted to test the influence of Cl−. As shown in Fig. S11,† the kobs values of BPA degradation were decreased as 0.0176 min−1 for Fe3O4@β-CD and 0.0098 min−1 for Fe3O4 in the presence of 20 mM Cl−. The kobs values for the BPA degradation decreased obviously as the Cl− concentration increased from 5 to 20 mM, which meant Cl− scavenged ˙OH and SO4˙− more effectively, forming a lower steady state radical concentration. At the same time, comparison of normalized first order rate constants of Fe3O4@β-CD and Fe3O4 for BPA degradation in the presence of Cl− was performed (Fig. 6). When β-CD was present in the system, less interference from added Cl− was observed. The possible reason may be that a plenty of iron is bound to β-CD, and a majority of BPA molecules are bound to iron bearing β-CD. Further, the generated ˙OH and SO4˙− was also in close distance to BPA. As a result, Cl− had less probability to effectively scavenge this generated radicals.48 The chemical reaction involved in Fe3O4@β-CD/PS system in the presence of Cl− can be expressed as following reactions.46,47,49| | |
SO4˙− + Cl− ↔ SO42− + Cl˙
| (7) |
| | |
Cl2˙− + Cl2˙− → Cl2 + 2Cl−
| (9) |
| | |
Cl2˙− + H2O → ClOH˙− + H+ + Cl−
| (10) |
| | |
ClOH˙− + H+ ↔ Cl˙ + H2O
| (11) |
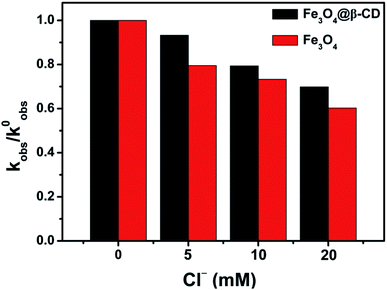 |
| | Fig. 6 Normalized first order rate constants (kobs/k0obs) for BPA degradation in the presence of Cl− (added as NaCl). Conditions: [BPA] = 0.1 mM, [PS] = 5 mM, [Fe3O4@β-CD] = 1.0 g L−1, pH 3.0 and T = 25 °C. | |
3.7 Possible degradation pathway of BPA
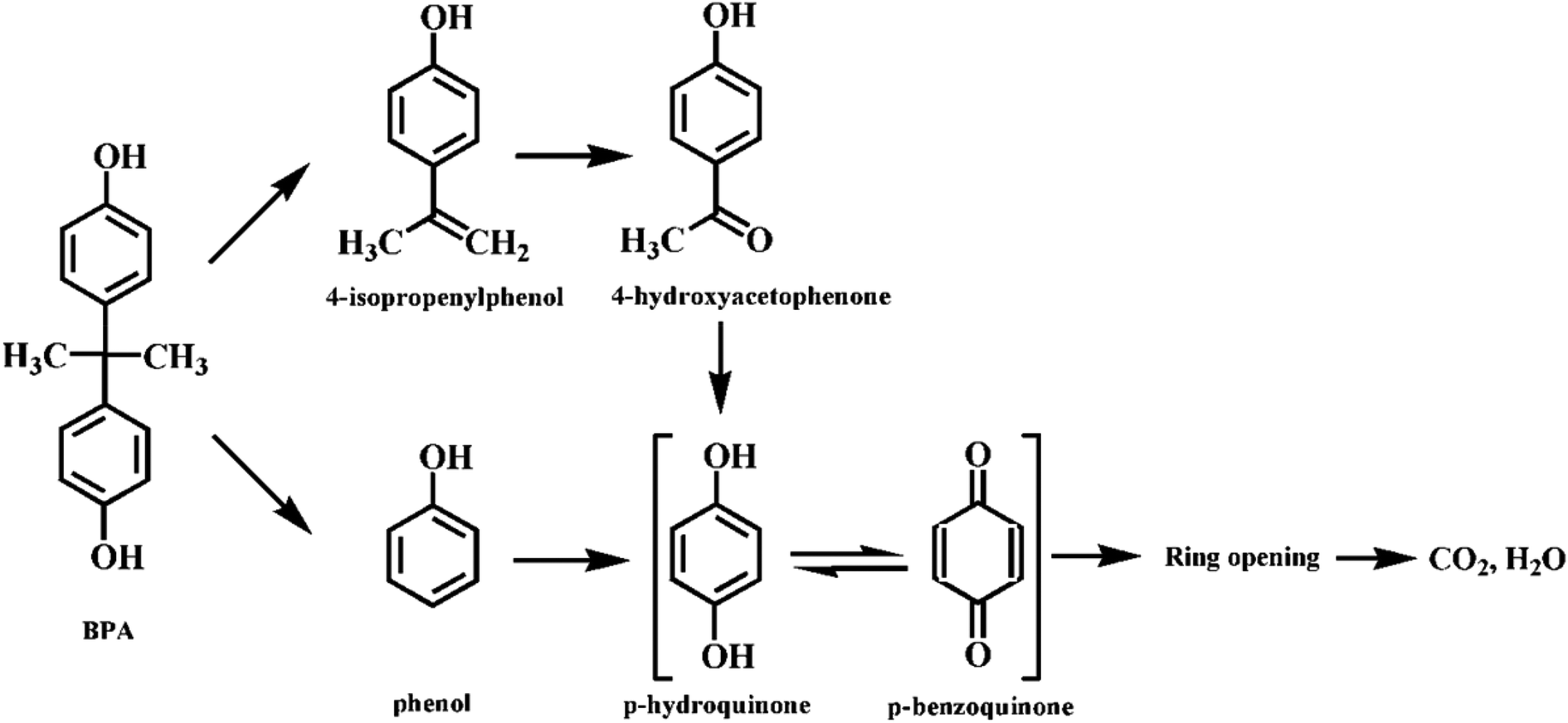
The degradation intermediates were identified by GC-MS analysis in order to understand the degradation pathways of BPA in the Fe3O4@β-CD/PS system (Fig. S12, ESI†). Four main aromatic intermediates, such as phenol, p-benzoquinone, 4-isopropenylphenol, and 4-hydroxyacetophenone were determined in the present study. The determined intermediates suggest that several degradation mechanisms may occur during BPA degradation process. The mechanism can be explained as follows; firstly, BPA was attacked by the radicals generated from the activation of PS and lost one electron, which resulted in the formation of BPA radicals.50 Secondly, the C–C bond of BPA radicals between the isopropyl and benzene ring may be attacked by sulfate radicals due to the higher electron density.51 As a result, BPA radicals were decomposed into phenol and 4-isopropenylphenol by β-scission, which were further converted to p-hydroquinone and 4-hydroxyacetophenone by chemical oxidation reaction. Further, 4-hydroxyacetophenone could be oxidized to p-hydroquinone, which was easily transformed to p-benzoquinone. Finally, the aromatic intermediates were degraded into ring-opening compounds and then further mineralized into CO2 and H2O.
The degree of BPA degradation is investigated by testing solution TOC in the Fe3O4@β-CD/PS system. As shown in Fig. S13,† the results showed that the TOC removal obtained after 120 min was 49%, indicating that a residual amount of BPA intermediates remained in the reaction solution.
Therefore, on basis of the current experimental results and the previous reports for BPA degradation,50 the possible degradation pathway of BPA induced by activation of PS over Fe3O4@β-CD nanocomposite was presented in Fig. 7.
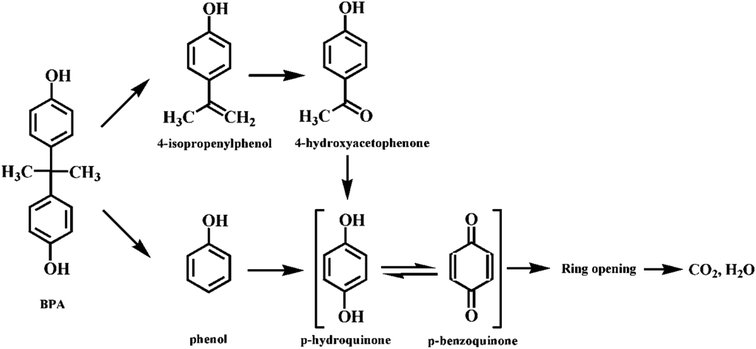 |
| | Fig. 7 Possible pathway for BPA degradation in the Fe3O4@β-CD/PS system. | |
4. Conclusions
In this study, Fe3O4@β-CD nanocomposite prepared via one-step coprecipitation method was first used as a heterogeneous catalyst to activate PS. The results indicated that Fe3O4@β-CD catalyst posed much higher activity in activating PS into radicals, which could further decompose BPA into small organic intermediates or even inorganic compounds. The various operating parameters including pH value, PS concentration, and Fe3O4@β-CD load were investigated systematically. At a PS concentration of 5 mM, complete degradation of 0.1 mM BPA was achieved in 120 min by utilizing 1.0 g L−1 Fe3O4@β-CD nanocomposite at pH 3.0. Both SO4˙− and ˙OH were demonstrated to be involved in this degradation process and SO4˙− was found to have a more crucial effect during the activation process of PS via quenching agents (TBA, EtOH) and ESR/DMPO experiments. Through recycling experiments, the catalyst was proved as recyclable and stable for a longer period of time. The influence of Cl− on the degradation activity showed that β-CD modified Fe3O4 nanocomposite could reduce interference from Cl− and enhance degradation efficiency for BPA. Additionally, the possible degradation pathway of BPA was proposed on the basis of intermediates identified through GC-MS analysis. Therefore, the activation of PS by Fe3O4@β-CD has great potential as an effective catalytic material for environmental applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NSFC 21377068 and 21575077), the National Basic Research Program of China (973 Program 2013CB934301), Natural Science Foundation of Shandong Province of China (No. ZR2014BM027) and the Fundamental Research Funds of Shandong University (2016JC030).
References
- C. A. Staples, P. B. Dorn, G. M. Klecka, S. T. O'Block and L. R. Harris, Chemosphere, 1998, 36, 2149–2173 CrossRef CAS PubMed.
- X. Yang, P. F. Tian, C. Zhang, Y. Q. Deng, J. Xu, J. Gong and Y. F. Han, Appl. Catal., B, 2013, 134–135, 145–152 CrossRef CAS.
- T. Suzuki, Y. Nakagawa, I. Takano, K. Yaguchi and K. Yasuda, Environ. Sci. Technol., 2004, 38, 2389–2396 CrossRef CAS.
- C. Guo, M. Ge, L. Liu, G. Gao, Y. Feng and Y. Wang, Environ. Sci. Technol., 2010, 44, 419–425 CrossRef CAS PubMed.
- I. Bautista-Toledo, M. A. Ferro-Garcia, J. Rivera-Utrilla, C. Moreno-Castilla and F. J. V. Fernandez, Environ. Sci. Technol., 2005, 39, 6246–6250 CrossRef CAS PubMed.
- A. Alsbaiee, B. J. Smith, L. Xiao, Y. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef CAS.
- Y. T. Xie, H. B. Li, L. Wang, Q. Liu, Y. Shi, H. Y. Zheng, M. Zhang, Y. T. Wu and B. Lu, Water Res., 2011, 45, 1189–1198 CrossRef CAS.
- A. Svenson, A. S. Allard and M. Ek, Water Res., 2003, 37, 4433–4443 CrossRef CAS PubMed.
- F. Zaviska, P. Drogui, J. F. Blais and G. Mercier, J. Appl. Electrochem., 2010, 42, 95–109 CrossRef.
- B. Darsinou, Z. Frontistis, M. Antonopoulou, I. Konstantinou and D. Mantzavinos, Chem. Eng. J., 2015, 280, 623–633 CrossRef CAS.
- X. Tan, Y. Wan, Y. Huang, C. He, Z. Zhang, Z. He, L. Hu, J. Zeng and D. Shu, J. Hazard. Mater., 2017, 321, 162–172 CrossRef CAS.
- P. Xie, J. Ma, W. Liu, J. Zou, S. Yue, X. Li, M. R. Wiesner and J. Fang, Water Res., 2015, 69, 223–233 CrossRef CAS.
- N. Wang, L. Zhu, M. Lei, Y. She, M. Cao and H. Tang, ACS Catal., 2011, 1, 1193–1202 CrossRef CAS.
- M. Antonopoulou, E. Evgenidou, D. Lambropoulou and I. Konstantinou, Water Res., 2014, 53, 215–234 CrossRef CAS PubMed.
- A. Tsitonaki, B. Petri, M. Crimi, H. Mosbæk, R. L. Siegrist and P. L. Bjerg, Crit. Rev. Environ. Sci. Technol., 2010, 40, 55–91 CrossRef CAS.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- L. Xu and J. Wang, Appl. Catal., B, 2012, 123–124, 117–126 CrossRef CAS.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef CAS.
- C. Liang, Z. S. Wang and C. J. Bruell, Chemosphere, 2007, 66, 106–113 CrossRef CAS.
- S. Yang, P. Wang, X. Yang, L. Shan, W. Zhang, X. Shao and R. Niu, J. Hazard. Mater., 2010, 179, 552–558 CrossRef CAS PubMed.
- Y. H. Guan, J. Ma, X. C. Li, J. Y. Fang and L. W. Chen, Environ. Sci. Technol., 2011, 45, 9308–9314 CrossRef CAS PubMed.
- Y. C. Lee, S. L. Lo, J. Kuo and C. P. Huang, J. Hazard. Mater., 2013, 261, 463–469 CrossRef CAS PubMed.
- S. Yang, T. Xiao, J. Zhang, Y. Chen and L. Li, Sep. Purif. Technol., 2015, 143, 19–26 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS.
- A. Rastogi, S. R. Al-Abed and D. D. Dionysiou, Water Res., 2009, 43, 684–694 CrossRef CAS.
- A. Romero, A. Santos, F. Vicente and C. Gonzalez, Chem. Eng. J., 2010, 162, 257–265 CrossRef CAS.
- J. Yan, M. Lei, L. Zhu, M. N. Anjum, J. Zou and H. Tang, J. Hazard. Mater., 2011, 186, 1398–1404 CrossRef CAS PubMed.
- C. Tan, N. Gao, Y. Deng, J. Deng, S. Zhou, J. Li and X. Xin, J. Hazard. Mater., 2014, 276, 452–460 CrossRef CAS PubMed.
- H. Ren, Y. Su, X. Han and R. Zhou, J. Chem. Technol. Biotechnol., 2017, 92, 1421–1427 CrossRef CAS.
- R. Zhou, N. Shen, J. Zhao, Y. Su and H. Ren, J. Mater. Chem. A., 2018, 6, 1275–1283 CAS.
- C. Sun, R. Zhou, E. Jianan, J. Sun, Y. Su and H. Ren, RSC Adv., 2016, 6, 10633–10640 RSC.
- M. Wang, G. Fang, P. Liu, D. Zhou, C. Ma, D. Zhang and J. Zhan, Appl. Catal., B, 2016, 188, 113–122 CrossRef CAS.
- A. Z. M. Badruddoza, Z. B. Z. Shawon, W. J. D. Tay, K. Hidajat and M. S. Uddin, Carbohydr. Polym., 2013, 91, 322–332 CrossRef CAS PubMed.
- C. Fu, G. Zhao, H. Zhang and S. Li, Int. J. Electrochem. Sci., 2014, 9, 46–60 Search PubMed.
- F. Han, L. Ma, Q. Sun, C. Lei and A. Lu, Nano Res., 2014, 7, 1706–1717 CrossRef CAS.
- C. Liang, C. F. Huang, N. Mohanty, C. J. Lu and R. M. Kurakalva, Ind. Eng. Chem. Res., 2007, 46, 6466–6479 CrossRef CAS.
- L. Xu and J. Wang, Environ. Sci. Technol., 2012, 46, 10145–10153 CrossRef CAS PubMed.
- Y. Lei, C. S. Chen, Y. J. Tu, Y. H. Huang and H. Zhang, Environ. Sci. Technol., 2015, 49, 6838–6845 CrossRef CAS PubMed.
- L. Bu, Z. Shi and S. Zhou, Sep. Purif. Technol., 2016, 169, 59–65 CrossRef CAS.
- G. V. Buxton, M. Bydder and G. A. Salmon, Phys. Chem. Chem. Phys., 1999, 1, 269–273 RSC.
- H. Liu, T. A. Bruton, W. Li, J. Van Buren, C. Prasse, F. M. Doyle and D. L. Sedlak, Environ.
Sci. Technol., 2016, 50, 890–898 CrossRef CAS PubMed.
- C. Liang and H. W. Su, Ind. Eng. Chem. Res., 2009, 48, 558–5562 Search PubMed.
- J. Sharma, I. M. Mishra, D. D. Dionysiou and V. Kumar, Chem. Eng. J., 2015, 276, 193–204 CrossRef CAS.
- T. Zhang, H. Zhu and J. P. Croue, Environ. Sci. Technol., 2013, 47, 2784–2791 CrossRef CAS PubMed.
- C. Liang, Z. S. Wang and N. Mohanty, Sci. Total Environ., 2006, 370, 271–277 CrossRef CAS PubMed.
- G. P. Anipsitakis, T. P. Tufano and D. D. Dionysiou, Water Res., 2008, 42, 2899–2910 CrossRef CAS PubMed.
- M. E. Lindsey, G. Xu, J. L and M. A. Tarr, Sci. Total Environ., 2003, 307, 215–229 CrossRef CAS.
- X. Y. Yu, Z. C. Bao and J. R. Barker, J. Phys. Chem. A, 2004, 108, 295–308 CrossRef CAS.
- J. Sharma, I. M. Mishra and V. Kumar, J. Environ. Manage., 2016, 166, 12–22 CrossRef CAS.
- R. A. Torres-Palma, J. I. Nieto, E. Combet, C. Petrier and C. Pulgarin, Water Res., 2010, 44, 2245–2252 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01696h |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Long Mac,
Yuqiang Zhangc,
Quanqin Zhao*a and
Jinhua Zhan
a,
Long Mac,
Yuqiang Zhangc,
Quanqin Zhao*a and
Jinhua Zhan