
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization and evaluation of bulky bis(pyrazolyl)palladium complexes in Suzuki–Miyaura cross-coupling reactions†
Edward Ocansey ,
James Darkwa* and
Banothile C. E. Makhubela*
,
James Darkwa* and
Banothile C. E. Makhubela*
Department of Chemistry, University of Johannesburg, PO Box 524, Auckland Park, 2006, South Africa. E-mail: bmakhubela@uj.ac.za
First published on 12th April 2018
Abstract
Pyrazole-containing compounds have been used in recent times as ligands to stabilize metal complexes used as pre-catalysts in cross-coupling reactions. With various substituents at various positions in the pyrazole ring, the overall electrophilic and steric properties of the metal complexes can be fine-tuned. Herein, we report the synthesis of bulky pyrazole-based ligands by condensation of methyl 4-(bromomethyl)benzoate or benzyl bromide with various substituted pyrazole compounds. These ligands were utilised in the synthesis of bis(pyrazolyl)palladium(II) complexes. The complexes' catalytic activity in Suzuki–Miyaura cross-coupling reactions was evaluated. Phenyl bearing pre-catalyst 7, at a catalyst loading of 0.33 mol%, successfully converted 98% of bromobenzene and phenylboronic acid to biphenyl in 4 h at 140 °C, while the tertiary butyl bearing pre-catalyst 8 converted up to 81% of the same substrates to biphenyl. An increase in conversion was seen for all pre-catalysts when an electron-withdrawing substituent was present on the aryl halide substrate, and the opposite was observed when the electron-withdrawing group was present on the phenyl boronic acid.
Introduction
Synthesis of biaryl compounds, which are the building blocks for the preparation of the Merck and Novartis hypertensive drugs losartan and valsartan, involves the use of transition metal catalysts such as palladium and copper.1–5 Most notably, palladium complexes have gained widespread use as catalysts in both industry and academia for performing cross-coupling reactions mainly due to their thermal, oxidative and moisture stability, facile synthesis and the ability to modulate the steric and electronic properties of these palladium complexes.6 Bis-ligated palladium complexes, especially those with bulky monodentate ligands are well known for their anticancer properties7,8 and as effective cross-coupling catalysts.9,10Palladium catalysts coordinated in a trans fashion have been reported to demonstrate increased activity for cross-coupling between a wide array of aryl chlorides and amines at low catalyst loading, with a varied substrate scope.11 Similar observations have been made with the phosphine-ferrocenyl ligands reported by Štěpnička et al.12 Despite the extreme bulk of these ligands, they have insignificant distortion of the geometry around the metal from square planar as confirmed by X-ray crystallography. Although these palladium complexes contain bulky ligands, they do not in any way affect the formation of the active Pd(0) species that are used in the Suzuki–Miyaura cross-coupling reactions. Bulky ligands, might, in fact, contribute to stabilization of the active Pd(0) species, and in spite of their steric bulk is able to isomerize to the cis conformation during the catalysis.12
The most popular ligand system in cross-coupling reactions are ligands that have phosphorus donor atoms. Phosphorus-containing ligands facilitate the formation of bulky and electron rich complexes which allow for the effective cross-coupling of aryl halides and boronic acids.11 These ligands with phosphorus donor atoms have the ability to impart improved catalytic activity when used to form transition metal complexes. However, the drawback of phosphorus-containing ligands is their extreme sensitivity to air and moisture. Nitrogen donor ligands are providing alternatives to phosphorus donor ligands in that they can be tuned to obtain desired electronic and steric properties, in order to enhance the stability of the active Pd(0) species.13 An interesting class of nitrogen donor ligands are ligands that have pyrazolyl units. Pyrazolyl palladium complexes are ideal for cross-coupling reaction because the pyrazole nitrogen is a weaker σ-donor compared to imine and pyridines, and hence provides highly electrophilic metal centres. Additionally, the ability to vary substituents on the pyrazolyl ring allows the overall electrophilic and steric properties of resultant metal complexes to be adjusted.14–16 This is because the type of substituent affects the nucleophilicity of the nitrogen atoms in the ring and the bond strength of the subsequent ligated metal. Indeed several pyrazolyl palladium complexes have been used in recent times as pre-catalysts in cross-coupling reactions.10,17,18 Also with an increase in electrophilicity at the metal centre, the catalytic activity is enhanced.14,19,20 Mukherjee and Sarkar21 have demonstrated that pyrazolylimine palladium complexes have catalytic activities for Suzuki–Miyaura cross-coupling reaction that is dependent on the donor atoms and their steric environment.21 These pyrazolylimine ligands with low steric bulk and hard donor group such as NMe2 failed to catalyze the Suzuki–Miyaura cross-coupling reactions. This indicates that the ligands could not stabilize the Pd(0) species that is expected to catalyze the cross-coupling reaction. However, larger aryl groups on the imine nitrogen in the pyrazolylimine ligands were efficient catalysts for the Suzuki–Miyaura cross-coupling reaction.21
We herein report bis(pyrazolyl)palladium complexes with bulky substituents on the ligands and their applications as catalysts in the Suzuki–Miyaura cross-coupling reaction that clearly support how steric bulk of ligands enhance catalytic activities in cross-coupling reactions.
Experimental
Materials
All reactions were carried out in air unless otherwise stated. All solvents used were reagent grade, purchased from Sigma Aldrich and dried under nitrogen before use. 4-(Chloromethyl)benzoic acid, thionyl chloride, 1,3-diphenyl-1,3-propanedione, hydrazine monohydrate, sodium hydride, 2,2,6,6-tetramethyl-3,5-heptanedione, potassium hydroxide, benzyl bromide, benzyl chloride, sodium hydroxide, sodium bicarbonate, potassium tert-butoxide, ethyl acetate and glacial acetic acid were purchased from Sigma Aldrich and used without further purification. PdCl2 was purchased from Heraeus South Africa and used as received. [PdCl2(MeCN)2],22 3,5-di-tert-butylpyrazole,23 3,5-diphenylpyrazole24 and methyl-4-(chloromethyl)benzoate25 were synthesized according to literature procedures.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 diethylether:hexane mixture. The solvent was then removed by rotary evaporation followed by drying in vacuo for 6 hours to obtain the product. Appearance white solid (yield = 603.00 mg, 43%). Solubility: soluble in chloroform, methanol, DCM, hexane and diethylether, insoluble in water. Melting point range 122–123 °C. FT-IR (νmax/cm−1): 1712 (C
:3 diethylether:hexane mixture. The solvent was then removed by rotary evaporation followed by drying in vacuo for 6 hours to obtain the product. Appearance white solid (yield = 603.00 mg, 43%). Solubility: soluble in chloroform, methanol, DCM, hexane and diethylether, insoluble in water. Melting point range 122–123 °C. FT-IR (νmax/cm−1): 1712 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 1H NMR (100 MHz, CDCl3, 30 °C) (ppm) = 7.95 (d, 2H, JHH = 8 Hz, Harom), 7.86 (d, JHH = 8 Hz, 2H, Harom), 7.42–7.31 (m, 8H, Harom), 7.15 (d, 2H, JHH = 8 Hz, Harom), 6.67 (s, 1H, Hpz), 5.42 (s, 2H, HCH2), 3.87 (s, 3H, HCH3). 13C{1H} NMR (400 MHz, CDCl3, 30 °C) (ppm) = 166.8; 151.3; 145.6; 142.8; 133.3; 130.5; 130.4; 130.2; 129.9; 129.3; 128.8; 128.7; 127.8; 126.7; 125.7; 103.9; 53.0; 52.1; ESI-MS [M + H]+ = 369.16. Elemental analysis; anal. calcd for C24H20N2O2: C, 78.24; 5.47; N, 7.60%, found C, 78.42; H, 5.94; N, 7.31%.:3 diethylether:hexane mixture. The first band was collected and identified as of methyl-4-(chloromethyl)benzoate after TLC comparison. The desired product eluted as the second band. The solvent was then removed by rotary evaporation followed by drying in vacuo for 6 h to obtain the product. (Yield = 501.22 mg, 40%). Solubility: soluble in chloroform, methanol, DCM, hexane and diethyl ether, insoluble in water. Melting point range 138–139 °C. FT-IR (νmax/cm−1): 1713.12 (CO). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.90 (d, 2H, JHH = 8 Hz, Harom), 6.90 (d, JHH = 8 Hz, 2H, Harom), 5.92 (s, 1H, Hpz), 5.49 (s, 1H, HCH2), 3.86 (s, 3H, HCH3), 1.29 (s, 9H,
O). 1H NMR (100 MHz, CDCl3, 30 °C) (ppm) = 7.95 (d, 2H, JHH = 8 Hz, Harom), 7.86 (d, JHH = 8 Hz, 2H, Harom), 7.42–7.31 (m, 8H, Harom), 7.15 (d, 2H, JHH = 8 Hz, Harom), 6.67 (s, 1H, Hpz), 5.42 (s, 2H, HCH2), 3.87 (s, 3H, HCH3). 13C{1H} NMR (400 MHz, CDCl3, 30 °C) (ppm) = 166.8; 151.3; 145.6; 142.8; 133.3; 130.5; 130.4; 130.2; 129.9; 129.3; 128.8; 128.7; 127.8; 126.7; 125.7; 103.9; 53.0; 52.1; ESI-MS [M + H]+ = 369.16. Elemental analysis; anal. calcd for C24H20N2O2: C, 78.24; 5.47; N, 7.60%, found C, 78.42; H, 5.94; N, 7.31%.:3 diethylether:hexane mixture. The first band was collected and identified as of methyl-4-(chloromethyl)benzoate after TLC comparison. The desired product eluted as the second band. The solvent was then removed by rotary evaporation followed by drying in vacuo for 6 h to obtain the product. (Yield = 501.22 mg, 40%). Solubility: soluble in chloroform, methanol, DCM, hexane and diethyl ether, insoluble in water. Melting point range 138–139 °C. FT-IR (νmax/cm−1): 1713.12 (CO). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.90 (d, 2H, JHH = 8 Hz, Harom), 6.90 (d, JHH = 8 Hz, 2H, Harom), 5.92 (s, 1H, Hpz), 5.49 (s, 1H, HCH2), 3.86 (s, 3H, HCH3), 1.29 (s, 9H, 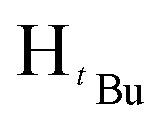 ), 1.22 (s, 9H,
), 1.22 (s, 9H, 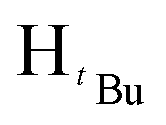 ). 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 166.8; 160.6; 151.9; 144.6; 129.7; 129.6; 128.8; 125.8; 125.7; 100.3; 54.0; 51.9; 31.2; 30.1: ESI-MS [M + H]+ = 329.22; elemental analysis; anal. calcd for C20H28N2O2: C, 73.14; H, 8.59; N, 8.53%, found C, 73.22; H, 9.16; N, 7.85%.:7 hexane:diethyl ether mixture. The second band was collected and identified as the product after TLC comparison with the starting materials. The solvent was then removed by rotary evaporation to obtain the product. Appearance; white crystals (yield 96.000 mg, 68%). Solubility: soluble in chloroform, DCM, and diethylether, insoluble in hexane, methanol and water. Melting point range 94–96 °C. FT-IR (νmax/cm−1): 1460 (CN). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.71 (d, 3JHH = 7.2 Hz, 2H, Harom), 7.35–7.23 (m, 11H, Harom), 7.03 (d, 3JHH = 6.8 Hz, 2H, Harom), 6.58 (s, 1H, Hpz), 5.31 (s, 2H, HCH2). 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 150.9; 145.4; 137.7; 133.4; 130.6; 128.8; 128.7; 128.4; 128.3; 127.5; 127.4; 126.9; 126.8; 126.7; 103.7; 53.2: ESI-MS [M + H]+ = 311.15 (100%). Elemental analysis; anal. calcd for C22H18N2: C, 85.13; H, 5.85; N, 9.03%, found C, 85.40; H, 5.65; N, 9.41%.:7 hexane:diethylether mixture. The second band was collected and identified as the product after TLC comparison with the starting materials. The solvent was then removed by rotary evaporation to obtain the product. Appearance; white crystals (yield = 110.10 mg, 73%). Solubility: soluble in chloroform, insoluble in water at room temperature. Solubility: soluble in chloroform, methanol, DCM, hexane and diethyl ether, insoluble in water. Melting point range 60–62 °C. FT-IR (νmax/cm−1): 1456 (CN). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.25 (t, 3JHH = 5.6 Hz, 3JHH = 9.2 Hz, 3H, Harom), 6.85 (d, 3JHH = 7.2 Hz 2H, Harom), 5.89 (s, 1H, Hpz), 5.45 (s, 2H, HCH2), 1.29 (s, 9H,
). 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 166.8; 160.6; 151.9; 144.6; 129.7; 129.6; 128.8; 125.8; 125.7; 100.3; 54.0; 51.9; 31.2; 30.1: ESI-MS [M + H]+ = 329.22; elemental analysis; anal. calcd for C20H28N2O2: C, 73.14; H, 8.59; N, 8.53%, found C, 73.22; H, 9.16; N, 7.85%.:7 hexane:diethyl ether mixture. The second band was collected and identified as the product after TLC comparison with the starting materials. The solvent was then removed by rotary evaporation to obtain the product. Appearance; white crystals (yield 96.000 mg, 68%). Solubility: soluble in chloroform, DCM, and diethylether, insoluble in hexane, methanol and water. Melting point range 94–96 °C. FT-IR (νmax/cm−1): 1460 (CN). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.71 (d, 3JHH = 7.2 Hz, 2H, Harom), 7.35–7.23 (m, 11H, Harom), 7.03 (d, 3JHH = 6.8 Hz, 2H, Harom), 6.58 (s, 1H, Hpz), 5.31 (s, 2H, HCH2). 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 150.9; 145.4; 137.7; 133.4; 130.6; 128.8; 128.7; 128.4; 128.3; 127.5; 127.4; 126.9; 126.8; 126.7; 103.7; 53.2: ESI-MS [M + H]+ = 311.15 (100%). Elemental analysis; anal. calcd for C22H18N2: C, 85.13; H, 5.85; N, 9.03%, found C, 85.40; H, 5.65; N, 9.41%.:7 hexane:diethylether mixture. The second band was collected and identified as the product after TLC comparison with the starting materials. The solvent was then removed by rotary evaporation to obtain the product. Appearance; white crystals (yield = 110.10 mg, 73%). Solubility: soluble in chloroform, insoluble in water at room temperature. Solubility: soluble in chloroform, methanol, DCM, hexane and diethyl ether, insoluble in water. Melting point range 60–62 °C. FT-IR (νmax/cm−1): 1456 (CN). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.25 (t, 3JHH = 5.6 Hz, 3JHH = 9.2 Hz, 3H, Harom), 6.85 (d, 3JHH = 7.2 Hz 2H, Harom), 5.89 (s, 1H, Hpz), 5.45 (s, 2H, HCH2), 1.29 (s, 9H, 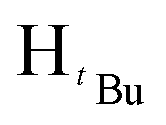 ), 1.23 (s, 9H,
), 1.23 (s, 9H, 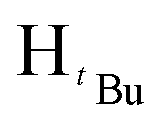 ). 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 160.3; 151.2; 139.2; 128.3; 128.1; 126.7; 126.5; 125.8; 100.1; 54.1; 31.9; 30.3; ESI-MS [M + H]+ = 271.21; elemental analysis; anal. calcd for C18H26N2: C, 79.95; H, 9.69; N, 10.36%, found C, 80.08; H, 9.34; N 10.14%.O). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 8.15 (d, JHH = 6.8 Hz, 2H, Harom), 7.92(d, JHH = 8 Hz, 2H, Harom), 7.33–7.28 (m, 6H, Harom), 6.99 (d, JHH = 5.6 Hz, 4H, Harom), 6.37 (s, Hpz, 1H), 6.16 (s, HCH2, 2H), 3.90 (s, HCH3, 3H); 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 167.4, 156.2; 149.9; 138.3; 131.8; 129.4; 129.2; 128.9; 128.8; 128.7; 128.5; 128.4; 128.3; 128.0; 127.9; 105.1; 57.1; 52.1; ESI-MS [M]+ = 913.15 (10%); [M − Cl]+ = 878.18 (5%); [M − 2Cl]+ = 840.21 (5%); elemental analysis; anal. calcd for C48H40Cl2N4O4Pd: C, 63.06; H, 4.41; N, 6.13% found C, 63.18; H 4.71 N, 5.83%.O). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 8.10 (d, JHH = 8 Hz, 2H, Harom), 8.05 (d, JHH = 7.6 Hz, 2H, Harom), 7.21 (d, JHH = 8 Hz, 2H, Harom), 7.06 (d, JHH = 8 Hz, 2H, Harom), 6.02 (m, 6H, HCH2, Hpz), 3.93 (s, 3H, HCH3), 3.88 (s, 3H, HCH3), 1.93 (s,
). 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 160.3; 151.2; 139.2; 128.3; 128.1; 126.7; 126.5; 125.8; 100.1; 54.1; 31.9; 30.3; ESI-MS [M + H]+ = 271.21; elemental analysis; anal. calcd for C18H26N2: C, 79.95; H, 9.69; N, 10.36%, found C, 80.08; H, 9.34; N 10.14%.O). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 8.15 (d, JHH = 6.8 Hz, 2H, Harom), 7.92(d, JHH = 8 Hz, 2H, Harom), 7.33–7.28 (m, 6H, Harom), 6.99 (d, JHH = 5.6 Hz, 4H, Harom), 6.37 (s, Hpz, 1H), 6.16 (s, HCH2, 2H), 3.90 (s, HCH3, 3H); 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 167.4, 156.2; 149.9; 138.3; 131.8; 129.4; 129.2; 128.9; 128.8; 128.7; 128.5; 128.4; 128.3; 128.0; 127.9; 105.1; 57.1; 52.1; ESI-MS [M]+ = 913.15 (10%); [M − Cl]+ = 878.18 (5%); [M − 2Cl]+ = 840.21 (5%); elemental analysis; anal. calcd for C48H40Cl2N4O4Pd: C, 63.06; H, 4.41; N, 6.13% found C, 63.18; H 4.71 N, 5.83%.O). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 8.10 (d, JHH = 8 Hz, 2H, Harom), 8.05 (d, JHH = 7.6 Hz, 2H, Harom), 7.21 (d, JHH = 8 Hz, 2H, Harom), 7.06 (d, JHH = 8 Hz, 2H, Harom), 6.02 (m, 6H, HCH2, Hpz), 3.93 (s, 3H, HCH3), 3.88 (s, 3H, HCH3), 1.93 (s, 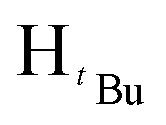 , 6H), 1.89 (s,
, 6H), 1.89 (s, 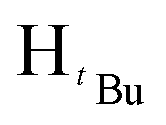 , 1H), 1.81 (s,
, 1H), 1.81 (s, 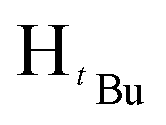 , 6H), 1.61 (s,
, 6H), 1.61 (s, 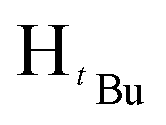 , 2H), 1.54 (s,
, 2H), 1.54 (s, 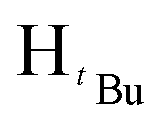 , 3H), 1.29 (s,
, 3H), 1.29 (s, 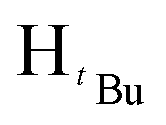 , 1H), 1.24 (s,
, 1H), 1.24 (s, 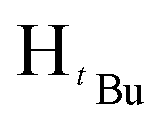 , 7H), 1.22 (s,
, 7H), 1.22 (s, 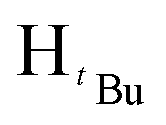 , 7H), 1.17 (s,
, 7H), 1.17 (s,  , 3H), 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 167.3; 167.2; 167.1; 167.0; 165.5; 160.9; 157.2; 153.4; 144.2; 141.9; 141.7; 130.7; 130.6; 130.5; 130.3; 130.2; 130.1; 130.0; 129.9; 129.7; 127.2; 126.9; 125.8; 105.4; 105.1; 56.6; 51.9; 33.0; 32.8; 32.3; 31.8; 31.7; 31.6; 31.5; 31.4; 31.3; 31.2; 30.5; 30.3; 29.9; ESI-MS [M]+ = 835.28 (40%). Elemental analysis; anal. calcd for C40H56Cl2N4O4Pd: C, 57.59; H, 6.77, N, 6.72% found C, 57.06; H, 6.77; N, 6.69%.N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 8.20 (d, JHH = 7.2 Hz, 2H, Harom), 7.29–7.13 (m, 9H, Harom), 6.99 (d, JHH = 7.2 Hz, 2H, Harom), 6.94 (t, JHH = 8 Hz, J1HH = 7.6 Hz, 2H, Harom), 6.30 (s, Hpz, 1H), 6.10 (s, HCH2, 2H); 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 155.3; 149.1; 137.3; 131.0; 129.1; 129.0; 128.9; 128.8; 128.7; 128.6; 128.5; 128.4; 128.3; 128.2; 110.1; 56.4; ESI-MS [M + Na]+ = 823.12 (50%); [M − Cl]+ = 765.17 (50%); [M − 2Cl]+ = 725.18 (5%) elemental analysis; anal. calcd for C44H36Cl2N4Pd: C, 66.22; H, 4.55, N, 7.02% found C, 66.46; H, 4.51; N, 6.78%.N). 1H NMR (100 MHz, CDCl3, 30 °C) (ppm) = 7.4–7.2 (m, 6H, Harom), 7.04 (d, JHH = 4.8 Hz, 4H, Harom), 6.00 (m, 6H, Hpz, HCH2), 1.94 (s,
, 3H), 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 167.3; 167.2; 167.1; 167.0; 165.5; 160.9; 157.2; 153.4; 144.2; 141.9; 141.7; 130.7; 130.6; 130.5; 130.3; 130.2; 130.1; 130.0; 129.9; 129.7; 127.2; 126.9; 125.8; 105.4; 105.1; 56.6; 51.9; 33.0; 32.8; 32.3; 31.8; 31.7; 31.6; 31.5; 31.4; 31.3; 31.2; 30.5; 30.3; 29.9; ESI-MS [M]+ = 835.28 (40%). Elemental analysis; anal. calcd for C40H56Cl2N4O4Pd: C, 57.59; H, 6.77, N, 6.72% found C, 57.06; H, 6.77; N, 6.69%.N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 8.20 (d, JHH = 7.2 Hz, 2H, Harom), 7.29–7.13 (m, 9H, Harom), 6.99 (d, JHH = 7.2 Hz, 2H, Harom), 6.94 (t, JHH = 8 Hz, J1HH = 7.6 Hz, 2H, Harom), 6.30 (s, Hpz, 1H), 6.10 (s, HCH2, 2H); 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 155.3; 149.1; 137.3; 131.0; 129.1; 129.0; 128.9; 128.8; 128.7; 128.6; 128.5; 128.4; 128.3; 128.2; 110.1; 56.4; ESI-MS [M + Na]+ = 823.12 (50%); [M − Cl]+ = 765.17 (50%); [M − 2Cl]+ = 725.18 (5%) elemental analysis; anal. calcd for C44H36Cl2N4Pd: C, 66.22; H, 4.55, N, 7.02% found C, 66.46; H, 4.51; N, 6.78%.N). 1H NMR (100 MHz, CDCl3, 30 °C) (ppm) = 7.4–7.2 (m, 6H, Harom), 7.04 (d, JHH = 4.8 Hz, 4H, Harom), 6.00 (m, 6H, Hpz, HCH2), 1.94 (s,  , 7H), 1.91 (s,
, 7H), 1.91 (s,  , 2H), 1.83 (s,
, 2H), 1.83 (s,  , 9H), 1.69 (s,
, 9H), 1.69 (s, 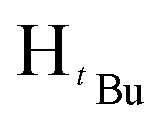 , 2H), 1.26 (d, JHH = 14.4 Hz,
, 2H), 1.26 (d, JHH = 14.4 Hz, 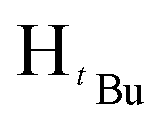 , 7H), 1.19 (d, JHH = 6 Hz,
, 7H), 1.19 (d, JHH = 6 Hz, 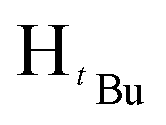 , 7H), 1.18 (s,
, 7H), 1.18 (s, 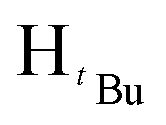 , 2H), 0.86 (s,
, 2H), 0.86 (s,  , 2H). 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 164.1; 164.0; 157.0; 156.9; 135.8; 135.6; 129.0; 128.8; 128.6; 128.2; 128.1; 127.9; 127.0; 126.8; 125.8; 105.2; 105.1; 105.0; 55.9; 55.7; 33.0; 32.9; 30.5; 30.3; ESI-MS [M]+ = 719.27 (100%); elemental analysis; anal. calcd for C36H52Cl2N4Pd: C, 60.21; H, 7.30, N, 7.80% found C, 60.35; H, 7.60; N, 7.70%.
, 2H). 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 164.1; 164.0; 157.0; 156.9; 135.8; 135.6; 129.0; 128.8; 128.6; 128.2; 128.1; 127.9; 127.0; 126.8; 125.8; 105.2; 105.1; 105.0; 55.9; 55.7; 33.0; 32.9; 30.5; 30.3; ESI-MS [M]+ = 719.27 (100%); elemental analysis; anal. calcd for C36H52Cl2N4Pd: C, 60.21; H, 7.30, N, 7.80% found C, 60.35; H, 7.60; N, 7.70%.Characterization of compounds
NMR spectra was obtained using Bruker-400 MHz NMR spectrometer (1H at 400 MHz and 13C{1H} at 100 MHz). Spectrometer chemical shifts were reported relative to the internal standard tetramethylsilane (δ 0.00 ppm) and referenced to the residual proton and carbon signals at 7.24 and 77.0 ppm respectively of CDCl3. Infrared spectra were obtained neat using a Perkin Elmer Spectrum BX II fitted with an ATR probe. Melting points were obtained using a Gallenkamp Digital Melting-point Apparatus 5A 6797. Elemental analysis was performed on a Thermos Scientific FLASH 2000 CHNS-O Analyzer. Mass spectrometry was performed using Waters Synapt G2 mass spectrometer with both ESI positive and cone voltage 15 V. XRD spectra were obtained from a Bruker APEX-II CCD Diffractometer. Analytical thin layer chromatography (TLC) was performed on silica gel coated aluminium plates (0.2 mm). Developed plates were visualized with UV light or under iodine staining. Silica gel column chromatography was performed using silica gel 60 (70–230 mesh).General experimental description of Suzuki cross-coupling reaction
The Suzuki–Miyaura cross-coupling reaction was performed using a Radleys Carousel 12 Plus Reaction Station™ using 24 × 150 mm quick-thread glass reaction tubes. The general procedure involved charging the reaction vessel with required amounts of aryl halide, boronic acid, base, internal standard (n-decane) and solvent. A sample of the reaction mixture in the reaction vessel was taken at time zero and analyzed by GC to determine the ratios of various components. The required catalyst amount (catalyst loading in mol%) was then added and the reaction left to proceed with a stir rate of 600 rpm. After reaction time had elapsed, aryl halide conversion to biphenyl was calculated by analyzing retention peak areas of aryl halide with reference to internal standard n-decane. GC analyses were performed using a Scion 456 GC with 30 m × 0.25 mm cyanopropylphenylmethylpolysiloxane phase column set to an initial temperature of 80 °C and then increased to 300 °C at 10 °C min−1.Results and discussion
Synthesis and characterization of pyrazolyl ligands (1–4)
Compounds 1–4 were synthesized in a substitution reaction involving substituted pyrazoles with the respective benzylic halides in the presence of excess base (Scheme 1). These compounds were obtained in moderate to good yields. The substitution reactions could readily occur at room temperature using DMSO, DMF or acetonitrile as solvents. The compounds display excellent solubility in organic solvents such as chloroform and DCM. They were extracted from either DMSO or DMF aqueous mixtures using diethyl ether. The substituted pyrazolyl ligands were characterized by 1H NMR spectroscopy, mass spectrometry, elemental analysis and infrared spectroscopy. Downfield shifts in the 1H NMR signals of the benzylic CH2 linker protons from around 4.50 ppm to around 5.50 ppm was observed indicating successful formation of the ligands. Further evidence for the synthesis of these ligands was confirmed via mass spectrometry and elemental analysis.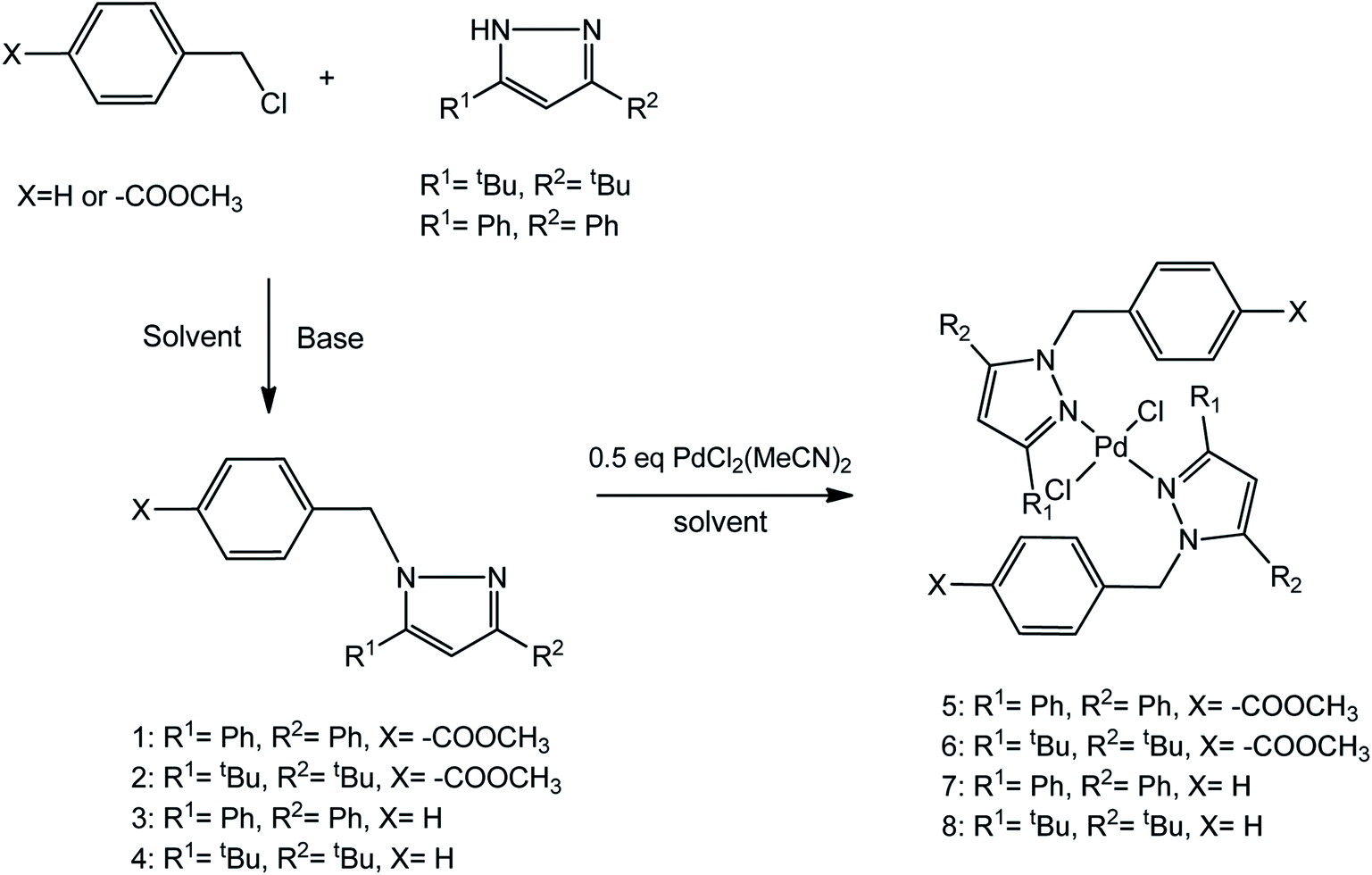 | ||
| Scheme 1 Synthesis of pyrazolyl ligands and complexes. | ||
Synthesis and characterization of bis(pyrazolyl)palladium(II) complexes (5–8)
Complexes 5–8 were obtained by reacting the appropriate ligands in a 2:1 ratio with [PdCl2(MeCN)2] in DCM at room temperature for 24 h. Over the course of the reaction, the resulting complexes 6, 7 and 8 remained soluble in DCM while 5 formed a yellow colloidal suspension. For all the complexes, the reaction mixtures were concentrated in vacuo and n-hexane or diethyl ether was added to the concentrated solution to encourage precipitation. The precipitates were obtained as varying shades of yellow solids in moderate to good yields. The complexes were also soluble in organic solvents such as DCM and chloroform. Characterization of these complexes by 1H NMR spectroscopy showed a shift in the benzylic (C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2) protons from around 5.40–5.50 ppm in the ligands to around 6.00–6.10 ppm in the complexes. This was as a result of coordination of the pyrazolyl nitrogen atoms to the palladium, resulting in a reduced electron density around the C2 protons. Mass spectra for all the complexes also confirmed their synthesis by showing the m/z of the molecular ion peak or fragmentation patterns corresponding to the loss of chloride ions or gain of sodium ions.
2) protons from around 5.40–5.50 ppm in the ligands to around 6.00–6.10 ppm in the complexes. This was as a result of coordination of the pyrazolyl nitrogen atoms to the palladium, resulting in a reduced electron density around the C2 protons. Mass spectra for all the complexes also confirmed their synthesis by showing the m/z of the molecular ion peak or fragmentation patterns corresponding to the loss of chloride ions or gain of sodium ions.
Molecular structure of compound 7
The solid state structure of complex 7 was obtained using single-crystal X-ray diffraction. Crystals for complex 7 were grown by slow evaporation of a DCM solution of 7 at room temperature and were obtained as yellow crystals. Complex 7 crystallized in a P1 space group with a distorted square planar geometry at the palladium centre. The pyrazolyl ligands coordinated to the palladium in a trans fashion (Fig. 1). The bond angles around the Pd are between 89.61(4)° and 90.39(4)° which is in agreement of a square planar geometry. The crystal data and structure refinement parameters for 7 are presented in Table 1. With only a slight distortion at the palladium centre, 1H NMR spectrum for 7 showed that all phenyl protons on the pyrazole ring resided in the same chemical environments. Pd–Npz and Pd–Cl bond lengths as well as bond angles around the palladium centre for 7 were within reported ranges for similar bis(pyrazolyl)palladium complexes in literature.10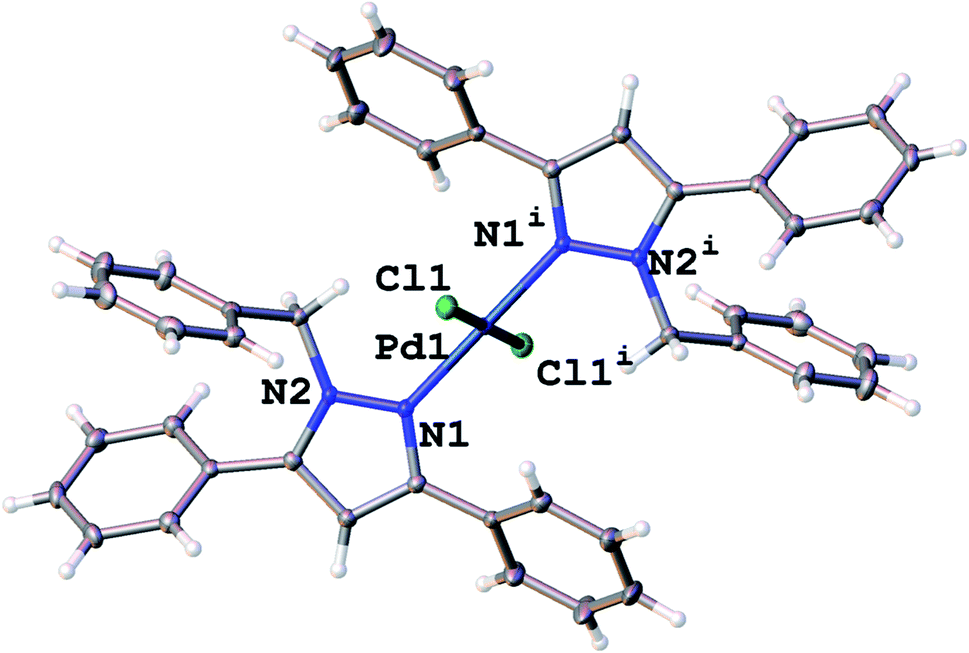 | ||
| Fig. 1 Molecular structure of 7. Selected bond distance (Å) and bond angles (°); Pd(1)–Cl(1) 2.2924(4), Pd(1)–N(1) 2.0135(15), N(2)–N(1) 1.363(2); N(1)–Pd(1)–Cl(1) 90.39(4), N(1)–Pd(1)–Cl(1i) 89.61(4). | ||
| Identification code | Complex 7 |
| Empirical formula | C44H36Cl2N4Pd |
| Formula weight | 798.06 |
| Temperature/K | 100(2) |
| Crystal system | Triclinic |
| Space group | P-1 |
| a/Å | 6.7767(6) |
| b/Å | 10.2586(9) |
| c/Å | 13.5319(13) |
| α/° | 102.123(2) |
| β/° | 98.442(2) |
| γ/° | 94.970(2) |
| Volume/Å3 | 903.05(14) |
| Z | 2 |
| ρcalc. g cm−3 | 1.467 |
| μ/mm−1 | 0.700 |
| F(000) | 408.0 |
| Crystal size/mm3 | 0.364 × 0.125 × 0.12 |
| Radiation | Mo Kα (λ = 0.71073) |
| 2θ range for data collection/° | 4.092 to 56.714 |
| Index ranges | −8 ≤ h ≤ 9, −13 ≤ k ≤ 13, −18 ≤ l ≤ 18 |
| Reflections collected | 12765 |
| Independent reflections | 4497 [Rint = 0.0346, Rsigma = 0.0391] |
| Data/restraints/parameters | 4497/0/304 |
| Goodness-of-fit on F2 | 1.079 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0287, wR2 = 0.0675 |
| Final R indexes [all data] | R1 = 0.0316, wR2 = 0.0688 |
| Largest diff. peak/hole/e Å−3 | 0.46/−0.31 |
Catalytic studies
Carrying out the Suzuki–Miyaura cross–coupling reaction at 140 °C resulted in the formation of Pd-black when using 5, 6, 7 and 8 irrespective of the solvent used. Similar observations have been reported in literature for pyrazolyl palladium complexes,21,27 which were used in cross-coupling reactions carried out at temperatures higher than 65 °C. However, carrying out the Suzuki–Miyaura cross-coupling reaction using 7, at the established optimum reaction conditions, in toluene (using bromobenzene and phenylboronic acid as cross-coupling partners) with a large excess of mercury (300:1, Hg:Pd) resulted in a drop in conversion from 98% to 84%. This appreciably high conversion obtained using 7, in the presence of mercury indicates in situ generated multi-species catalysts responsible for the catalysis.28–30
Similar observations were made when 8 was used as a pre-catalysts in the presence of metallic mercury, where conversion dropped from 81% to 77%.
This is in contrast to other palladium catalyzed Suzuki–Miyaura cross-coupling for which there was no aryl halide conversion after introducing mercury to the reaction mixture.
Generally phenyl bearing pre-catalysts 5 and 7 outperformed their tertiary butyl counterparts 6 and 8 over the course of these reactions. The presence of ester groups in 5 and 6 did not contribute any steric effect but lowered the electron-donation towards the palladium centre. This resulted in slightly lowered aryl halide conversions.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | X | Conv. (%) | Selectivity (%) | TON | TOF (h−1) |
| a Reactions carried out in toluene (4.27 mL) with 5 mmol of aryl halide, 8 mmol of phenyl boronic acid, 10 mmol of KOH and 0.3 mol% Pd catalyst loading using n-decane as internal standard at 140 °C for 4 h. Cat. = catalyst, Conv. = conversion. Average error estimates; 5 (±0.6210), 6 (±0.5919), 7 (±0.4987), 8 (±0.4331). | ||||||
| 1 | 5 | H | 98 | 100 | 298 | 74 |
| 2 | 6 | H | 87 | 100 | 263 | 66 |
| 3 | 7 | H | 98 | 100 | 298 | 74 |
| 4 | 8 | H | 81 | 100 | 245 | 61 |
| 5 | 5 | 4-CH3 | 87 | 100 | 262 | 66 |
| 6 | 6 | 4-CH3 | 68 | 99 | 206 | 51 |
| 7 | 7 | 4-CH3 | 100 | 99 | 303 | 76 |
| 8 | 8 | 4-CH3 | 47 | 97 | 141 | 35 |
| 9 | 5 | 2-CH3 | 64 | 100 | 195 | 49 |
| 10 | 6 | 2-CH3 | 39 | 99 | 117 | 29 |
| 11 | 7 | 2-CH3 | 98 | 99 | 298 | 75 |
| 12 | 8 | 2-CH3 | 70 | 97 | 212 | 53 |
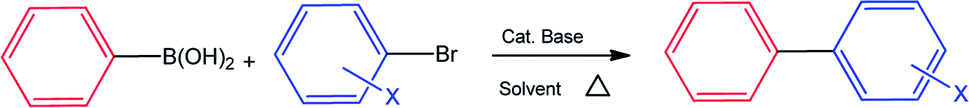
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | X | Y | Conv. (%) | Selectivity (%) | TON | TOF (h−1) |
| a Reactions carried out in DMF (1.5 mL) with 1 mmol of aryl halide, 1.6 mmol of phenyl boronic acid, 2 mmol of KOH and varying 0.3 mol% Pd using n-decane as internal standard at 140 °C for 4 h. Reactions carried out in toluene (1.5 mL). Cat. = catalyst, Conv. = conversion. Average error estimates; 5 (±0.5661), 6 (±0.4941), 7 (±0.5374), 8 (±0.4821). | |||||||
| 1 | 5 | 4-CHO | 4-CH3 | 53 | 97 | 162 | 40 |
| 2 | 6 | 4-CHO | 4-CH3 | 22 | 77 | 66 | 17 |
| 3 | 7 | 4-CHO | 4-CH3 | 63 | 97 | 190 | 47 |
| 4 | 8 | 4-CHO | 4-CH3 | 21 | 93 | 63 | 16 |
| 5 | 5 | 4-Cl | 4-CH3 | 73 | 89 | 222 | 55 |
| 6 | 6 | 4-Cl | 4-CH3 | 73 | 97 | 222 | 55 |
| 7 | 7 | 4-Cl | 4-CH3 | 76 | 75 | 231 | 58 |
| 8 | 8 | 4-Cl | 4-CH3 | 78 | 94 | 237 | 59 |
| 9 | 5 | 4-Cl | H | 63a | 99 | 190 | 48 |
| 10 | 6 | 4-Cl | H | 79a | 99 | 238 | 60 |
| 11 | 7 | 4-Cl | H | 75a | 99 | 227 | 57 |
| 12 | 8 | 4-Cl | H | 62a | 99 | 189 | 47 |
| 13 | 5 | 4-Cl | 2-CN | 100a | 100 | 303 | 76 |
| 14 | 6 | 4-Cl | 2-CN | 91a | 79 | 276 | 69 |
| 15 | 7 | 4-Cl | 2-CN | 100a | 100 | 303 | 76 |
| 16 | 8 | 4-Cl | 2-CN | 100a | 72 | 303 | 76 |
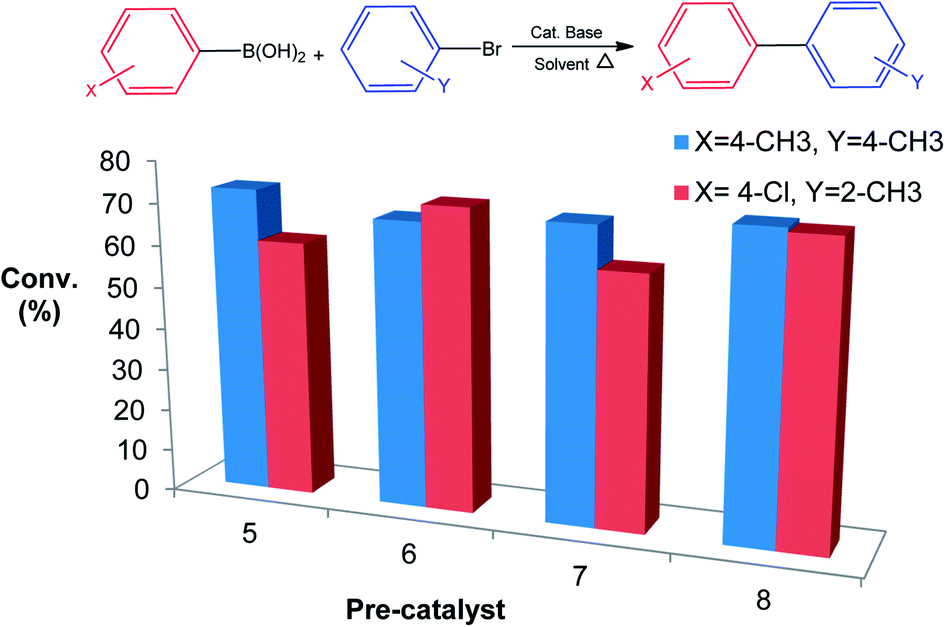 | ||
| Fig. 2 Effect of steric bulk on phenyl boronic acids and aryl halides. Reactions carried out in DMF (1.5 mL) with 1 mmol of aryl halide, 1.6 mmol of phenyl boronic acid, 2 mmol of KOH and varying 0.3 mol% Pd using n-decane as internal standard at 140 °C for 4 h. | ||
The influence of electron-withdrawing groups on the aryl halides was also investigated. When chlorobenzene was used as a coupling partner with phenylboronic acid, there were no conversions with any of the pre-catalysts. However, in the presence of an electron-withdrawing cyano group in 2-chlorobenzonitrile, we observed moderate conversions (46% for 5 and 6, 44% for 7 and 30% for 8) with all the pre-catalysts despite the steric limitations that might have been imposed by the nitrile group. This was probably as a result of the decrease in the bond dissociation energy of the C–Cl bond caused by the presence of the cyano group. Due to the electron-withdrawing nature of the cyano group from the C–Cl bond, the 2-chlorobenzonitrile is activated, thus the energy required in the formation of the oxidative addition intermediate is lowered.
A series of phenyl boronic acids with electron-withdrawing substituents were investigated in order to evaluate their effect on the Suzuki–Miyaura cross-coupling reactions. A typical example of such phenyl boronic acid that was investigated was 4-chlorophenyl boronic acid. With the presence of an electron-withdrawing group on the phenyl boronic acid, the results obtained were contrary to having the electron-withdrawing group on the aryl halide. For example, when phenyl boronic acid and bromobenzene were used as substrates (entry 3, Table 2) 98% conversion was obtained for 7. When 4-chlorophenyl boronic acid and bromobenzene were used as substrates using 7, we observed 75% conversion (entry 11, Table 3). Similar observations were also made for the other pre-catalysts. When 4-chlorophenyl boronic acid and 4-bromo toluene were used as substrates, conversion obtained for 7 was 76% whereas 63% was obtained when 4-formylphenyl boronic acid and 4-bromo toluene were used. The presence of the lesser electron-withdrawing chloride resulted in increased aryl halide conversions as compared to the more electron-withdrawing formyl substituent (entries 1–8, Table 3). The low conversions with the phenyl boronic acid bearing electron-withdrawing group could be due to the decrease in the rate of the transmetallation step of the catalytic cycle as has been reported in literature.31
High conversions were observed for all four pre-catalysts when 2-bromobenzonitrile was coupled with 4-chlorophenylboronic acid. These high conversions were observed despite the presence of an electron-withdrawing chloro group on the phenyl boronic acid which would have been expected to reduce conversions (Table 3, entries 13–16). This was probably because the oxidative addition intermediate is expected to be formed relatively easily. Therefore, even if there is a decrease in the rate of transmetallation (with the presence of an electron-withdrawing group on the phenyl boronic acid), the reaction still gives excellent conversions.
We have observed that steric bulk around the carbon–boron bond or carbon–halide bond resulted in poor conversions. An example is the reaction between 2-nitroboronic acid or 3-nitroboronic and bromobenzene. This reaction resulted in the loss of the boronic acid during coupling to form nitrobenzene which was confirmed by mass spectrometry. Using 4-formylboronic acid and 2-bromobenzene as substrates resulted in no conversion for all four pre-catalysts. These poor conversions could be as a result of steric hindrance or due to the electron-withdrawing effect of the formyl group on the phenyl boronic acids. When 4-bromotoluene was used instead of 2-bromobenzene as coupling partner with 4-formylboronic acid, there was an increase in conversion possibly due to reduced steric hindrance (Table 3, entries 1–4). Similar observations were made when 4-chlorophenyl boronic acid was coupled with either 4-bromotoluene or 2-bromotoluene. Surprisingly, conversions for 6 and 8 were not significantly affected when an electron-withdrawing halide was present on the boronic acid as well as bulky group around the carbon bromide bonds (Fig. 2). This might indicate that carrying out the cross-coupling reaction in coordinating solvent (DMF) may result in a different catalytic pathway as compared to using a non-coordinating solvent such as toluene. This observation is also made where phenyl bearing pre-catalysts 5 and 7 were clearly outperformed by tertiary butyl counterparts 6 and 8 (Fig. 2) This suggests that catalytic activity is not solely dependent on the nature of the supporting ligands or resulting active species, but sometimes on the nature of the coupling reagents and/or solvent. In addition, with 8 as a pre-catalyst using p-tolylboronic acid and 4-bromotoluene as cross-coupling partners (in DMF and metallic mercury), there was a decrease in conversion from 73% to 52%. This slight drop in conversion suggests a cocktail of catalysts promoting the reaction.28,29
Conclusions
Four pyrazole containing ligands with tBu and phenyl substituents on the pyrazole ring were prepared and characterized using NMR, mass spectrometry, infrared and single crystal X-ray diffraction (for 7). Four new bis(palladium) complexes were synthesized and characterized from these ligands. The complexes were evaluated for their catalytic activity in Suzuki–Miyaura cross-coupling reactions. The pre-catalysts proved to be effective and the presence of steric bulk around the palladium centre did not inhibit conversions. In general, complexes bearing phenyl substituents performed better than those with tBu substituents. This was likely as a result of the conjugated system the phenyl substituents formed with the pyrazolyl ring, resulting in increased electron-donation to the palladium. This could also be as a result of steric bulk by the tBu substituents that inhibits interaction between the palladium centre and the substrates. The presence of ester groups on two of the pre-catalysts (5 and 6) resulted in slightly lowered conversions. Generally, all the pre-catalysts were selective towards the formation of the biaryl and no homo coupling products were observed. The presence of electron-withdrawing groups on the aryl halide served to increase conversions while an electron-withdrawing group on the phenyl boronic acid reduced conversion. Steric bulk around either the carbon–halide bond or the carbon–boron bond also lead to reduced conversions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Research Foundation of South Africa (NRF) (Grant Numbers: 95517 and 99269), Sasol and the University of Johannesburg Centre for Synthesis and Catalysis for financial support. We would also like to acknowledge Mr Gershon Amenuvor for his help in the solving of the crystal structure.References
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- M. E. Hanhan, Appl. Organomet. Chem., 2008, 22, 270–275 CrossRef CAS.
- Y. Lai, Z. Zong, Y. Tang, W. Mo, N. Sun, B. Hu, Z. Shen, L. Jin, W. Sun and X. Hu, Beilstein J. Org. Chem., 2017, 13, 213–221 CrossRef CAS PubMed.
- S. Ghosh, A. S. Kumar and G. N. Mehta, Beilstein J. Org. Chem., 2010, 4, 4–7 Search PubMed.
- L. J. Goossen and B. Melzer, J. Org. Chem., 2007, 7473–7476 CrossRef CAS PubMed.
- Y. Yang, G. Li, Z. Song, X. Yang and P. Liu, Lett. Org. Chem., 2010, 7, 533–538 CrossRef CAS.
- A. R. Kapdi and I. J. S. Fairlamb, Chem. Soc. Rev., 2014, 43, 4751–4777 RSC.
- F. K. Keter, S. Kanyanda, S. S. L. Lyantagaye, J. Darkwa, D. J. G. Rees and M. Meyer, Cancer Chemother. Pharmacol., 2008, 63, 127–138 CrossRef CAS PubMed.
- S. O. Ojwach and J. Darkwa, Inorg. Chim. Acta, 2010, 363, 1947–1964 CrossRef CAS.
- C. Chou, C. Yang, H. Syu and T. Kuo, J. Organomet. Chem., 2013, 745–746, 387–392 CrossRef CAS.
- Y. Zhang and G. Lavigne, Eur. J. Inorg. Chem., 2015, 2042–2050 CrossRef CAS.
- P. Štěpnička, M. Krupa, M. Lamač and I. Císařová, J. Organomet. Chem., 2009, 694, 2987–2993 CrossRef.
- Y. Tang, Y. Zeng, Q. Hu, F. Huang and L. Jin, Adv. Synth. Catal., 2016, 358, 2642–2651 CrossRef CAS.
- I. Guzei, K. Li, G. Bikzhanova, J. Darkwa and S. Mapolie, Dalton Trans., 2003, 715–722 RSC.
- S. Mohlala, M. Mohlala, I. A. Guzei, J. Darkwa and S. F. Mapolie, J. Mol. Catal. A: Chem., 2005, 241, 93 CrossRef.
- A. Budhai, B. Omondi, S. O. Ojwach, C. Obuah, E. Y. Osei-Twum and J. Darkwa, Catal. Sci. Technol., 2013, 3, 3130 CAS.
- N. M. Motsoane, I. A. Guzei and J. Darkwa, Z. Naturforsch., 2007, 62b, 323–330 Search PubMed.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed.
- A. N. J. Blok, P. H. M. Budzelaar and A. W. Gal, Organometallics, 2003, 22, 2564–2570 CrossRef CAS.
- D. Appavoo, B. Omondi, I. A. Guzei, J. L. Van Wyk, O. Zinyemba and J. Darkwa, Polyhedron, 2014, 69, 55–60 CrossRef CAS.
- A. Mukherjee and A. Sarkar, Tetrahedron Lett., 2005, 46, 15–18 CrossRef CAS.
- G. K. Anderson, M. Lin, A. Sen and E. Gretz in Inorganic Syntheses, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2007, pp. 60–63 Search PubMed.
- G. Yang and R. G. Raptis, Inorg. Chim. Acta, 2003, 352, 98–104 CrossRef CAS.
- F. Wu, H. Tong, K. Wang, X. Zhang and J. Zhang, J. Coord. Chem., 2016, 69, 1–5 CrossRef.
- M. Jung, Y. Lee, M. Park, H. Kim, H. Kim, E. Lim, J. Tak, M. Sim, D. Lee, N. Park, W. K. Oh, Y. Hur, S. Kang and H. Lee, Bioorg. Med. Chem. Lett., 2007, 17, 4481–4486 CrossRef CAS PubMed.
- K. Li, J. Darkwa, I. A. Guzei and S. F. Mapolie, J. Organomet. Chem., 2002, 660, 108–115 CrossRef CAS.
- A. Mukherjee and A. Sarkar, ARKIVOC, 2003, 2003, 87–95 Search PubMed.
- A. S. Kashin and V. P. Ananikov, Russ. Chem. Rev., 2013, 82, 648–685 CrossRef.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef CAS.
- Z. Li, C. Gelbaum, J. S. Fisk, B. Holden, A. Jaganathan, G. T. Whiteker, P. Pollet and C. L. Liotta, J. Org. Chem., 2016, 81, 8520–8529 CrossRef CAS PubMed.
- K. W. Quasdorf, A. Antoft-finch, P. Liu, A. L. Silberstein, A. Komaromi, T. Blackburn, S. D. Ramgren, K. N. Houk, V. Snieckus and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 6352–6363 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC number 1555880 contains the supplementary crystallographic data for complex 7. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra01430b |
| This journal is © The Royal Society of Chemistry 2018 |