
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynchronous, efficient and fast removal of phosphate and organic matter by carbon-coated lanthanum nanorods†
Xintong Zhanga,
Wei Wang *a,
Shiyu Daia and
Fuyi Cui*ab
*a,
Shiyu Daia and
Fuyi Cui*ab
aSchool of Environment, Harbin Institute of Technology, Harbin 150090, P. R. China. E-mail: wangweirs@hit.edu.cn; cuifuyi@hit.edu.cn
bCollege of Urban Construction and Environmental Engineering, Chongqing University, Chongqing, China
First published on 27th March 2018
Abstract
Both phosphate and organic carbon can serve as nutrients for microorganism growth. Simultaneous removal of both nutrients would realize the antibacterial strategy of nutrient starvation better to ensure water quality safety. In addition, a short treatment time is the premise for the application of a material in water treatment. Herein, carbon-coated lanthanum nanorods with a uniform distribution of La and C (C–La-MOF) were rationally prepared through glucose and La-MOF hydrothermal treatment and further carbonization to synchronously and rapidly remove phosphate and organic matter. The carbon layer thickness was tuned by varying the hydrothermal time to find the optimal balance between excellent phosphate intake and low lanthanum leakage. C–La-MOF had a strong anti-interference ability and high phosphate capture capacity over a wide pH range of 2–12. Impressively, when phosphate and organic carbon coexisted in solution, their removal performances remained relatively unchanged compared with that when the two nutrients existed independently, and their adsorption equilibriums could be easily reached within 10 min. All of the above results prove that C–La-MOF is a promising material for practical drinking water treatment.
1. Introduction
To tackle the problem of microbiological contamination in water, especially drinking water, thus far many methods, such as the application of disinfectant and antibacterial materials, have been employed.1–5 Thereof, nutrient starvation, i.e., the direct removal of nutrients essential for microbial growth in order to starve microorganisms, is expected to be a green and effective strategy. This method could bypass the side effects caused by other methods, such as cancer risk and biotoxicity.3,6,7 Phosphorus (P) and carbon (C) are both essential elements in microbial metabolic processes and growth.8 Accordingly, they both can be the limiting nutrient for microbial growth in drinking water. Ensuring the biological stability of drinking water through reduction of AOC (assimilable organic carbon) and managing water eutrophication through the removal of P belong to nutrient starvation strategy. However, it is difficult to reduce AOC to trace levels (<10 μg C L−1) to inhibit bacterial regrowth in drinking water.9,10 Hence, extensive studies have focused on P and attempted to replace AOC with P as the indicator of the biological stability of drinking water10–14 because P can be removed to low concentrations by conventional coagulation compared with AOC.15 However, when C and P coexisted, microorganisms could grow even though either C or P was reduced to trace levels.16,17 In our previous study, we found that when C and P were removed concurrently, the potential for microbial growth in water could be largely reduced.18 As a consequence, it is imperative to develop a material to synchronously remove organic matter and phosphorus. Although carbon nanofibers with embedded LaCO3OH nanoparticles (LCNFs) could achieve this goal, it took several hours to do so,18 which was unacceptable for water treatment engineering because a long treatment time means a high cost. Therefore, an efficient adsorbent that can synchronously and rapidly remove organic matter and phosphorus should be developed.Since phosphate is the only form of phosphorus that can be directly assimilated by organisms,19,20 microbial growth could be limited by removing phosphate. Tremendous efforts have been made to remove phosphate from water.21–25 Thereof, lanthanum-based adsorbents stand out due to their very strong binding affinity for phosphate (solubility of lanthanum phosphate pKsp = 26.17).26 Lanthanum (La) is an abundant and inexpensive rare earth element and considered to be environmentally benign and biocompatible.27–29 La has been employed to remove excess phosphate in the human body30,31 and control eutrophication in lakes (Phoslock).32 In our previous study, inorganic La-MOF-500 composed of La2O2CO3 was fabricated by calcining La-MOF (lanthanum metal organic frameworks), which provided an appealing phosphate intake of 173.8 mg P g−1.33
In terms of organic carbon removal, carbon materials adsorption is preferable owing to the high adsorption capacity and simple operation. To obtain the simultaneous removal of organic carbon and phosphate, a carbon material and lanthanum material need to be integrated into one material. Among multitudinous carbon–metal composites, carbon-coated materials attracted our interest because the outer carbon layer might not only adsorb organic matter but also prevent lanthanum from leaking into water. To produce such materials, the glucose hydrothermal method is a good choice,34 and much research has used this method to synthesize core–shell structures.35–40 In addition, glucose is accessible and nontoxic when serving as a carbon source. As for a lanthanum source, La-MOF-500 is a candidate owing to its intriguing phosphate intake. However, inorganic La-MOF-500 might dissolve in the acid produced during glucose hydrothermal treatment, which might affect the structural uniformity and will probably further impact the phosphate adsorption performance. The relevant results can be found in the following experimental results. As a result, we chose La-MOF, the precursor of La-MOF-500, as the lanthanum source due to its acid resistance.
For these reasons, we coated La-MOF with glucose through a hydrothermal reaction and further carbonization. Finally, we obtained carbon-coated lanthanum nanorods with a uniform distribution of La and C (C–La-MOF), implying abundant C–La micro-interfaces, which would improve the removal performances of phosphate and organic matter. As we expected, C–La-MOF exhibited a robust phosphate intake and low lanthanum leakage. Besides, it could effectively and rapidly remove organic matter and trace levels of phosphate. Even when phosphate and organic matter coexisted, C–La-MOF could still completely remove the phosphate and achieve the highest organic matter removal efficiency in 10 min concurrently. This result is promising for water treatment engineering. These characteristics make this capture agent applicable to microbiological growth control and bring this technology another step closer to water treatment engineering applications.
2. Experimental
2.1. Materials
Lanthanum nitrate (La(NO3)3·6H2O) and La2O3 were provided by Aladdin Chemical Reagent Co., Ltd. (Shanghai, China). 1,3,5-Benzentricarboxylate (1,3,5-BTC or H3BTC) was purchased from Hans Chemical Reagent Co., Ltd. (Beijing, China). Glucose was purchased from Tianjin Kermel Chemical Reagent Co., Ltd. Sodium hydroxide (NaOH) and potassium dihydrogen phosphate (KH2PO4) were purchased from Xilong Chemical Co., Ltd. (Guangdong, China). All of the chemical reagents used were analytical grade and used as received without further purification. HCl and HNO3, used to prepare aqua regia and perchloric acid (HClO4), were of MOS grade (metal-oxide-semiconductor grade), and were purchased from Tianjin Guangfu Fine Chemical Research Institute (Tianjin, China).2.2. Fabrication of La-MOF and La-MOF-500
La-MOF was prepared as described in our previous work.33 In brief, the same volumes of 50 mM La(NO3)3·6H2O water solution and 50 mM H3BTC water/ethanol solution (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were mixed together at 25 °C, and the reaction proceeded at 135 rpm for 1.5 h. After 0.5 h of settling, the as-prepared products were rinsed with water/ethanol mixture (v/v, 1:1) 5 times and then dried in an oven at 60 °C. La-MOF-500 was the product of La-MOF after thermal pyrolysis at 500 °C for 3 h in a furnace in air.
:1) were mixed together at 25 °C, and the reaction proceeded at 135 rpm for 1.5 h. After 0.5 h of settling, the as-prepared products were rinsed with water/ethanol mixture (v/v, 1:1) 5 times and then dried in an oven at 60 °C. La-MOF-500 was the product of La-MOF after thermal pyrolysis at 500 °C for 3 h in a furnace in air.
2.3. Fabrication of the carbon-coated lanthanum composite
For the production of the carbon-coated lanthanum composite, a simple hydrothermal method was used. In a typical synthesis, 1 g of La-MOF and 8 g of glucose were mixed in 40 mL of deionized water. After magnetic stirring for 30 min, the mixture was transferred to a 100 mL Teflon-lined stainless steel autoclave and then kept at 180 °C for a certain time. After cooling down naturally, the precipitate was harvested by centrifugation and washed thoroughly with water before being dried at 60 °C. The as-prepared powder (G–La-MOF) was stabilized in air by heating to 280 °C at a rate of 1 °C min−1 and maintaining this temperature for 1 h, and then carbonized by heating to 500 °C at a rate of 5 °C min−1 in a N2 atmosphere and maintaining this temperature for 3 h. C–La-MOF, C–La-MOF10 and C–La-MOF12 were short for the carbonized samples with hydrothermal times of 6 h, 10 h and 12 h, respectively.For comparison, the same procedure was used to fabricate GC (pure carbon derived from glucose), C–La2O3, C–La and C–La-MOF-500 by excluding La-MOF or by changing La-MOF to La2O3, La(NO3)3·6H2O or La-MOF-500, respectively.
2.4. Characterization
The crystal structure of the samples was investigated using an X-ray diffraction (XRD) by Bruker D8 Advance diffractometer in the range of 2θ = ∼90° using Cu KR radiation as X-ray source. FT-IR spectra of the samples were recorded on a PerkinElmer Spectrum One B spectrometer with KBr as the reference. The microstructures of the products were observed on a scanning electron microscope (SEM, Quanta FEG 250). TEM, HRTEM, and SAED measurements were carried out on a FEI Tecnai G2 F30. The lanthanum content in C–La-MOF was detected by using an inductively coupled plasma optical emission spectrometer (ICP-OES, Optima 5300 DV, PerkinElmer). The samples for ICP-OES needed to be digested before sample injection and the digestion method is described in the ESI.†2.5. Lanthanum leakage experiments
A lanthanum leakage experiment was conducted in 50 mL of DI water. 0.2 g L−1 La-MOF-500 and 0.5 g L−1 C–La-MOF were added into the water, respectively. The solution pH was then adjusted to ∼7 with NaOH and HCl. The experiment was conducted at 25 °C for 3 days at 150 rpm. An aliquot of the solution was sampled every day, and the samples were filtered through a 0.45 μm membrane syringe filter prior to analysis. The lanthanum concentration in water was determined by ICP-OES and converted into mg La (g La)−1 according to the lanthanum content in the materials.2.6. Phosphate adsorption experiments
A series of batch tests were conducted to investigate the phosphate adsorption performances of our prepared adsorbents. The phosphate solutions in studies were prepared with KH2PO4 and DI water. All adsorption experiments were conducted at 25 °C for 24 h at 150 rpm and all solution samples were filtered through a 0.45 μm membrane syringe filter prior to analysis. The phosphate concentration in the filtrate was determined by Mo–Sb anti-spectrophotometer method using a UV-vis spectrophotometer (HACH, DR5000, America). However, when the initial phosphate concentration was less than 1 mg P L−1, the phosphate concentration was determined by inductively coupled plasma mass spectrometry (ICP-MS, 7700 Series, Agilent Technologies, USA).The effect of the hydrothermal time on the phosphate adsorption capacity was evaluated in 50 mg P L−1 KH2PO4 solutions: 0.5 g L−1 C–La-MOF, C–La-MOF10 and C–La-MOF12 were added into separate solutions.
A comparison of the phosphate adsorption capacity of C–La-MOF, GC, C–La2O3, C–La and C–La-MOF-500 was carried out by adding 0.5 g L−1 of the adsorbent into a 50 mg P L−1 solution.
Adsorption isotherms of phosphate were determined in 150 mL conical flasks. C–La-MOF (0.5 g L−1) were added into 50 mL of phosphate solutions with different concentrations of 1 to 80 mg P L−1. Langmuir and Freundlich models were applied to describe the adsorption isotherm data in nonlinear regression forms.41
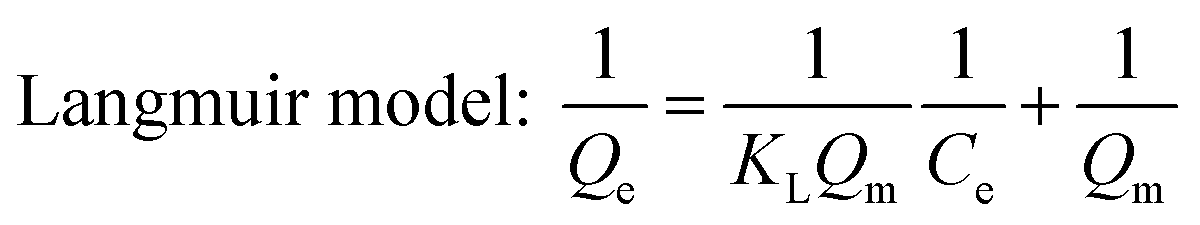 | (1) |
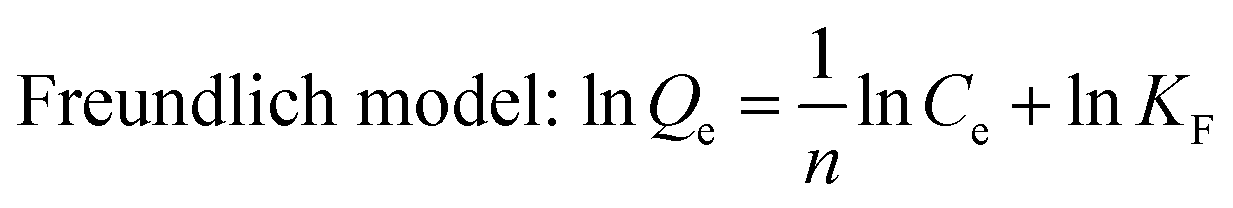 | (2) |
In kinetic experiments, 0.5 g L−1 C–La-MOF was added to a 50 mg P L−1 phosphate solution. An aliquot of solution was sampled at specified time intervals. To analyse the kinetic mechanism of the adsorption process, the experimental data were fitted to pseudo-first-order and pseudo-second-order models which are described as the following equations:42
|
Pseudo-first-order equation: ln(qe − qt) = lnqe − k1t
| (3) |
| Pseudo-second-order equation: t/qt = 1/(k2qe2) + t/qe | (4) |
To study the effect of pH on phosphate removal, 0.5 g L−1 C–La-MOF was added to a 50 (mg P L−1) phosphate solution. The initial pH, ranging from 2.0 to 12.0, was adjusted by adding NaOH and/or HCl solutions. The effect of coexisting anions on the phosphate adsorption capacity was evaluated by dissolving NO3−, SO42−, Cl− and CO32− into a 50 (mg P L−1) phosphate solution with a molar ratio of the anion to phosphate of 1:1. The experiments were conducted either without pH adjustment or at pH ∼7, respectively. Then, 0.5 g L−1 C–La-MOF was added to these solutions.
The experiment to assess the adsorption of a low concentration of phosphate was conducted in a 100 μg P L−1 solution. The dosage of C–La-MOF was 10–100 mg L−1 in this experiment. Aliquots (5 mL) were sampled at different time intervals.
2.7. Organic carbon removal
100 mg L−1 C–La-MOF, GC, C–La2O3, La-MOF, La-MOF-500 and G–La-MOF were used to remove 3 (mg TOC L−1) HA, respectively, to investigate the HA removal characteristics of C–La-MOF. The HA solutions in studies were acquired by diluting the HA stock solution according to the standard curve. The initial pH of the solutions was adjusted to ∼7 with NaOH and HCl. In this section, the operational conditions and sample pre-treatment were identical with those in phosphate removal experiments. The samples were taken out of the containers at different time intervals. The HA concentration was determined by a UV-vis spectrophotometer and expressed as TOC, and the determination method of HA was detailed in the ESI.†2.8. Simultaneously removal of phosphate and organic carbon
To investigate the simultaneously removal efficiency of HA and phosphate by C–La-MOF, 100 mg L−1 C–La-MOF was mixed with the solution containing 100 μg P L−1 phosphate and different concentrations of HA ranging from 0 to 5 mg TOC L−1. The initial pH of the solutions was adjusted to ∼7 with NaOH and HCl. In this section, the operational conditions, sampling method and sample pre-treatment were identical with those in HA removal experiments. The concentrations of phosphate and HA were determined by ICP-MS and a UV-vis spectrophotometer, respectively.2.9. Separation characteristics of C–La-MOF
To study the separation characteristics of C–La-MOF, 0.5 g L−1 C–La-MOF was added to 50 mL of DI water at different pH values and the mixtures were shaken at 25 °C for 0.5 h at 150 rpm. Then, the solutions were allowed to settle for 20 min and the supernatants of the solutions were taken out for measurement of the turbidity via a turbidimeter (HACH, 2100AN, America). The supernatants were then filtered through a 0.45 μm membrane syringe filter, and their turbidity was measured with turbidimeter.3. Results and discussion
3.1. Preparation and characterization
To obtain C–La-MOF, the carbon-coated lanthanum composite that can synchronously capture phosphate and organic carbon, a short hydrothermal treatment was first performed with glucose and La-MOF serving as the carbon source and lanthanum source, respectively. During the hydrothermal treatment, the glucose molecules absorbed on La-MOF nanorods were first polymerized and carbonized.43 The precursors in solution then gradually diffused to the La-MOF nanorods and continued coating the nanorods with newly formed polysaccharide.35 In this process, the carbonaceous layer thickness increased, and the structure of the inner carbon layer and outer polysaccharide layer formed, which can be seen in Fig. S2 in the ESI.†34 However, the outer polysaccharide carbon layer and the organic ligand of La-MOF, H3BTC, might be unstable and thus leak into water, consequently increasing the organic carbon content. Therefore, further carbonization was necessary. The urchin-like La-MOF, the La source, is composed of numerous nanorods, and these nanorods with very high density pointed toward the centre of the sphere.33 However, the SEM image of C–La-MOF shown in Fig. 1a reveals only single nanorods rather than urchin-like microspheres. It was believed that the hydrothermal treatment caused the nanorods constituting urchin-like microspheres to disperse due to the unstable urchin-like structure. In the TEM image of C–La-MOF (Fig. 1b), the carbon coating layer can be easily identified (indicated by the two red arrows), even after further carbonization. From HRTEM image of C–La-MOF shown in Fig. S3a,† no lattice fringe was observed, indicating that C–La-MOF was amorphous, which could also be verified by the diffusive ring pattern in SAED (Fig. S3b†) and the XRD pattern (Fig. S3c†) of C–La-MOF. C–La-MOF was composed of three elements C, O and La because there were only these three elements in the reactants (glucose, La(1,3,5-BTC)(H2O)6). From the elemental mapping of C–La-MOF shown in Fig. 1c, the elements C, O and La were uniformly distributed in the nanorod, indicating the existence of abundant C–La micro-interfaces in C–La-MOF. In addition, the lanthanum content in C–La-MOF was determined by ICP-OES to be 44.93 wt%. | ||
| Fig. 1 (a) SEM image of C–La-MOF. (b) TEM image of C–La-MOF and a detailed enlargement. (c) High-angle annular dark field (HAADF) image of C–La-MOF and the corresponding elemental mapping of C, O and La. | ||
From Fig. 2a, the daily lanthanum leakage from La-MOF-500 was much higher than that from C–La-MOF because the La in La-MOF-500 (La2O2CO3) contacted the water directly, while the carbon layer of C–La-MOF could prevent La from leaking into water because of its dense structure.34 However, when the carbon layer was too thick, it would hamper the contact of phosphate with La sites. According to the formation mechanism of the carbon layer, the longer the hydrothermal time, the more polysaccharide deposited on the nanorod, and the thicker the carbonaceous layer. Thus, the hydrothermal time is an important factor that influences the carbon layer thickness.44 In addition, the thickness of the carbonaceous layer formed in the hydrothermal treatment determined the final carbon layer thickness of C–La-MOF. Therefore, we first studied the influence of hydrothermal time on the final carbon layer thickness of the adsorbents. The TEM images and carbon layer thickness distribution of the products hydrothermally treated for different amounts of time are shown in Fig. 1b and S4.† Irrespective of the hydrothermal time (6 h, 10 h or 12 h), the samples maintained the nanorod structure. However, the carbon shell thickness increased from 5.03 nm to 8.64 nm to 11.82 nm, when the hydrothermal reaction time increased from 6 h to 10 h to 12 h, respectively. Next, the phosphate adsorption capacities of the above products were investigated to understand the relationship between the carbon layer thickness and the performance. It is evidenced in Fig. 2b that the phosphate adsorption capacity of C–La-MOF12, the sample treated for 12 h, was lowest, corresponding to the thickest carbon layer. A thick carbon layer hindered the contact between the La species and phosphate. C–La-MOF10 presented a similar phosphate capture ability to C–La-MOF, but the fabrication of C–La-MOF10 took four hours more than C–La-MOF. Thus, in the subsequent study, C–La-MOF was selected as the candidate to investigate the feasibility to adsorb phosphate and organic carbon from water.
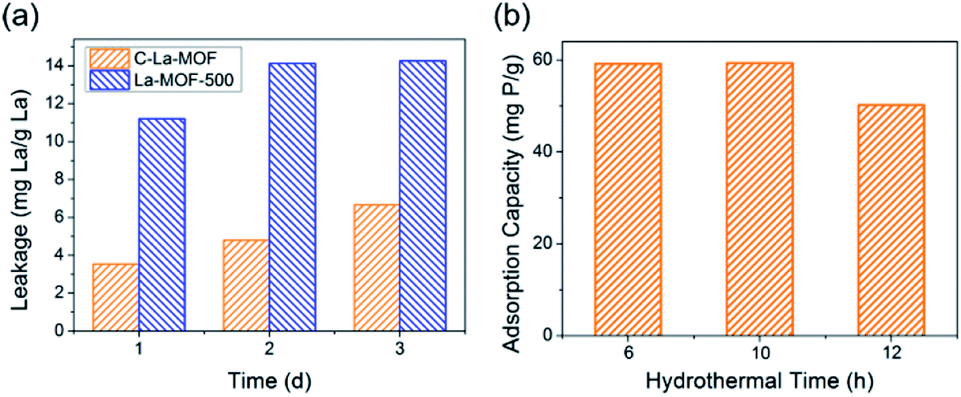 | ||
| Fig. 2 (a) Lanthanum leakage from C–La-MOF and La-MOF-500. (b) The effect of the hydrothermal time on the phosphate adsorption capacity. | ||
To study the effect of different La sources on the structure and phosphate capture performance, various carbon-coated lanthanum materials were fabricated. Fig. 3 presents the phosphate intake of different materials. Obviously, C–La-MOF presented the best phosphate capture property. However, GC and C–La had no phosphate capture capacity. The significant performance gap between C–La-MOF and GC indicated that the La species in C–La-MOF was the active ingredient for phosphate adsorption. From the TEM and SEM images of C–La shown in Fig. S5a and b,† there was no La species in/on C–La, and C–La formed only carbon spheres. Therefore, C–La did not exhibit any phosphate adsorption capacity.
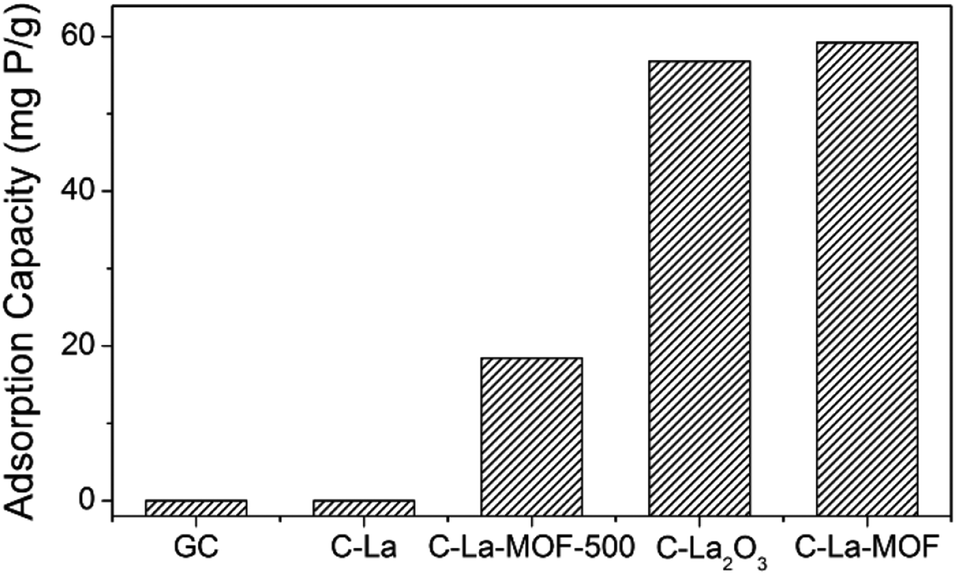 | ||
| Fig. 3 Phosphate adsorption capacities of various materials. | ||
The phosphate adsorption capacity of C–La-MOF-500 was 18.41 mg P g−1, less than that of C–La-MOF. However, their adsorption capacities were much less than that of La-MOF-500 (173.8 mg P g−1).33 There are spheres and nanorods in the SEM image of C–La-MOF-500 shown in Fig. S5c,† and the nanorods were damaged to some extent. During the hydrothermal treatment of aqueous glucose, the pH of the solution changed from approximately neutral to 3–4,45 which would lead to the dissolution of La-MOF-500 nanorods. However, the addition of La-MOF-500 was too high for complete dissolution. Thus, the damaged La nanorods were coated with a carbon layer and adsorbed phosphate as C–La-MOF did. The dissolved lanthanum together with glucose formed La-carbon spheres, and the elements C, O and La were uniformly distributed in the spheres (Fig. S5d†). The diameter of the spheres (approximately 6 μm) was much larger than that of C–La-MOF nanorods (approximately 130 nm). Compared to C–La-MOF, much La sealed in the C–La-MOF-500 spheres became inactive towards phosphate, leading to the smaller phosphate adsorption capacity of C–La-MOF-500. Compared with La-MOF-500, the La in carbon-coated La nanorods and La-carbon spheres was more difficult to contact phosphate, resulting in a much lower phosphate intake of C–La-MOF-500 and C–La-MOF.
The phosphate adsorption capacity of C–La2O3 was close to that of C–La-MOF and higher than that of C–La-MOF-500. Like C–La-MOF-500, C–La2O3 also consisted of two parts: spheres and bulks (Fig. S5e†). In addition, from the SEM image of C–La2O3 after phosphate adsorption shown in Fig. S5f,† the surface of both spheres and bulks became rough, indicating that phosphate was adsorbed on both spheres and bulks; in other words, lanthanum existed in both the spheres and bulks. Additionally, La species were not uniformly distributed in every sphere, which was verified by the roughness comparison of different spheres after phosphate adsorption shown in Fig. S5f and g.† The explanation of the relations between the structures and phosphate adsorption capacity was similar to that for C–La-MOF-500. However, the La content in La2O3 (85.3%) was higher than that in La(1,3,5-BTC)(H2O)6 (30.4%) and La-MOF-500 (La2O2CO3, 75.1%). The high lanthanum content resisted the unfavourable effect of the structures on phosphate adsorption performance. Hence, the structures and lanthanum content of C–La2O3 together determined its phosphate adsorption capacity.
La-MOF could be formed at pH∼3,33 which implies that La-MOF was resistant to acid, different from the inorganic lanthanum source, La2O3 and La-MOF-500 (La2O2CO3). Hence, during the hydrothermal treatment, the amount of dissolved La from La-MOF nanorods was too low to form La-carbon spheres; thus, C–La-MOF still maintained the uniform nanorod structure similar to La-MOF. If the total La content was the same, the La content for phosphate capture in either C–La-MOF-500 or C–La2O3 was less than that in C–La-MOF because the deeply sealed La in spheres was inactive towards phosphate adsorption. In other words, the uniform nanorod structure of C–La-MOF provided more lanthanum active sites for phosphate than C–La-MOF-500 and C–La2O3, and thus, C–La-MOF achieved robust phosphate adsorption performance.
3.2. Characterization of phosphate removal by C–La-MOF
Both Langmuir and Freundlich isotherms were applied to determine the maximum adsorption capacity of phosphorus on C–La-MOF (Fig. 4a), and the corresponding parameters are listed in Table S1.† The experimental data could be well fitted to the Freundlich isotherm model rather than the Langmuir model on the basis of the correlation coefficients (R2, see Table S1†). The adsorption capacity of C–La-MOF estimated by the Freundlich model was 56.02 mg P g−1 at the initial phosphate concentration of 50 mg P L−1, and the value of 1/n was less than 1.0 (Table S1†), representing a favourable adsorption condition.46 The time-dependent adsorption for high concentration phosphate is shown in Fig. 4b. The phosphate adsorption could reach equilibrium within 6 h. The pseudo-first-order model and pseudo-second-order model are shown in Fig. S6,† and the corresponding parameters and correlation coefficients (R2) are provided in Table S2.† According to the R2 values, the results better matched the pseudo-second-order model (R2 > 0.99), suggesting a chemisorption process.42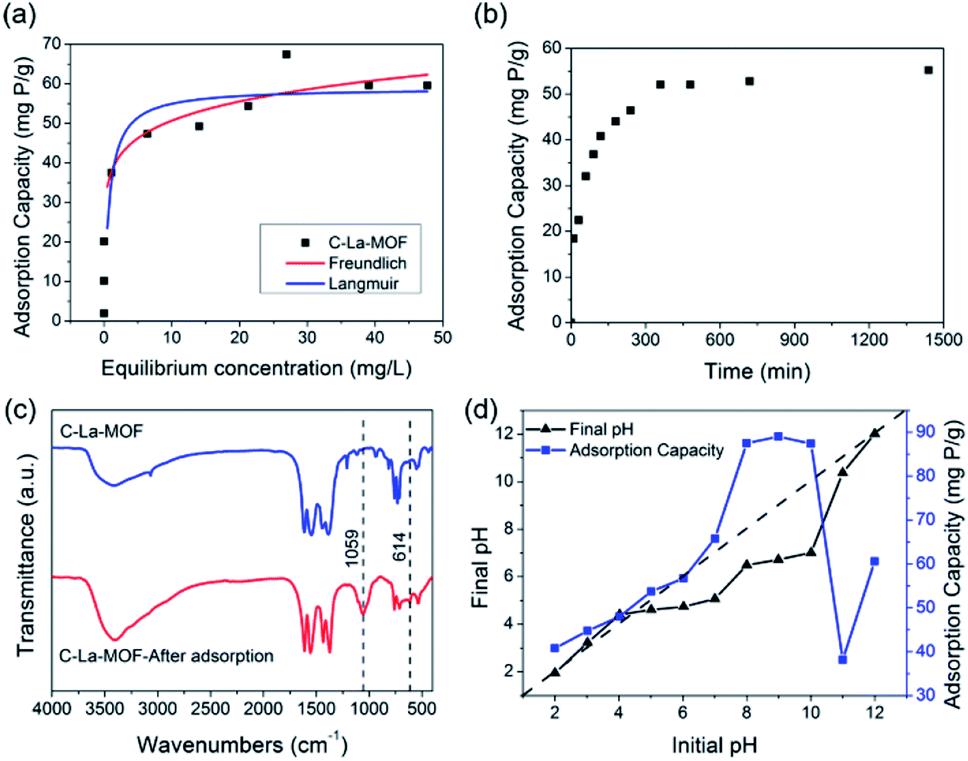 | ||
| Fig. 4 (a) Adsorption isotherms of phosphate on C–La-MOF. (b) Effect of the contact time on the phosphate adsorption capacity of C–La-MOF (initial P concentration = 50 mg P L−1). (c) FTIR spectra of C–La-MOF before and after adsorption. (d) Effect of the initial pH on the phosphate adsorption of C–La-MOF and the final pH variation of the solution. | ||
To understand the interaction between the adsorbent and phosphate, the FT-IR spectra of C–La-MOF before and after adsorption of phosphate were compared and are shown in Fig. 4c. Two distinct new bands were observed at 614 and 1059 cm−1. The first band was attributed to the bend vibration of O–P–O in PO43− groups.47 In addition, the absorption peak at approximately 1059 cm−1 was attributed to the typical asymmetric stretch vibration of P–O in PO43− groups.48 In the SEM image of C–La-MOF after phosphate capture (Fig. S7†), the surface of C–La-MOF became rough. These results proved that PO43− groups were adsorbed onto C–La-MOF.
To further shed light on the mechanism of phosphate adsorption of C–La-MOF, the effect of the pH on the phosphate uptake capacity of C–La-MOF was studied and is presented in Fig. 4d. The point of zero charge (pHzpc) of the sorbent was approximately 7.71, as identified from the zeta potentials at different pH values (Fig. S8†). The phosphate capacity could be maintained at a high level in the pH range of 2–12, and even the lowest adsorption capacity could reach 38.2 mg P g−1, indicating a wide application range. The phosphate removal efficiencies were highest in the pH range of 8–10, and the highest phosphate adsorption capacity was 89 mg P g−1.
When the pH was lower than 2.13, the predominant species of phosphate was the neutral H3PO4,49 which is weakly attached to the sites of C–La-MOF. At pH 2.13–7.71, the protonation of the samples made the surface of C–La-MOF positively charged, consequently facilitating the interaction with anionic groups, H2PO4− and HPO42−, via electrostatic attraction.48,50 Meanwhile, La active sites could complex with phosphate, forming La–O coordination bonds.51 The proportion of HPO42− increased with the increase of pH, and HPO42− has a higher affinity than H2PO4− for the La(III) centre.52 Therefore, the phosphate adsorption capacity increased with the pH increase. When the pH increased further from 8.0 to 11.0, the predominant species was HPO42−, and the surface of C–La-MOF became negatively charged. Electrostatic repulsion worked, and hydroxyl ions would compete with phosphate for adsorption sites, which hindered the adsorption of phosphate on C–La-MOF. However, the C–La micro-interfaces in C–La-MOF provided abundant adsorption sites to reduce the competition. In addition, in this pH range, the pH values after 24 h of adsorption (final pH) were 6.49–7.01, which were higher than that in the initial pH range of 3–7 (3.23–5.07) (Fig. 4d). It means that the proportion of HPO42− in the initial pH range of 8–10 was higher than that in the initial pH range of 3–7. Thus, the complexation between La and phosphate together with the abundant adsorption sites of C–La-MOF overwhelmingly overcame the adverse effects of electrostatic repulsion and hydroxyl ion competition. In consequence, C–La-MOF achieved high phosphate adsorption capacities in this pH range. At pH > ∼10.0, the more negatively charged C–La-MOF surface (Fig. S8†) and high concentration of hydroxyl ions allowed the electrostatic repulsion and hydroxyl ion competition to become dominant, leading to a sharp reduction in phosphate adsorption capacity. Even so, the complexation between La and phosphate could still maintain a phosphate adsorption capacity of over 38 mg P g−1.
The studies of competitive adsorption of phosphate on C–La-MOF were carried out in the presence of common ions such as Cl−, SO42−, NO3− and CO32− in water. The initial phosphate concentration was 50 mg P L−1, and the molar ratio of coexisting ions to phosphate was fixed at 1:1. First, the experiment without pH adjustment was conducted. It is found from Fig. 5a that Cl−, SO42− and NO3− had trivial influence on the phosphate adsorption capacity. In the previous study, coexisting CO32− would compete with phosphate for the adsorptive sites, which was attributed to the small Ksp (3.98 × 10−34) of La2(CO3)3, thereby leading to a decrease in the phosphate adsorption capacity.33,50,53–55 However, the experimental results in this study was opposite. CO32− improved the phosphate adsorption capacity significantly, and the phosphate adsorption capacity was 82.97 mg P g−1. The reason for the differing results was the different pH of the solutions. The initial pH of the solution containing CO32− was 8.04, and the initial pH of the solutions containing other anions were approximately 5.2. In addition, in the study of the pH effect (Fig. 4d), the phosphate adsorption capacity at pH ∼ 8 was higher than that at pH ∼ 5, and the adsorption capacity at the two pH values was 87.52 mg P g−1 and 53.74 mg P g−1, respectively, which agreed with the experimental results of the anion effect. To eliminate the initial pH effect, an experiment with initial pH of ∼7 was performed, and the results are shown in Fig. 5b. Cl−, SO42− and NO3− still had trivial influence on the adsorption capacity, but the adsorption capacity was improved to approximately 68 mg P g−1, which agreed with the result of the pH effect (Fig. 4d). The adsorption capacity in the presence of CO32− was 76.99 mg P g−1, which was less than that without pH adjustment due to the reduced initial pH value. Nonetheless, CO32− still had positive influence on the phosphate adsorption capacity, attributed to the buffering of CO32−. The solution pH would decrease as phosphate was adsorbed onto the adsorbents (Fig. 4d). When the initial pH was ∼7, the pH value after 24 h of adsorption was generally approximately 5.1 but was 6.51 when CO32− existed, due to the buffering of CO32−. From Fig. 4d, before pH ∼ 9, the higher the pH, the higher the phosphate adsorption capacity. Therefore, the adsorption capacity in the presence of CO32− was higher than the others. These coexisting anions in water had trivial or positive influence on the phosphate adsorption capacity, suggesting a high selective phosphate removal capacity of C–La-MOF with a strong anti-interference ability.
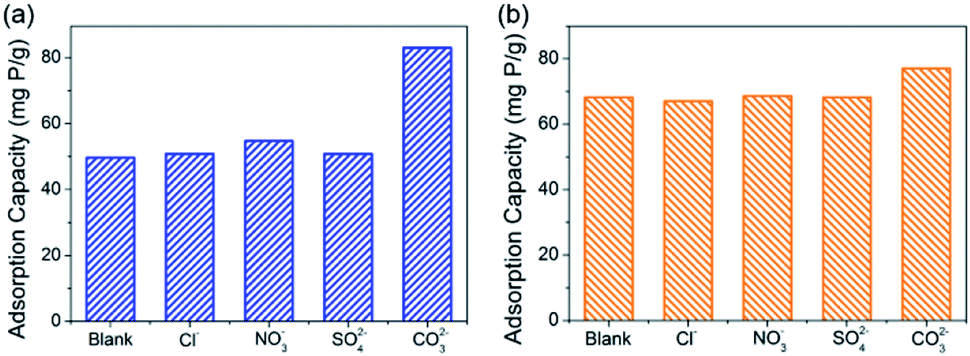 | ||
| Fig. 5 (a) and (b) show the effect of coexisting anions on the phosphate adsorption capacity of C–La-MOF without pH adjustment and at pH ∼ 7, respectively. | ||
3.3. Low-concentration phosphate removal
The treatment of phosphate faces a thorny problem in the case that possible abnormal growth of microorganism occurs at trace phosphate concentrations. Some countries use 30 μg P L−1 to prevent eutrophication,56 and more harsh limitations are adopted to reduce microbial growth in water supply pipe networks.10,11 Thus, it is necessary to assess the removal ability of our adsorbents at trace phosphate concentrations. Herein, an adsorption test was conducted in synthetic solution with an initial phosphate concentration of 100 μg P L−1. The equilibrium concentrations of phosphate associated with the adsorbent dosage at different contact times are shown in Fig. 6. On the whole, the residual phosphate concentration reduced as the adsorbent dosage and contact time increased. Compared with LCNFs (carbon nanofiber with embedded LaCO3OH nanoparticles) in our previous study,18 C–La-MOF removed trace amounts of phosphate faster. The residual phosphate concentrations for C–La-MOF and LCNFs were 42.38 μg P L−1 and 79.56 μg P L−1, respectively, when the dosage was 10 mg L−1 and the contact time was 30 min. 20 mg L−1 C–La-MOF could remove phosphate to less than 30 μg P L−1 in 20 min to satisfy the criterion for eutrophication prevention. The residual phosphate concentration dropped below 10 μg P L−1 in 20 min when the adsorbent dosage was greater than or equal to 30 mg L−1. If the contact time could be extended to 1 h, 30 mg L−1 of adsorbent could remove phosphate completely to fulfil harsher standards.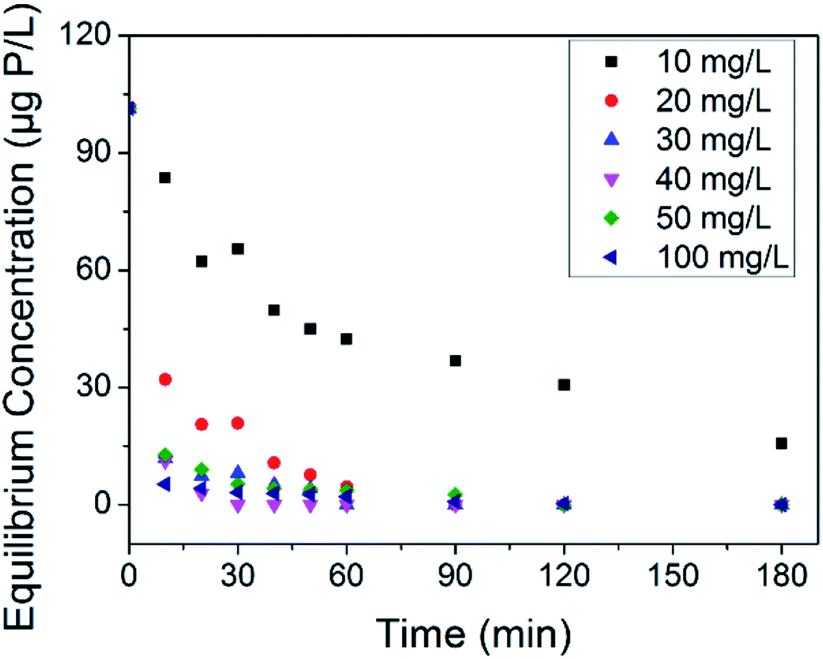 | ||
| Fig. 6 Effect of the adsorbent dosage on trace concentrations of phosphate removal. | ||
3.4. Organic carbon removal
In addition to phosphorus, there are organic matters in water. Hence, the removal characteristics of organic matters by C–La-MOF should also be studied. Humic acid (HA) was used as representative organic matter in this work because of its extensive existence in the aquatic environment and its bioavailability.57–59 The HA concentration was expressed as TOC, i.e., the HA removal rate was actually the TOC removal rate. The TOC removal rates of different materials in 24 h and 1 h are displayed in Fig. 7a and b, respectively. The HA removal rate gap between C–La-MOF and G-La-MOF demonstrated the significance of carbonization. The outer polysaccharide layer of G–La-MOF and H3BTC in La-MOF could be released into water, consequently increasing the TOC content. In Fig. 7a, HA could be removed by GC and La-MOF-500, reflecting the role of the carbon and La species in the adsorption of HA, respectively. The adsorption of HA by carbon (GC) belonged to physical adsorption, and La species could complex with HA.60 Thus, 73.4% of HA removed by C–La-MOF could be attributed to the synchronous adsorption by the carbon and La species in C–La-MOF. In addition, the surface of C–La-MOF was positively charged at pH 7 (Fig. S8†), and HA was negatively charged.61 Therefore, the electrostatic attraction also facilitated the HA adsorption on C–La-MOF.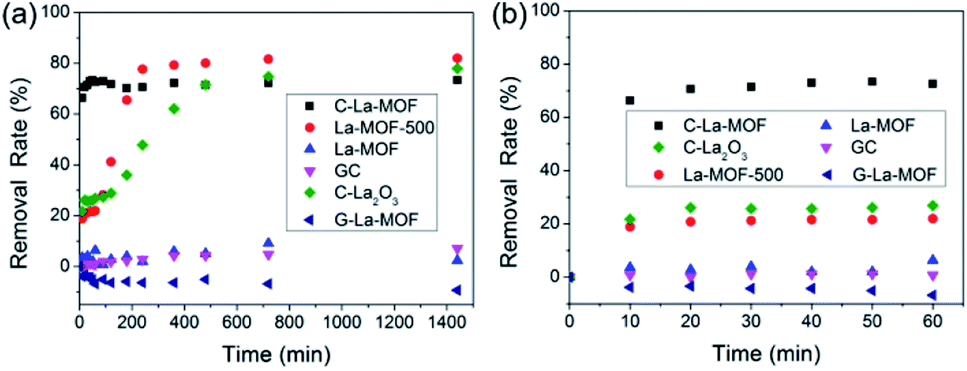 | ||
| Fig. 7 (a) and (b) show the TOC removal rates of different materials in 24 h and 1 h, respectively. The initial TOC concentration and the dosage of sorbent were 3 mg TOC L−1 and 100 mg L−1, respectively, pH = 7. | ||
La-MOF could not stably reduce TOC in water because of its organic ligand, H3BTC. The removal of HA was achieved by complexation with La sites. However, the La sites in the MOFs were occupied in advance by organic bridging linkers (H3BTC) also via complexation. Therefore, if the combining ability of La sites with HA was much higher than that with H3BTC, H3BTC would be released into water. If it was lower than or comparable to that with H3BTC, HA could not be reduced. In either case, TOC in water would not decrease.
The remaining three lanthanum-containing materials, La-MOF-500, C–La2O3 and C–La-MOF, achieved a high removal rate, and the gap between their rates was small (Fig. 7a). However, La-MOF-500 and C–La2O3 required a long time to achieve their high removal rates. In water treatment engineering, a longer contact time means increased cost. Hence, it is a great advantage that one material could obtain a high removal rate in a short time. In this regard, C–La-MOF showed its superiority. It achieved its highest removal rate in 50 min (Fig. 7b), and subsequent variation of the removal rate over time remained stable. The rich and accessible C–La micro-interfaces in C–La-MOF provided abundant adsorption sites and could help HA rapidly reach the active sites, consequently shortening the adsorption time. In comparison, the active sites in only carbon (GC) or only La species (La-MOF-500) was less because of the lack of the C–La micro-interfaces. Although C–La2O3 had C–La micro-interfaces, they might be not abundant because of its unevenly distributed La species in every sphere (Fig. S5f and g†). In some C–La2O3 spheres, the La species were uniformly distributed in and on the sphere. However, in other C–La2O3 spheres, the La species were deeply sealed in the sphere. In either case, the C–La micro-interfaces that were far away from the surface of the sphere were inactive for HA adsorption. Therefore, C–La2O3 did not show rapid removal of HA.
3.5. Simultaneous removal of phosphate and organic carbon
After studying the removal characteristics of HA, simultaneous removal of HA and phosphorous was evaluated. The effects of different HA concentrations on both phosphate removal and HA removal were assessed at a constant phosphate concentration, and the results are shown in Fig. 8. It can be observed in Fig. 8a that the initial HA concentration had little effect on the phosphate removal rate, and 100 mg L−1 C–La-MOF could remove phosphate completely in 10 min at various HA initial concentrations, due to the stronger bond between the La species and phosphate. Similarly, the coexisting phosphate did not affect HA removal, which can be seen from the data comparison at an initial HA concentration of 3 mg TOC L−1 shown in Fig. 7b and 8b. In addition, the highest HA removal rates at various initial concentrations were reached in 10 min. The low mutual influence of phosphate and HA can be attributed to the rich C–La micro-interfaces that could provide sufficient adsorption sites for both HA and trace amounts of phosphate. In Fig. 8b, the HA removal rate increased with the initial HA concentration increase due to the low initial concentration of HA. The initial HA concentration in most of studies was >10 mg L−1 or even hundreds of mg L−1.62–64 The removal rules and mechanisms valid at high concentration would not apply to conditions of low concentrations. At high concentrations, the removal rate decreased with an increase in the initial concentration, which may be due to the saturation of adsorption sites on the adsorbent surface.65 However, the abundant adsorption sites of C–La-MOF were sufficient for adsorbates at low initial concentration, and thus, the adsorption sites would not be the limiting factor of adsorption. Therefore, in the low concentration range, the higher the initial concentration, the greater the collision probability and the higher driving force for mass transfer, consequently leading to the increase in the HA removal rate. | ||
| Fig. 8 Removal rates of (a) phosphate and (b) HA by C–La-MOF with different concentrations of HA. The initial phosphate concentration was 100 μg P L−1, and the initial HA concentrations ranged from 0 to 5 mg TOC L−1. | ||
3.6. Separation characteristics of C–La-MOF
In water treatment engineering, it is important to separate the adsorbents from water. Therefore, the separation characteristics of C–La-MOF at different pH values was studied. Most C–La-MOF sank to the bottom of bottles when the solutions were shaken at 150 rpm. From Table S3,† the turbidity of the solutions after sedimentation for 20 min at pH ∼10 and ∼11 was 95.87 NTU and 31.67 NTU, respectively, higher than that at other pH values (2.89–13.27 NTU). In general, the raw water pH was 6.5–8.5.66 The turbidity after sedimentation in this pH range was close to or even less than that of source water.67–69 When further filtration was carried out, the turbidity of solutions at pH 2–12 was less than 1 NTU, which could satisfy the turbidity requirement regulated in the Chinese National Standards for Drinking Water Quality (GB5749-2006).4. Conclusions
Carbon-coated lanthanum nanorods with a uniform distribution of La and C were rationally prepared through glucose and La-MOF hydrothermal treatment and further carbonization. The tuned carbon layer thickness of C–La-MOF by the hydrothermal time coordinated the balance between excellent phosphate intake and low lanthanum leakage. Compared with the carbon-coated lanthanum composites fabricated by the same method but with different lanthanum precursors, C–La-MOF exhibited a superior phosphate adsorption capacity owing to its structural uniformity. C–La-MOF revealed a strong anti-interference ability and achieved a high phosphate capture capacity over a wide initial pH range of 2–12 due to the strong complexation between La and phosphate. Besides, it could completely remove trace concentrations of phosphate and achieve the highest HA removal rate within 1 h. Even when phosphate and HA coexisted, the removal performance of both species remained intact. Thus, C–La-MOF has remarkable prospects for bacterial contamination control in practical water treatment through the simultaneous removal of phosphate and organic carbon.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge National Natural Science Foundation of China (Grant no. 51678181), State Key Laboratory of Urban Water Resource and Environment in HIT of China (No. 2016DX02), HIT Environment and Ecology Innovation Special Funds (No. HSCJ201606), Postdoctoral Science Foundation of Heilongjiang Prov. (LBH-TZ0606 and LBH-Q16012). Scientific Research Foundation for Returned Scholars, Heilongjiang of China (LC2017023).References
- I. Irwansyah, Y. Q. Li, W. Shi, D. Qi, W. R. Leow, M. B. Tang, S. Li and X. Chen, Adv. Mater., 2015, 27, 648–654 CrossRef CAS PubMed.
- J. Gehring, B. Trepka, N. Klinkenberg, H. Bronner, D. Schleheck and S. Polarz, J. Am. Chem. Soc., 2016, 138, 3076–3084 CrossRef CAS PubMed.
- M. Mahmoudi and V. Serpooshan, ACS Nano, 2012, 6, 2656–2664 CrossRef CAS PubMed.
- C. A. Strassert, M. Otter, R. Q. Albuquerque, A. Höne, Y. Vida, B. Maier and L. De Cola, Angew. Chem., Int. Ed., 2009, 48, 7928–7931 CrossRef CAS PubMed.
- S. Purwajanti, L. Zhou, Y. Ahmad Nor, J. Zhang, H. Zhang, X. Huang and C. Yu, ACS Appl. Mater. Inter., 2015, 7, 21278–21286 CrossRef CAS PubMed.
- S. Regli, J. Chen, M. Messner, M. S. Elovitz, F. J. Letkiewicz, R. A. Pegram, T. J. Pepping, S. D. Richardson and J. M. Wright, Environ. Sci. Technol., 2015, 49, 13094–13102 CrossRef CAS PubMed.
- M. H. Xiong, Y. J. Li, Y. Bao, X. Z. Yang, B. Hu and J. Wang, Adv. Mater., 2012, 24, 6175–6180 CrossRef CAS PubMed.
- A. C. C. Rotzetter, C. R. Kellenberger, C. M. Schumacher, C. Mora, R. N. Grass, M. Loepfe, N. A. Luechinger and W. J. Stark, Adv. Mater., 2013, 25, 6057–6063 CrossRef CAS PubMed.
- D. van der Kooij in Drinking Water Microbiology, ed. G. A. McFeters, Springer, New York, 1990, vol. 3, pp. 57–87 Search PubMed.
- A. Sathasivan and S. Ohgaki, Water Res., 1999, 33, 137–144 CrossRef CAS.
- I. T. Miettinen, T. Vartiainen and P. J. Martikainen, Appl. Environ. Microb., 1997, 63, 3242–3245 CAS.
- W. Fang, J. Y. Hu and S. L. Ong, J. Appl. Microbiol., 2009, 106, 1328–1335 CrossRef CAS PubMed.
- L. C. Gerber, N. Moser, N. A. Luechinger, W. J. Stark and R. N. Grass, Chem. Commun., 2012, 48, 3869–3871 RSC.
- I. T. Miettinen, T. Vartiainen and P. J. Martikainen, Nature, 1996, 381, 654–655 CrossRef CAS PubMed.
- A. Sathasivan, S. Ohgaki and M. Otaki, Evaluation of water treatment processes in terms of bacterial regrowth control, 19th biennial conference of IAWQ, 1997 Search PubMed.
- M. J. Lehtola, I. T. Miettinen, T. Vartiainen and P. J. Martikainen, Water Res., 2002, 36, 3681–3690 CrossRef CAS PubMed.
- J. Lou, C. Chang, W. Tseng and J. Han, Urban Water J., 2015, 12, 672–677 CrossRef CAS.
- X. Zhang, W. Wang, W. Shi, J. He, H. Feng, Y. Xu, F. Cui and C. Wang, J. Mater. Chem. A, 2016, 4, 12799–12806 CAS.
- D. L. Correll, J. Environ. Qual., 1998, 27, 261–266 CrossRef CAS.
- L. Dithmer, A. S. Lipton, K. Reitzel, T. E. Warner, D. Lundberg and U. G. Nielsen, Environ. Sci. Technol., 2015, 49, 4559–4566 CrossRef CAS PubMed.
- J. He, W. Wang, F. Sun, W. Shi, D. Qi, K. Wang, R. Shi, F. Cui, C. Wang and X. Chen, ACS Nano, 2015, 9, 9292–9302 CrossRef CAS PubMed.
- I. Emmanuelawati, J. Yang, J. Zhang, H. Zhang, L. Zhou and C. Yu, Nanoscale, 2013, 5, 6173–6180 RSC.
- J. Yang, L. Zhou, J. Zhang, J. Zou, Z. Yuan and C. Yu, Chem.–Eur. J., 2013, 19, 5578–5585 CrossRef CAS PubMed.
- L. Chen, X. Zhao, B. Pan, W. Zhang, M. Hua, L. Lv and W. Zhang, J. Hazard. Mater., 2015, 284, 35–42 CrossRef CAS PubMed.
- D. Guaya, C. Valderrama, A. Farran, C. Armijos and J. L. Cortina, Chem. Eng. J., 2015, 271, 204–213 CrossRef CAS.
- F. H. Firsching and S. N. Brune, J. Chem. Eng. Data, 1991, 36, 93–95 CrossRef CAS.
- S. Tokunaga, S. A. Wasay and S. W. Park, Water Sci. Technol., 1997, 35, 71–78 CAS.
- E. W. Shin, K. G. Karthikeyan and M. A. Tshabalala, Environ. Sci. Technol., 2005, 39, 6273–6279 CrossRef CAS PubMed.
- Phoslock Water Solutions Ltd., http://www.phoslock.eu/media/7464/lanthanum-and-human-toxicity.pdf, (accessed March 2017).
- M. R. Chan, F. Ghandour, N. S. Murali, M. Washburn and B. C. Astor, Journal of Nephrology and Therapeutics, 2014, 4, 1000162 Search PubMed.
- F. Chen, Z. Gu, J. Yi, X. Xu, P. Kulkarni and Q. Tang, New J. Chem., 2016, 40, 2644–2648 RSC.
- S. Meis, B. M. Spears, S. C. Maberly and R. G. Perkins, Water Res., 2013, 47, 4460–4473 CrossRef CAS PubMed.
- X. Zhang, F. Sun, J. He, H. Xu, F. Cui and W. Wang, Chem. Eng. J., 2017, 326, 1086–1094 CrossRef CAS.
- L. Kong, X. Lu, X. Bian, W. Zhang and C. Wang, ACS Appl. Mater. Inter., 2011, 3, 35–42 CrossRef CAS PubMed.
- H. Qian, S. Yu, L. Luo, J. Gong, L. Fei and X. Liu, Chem. Mater., 2006, 18, 2102–2108 CrossRef CAS.
- X. M. Sun and Y. D. Li, Langmuir, 2005, 21, 6019–6024 CrossRef CAS PubMed.
- X. W. D. Lou, J. S. Chen, P. Chen and L. A. Archer, Chem. Mater., 2009, 21, 2868–2874 CrossRef CAS.
- Y. Lin, J. Duh and M. Hung, J. Phys. Chem. C, 2010, 114, 13136–13141 CAS.
- Z. Wang, H. Guo, Y. Yu and N. He, J. Magn. Magn. Mater., 2006, 302, 397–404 CrossRef CAS.
- Z. Zhang, H. Duan, S. Li and Y. Lin, Langmuir, 2010, 26, 6676–6680 CrossRef CAS PubMed.
- J. Liu, L. Wan, L. Zhang and Q. Zhou, J. Colloid Interf. Sci., 2011, 364, 490–496 CrossRef CAS PubMed.
- S. Yoon, C. Lee, J. Park, J. Kim, S. Kim, S. Lee and J. Choi, Chem. Eng. J., 2014, 236, 341–347 CrossRef CAS.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- P. Wu, N. Du, H. Zhang, J. Yu and D. Yang, J. Phys. Chem. C, 2010, 114, 22535–22538 CAS.
- S. Ikeda, K. Tachi, Y. H. Ng, Y. Ikoma, T. Sakata, H. Mori, T. Harada and M. Matsumura, Chem. Mater., 2007, 19, 4335–4340 CrossRef CAS.
- Z. Z. Ismail and H. N. AbdelKareem, Waste Management, 2015, 45, 66–75 CrossRef CAS PubMed.
- S. Gallini, J. R. Jurado and M. T. Colomer, Chem. Mater., 2005, 17, 4154–4161 CrossRef CAS.
- L. Zhang, Q. Zhou, J. Liu, N. Chang, L. Wan and J. Chen, Chem. Eng. J., 2012, 185, 160–167 CrossRef.
- L. Zhang, M. Li, Y. Gao, J. Liu and Y. Xu, Desalin. Water Treat., 2014, 57, 4671–4680 Search PubMed.
- W. Huang, Y. Zhu, J. Tang, X. Yu, X. Wang, D. Li and Y. Zhang, J. Mater. Chem. A, 2014, 2, 8839 CAS.
- L. Zhang, L. Wan, N. Chang, J. Liu, C. Duan, Q. Zhou, X. Li and X. Wang, J. Hazard. Mater., 2011, 190, 848–855 CrossRef CAS PubMed.
- R. S. S. Wu, K. H. Lam, J. M. N. Lee and T. C. Lau, Chemosphere, 2007, 69, 289–294 CrossRef CAS PubMed.
- J. Yang, L. Zhou, L. Zhao, H. Zhang, J. Yin, G. Wei, K. Qian, Y. Wang and C. Yu, J. Mater. Chem., 2011, 21, 2489 RSC.
- W. Huang, D. Li, Z. Liu, Q. Tao, Y. Zhu, J. Yang and Y. Zhang, Chem. Eng. J., 2014, 236, 191–201 CrossRef CAS.
- J. Liu, Q. Zhou, J. Chen, L. Zhang and N. Chang, Chem. Eng. J., 2013, 215, 859–867 CrossRef.
- M. Zamparas, A. Gianni, P. Stathi, Y. Deligiannakis and I. Zacharias, Appl. Clay Sci., 2012, 62–63, 101–106 CrossRef CAS.
- R. Cusnir, M. Jaccard, C. Bailat, M. Christl, P. Steinmann, M. Haldimann, F. Bochud and P. Froidevaux, Environ. Sci. Technol., 2016, 50, 5103–5110 CrossRef CAS PubMed.
- S. Van Geluwe, L. Braeken and B. Van der Bruggen, Water Res., 2011, 45, 3551–3570 CrossRef CAS PubMed.
- E. Lorenc-Grabowska and G. Gryglewicz, J. Colloid Interf. Sci., 2005, 284, 416–423 CrossRef CAS PubMed.
- O. Pourret, M. Davranche, G. Gruau and A. Dia, Chem. Geol., 2007, 243, 128–141 CrossRef CAS.
- J. Wang, X. Han, H. Ma, Y. Ji and L. Bi, Chem. Eng. J., 2011, 173, 171–177 CrossRef CAS.
- X. Wang, S. Tao and B. Xing, Environ. Sci. Technol., 2009, 43, 6214–6219 CrossRef CAS PubMed.
- S. Wang and Z. H. Zhu, J. Colloid Interf. Sci., 2007, 315, 41–46 CrossRef CAS PubMed.
- S. Vreysen and A. Maes, Appl. Clay Sci., 2008, 38, 237–249 CrossRef CAS.
- M. T. Yagub, T. K. Sen, S. Afroze and H. M. Ang, Adv. Colloid Interface Sci., 2014, 209, 172–184 CrossRef CAS PubMed.
- USGS science for a changing world, https://pubs.usgs.gov/wsp/wsp2254/pdf/wsp2254a.pdf, (accessed February 2018).
- D. Cook, G. Newcombe and P. Sztajnbok, Water Res., 2001, 35, 1325–1333 CrossRef CAS PubMed.
- S. J. Xia, J. Nan, R. P. Liu and G. B. Li, Desalination, 2004, 170, 41–47 CrossRef CAS.
- A. Koohestanian, M. Hosseini and Z. Abbasian, Am.-Eurasian J. Agric. Environ. Sci., 2008, 4, 266–273 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental method, SEM, TEM, HRTEM, SAED, XRD, elemental mapping, adsorption kinetic model, thickness distribution, zeta potential, equilibrium isotherm model parameters, kinetic model parameters and turbidity. See DOI: 10.1039/c8ra01519h |
| This journal is © The Royal Society of Chemistry 2018 |