
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile one-pot green synthesis of gold nanoparticle-graphene-PEDOT:PSS nanocomposite for selective electrochemical detection of dopamine†
Paweena Pananona,
Chakrit Sriprachuabwongab,
Anurat Wisitsoraatb,
Piyachat Chuysinuanc,
Adisorn Tuantranontb,
Patchareenart Saparpakornd and
Decha Dechtrirat *ae
*ae
aDepartment of Materials Science, Faculty of Science, Kasetsart University, Bangkok, Thailand. E-mail: fscidcd@ku.ac.th
bNational Electronics and Computer Technology Center (NECTEC), National Science and Technology Development Agency (NSTDA), Pathumthani, Thailand
cLaboratory of Organic Synthesis, Chulabhorn Research Institute, Bangkok, Thailand
dDepartment of Chemistry, Faculty of Science, Kasetsart University, Bangkok, Thailand
eSpecialized Center of Rubber and Polymer Materials for Agriculture and Industry (RPM), Faculty of Science, Kasetsart University, Bangkok, Thailand
First published on 3rd April 2018
Abstract
A facile one-pot and green method was developed to prepare a nanocomposite of gold nanoparticle (AuNP), graphene (GP) and poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS). Graphene was first electro-exfoliated in a polystyrene sulfonate solution, followed by a one-step simultaneous in situ formation of gold nanoparticle and PEDOT. The as-synthesized aqueous dispersion of AuNP-GP-PEDOT:PSS was thereafter used to modify the glassy carbon electrode (GCE). For the first time, the quaternary composite between AuNP, GP, PEDOT and PSS was used for selective determination of dopamine (DA) and uric acid (UA) in the presence of ascorbic acid (AA). In comparison to a bare GCE, the nanocomposite electrode shows considerably higher electrocatalytic activities toward the oxidation of DA and UA due to a synergistic effect between AuNP, GP, PEDOT and PSS. Using differential pulse voltammetry (DPV), selective determination of DA and UA in the presence of AA could be achieved with a peak potential separation of 110 mV between DA and UA. The sensor exhibits wide linear responses for DA and UA in the ranges of 1 nM to 300 μM and 10 μM to 1 mM with detection limits (S/N = 3) of 100 pM and 10 μM, respectively. Furthermore, the proposed sensor was also successfully used to determine DA in a real pharmaceutical injection sample as well as DA and UA in human serum with satisfactory recovery results.
Introduction
During the last decade, nanomaterials have made a major impact on the field of electrochemical biosensors due to their distinctive physical, chemical and electronic properties. These inherent features have made them the best fit for building up novel sensing platforms with enhanced signal generation and amplification.1 Among them, graphene is one of the most widely used materials. Graphene has extraordinary properties including remarkable electron mobility, high electronic conductivity, a large specific surface area and unexpected mechanical stability. The π-conjugated electrons of graphene can also efficiently delocalize along its 2D surface making graphene very sensitive to local changes in the chemical environment. These unique properties have enabled graphene to become an excellent sensing material.2Graphene can be synthesized either by physical or chemical means.3 Among those various existing protocols, electrolytic exfoliation in an aqueous polyelectrolyte solution is of particular interest. This is because the obtained graphene product is stable and almost oxide-free.4 As a result, it requires no additional reduction step, no reducing agent and no chemical waste making this protocol a green alternative method. However, it is well known that each of these graphene layers tend to agglomerate through van der Waals and π–π stacking interactions. To address this problem, the decoration of graphene sheets with noble metal nanoparticles has proved to be a good strategy to prevent the restacking process.
Besides increasing stability, metal nanoparticles decorated graphene sheets also exhibit significantly enhanced performance including outstanding electrocatalytic activity and high electrical conductivity. These features have led to an increasing use of graphene/metal nanoparticle composites in the fabrication of electrochemical biosensors.5 Among the more commonly used metal nanoparticles, gold is the most widely studied metal. Gold nanoparticles (AuNPs) have been broadly reported for their high catalytic activity which could facilitate the electron transfer rate and decrease the electrode overpotentials.6 A conventional synthesis of AuNPs usually involves chemicals which pose health and environmental risks. As a consequence, the need for more green methods to reduce the metal salts to metallic nanoparticles is in high demand.
Conducting polymers have been used as electrode-modifying materials to enhance the physico-chemical properties of bare electrodes. They behave in particular as a redox mediator toward several analytes, significantly improving sensitivity of the analytical detection and even lowering the detection limit.7 Of all known conducting polymers, PEDOT:PSS has proven itself as a promising candidate for sensor applications not only due to its excellent anti-fouling property, but also its high electrical conductivity. Furthermore, it also possesses good mechanical and chemical stability.
Dopamine (DA) and uric acid (UA) are small biological molecules normally found in body fluids such as urine and blood plasma. DA is a catecholamine neurotransmitter which plays a crucial role in regulating neuronal functions such as cognition, attention span, emotion and movement.8 Therefore, a deficiency of DA in the human body could lead to neurological disorders including Parkinson's disease,9 Alzheimer's disease,10 schizophrenia,11 restless leg syndrome (RLS)12 and attention deficit hyperactivity disorder (ADHD).13 In contrast, a high level of DA could cause an increased risk of depression14 and could also be a sign of drug addiction.15 UA is a major end product of purine metabolism. A noticeably elevated UA level could be associated with clinical disorders such as gout, hyperuricemia, Lesch–Nyhan syndrome and renal failure.16 The significance of dopamine and uric acid in the clinical diagnostic perspectives has drawn great awareness for the development of sensitive and reliable techniques for their detection. Over the past decade, various analytical techniques have been established including liquid chromatography,17,18 capillary electrophoresis,19 fluorescence spectroscopy,20,21 colorimetry,22,23 UV-Vis spectrophotometry24 and electrochemical analysis.25,26
Due to the electroactive nature of DA and UA, electrochemical techniques are of high interest because they offer certain benefits such as cost effectiveness, rapid response, high sensitivity, simple instrumentation and field portability. As DA and UA typically coexist in biological samples, simultaneous detection of both analytes is highly desired. Besides DA and UA, the co-occurrence of ascorbic acid (AA) in the physiological fluids also poses problems. Using conventional bare carbon and metal electrodes, these three molecules are oxidized at nearly the same potential, yielding to overlapping oxidation peaks.27 As a result, simultaneous electrochemical determination of DA and UA in the presence of AA is not possible. Furthermore, the electrode fouling occurring by adsorption of electrochemical oxidation products at the bare electrode surface also causes relatively poor selectivity and sensitivity.28 To overcome these problems, various materials including polymer films (chitosan, Nafion, PEDOT),29–31 carbon nanomaterials (carbon nanotube, graphene),32,33 metal nanoparticles (Au, Pt, Pd),34–36 metal oxides (TiO2, SnO2, ZnO, CuO)37–39 and their composites have been used to modify the electrode surfaces.
PEDOT modified electrodes40,41 have been reported for their selective determination of DA in the presence of AA and UA. In comparison to the broad and overlapped voltammetric responses of the bare electrodes, the PEDOT modified surfaces showed well-resolved peaks which could clearly validate its catalytic activity toward the oxidation of DA, AA and UA by lowering their oxidation potentials and increasing their currents. PEDOT behaves in this case as a redox mediator which is responsible for promoting electron shuttling between the electrode and the electroactive species.41
Graphene oxide (GO) was integrated into the PEDOT matrix and its composite was successfully used by Weaver et al.42 for selective determination of DA in the presence of AA and UA. It was found that GO in its unreduced form could create the sensitive and selective DA sensor by increasing the active surface area of the electrode and selectively amplifying the DA oxidation current through the electrostatic attraction between the cationic DA molecules and the negatively charged groups presented on the GO surface. While the DA signal could be greatly enhanced, the oxidation currents of anionic AA and UA remained unchanged. In addition to electrostatic forces, π–π interactions between the graphitic surface of GO and the phenol ring of DA also play a crucial role in the improved sensitivity toward DA. Since the electrical conductivity of GO is rather poor, Wang et al.43 has used reduced graphene oxide (RGO) to modify the electrode instead. In combination with PEDOT, the resulting RGO nanocomposite exhibited significantly enhanced catalytic activity as could be realized by a considerable improvement in the oxidative current of DA.
Synergism between the PEDOT matrix and gold nanoparticles for DA sensing was also demonstrated by Kumar et al.44 Incorporation of AuNP into PEDOT could substantially enhance the oxidation current of DA. This could be attributed to AuNP which functioned in this case as electron antennae responsible for improving the electron transfer rate between the electrode and DA. Furthermore, favourable weak adsorption of DA on AuNP through the interaction between the –NH2 group and Au also involved in increasing the DA oxidation current.
To suppress the response of AA and selectively amplify the signal of DA, negatively charged compounds such as sulfonated chitosan,20 Nafion,21 poly(p-aminobenzene sulfonic acid) (p-ABSA),45 polystyrene sulfonate (PSS)31 and sodium dodecyl sulfate (SDS)46,47 have been used to modify the electrodes. As reported by Atta et al.,46,47 a negatively charged layer of SDS was formed by adsorption of the anionic surfactant on top of the PEDOT modified electrode. While AA was expelled from the SDS layer via the repulsive force, accumulation of DA at the electrode surface was on the other hand driven by the electrostatic attraction.
According to a literature survey, there are no previous reports using a composite between AuNP, GP, PEDOT and PSS for DA detection. Therefore, we herein combine those materials to prepare a highly sensitive electrochemical sensor for selective determination of DA. In this work, we demonstrate a facile synthesis of AuNP-GP-PEDOT:PSS nanocomposite under ambient condition comprising of two simple steps as depicted in Scheme 1. Firstly, graphene is prepared by electrolytic exfoliation of graphite in an aqueous polystyrene sulfonate solution. The roles of PSS are not only to intercalate and trigger the exfoliation but also to disperse and stabilize the exfoliated graphene flakes, as well as to prevent the oxidation of graphene during the peel-off process.4 PSS will also assist the dissolution of EDOT monomer, facilitate the polymerization and then behave as a counter anion to compensate the positively charged PEDOT. In the second step after adding HAuCl4 and EDOT monomer to the GP-PSS dispersion, gold nanoparticles and PEDOT are formed simultaneously via in situ reduction of HAuCl4 and oxidative polymerization of EDOT. Without using any other chemical reagents, this step is thus considered green. The resulting AuNP-GP-PEDOT:PSS dispersion is drop casted on a glassy carbon electrode and the modified electrode is then used as an electrochemical sensor to detect DA and UA. This is for the first time that a quaternary composite between AuNP, GP, PEDOT and PSS is used for the sensitive and selective detection of DA in the presence of UA and AA. Combining the advantageous features of AuNP, GP, PEDOT and PSS, the sensitivity of the sensor can be greatly enhanced and the electrode overpotentials are significantly minimized.
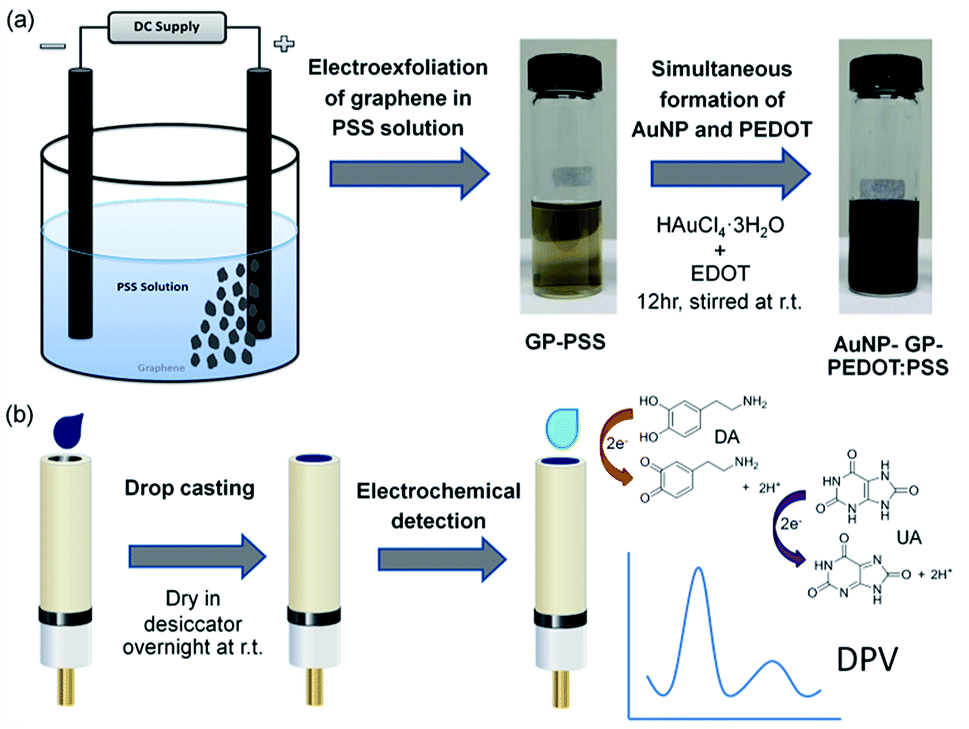 | ||
| Scheme 1 Schematic representation of nanocomposite based electrochemical sensor: (a) synthesis of AuNP-GP-PEDOT:PSS aqueous dispersion, (b) preparation of nanocomposite modified electrode and electrochemical determination of DA and UA. | ||
Experimental
Materials
Dopamine, ascorbic acid and uric acid were purchased from TCI Chemicals. Hydrogen tetrachloroaurate (III) (HAuCl4·3H2O), 3,4-ethylenedioxythiophene (EDOT), polystyrenesulfonate (PSS), potassium hexacyanoferrate (III) (K3Fe(CN)6) and potassium hexacyanoferrate (II) trihydrate (K4Fe(CN)6·3H2O) were obtained from Sigma-Aldrich. A human serum assayed control (Liquid Assayed Multiqual® Levels 1) was purchased from BIO-RAD. All buffer salts and organic solvents were acquired from Merck. Ultrapure water was used throughout this work.Apparatus
The morphology of the gold nanoparticles in the dispersion was examined by a transmission electron microscope (TEM, JEOL JEM-2010). The surface morphology and functional groups of the nanocomposite film were characterized by a scanning electron microscope (SEM, Hitachi S-3400N), a Fourier transform infrared spectrometer (Perkin Elmer System 2000) and a confocal Raman spectrometer (Perkin Elmer System 2000). XRD was carried out on an X-ray diffractometer (PANalytical – X'Pert PRO). All electrochemical experiments were performed using a potentiostat from PalmSens.Synthesis of AuNP-GP-PEDOT:PSS composite
Two graphite rods were immersed in 1% (w/v) of poly(sodium 4-styrenesulfonate) solution and a constant potential of 8 V was applied between electrodes for 5 hours. AuNP-GP-PEDOT:PSS was subsequently synthesized by in situ reduction of HAuCl4 with EDOT monomer in the preformed GP-PSS dispersion. HAuCl4·3H2O was dissolved in 25 mL of GP-PSS solution under continuous stirring and an EDOT aqueous solution was then slowly added. After 12 hours, AuNP-GP-PEDOT:PSS aqueous dispersion was obtained.Preparation of nanocomposite electrode
The glassy carbon electrode (GCE) was sequentially polished with different sizes of alumina slurry including 1.0, 0.3 and 0.05 μm, thoroughly rinsed with ultrapure water after each step and dried under a nitrogen stream. The AuNP-GP-PEDOT:PSS nanocomposite (1.5 μL) was casted on the pre-clean GCE and dried in a desiccator at room temperature overnight.Electrochemical measurements
All electrochemical experiments were performed in a three-electrode system using a PalmSens electrochemical workstation with a bare or a modified glassy carbon electrode as the working electrode, a Pt wire as the counter electrode and a Ag/AgCl (3 M NaCl) electrode as the reference electrode. The supporting electrolyte in all experiments was 100 mM phosphate buffer (pH 7.0). Prior to the measurement, the electrolytes were deoxygenated and kept under nitrogen atmosphere. CV was recorded within the potential range from −0.3 to 0.6 V at a scan rate of 50 mV s−1. DPV was performed within the potential range from −0.3 to 0.6 V, with a pulse amplitude of 100 mV and a scan rate of 10 mV s−1. A baseline correction of the resulting voltammograms was performed and the peak current was recorded. Solutions of dopamine, uric acid and ascorbic acid were freshly prepared prior to the measurement.Determination of DA and UA in real samples
Determination of DA and UA in real samples was carried out by DPV using the modified electrode. For a dopamine hydrochloride injection (250 mg/10 mL), solutions of different concentrations were prepared by appropriate dilution with 100 mM phosphate buffer (pH 7.0). For a human serum sample, prior to the measurement the sample was filtrated and diluted with 100 mM phosphate buffer (pH 7.0). Known amounts of DA and UA were added to the real samples and the recovery results of the spiked samples were calculated.Results and discussion
Synthesis and characterization of AuNP-GP-PEDOT:PSS composite
The synthesis of AuNP-GP-PEDOT:PSS aqueous dispersion could be achieved by two mild simple steps. Graphene was first electrolytic exfoliated in PSS solution. The light brown homogeneous dispersion of GP-PSS was obtained (Fig. S1a†) and found to be stable for several months without any sign of precipitation. The morphology of the GP-PSS was confirmed using TEM. As shown in Fig. S1b,† a sheet-like structure with an irregular shape of graphene was observed. The obtained GP-PSS was also characterized by Raman spectroscopy. Fig. S1c† shows Raman spectra of the GP-PSS composite in comparison to the neat PSS. As expected, two prominent D and G bands were observed in GP-PSS at 1348 and 1601 cm−1, respectively. The D band is typically ascribed to the structural defects or edged areas in graphene, whereas the G band is attributed to the vibration of sp2 carbon atoms. The intensity ratio of the D and G bands (ID/IG) could therefore reflect the average size of the sp2 domains and the degree of disorder of the graphene sheets. In this work, the ID/IG ratio of GP-PSS is equal to 1.08, suggesting a higher extent of defects in the graphene sheets typically caused by the exfoliation process48,49 or a more disordered structure of graphene stemming from the adsorption-induced charge transfer between graphene and PSS.50To the solution of GP-PSS, HAuCl4 was added. Under continuous stirring, EDOT was subsequently added, yielding to a formation of PEDOT via the in situ oxidative polymerization process. This was confirmed by a gradual change in solution color from light brown to deep dark blue (Fig. 1a). At the same time, AuNPs were also formed through the in situ reduction of HAuCl4 using EDOT monomer as a reducing agent. This could be directly observed in the TEM image (Fig. 1b and c), showing AuNPs as black spots decorated on the surface of the graphene sheet.
 | ||
| Fig. 1 (a) Photograph, (b) TEM image with low magnification and (c) TEM image with high magnification of the as-prepared AuNP-GP-PEDOT:PSS. | ||
The presence of PEDOT in the as-synthesized nanocomposite could be confirmed by the FTIR spectrum. As shown in Fig. S2a,† functional groups of PEDOT and PSS were observed. The vibrations at 1506 and 1324 cm−1 were ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C bonds of thiophene, respectively. The adsorption bands at 983, 845 and 691 cm−1 were correlated to the C–S bond of the thiophene moiety. The peaks at 1183, 1122 and 1065 cm−1 were attributed to the C–O–C bond of the ethylenedioxide unit. The S
C and C–C bonds of thiophene, respectively. The adsorption bands at 983, 845 and 691 cm−1 were correlated to the C–S bond of the thiophene moiety. The peaks at 1183, 1122 and 1065 cm−1 were attributed to the C–O–C bond of the ethylenedioxide unit. The S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) O vibration at 1209 cm−1 and the O–S–O signal at 1031 cm−1 were accredited to the sulfonic group of PSS. The nanocomposite was further characterized by XRD and its pattern was shown in Fig. S2b.†
O vibration at 1209 cm−1 and the O–S–O signal at 1031 cm−1 were accredited to the sulfonic group of PSS. The nanocomposite was further characterized by XRD and its pattern was shown in Fig. S2b.†
The diffraction features at 38.18°, 44.39°, 64.58° and 77.55° are attributed to the (111), (200), (220), and (311) planes of the cubic lattice structure of Au. The average particle size of AuNP could be estimated by the Scherrer equation:
| D = κλ/βcosθ |
 | ||
| Fig. 2 SEM image of the AuNP-GP-PEDOT:PSS nanocomposite modified electrode. | ||
Electrochemical characteristics of the composite electrodes
Electrochemical properties of AuNP-GP-PEDOT:PSS/GCE and other related electrodes including AuNP-PEDOT:PSS/GCE, GP-PEDOT:PSS/GCE, PEDOT:PSS/GCE and bare GCE were initially assessed using [Fe(CN)6]3−/4−. As shown in Fig. 3, all the composite electrodes showed increased oxidation/reduction peak currents compared with the bare GCE. The highest current response was observed on AuNP-GP-PEDOT:PSS/GCE. Meanwhile, the smallest peak potential separation (ΔEp) of 113 mV could also be observed on AuNP-GP-PEDOT:PSS/GCE in comparison with those on AuNP-PEDOT:PSS/GCE (116 mV), GP-PEDOT:PSS/GCE (160 mV), PEDOT:PSS/GCE (187 mV) and GCE (206 mV). These electrochemical behaviours indicated that the AuNP-GP-PEDOT:PSS composite possessed the highest catalytic activity and the fastest electron transfer. This is possibly due to a profound synergistic effect between AuNP, GP and PEDOT. | ||
| Fig. 3 CVs of AuNP-GP-PEDOT:PSS/GCE, AuNP-PEDOT:PSS/GCE, GP-PEDOT:PSS/GCE, PEDOT:PSS/GCE and GCE in 5 mM [Fe(CN)6]3−/4−. | ||
Electrochemical behaviors of AA, DA and UA in a mixture solution
To investigate the reliability of the sensor for selective determination of DA and UA in the presence of AA, cyclic voltammograms (Fig. 4) of a ternary mixture solution at different composite electrodes (i.e., AuNP-GP-PEDOT:PSS/GCE, AuNP-PEDOT:PSS/GCE, GP-PEDOT:PSS/GCE and PEDOT:PSS/GCE) were recorded. As shown, the voltammetric peaks of these three analytes severely overlapped at the bare GCE, while well separated oxidation peaks with higher current responses were observed under identical conditions at the composite electrodes. Among them, AuNP-GP-PEDOT:PSS/GCE showed the highest current response and peak separation. Such an enhancement in the peak current by the AuNP-GP-PEDOT:PSS composite could be explained by the effective movement of electrons through graphene, AuNPs and the highly conductive PEDOT matrix. On the other hand, the improvement in the peak separation could be primarily described by a decrease in the oxidation peak width and a shift in the peak location of each analyte. As shown in the individual oxidation of AA and DA at the AuNP-GP-PEDOT:PSS electrode, a significant leftward shift of 198 mV in the oxidation potential of AA (from 0.241 V to 0.043 V, Fig. S3a†) and a moderate negative shift of 82 mV in the oxidation peak of DA (from 0.266 V to 0.184 V, Fig. S3b†) were observed due to the electrocatalytic effect of the nanocomposite on the oxidation reactions. These changes in peak position were responsible for the peak separation between AA–DA and DA–UA in the voltammogram of the mixture solution (Fig. S3d†). Unlike AA and DA, a slightly shifted peak position of UA (from 0.344 V to 0.304 V, Fig. S3c†) only had a small effect on the peak separation.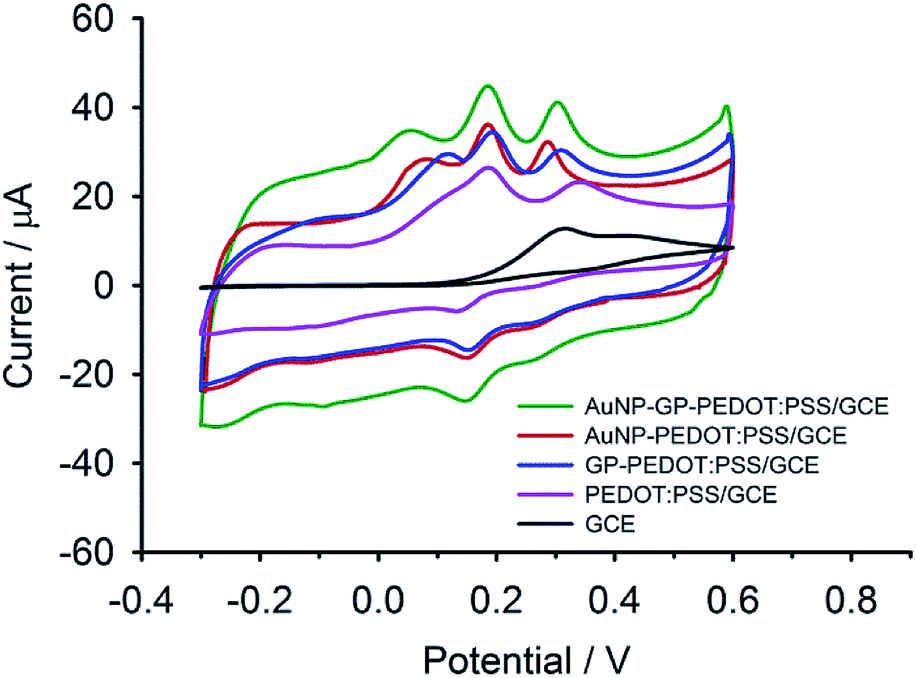 | ||
| Fig. 4 CVs of a ternary mixture solution containing 1 mM of AA, 50 μM of DA and 200 μM of UA at AuNP-GP-PEDOT:PSS/GCE, AuNP-PEDOT:PSS/GCE, GP-PEDOT:PSS/GCE, PEDOT:PSS/GCE and bare GCE. | ||
Using differential pulse voltammetry (DPV), the capacitive current due to the charging of PEDOT:PSS could be minimized and thereby enhancing the accuracy of the faradaic current measurement.22 As displayed in Fig. 5, only two broad oxidation peaks were observed at the bare GCE. The overlapped peaks could be resolved into three peaks with enhanced oxidative currents at the PEDOT:PSS modified electrode. These improved electrochemical behaviours were observed in previous reports31,40,41 and were attributed to PEDOT which could facilitate electron shuttling between the electrode surface and the electroactive species through its redox mediation.41 The peak currents were even higher when GP was integrated into the composite. This is especially for DA where the increment of the peak height was almost double compared with AA and UA. This favourable enhancement in the peak current of DA at the GP-PEDOT:PSS electrode was due in part to the π–π interactions between the graphitic surface of GP and the phenol ring of DA.42 In addition, DA which had a pKa of 8.9 (aminoethyl group) was present in a cationic form at the neutral pH of the measuring buffer, while the nanocomposite carried a net negative charge of PSS. This would cause the electrostatic attraction of DA to the negatively charged electrode surface and thus even enhanced the oxidative current of DA.31 In the case of AuNP-PEDOT:PSS/GCE, the peak currents of DA and UA increased, while the AA peak signal remained unchanged in relative to that of the GP-PEDOT:PSS composite. The increase in oxidation currents of DA and UA at the AuNP-PEDOT:PSS electrode was ascribed to the adsorption of DA and UA on the AuNP surfaces through favourable interactions of nitrogen bases (i.e., –NH2, NH) with Au.44,47 When the AuNP-GP-PEDOT:PSS/GCE was used, the highest current responses of DA and UA were obtained suggesting that AuNP together with GP, PEDOT and PSS could efficiently cooperate with each other and thereby could maximize the signal currents through a synergistic effect. In addition, the AuNP-GP-PEDOT:PSS modified electrode also exhibited the highest peak resolution with the peak-to-peak separation of 150 mV and 110 mV between AA–DA and DA–UA, respectively. Therefore, AuNP-GP-PEDOT:PSS was considered within this work as the most promising composite material and was further used to fabricate a highly sensitive and selective electrochemical sensor for DA.
 | ||
| Fig. 5 DPVs of a ternary mixture solution containing 1 mM of AA, 50 μM of DA and 200 μM of UA at AuNP-GP-PEDOT:PSS/GCE, AuNP-PEDOT:PSS/GCE, GP-PEDOT:PSS/GCE, PEDOT:PSS/GCE and bare GCE. | ||
Electrochemical detection of DA and UA
DPV was used to investigate the performance of the modified electrode, since it showed better sensitivity and higher peak resolution than CV. The individual determination of DA and UA at the composite electrode was carried out in the potential window of −0.3 to 0.6 V in phosphate buffer (pH 7.0). As shown in Fig. 6a, the electrochemical response of DA increases with the increase in DA concentration. A linear correlation between the peak current and the DA concentration (Fig. 6c) was observed from 1 nM to 300 μM with a correlation coefficient (R2) of 0.997 and a detection limit of 100 pM based on the signal-to-noise ratio (S/N = 3). Such a low limit of detection could be clearly realized by a highly electrocatalytic activity and a fast electron transfer occurring in the nanocomposite. These are principally due to a synergistic effect between the increased surface area and high catalytic activity of AuNP, as well as the superior conductivity of GP and PEDOT. As illustrated in Fig. 6b and d, the peak current increases linearly with the increase of UA concentration over the range of 10 μM to 1 mM (R2 = 0.994). A detection limit of 10 μM based on the signal-to-noise characteristics (S/N = 3) could be achieved. Although the detection limit of UA found in this work is not as low as those obtained from the literature reports, it is still lower than the normal UA level in serum, making this material possible for real sample analysis. | ||
| Fig. 6 DPVs of different concentrations of (a) DA and (b) UA and their corresponding linear relationship between currents and concentrations of (c) DA and (d) UA, respectively (n = 9). | ||
In the ternary mixture, the concentration of one analyte was varied, while those of the other two species were kept constant. As the concentration of DA increased, those of AA and UA were fixed at 100 μM and 20 μM, respectively. Due to the unfavorable interactions between the modified electrode surface and AA, the anodic peak of AA was not clearly observed (Fig. 7a).
 | ||
| Fig. 7 DPVs of ternary mixtures containing (a) 100 μM of AA, 20 μM of UA and different concentrations of DA, (b) 100 μM of AA, 5 μM of DA and different concentrations of UA and their corresponding linear relationship between currents and concentrations of (c) DA and (d) UA (n = 9). | ||
As illustrated in Fig. 7a, the peak current of DA increases with the increasing concentration of DA, whereas that of UA remains unaltered. A linear response of DA was observed from 1 nM to 300 μM (R2 = 0.993) (Fig. 7c). Even in the presence of AA and UA in the solution, a detection limit of DA could still be as low as 1 nM (S/N = 3). As depicted in Fig. 7b, the concentration of UA was altered, while those of AA and DA were kept unchanged at 100 μM and 5 μM, respectively. A linear correlation between the concentration of UA and the current signal (Fig. 7d) was realized over a wide concentration range of 10 μM to 1 mM (R2 = 0.989) with a limit of detection of 10 μM (S/N = 3).
Reproducibility and stability
To investigate the inter-electrode reproducibility, nine independent composite electrodes were prepared under the same conditions and were used to determine DA and UA at different concentrations. It was found that the relative standard deviations (RSDs) of the peak currents were 2.32% and 3.30% (n = 9) for DA and UA, respectively, indicating excellent reproducibility of the prepared composite electrode. To further study the intra-electrode reproducibility, the modified electrodes (n = 3) were measured for 25 successive scans in a solution containing 500 μM DA. The current responses were recorded and the RSD was found to be 3.96%, suggesting exceptional reproducibility, as well as outstanding antifouling ability of the modified electrode. To evaluate the storage stability, the nanocomposite electrodes were stored in a dry state for 4 and 8 weeks and then used to measure the current response of 500 μM DA. As shown in Fig. S4,† the sensor responses remained 99.68% and 98.04% in comparison to its initial value after 4 and 8 weeks, respectively. This clearly indicates that the prepared sensor has good stability over a period of at least 8 weeks.Real sample analysis
To prove the feasibility of using the modified electrode for real sample analysis, a dopamine hydrochloride injection and human blood serum were used. The DA contents in the diluted injection samples at two different concentration levels were examined using the nanocomposite electrode. As shown in Table S1,† the recoveries were found to be in the range of 93.0–109.0% and the relative standard deviations (RSDs) were less than 4.00%. For the serum sample, a spike and recovery method was performed and the results are summarized in Table 1. Known amounts of both analytes were spiked into the sample solutions and the final concentrations were measured. The recoveries of DA and UA were in the range of 97.4–106.6% with excellent RSDs of less than 3.83%. The overall results could indicate a good performance of the sensor for real biological sample analysis.| Analyte | Spiked (μM) | Found (μM) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| DA | 10.0 | 10.3 | 103.0 | 3.83 |
| 100.0 | 104.1 | 104.1 | 1.99 | |
| 250.0 | 250.2 | 100.1 | 2.18 | |
| UA | 100.0 | 97.4 | 97.4 | 1.97 |
| 500.0 | 533.1 | 106.6 | 2.79 | |
| 1000.0 | 1000.0 | 100.0 | 2.03 |
Comparison with previously reported modified electrodes
The sensor performances in terms of a limit of detection and linear range of calibration, in addition to the simplicity of the fabrication process of the proposed sensor, were compared with other previously reported sensors for electrochemical determination of DA, as summarized in Tables S2† and S3.† In Table S2,† the reports published in the literature for selective determination of DA on PEDOT-based modified electrodes are listed. In comparison to the neat PEDOT coated electrodes,40,41 the graphene-PEDOT42,43 and AuNP-PEDOT44 modified sensors exhibited a significant improvement in limits of detection by taking the advantage of the signal enhancing characteristics of the embedded graphene and AuNP within the conducting PEDOT. In the presence of SDS, the detection limit of the PEDOT modified electrode was even one order of magnitude lower than that obtained from the assay without the anionic surfactant as reported by Atta et al.46 The same phenomenon was also observed on the AuNP-PEDOT modified sensor47 where the limit of detection could be lowered down to 390 pM by using SDS. This clearly confirms that the presence of the negatively charged compounds could efficiently enhance the diffusion and accumulation of the positively charged DA to and at the electrode surface via the electrostatic attraction and thus increasing the oxidation current of DA, as well as lowering its detection limit. Compared to all of those PEDOT-based modified electrodes, our sensor shows a broader concentration range and a lower limit of detection. This clearly indicates that a synergistic combination of AuNP, GP, PEDOT and PSS could significantly improve sensitivity of the analytical detection and thereby lowering the limit of detection.The detection limits of all the selected sensors in Table S3† are all lower than 10 nM and are among the best values reported so far for DA sensors. As far as the DA level in serum (sub-nanomolar) is concerned, only some of these reported materials are applicable. As shown, our established sensor has a better limit of detection and a wider linear range in comparison to most of the sensors. Although the detection limit of our modified electrode is not as low as what was reported by Qian et al. (2014),51 our sensor offers a wider linear response. In addition, it should be noted that our synthesis protocol is much greener, milder and simpler as compared to the others, which are mainly based on harsh chemical reactions, such as Hummers method, solvothermal technique and conventional chemical reduction via hydrazine and borohydride.
Conclusions
In this work, a facile strategy to prepare the AuNP-GP-PEDOT:PSS based electrochemical sensor was demonstrated. The synthesis protocol is considered as a green method because environmentally friendly water based reactions are used throughout all the preparation steps. In addition, there is no use of hazardous chemicals such as hydrazine and borohydride. This could significantly reduce the production of chemical waste. Moreover, the synthesis route is very versatile and could be used for the preparation of other nanocomposites comprising of graphene, metal nanoparticle (e.g., PtNP, PdNP, AgNP) and conductive polymer (e.g., polypyrrole, polyaniline). A synergistic effect of GP, AuNP and PEDOT:PSS could contribute to greatly enhanced sensitivity and reduced electrode overpotentials. Furthermore, the modified electrode could also provide well resolved oxidation peaks of DA and UA, as well as a significant suppression of AA signal. This could make a determination of DA and UA in the presence of AA feasible. An outstanding detection limit of 100 pM, as well as a wide linear response of 1 nM to 300 μM toward DA achieved in this work is among the greatest values reported for the nanocomposite based electrochemical sensors. The as-prepared electrode shows excellent reproducibility and high stability over at least 8 weeks without any significant change in its electrochemical response. There was also no electrode fouling observed, even after 25 successive scans. Additionally, such a sensor could be successfully demonstrated for real sample analysis with satisfactory accuracy and precision. This in turn indicates that our established electrochemical sensor could be a potential candidate for bioanalytical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Kasetsart University Research and Development Institute (KURDI), the Development and Promotion of Science and Technology Talents Project (DPST) and the Institute for the Promotion of Teaching Science and Technology (IPST, Grant No. 015/2558). This research is also supported in part by the Graduate Program Scholarship from The Graduate School, Kasetsart University. D. Dechtrirat also wishes to thank Specialized Center of Rubber and Polymer Materials for Agriculture and Industry (RPM), Faculty of Science, Kasetsart University for the publication support.Notes and references
- C. Zhu, G. Yang, H. Li, D. Du and Y. Lin, Anal. Chem., 2015, 87, 230–249 CrossRef CAS PubMed.
- E. B. Bahadır and M. K. Sezgintürk, Trends Anal. Chem., 2016, 76, 1–14 CrossRef.
- R. S. Edwards and K. S. Coleman, Nanoscale, 2013, 5, 38–51 RSC.
- J. M. Munuera, J. I. Paredes, S. Villar-Rodil, M. Ayán-Varela, A. Martínez-Alonso and J. M. D. Tascón, Nanoscale, 2016, 8, 2982–2998 RSC.
- M. Oyama, X. Chen and X. Chen, Anal. Sci., 2014, 30, 529–538 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- A. Malinauskas, Synth. Met., 1999, 107, 75–83 CrossRef CAS.
- D. Vallone, R. Picetti and E. Borrelli, Neurosci. Biobehav. Rev., 2000, 24, 125–132 CrossRef CAS PubMed.
- B. Scatton, L. Rouquier, F. Javoy-Agid and Y. Agid, Neurology, 1982, 32, 1039–1040 CrossRef CAS PubMed.
- A. Martorana and G. Koch, Front. Aging Neurosci., 2014, 6, 1–6 CAS.
- G. Chouinard and B. D. Jones, The Canadian Journal of Psychiatry, 1979, 24, 661–667 CrossRef CAS PubMed.
- R. Allen, Journal of Clinical Sleep Medicine, 2004, 5, 385–391 CrossRef PubMed.
- S. Sikström and G. Söderlund, Psychological Review, 2007, 114, 1047–1075 CrossRef PubMed.
- E. Dailly, F. Chenu, C. E. Renard and M. Bourin, Fundam. Clin. Pharmacol., 2004, 18, 601–607 CrossRef CAS PubMed.
- N. D. Volkow, J. S. Fowler, G. J. Wang, J. M. Swanson and F. Telang, Arch. Neurol., 2007, 64, 1575–1579 CrossRef PubMed.
- M. Jin, F. Yang, I. Yang, Y. Yin, J. J. Luo, H. Wang and X. F. Yang, Front. Biosci., 2012, 17, 656–669 CrossRef CAS.
- V. Carrera, E. Sabater, E. Vilanova and M. A. Sogorb, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 847, 88–94 CrossRef CAS PubMed.
- R. Kanďár, P. Drábková and R. Hampl, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 2834–2839 CrossRef PubMed.
- A. Zinellu, C. Carru, S. Sotgia and L. Deiana, Anal. Biochem., 2004, 330, 298–305 CrossRef CAS PubMed.
- L. Zhu, G. Xu, Q. Song, T. Tang, X. Wang, F. Wei and Q. Hu, Sens. Actuators, B, 2016, 231, 506–512 CrossRef CAS.
- D. Jin, M. H. Seo, B. T. Huy, Q. T. Pham, M. L. Conte, D. Thangadurai and Y. I. Lee, Biosens. Bioelectron., 2016, 77, 359–365 CrossRef CAS PubMed.
- Y. Lin, C. Chen, C. Wang, F. Pu, J. Ren and X. Qu, Chem. Commun., 2011, 47, 1181–1183 RSC.
- J. Lu, Y. Xiong, C. Liao and F. Ye, Anal. Methods, 2015, 7, 9894–9899 RSC.
- A. Abbaspour, A. Khajehzadeh and A. Ghaffarinejad, Analyst, 2009, 34, 1692–1698 RSC.
- K. Jackowska and P. Krysinski, Anal. Bioanal. Chem., 2013, 405, 3753–3771 CrossRef CAS PubMed.
- M. Sajid, M. K. Nazal, M. Mansha, A. Alsharaa, S. M. S. Jillani and C. Basheer, Trends Anal. Chem., 2016, 76, 15–29 CrossRef CAS.
- R. D. O'Neil, Analyst, 1994, 119, 767–779 RSC.
- Z. Gao and H. Huang, Chem. Commun., 1998, 2107–2108 RSC.
- C. S. R. Vusa, V. Manju, K. Aneesh, S. Berchmans and A. Palaniappan, RSC Adv., 2016, 6, 4818–4825 RSC.
- A. S. Kumar, P. Swetha and K. C. Pillai, Anal. Methods, 2010, 2, 1962–1968 RSC.
- I. Gualandi, D. Tonelli, F. Mariani, E. Scavetta, M. Marzocchi and B. Fraboni, Sci. Rep., 2016, 6, 35419 CrossRef CAS PubMed.
- D. M. Fernandes, M. Costa, C. Pereira, B. Bachiller-Baeza, I. Rodríguez-Ramos, A. Guerrero-Ruiz and C. Freire, J. Colloid Interface Sci., 2014, 432, 207–213 CrossRef CAS PubMed.
- A. Pandikumar, G. T. S. How, T. Peik-See, F. S. Omar, S. Jayabal, K. Z. Kamali, N. Yusoff, A. Jamil, R. Ramaraj, S. A. John, H. N. Lim and N. M. Huang, RSC Adv., 2014, 4, 63296–63323 RSC.
- N. Yusoff, A. Pandikumar, R. Ramaraj, H. N. Lim and N. M. Huang, Microchim. Acta, 2015, 182, 2091–2114 CrossRef CAS.
- N. F. Atta, M. F. El-Kady and A. Galal, Sens. Actuators, B, 2009, 141, 566–574 CrossRef CAS.
- Y. Liu, P. She, J. Gong, W. Wu, S. Xu, J. Li, K. Zhao and A. Deng, Sens. Actuators, B, 2015, 221, 1542–1553 CrossRef CAS.
- G. T. S. How, A. Pandikumar, H. N. Ming and L. H. Ngee, Sci. Rep., 2014, 4, 5044 CrossRef CAS PubMed.
- G. She, X. Huang, L. Jin, X. Qi, L. Mu and W. Shi, Small, 2014, 10, 4685–4692 CrossRef CAS PubMed.
- K. Khun, Z. H. Ibupoto, X. Liu, N. A. Mansor, A. P. Turner, V. Beni and M. Willander, J. Nanosci. Nanotechnol., 2014, 14, 6646–6652 CrossRef CAS PubMed.
- S. Senthil Kumar, J. Mathiyarasu, K. L. N. Phani and V. Yegnaraman, J. Solid State Electrochem., 2006, 10, 905–913 CrossRef.
- F. S. Belaidi, A. Civélas, V. Castagnola, A. Tsopela, L. Mazenq, P. Gros, J. Launay and P. Temple-Boyer, Sens. Actuators, B, 2015, 214, 1–9 CrossRef CAS.
- C. L. Weaver, H. Li, X. Luo and X. T. Cui, J. Mater. Chem. B, 2014, 2, 5209–5219 RSC.
- W. Wang, G. Xu, X. T. Cui, G. Sheng and X. Luo, Biosens. Bioelectron., 2014, 58, 153–156 CrossRef CAS PubMed.
- S. Senthil Kumar, J. Mathiyarasu and K. L. N. Phani, J. Electroanal. Chem., 2005, 578, 95–103 CrossRef.
- Y. Fang, Y. Liu, D. Shen, P. Li and Z. Yang, Nanosci. Nanotechnol. Lett., 2015, 7, 462–468 CrossRef.
- N. F. Atta, A. Galal and R. A. Ahmed, Bioelectrochemistry, 2011, 80, 132–141 CrossRef CAS PubMed.
- N. F. Atta, A. Galal and E. H. El-Ads, Electrochim. Acta, 2012, 69, 102–111 CrossRef CAS.
- W. Liu, C. Li, P. Zhang, L. Tang, Y. Gu, Y. Zhang, J. Zhang, Z. Liu, G. Sun and Z. Zhang, RSC Adv., 2015, 5, 73993–74002 RSC.
- T. Balamurugan and S. Berchmans, RSC Adv., 2015, 5, 50470–50477 RSC.
- S. Mayavan, H. S. Jang, M. J. Lee, H. S. Choi and S. M. Choi, J. Mater. Chem. A, 2013, 1, 3489–3494 CAS.
- T. Qian, C. Yu, X. Zhou, S. Wu and J. Shen, Sens. Actuators, B, 2014, 193, 759–763 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01564c |
| This journal is © The Royal Society of Chemistry 2018 |