
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroformylation of vinyl acetate and cyclohexene over TiO2 nanotube supported Rh and Ru nanoparticle catalysts†
Hongyuan Chuai a,
Xiaotong Liua,
Ya Chena,
Baolin Zhuab,
Shoumin Zhangab and
Weiping Huang*abc
a,
Xiaotong Liua,
Ya Chena,
Baolin Zhuab,
Shoumin Zhangab and
Weiping Huang*abc
aCollege of Chemistry, Nankai University, Tianjin 300071, China. E-mail: hwp914@nankai.edu.cn; Tel: +86-138-2009-6974
bThe Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China
cCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China
First published on 27th March 2018
Abstract
TiO2 nanotube (TNT) supported Rh and Ru nanoparticle catalysts were prepared via impregnation-photoreducing procedure and characterized with various methods. Their catalytic performances in hydroformylation were evaluated by using vinyl acetate and cyclohexene as substrates. The results indicate that the presence of Ru in the catalysts can enhance the catalytic activity of catalysts for the hydroformylation of vinyl acetate, but do not play the same role in the hydroformylation of cyclohexene; the sequence of loading metal has a significant effect on the catalytic performances of the title catalysts. Additionally, it is found that Ru/TNTs shows catalytic activity for the hydroformylation of vinyl acetate though it does not for the hydroformylation of cyclohexene.
1. Introduction
Nowadays the hydroformylation discovered by Otto Roelen in 1938 is still one of the most important catalytic reactions in the chemical industry.1–4 As for catalysts, including homogeneous and heterogeneous, the Co-,5,6 Ir-7,8 and Rh-based catalysts9–11 are widely reported and used for the hydroformylation, but the Rh-based catalysts are the dominant ones and have been increasingly being used.12,13 For functionalized alkenes, in which there is at least another functional group besides the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, e.g. vinyl acetate, their hydroformylation rate is affected by the nearby group or heteroatom as the group is adjacent to the CC bond because the group or heteroatom may coordinate to metal centers, and it is the chelation that affects the catalytic performances of catalysts.14,15 It is well known that the homogeneous catalyst used for hydroformylation reactions usually contains a type of single-active site, which may lose activity after being occupied or coordinated by the heteroatoms while catalyzing the hydroformylation of functionalized olefins. Therefore, it might not be the best option to use the catalyst with only one type of active site for the catalytic hydroformylation of functionalized olefins. When Rh is an only active site in the hydroformylation of functionalized alkenes, the efficiency of Rh should not be the highest one as the presence of chelation between Rh and functional group. To increase the efficiency of Rh, very expensive metal, in catalysts people have made great effort. Li et al. reported that using rhodium carbonyl and manganese carbonyl as catalyst simultaneously can significantly increase aldehyde formation in hydroformylation.16 Kunimori et al. reported that Mo-promoted Rh/SiO2 shows the good activity for hydroformylation.17 It should be a good scheme utilizing synergistic effect of two or more transition metals in catalysts to promote the conversion of substrate in hydroformylation,18,19 where the main active site can catalyze main reaction efficiently and the second active site decreases the influence of the nearby heteroatoms or groups. In our previous work, we used TiO2 nanotubes (TNTs)-supported Rh catalysts with different surface acidities for the hydroformylation of CN-containing olefin,20 the results showed that the higher the surface acidity is, the weaker the influence of –CN group, and the higher the linear-to-branched aldehyde ratio.
C bond, e.g. vinyl acetate, their hydroformylation rate is affected by the nearby group or heteroatom as the group is adjacent to the CC bond because the group or heteroatom may coordinate to metal centers, and it is the chelation that affects the catalytic performances of catalysts.14,15 It is well known that the homogeneous catalyst used for hydroformylation reactions usually contains a type of single-active site, which may lose activity after being occupied or coordinated by the heteroatoms while catalyzing the hydroformylation of functionalized olefins. Therefore, it might not be the best option to use the catalyst with only one type of active site for the catalytic hydroformylation of functionalized olefins. When Rh is an only active site in the hydroformylation of functionalized alkenes, the efficiency of Rh should not be the highest one as the presence of chelation between Rh and functional group. To increase the efficiency of Rh, very expensive metal, in catalysts people have made great effort. Li et al. reported that using rhodium carbonyl and manganese carbonyl as catalyst simultaneously can significantly increase aldehyde formation in hydroformylation.16 Kunimori et al. reported that Mo-promoted Rh/SiO2 shows the good activity for hydroformylation.17 It should be a good scheme utilizing synergistic effect of two or more transition metals in catalysts to promote the conversion of substrate in hydroformylation,18,19 where the main active site can catalyze main reaction efficiently and the second active site decreases the influence of the nearby heteroatoms or groups. In our previous work, we used TiO2 nanotubes (TNTs)-supported Rh catalysts with different surface acidities for the hydroformylation of CN-containing olefin,20 the results showed that the higher the surface acidity is, the weaker the influence of –CN group, and the higher the linear-to-branched aldehyde ratio.
In the present contribution, we report TNTs supported Rh and Ru nanoparticles catalysts and make comparison of the effect of Ru on the catalytic performances of catalysts for hydroformylation of vinyl acetate and cyclohexene.
2. Experimental
2.1 Preparation of catalyst
TNTs used were synthesized as the previous report.21Rh/TNTs was prepared via impregnation-photoreducing procedure as follows: 1.0 g of TNTs were dispersed in 20.0 mL of aqueous Rh2(Ac)4 solutions (0.043 g Rh2(Ac)4) and strong agitated for 24 h. After ultrasound for 2 h, the mixture was centrifuged. The green solid obtained was washed twice with ethanol, and then transferred into a quartz reactor with 50.0 mL ethanol–water solution (Vethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Vwater = 9:1). The mixture was irradiated with a 300 W high-pressure mercury lamp for 4 h under stirring at ambient temperature. After irradiation, the mixture was centrifuged, solid was washed with distilled water and ethanol, and dried at 40 °C for 12 h in vacuum. The obtained catalyst was labelled as Rh/TNTs. The other catalysts were prepared by the same procedure of preparing Rh/TNTs. For convenience of distinction, catalysts in this contribution were labelled as Rh/TNTs, Ru/TNTs, Rh–Ru/TNTs (Ru/TNTs was used as supporter to support Rh nanoparticles), Ru–Rh/TNTs (Rh/TNTs was used as supporter to support Ru nanoparticles), and Rh–Ru/TNTs (200 °C) (the catalyst Rh–Ru/TNTs was calcined at 200 °C for 2 h under N2), Rh–Ru/TNTs-U1 (the used catalyst), Rh–Ru/TNTs-U2 (the Rh–Ru/TNTs-U1 used for recycle).
:Vwater = 9:1). The mixture was irradiated with a 300 W high-pressure mercury lamp for 4 h under stirring at ambient temperature. After irradiation, the mixture was centrifuged, solid was washed with distilled water and ethanol, and dried at 40 °C for 12 h in vacuum. The obtained catalyst was labelled as Rh/TNTs. The other catalysts were prepared by the same procedure of preparing Rh/TNTs. For convenience of distinction, catalysts in this contribution were labelled as Rh/TNTs, Ru/TNTs, Rh–Ru/TNTs (Ru/TNTs was used as supporter to support Rh nanoparticles), Ru–Rh/TNTs (Rh/TNTs was used as supporter to support Ru nanoparticles), and Rh–Ru/TNTs (200 °C) (the catalyst Rh–Ru/TNTs was calcined at 200 °C for 2 h under N2), Rh–Ru/TNTs-U1 (the used catalyst), Rh–Ru/TNTs-U2 (the Rh–Ru/TNTs-U1 used for recycle).
2.2 Characterization of catalyst
The Rh and Ru contents in samples were measured by inductively coupled plasma mass spectrometer (X7, Thermo Electron Corporation). The specific surface area (SSA) of samples were determined at liquid N2 temperature with the BET method (BET, JW-K); phase structures of catalysts were characterized by X-ray Diffraction (XRD, Rigaku D/Max-2500 X-ray diffractometer with Cu Kα radiation). The morphology and microstructure of samples were observed with Transmission Electron Microscopy (TEM, 100 kV, JEM-2100) and scanning electron microscope (SEM, 25 kV, X-650). The chemical states of Rh and Ru in catalysts were determined by X-ray photoelectron spectroscopy (XPS) using an Al X-ray source (Al Kα-150 W, Kratos Axia Ultra DLA), and the binding energy was calibrated by taking C 1s peak at 284.6 eV as reference. The infrared spectra were recorded on an FT-IR spectrometer (USA, Bio-rad, FTS6000) in the spectral range 0–4000 cm−1, KBr wafers were used and the weight percentage of the samples in KBr was about 0.5%. Before testing, all samples were placed in a 250 mL stainless steel autoclave reactor pressurized with CO (1.5 MPa) for 12 h.2.3 Evaluation of catalytic performance
The hydroformylation reaction was used to evaluate catalytic performance of catalyst, and the activity of catalyst was compared by the percent conversion of olefin in the reaction. The hydroformylation reaction was carried out in a 250 mL stainless steel autoclave reactor with magnetic stirrer. In the process of experiment, the required amount of catalyst, substrate and solvent were put into the reactor in turn. The reactor was sealed, purged three times with CO or H2, and pressurized to a set pressure with syngas (CO and H2) while stirring, and then heated to the reaction temperature. The heating and stirring were stopped after the required reaction time. When the reactor was cooled down to room temperature, the pressure was released gradually, and the mixture of reaction was withdrawn, centrifuged and analyzed using GC analysis (Shimadzu GC-2014 gas chromatograph equipped with a 30 m × 0.53 mm × 1.0 μm SE-30 capillary column and a FID).3. Results and discussion
3.1 Characterization of the catalysts
The SSA of catalysts and contents of Rh or Ru in the catalysts determined by BET and ICP are listed in Table 1. The SSA of all catalysts are significantly lower than that of pure TNTs, which may be ascribed to that metal nanoparticles deposited on the outer and inner surface or occupied the interspace between walls of TNTs.22 The SSA of Rh–Ru/TNTs (200 °C) is lower than that of as-prepared Rh–Ru/TNTs because of calcination.| Entry | Catalyst | SSA (m2 g−1) | Rh content (wt%) | Ru content (wt%) |
|---|---|---|---|---|
| 1 | TNTs | 262.61 | — | — |
| 2 | Rh/TNTs | 228.69 | 0.10 | — |
| 3 | Ru/TNTs | 188.12 | — | 0.46 |
| 4 | Rh–Ru/TNTs | 247.61 | 0.09 | 0.42 |
| 5 | Ru–Rh/TNTs | 237.51 | 0.10 | 0.47 |
| 6 | Rh–Ru/TNTs (200 °C) | 234.24 | 0.10 | 0.51 |
To reveal the role of Ru, the contents of Rh in catalysts are purposefully kept in almost identical. The Rh nominal loading is 2% and Ru nominal loading is 0.5% for each case. From the ICP data, we can see that Ru exhibits high loading efficiency while Rh not. The difference might be caused by the starting material that chloride salt tends to be more efficiency than that of acetate salt. XRD was used to analyse the phase structure of catalysts. The XRD peaks at 25.24° and 48.1° are the diffractions of the (101) and (200) crystal planes of anatase TiO2 (JCPDS21-1272), respectively (Fig. 1). The phase does not change after calcination at 200 °C (Fig. 1F). In addition, there is no any diffraction related to Rh or Ru nanoparticles, which implies that Rh or Ru nanoparticles might be very small and well distributed.
 | ||
| Fig. 1 XRD patterns of TNTs (A), Rh/TNTs (B), Ru/TNTs (C), Rh–Ru/TNTs (D), Ru–Rh/TNTs (E), Rh–Ru/TNTs (200 °C) (F). | ||
The surface composition of Rh/TNTs was characterized by EDX and elemental mapping analysis (Fig. 2). In the marked red square (Fig. 2A), Ti and O display a homogeneous distribution (Fig. 2B–C), and Rh nanoparticles are uniformly dispersed on the surface of TNTs (Fig. 2D), so as on the other catalysts (S. 3†).
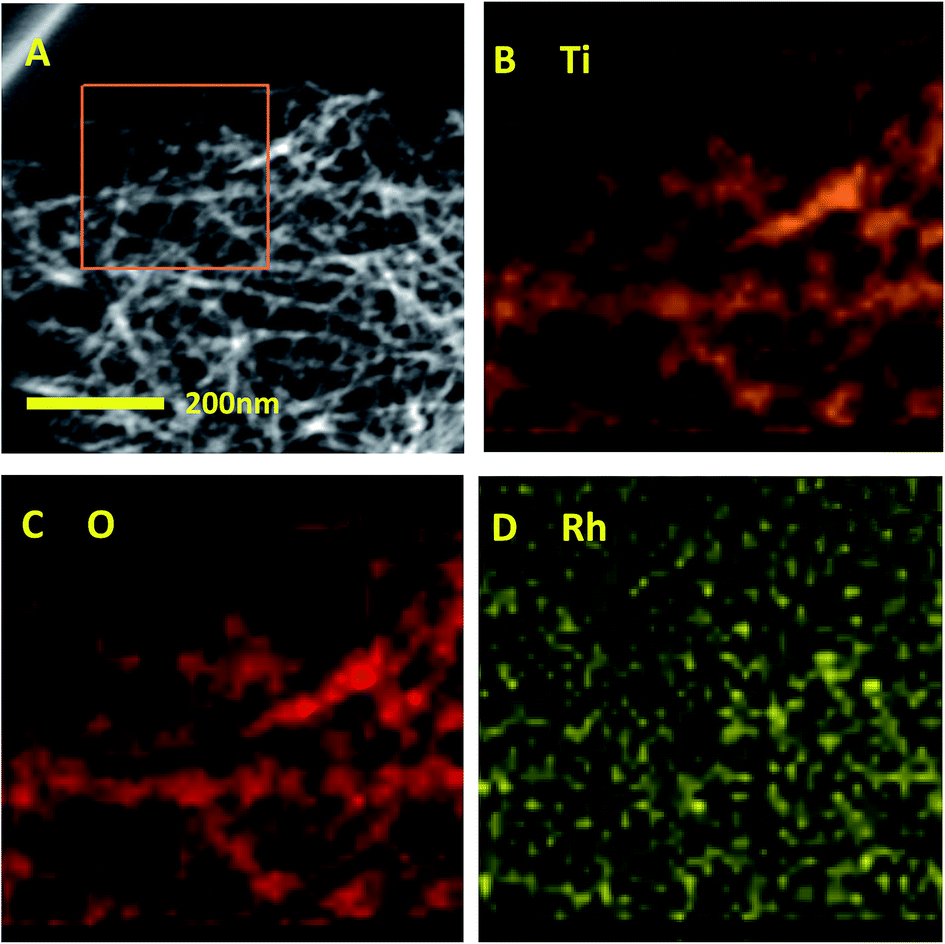 | ||
| Fig. 2 EDX elemental mapping analysis of Rh/TNTs. (A) HAADF-STEM image of Rh/TNTs, the marked area is scanned and analysed. (B–D) Mapping results for Ti (B), O (C), and Rh (D). | ||
The morphology of catalysts were further observed with TEM (Fig. 3). TNTs take on perfect tubular morphology and have the multiwall structure (Fig. 3A–D). There are some black spots in the as-prepared catalysts, which might be Rh or Ru nanoparticles. The size of particles is in the range of 1–2 nm. In the process of photo-reduction, the Rh or Ru atoms are apt to assemble on the new formed Rh or Ru nuclei to form Rh or Ru particles on the surface of TNTs for strong metal–metal interaction. The metal nanoparticles may grow up during calcination, as a result, the black spots in Rh–Ru/TNTs (200 °C) (Fig. 3E) are much larger than that in Rh–Ru/TNTs (Fig. 3C). The HRTEM image of the selected black spot in Fig. 3E shows that the lattice structure of the black spot is clearly different from that of TNTs (Fig. 3F). This further suggests that the black spots are metallic nanoparticles. We cannot determine the composition of nuclear particles accurately for the metallic nanoparticles are too small. The catalyst Rh–Ru/TNTs after catalysis used for once and twice still keeps nanotubular structure very well (Fig. 3G–H).
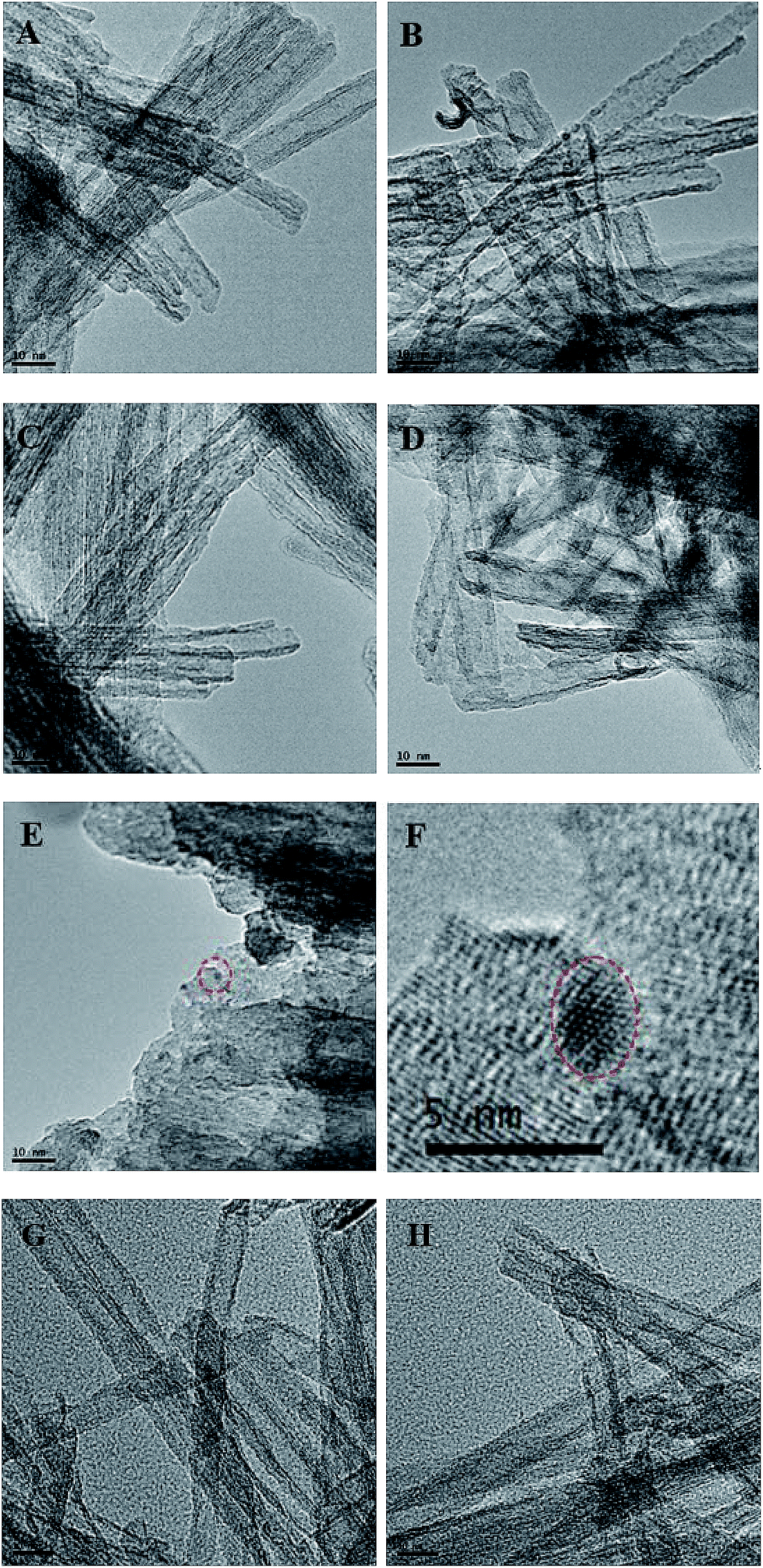 | ||
| Fig. 3 TEM images of Rh/TNTs (A), Ru/TNTs (B), Rh–Ru/TNTs (C), Ru–Rh/TNTs (D), Rh–Ru/TNTs (200 °C) (E), and HRTEM image of the selected area of E (F), Rh–Ru/TNTs-U1 (G), Rh–Ru/TNTs-U2 (H). | ||
XPS profiles of all Rh 3d in catalysts show broad doublets (Fig. 4(1A–1D)), the major Rh 3d 5/2 peaks appear at approximately 307.0 eV and 309.0 eV. These values indicate that the Rh in catalysts exists as two chemical states: BE at around 307.0 eV is associated with metallic Rh0, and 309.0 eV is originated from the oxidized Rh3+ species.15,23 Rh0 is the dominant component in as-prepared samples based on the peak area ratio. A small amount of Rh3+ species in catalysts might be due to the oxidation of surface layer in the process of characterization under air. However, the content of Rh0 in catalyst Rh–Ru/TNTs (200 °C) (Fig. 4(1D)) is lower than those in other samples based on the peak area ratio of Rh0 to Rh3+. In addition, we can find the variations in BE of Rh 3d, which means that there are interactions between metals or metal-supporter in the catalyst. These results may be understood by considering that the binding energy is affected not only by the metal itself, but also by the electronic interaction between the metal and other components in catalyst.
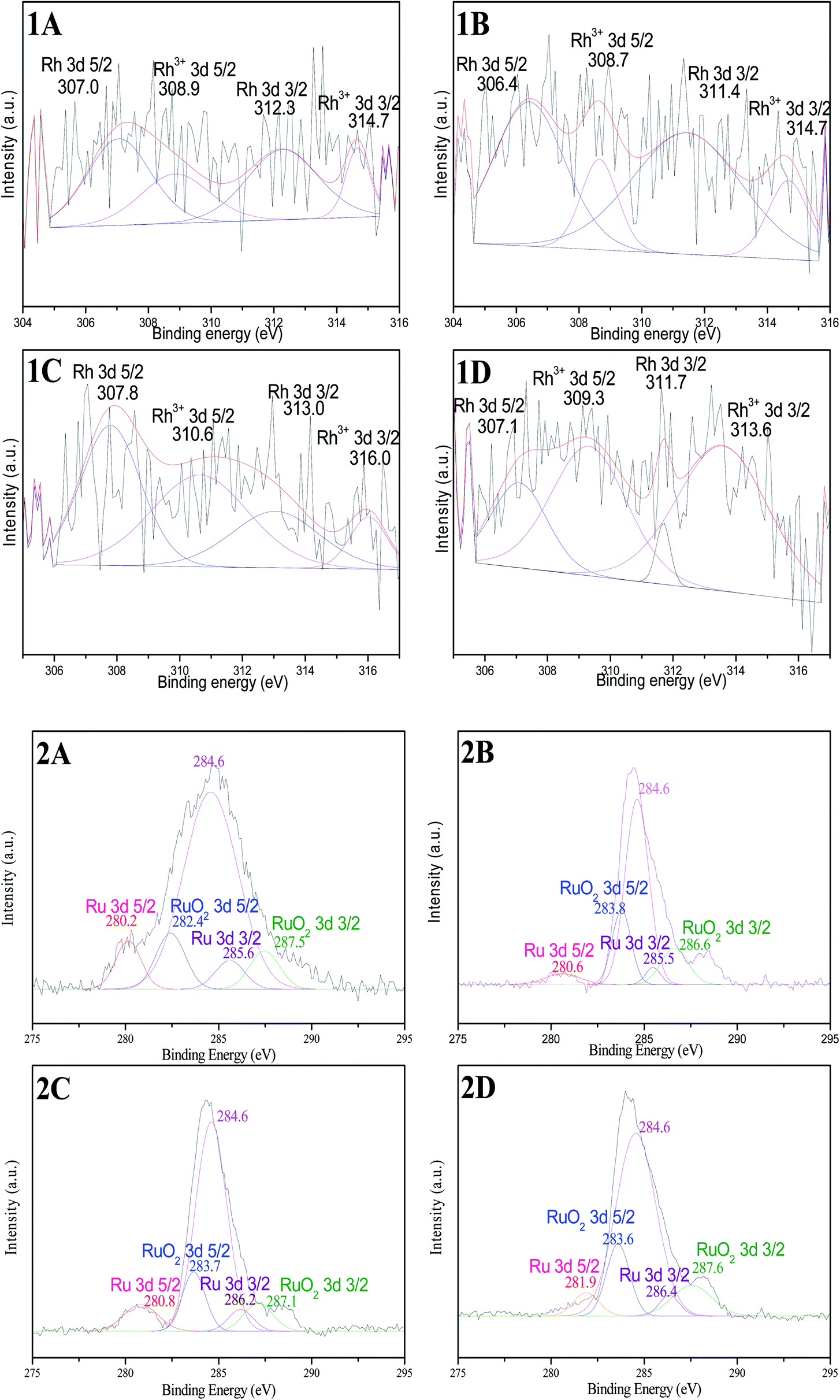 | ||
| Fig. 4 High-resolution XPS spectra of Rh 3d in Rh/TNTs (1A), Rh–Ru/TNTs (1B), Ru–Rh/TNTs (1C), Rh–Ru/TNTs (200 °C) (1D), and Ru 3d in Ru/TNTs (2A), Rh–Ru/TNTs (2B), Ru–Rh/TNTs (2C), Rh–Ru/TNTs (200 °C) (2D). | ||
It is well known that the XPS peaks of C 1s and Ru 3d suffer linear superposition.24 The broad peaks of samples can be fitted into several Gau ssian peaks (Fig. 4(2A–2D)). Peaks at 284.6 eV can be attributed to C 1s; peaks at around 280–281 and 285–286 eV are attributed to Ru0 3d 5/2 and Ru0 3d 3/2, respectively, which are consistent with the literature data of Ru0.23,25 The other peaks are attributed to component of RuO2. By contrast of XPS spectra of Rh 3d and Ru 3d of four samples, it can be concluded that there must be strong metal–metal interaction between Ru and Rh in the Rh–Ru/TNTs because the binding energies of Rh 3d and Ru 3d in the Rh–Ru/TNTs changed obviously.
FT-IR was used to explore the CO species adsorbed on catalysts (Fig. 5). All FT-IR spectra of Ru-containing catalysts absorbed CO display absorption peaks at 2066.22 and 1997.17 cm−1 (Fig. 5D–G), respectively, which are typical terminal (M–CO) and bridged (M2CO) CO adsorbed on the metals.26 However, the aforesaid peaks do not appear in the FT-IR spectra of Rh/TNTs (Fig. 5C), which means that the amount of CO adsorbed by Rh is quite small or the CO weakly bonded to the Rh/TNTs escapes away easily during characterization.1 It can be inferred that Ru is more favorable for absorbing CO than Rh.
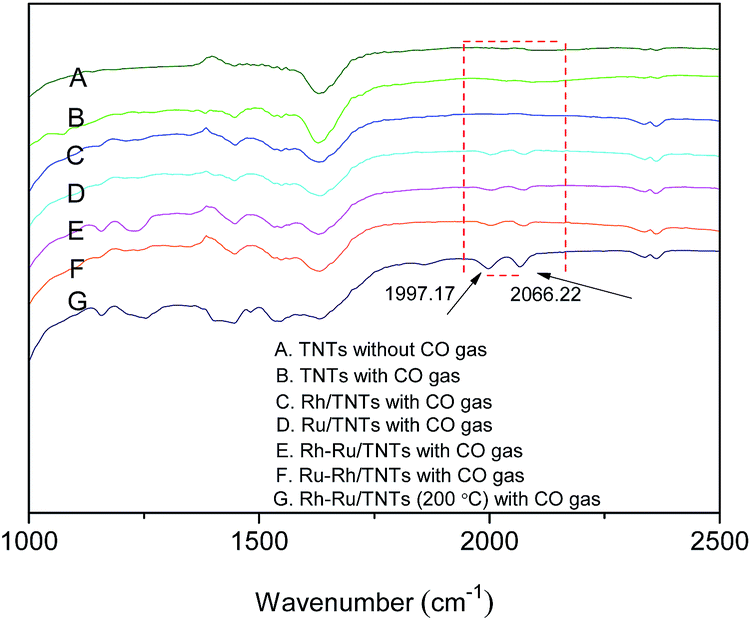 | ||
| Fig. 5 FT-IR spectra of TNTs without CO gas (A); samples with CO gas: TNTs (B), Rh/TNTs (C), Ru/TNTs (D), Rh–Ru/TNTs (E), Ru–Rh/TNTs (F), Rh–Ru/TNTs (200 °C) (G). | ||
3.2 Catalytic performances of the catalysts
Usually, the hydroformylation of vinyl acetate will provide two functional products: 2-acetoxy propanal and 3-acetoxy propanal (Scheme 1), however, in our experiment, the reaction has high regioselectivity for 2-acetoxy propanal (S. 1†). Table 2 shows the catalytic performance of as-prepared catalysts in the hydroformylation of vinyl acetate under different reaction time. It can be found that when the reaction time lasted for 1 h and 2 h (entry 1, 2, 4 and 5), the conversion of vinyl acetate over Rh–Ru/TNTs is higher than that over Rh/TNTs though the contents of Rh in two catalysts are almost identical and, on the contrary, the selectivity for aldehyde is lower, where 2-acetoxy propanal is the only product besides hydrogenation product. The result indicates that the addition of Ru does promote the activity of catalyst and accelerate the hydrogenation rate of the formed aldehyde,17 which results in the lower selectivity for aldehyde. Two possible factors may be responsible for the results. One is that the introduction of Ru enhances the adsorption capacity of catalyst for CO, as shown in FT-IR discussion; the other is that the Ru active center in the catalyst may act as Lewis acid attracting vinyl acetate through the carboxyl (Lewis base) and reducing the influence of the carboxyl on Rh active center, which should be advantageous to Rh catalysing the hydroformylation reaction and enhancing the reaction rate.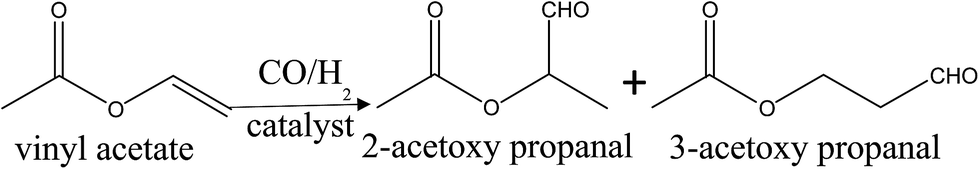 | ||
| Scheme 1 Hydroformylation of vinyl acetate. | ||
| Entry | Catalyst | Conversion of vinyl acetate (%) | Selectivity for aldehyde (%) | b:1b |
|---|---|---|---|---|
| a Reaction conditions: vinyl acetate = 5 mL, solvent (toluene) = 65 mL, catalyst = 0.40 g, syngas pressure 6 MPa (CO/H2 = 1), T = 100 °C.b b:l is 2-acetoxy propanal:3-acetoxy propanal. |
||||
| 1 | Rh/TNTs (1 h) | 39 | 82 | 100:0 |
| 2 | Rh/TNTs (2 h) | 68 | 60 | 100:0 |
| 3 | Rh/TNTs (8 h) | 100 | 57 | 100:0 |
| 4 | Rh–Ru/TNTs (1 h) | 45 | 57 | 100:0 |
| 5 | Rh–Ru/TNTs (2 h) | 71 | 57 | 100:0 |
| 6 | Rh–Ru/TNTs (8 h) | 100 | 55 | 100:0 |
| 7 | Ru/TNTs (8 h) | 25 | 16 | 100:0 |
| 8 | Ru–Rh/TNTs (8 h) | 83 | 45 | 100:0 |
| 9 | Rh–Ru/TNTs (200 °C, 8 h) | 85 | 70 | 100:0 |
| 10 | Rh–Ru/TNTs-U1 (8 h) | 37 | 51 | 100:0 |
To confirm the inference, we made contrast experiments by substituting cyclohexene which has no carboxyl group for vinyl acetate to further evaluate the catalytic performances of catalysts. In the hydroformylation process of cyclohexene, the main product is aldehyde (S. 2†), and a small quantity of aldehyde will be reduced to alcohol (Scheme 2). The results of cyclohexene hydroformylation over different catalysts are presented in Table 3. It is easily found that the difference in conversion of cyclohexene over Rh/TNTs and Rh–Ru/TNTs is obvious at 1 h and 2 h (entry 1–4). When the reaction lasted for 2 h, the conversion of cyclohexene over Rh/TNTs reaches up to 99.28%, while that over Rh–Ru/TNTs is much lower. These results indicate clearly that the presence of Ru in the catalysts does not enhance the hydroformylation reaction rate of cyclohexene though Ru has good ability to adsorb CO. This means that the function of Ru in Rh–Ru/TNTs for hydroformylation of vinyl acetate should be related to the coordination between Ru and carboxyl group, by which not only the vinyl acetate is attracted to catalyst, but also the effect of carboxyl group on Rh species is decreased, and then the catalytic activity of catalyst is enhanced. Kargbo et al. reported that the nearby heteroatoms of functionalized olefins are beneficial for catalytic reaction for coordination to metal centers.27 Because there is no carboxyl group in cyclohexene, Ru cannot play the same role and enhance the reaction rate in the hydroformylation of cyclohexene. Furthermore, by contrast, the negative effect of Ru on the hydroformylation of cyclohexene is obvious because Ru is not a better catalyst than Rh for the hydroformylation.
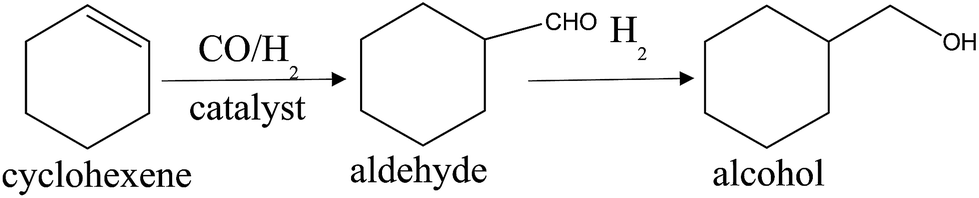 | ||
| Scheme 2 Hydroformylation of cyclohexene. | ||
| Entry | Catalyst | Conversion of cyclohexene (%) | Selectivity (%) | |
|---|---|---|---|---|
| Aldehyde | Alcohols | |||
| a Reaction conditions: cyclohexene = 5 mL, solvent (toluene) = 65 mL, catalyst = 0.40 g, syngas pressure 6 MPa (CO/H2 = 1), T = 100 °C.b T = 120 °C. | ||||
| 1 | Rh/TNTs (1 h) | 6 | 94 | 6 |
| 2 | Rh/TNTs (2 h) | 99 | 93 | 7 |
| 3 | Rh–Ru/TNTs (1 h) | 0 | 0 | 0 |
| 4 | Rh–Ru/TNTs (2 h) | 1 | 0 | 0 |
| 5 | Rh–Ru/TNTs (8 h) | 100 | 100 | 0 |
| 6 | Ru/TNTs (8 h)a,b | 0 | 0 | 0 |
| 7 | Ru–Rh/TNTs (8 h) | 0 | 0 | 0 |
| 8 | Ru–Rh/TNTs (8 h)b | 100 | 94 | 6 |
When the reaction time lasted for 8 h, the catalytic performance of the catalysts was also examined by hydroformylation of vinyl acetate and cyclohexene. The data of vinyl acetate hydroformylation are shown in Table 2, the catalytic activity of Rh/TNTs is far higher than that of Ru/TNTs. Compared with over Rh/TNTs, over Rh–Ru/TNTs the conversions of vinyl acetate are higher at 1 and 2 h, on the other hand, the catalyst Rh–Ru/TNTs is more active than Ru–Rh/TNTs and Rh–Ru/TNTs (200 °C). It means that the sequence of impregnation and calcination have a great influence on the catalytic activity of Rh–Ru catalysis system. It is worth noting that the selectivity of reaction for aldehyde over Rh–Ru/TNTs (200 °C) is much higher than that over other catalysts, which might come from the stronger interactions between Rh and Ru caused by calcination. For catalyst Ru–Rh/TNTs, (Rh/TNTs) is used as support to load Ru nanoparticles. It would take place certainly that the Rh nanoparticles are partly covered by Ru nanoparticles in the preparation process of Ru–Rh/TNTs, which may lead to the decrease in main active species and the catalytic activity. The larger size of Rh nanoparticles and the lower content of Rh0 in Rh–Ru/TNTs (200 °C) are responsible for that the Rh–Ru/TNTs (200 °C) shows much poor catalytic activity in comparison with Rh–Ru/TNTs. In order to examine the catalytic performance of the used catalyst Rh–Ru/TNTs, we collect the mixture by centrifuge after reaction. The obtained solid was washed with ethanol for several times and dried in vacuum. After treated, we carried experiment for recycle test. As shown in Table 2, the conversion of vinyl acetate decreased greatly while the selectivity of aldehyde decreased slightly (entry 10). The decrease in the catalytic activity in recycling might be due to the loss of Rh and Ru nanoparticles during the recovery and reuse.
The experiment results of cyclohexene hydroformylation over different catalysts lasted for 8 h are shown in Table 3. One can see from the data that Ru/TNTs does not shows any catalytic activity though Ru-complexes-catalyzed hydroformylation was reported28,29 (entry 6), however, Ru/TNTs shows catalytic activity for the hydroformylation of vinyl acetate. Due to the negative effect of Ru in cyclohexene hydroformylation, the catalytic performance of Rh–Ru/TNTs is much poor compared with Rh/TNTs in the first 2 h while it can also reach up to 99.93% when the reaction time rises to 8 h (entry 5). It can be inferred that the catalytic activity of Rh–Ru catalysis system will get worse when Ru nanoparticles is outside. Ru–Rh/TNTs has almost no catalytic activity at 100 °C even when the reaction lasted for 8 h (entry 7). However, Ru–Rh/TNTs shows good catalytic activity at 120 °C (entry 8). The results of entry 5 and 7 imply the sequence of loading metal has a great influence on the activity of the title catalyst and this is accord with the result of vinyl acetate hydroformylation.
Based on the experiment results and discussion above, we can draw a conclusion that Rh–Ru catalysis system has its limitation that it has influence on functionalized terminal olefins while not on the normal olefins. Besides, the fine structure of Rh–Ru catalysis system need to be further studied. However, it provides a new view to make functional catalyst. There are many researchers focus on multi-active centre catalyst system, especially Rh–Ru system.30–34 In current report, their research hotspot is the tandem reaction of Rh/Ru hydroformylation/hydrogenation which combines Rh-catalyzed hydroformylation and Ru-catalyzed hydrogenation in order to get higher efficiency. As for catalysis hydroformylation of vinyl acetate, most of the catalysts reported are rhodium complex modified with ligand while Rh–Ru system is rarely reported.35–37
4. Conclusions
The TiO2 nanotubes supported Rh and Ru nanoparticles catalysts show different catalytic performances in the hydroformylation of vinyl acetate and cyclohexene. Ru itself is not a good catalyst for the hydroformylation of vinyl acetate and cyclohexene, but it can enhance the hydroformylation rate of vinyl acetate by reacting with carboxyl group in Rh–Ru catalysis system. This opens a new way to prepare effective functional catalysts for hydroformylation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21373120, 21301098, 21406120 and 21071086).Notes and references
- L. A. Rupflin, J. Mormul, M. Lejkowski, S. Titlbach, R. Papp, R. Gläser, M. Dimitrakopoulou, X. Huang, A. Trunschke, M. G. Willinger, R. Schlögl, F. Rosowski and S. A. Schunk, ACS Catal., 2017, 7, 3584–3590 CrossRef.
- K. Cousin, S. Menuel, E. Monflier and F. Hapiot, Angew. Chem., Int. Ed., 2017, 56, 10564–10568 CrossRef CAS PubMed.
- B. Wang, J. F. Chen and Y. Zhang, RSC Adv., 2015, 5, 22300–22304 RSC.
- Y. P. Zhao, X. M. Zhang, J. Sanjeevi and Q. H. Yang, J. Catal., 2016, 334, 52–59 CrossRef CAS.
- W. L. Peddie, J. N. van Rensburg, H. C. M. Vosloo and P. van der Gryp, Chem. Eng. Res. Des., 2017, 121, 219–232 CrossRef CAS.
- G. Achonduh, Q. Yang and H. Alper, Tetrahedron, 2015, 71, 1241–1246 CrossRef CAS.
- C. Martinez-Macias, P. Serna and B. C. Gates, ACS Catal., 2015, 5, 5647–5656 CrossRef CAS.
- A. Kämper, S. J. Warrelmann, K. Reiswich, R. Kuhlmann, R. Franke and A. Behr, Chem. Eng. Sci., 2016, 144, 364–371 CrossRef.
- Y. K. Shi, X. J. Hu, L. Chen, Y. Lu, B. L. Zhu, S. M. Zhang and W. P. Huang, New J. Chem., 2017, 14, 6120–6126 RSC.
- M. Chevry, T. Vanbésien, S. Menuel, E. Monflier and F. Hapiot, Catal. Sci. Technol., 2017, 7, 114–123 CAS.
- Y. K. Shi, X. J. Hu, B. L. Zhu, S. R. Wang, S. M. Zhang and W. P. Huang, RSC Adv., 2014, 4, 62215–62222 RSC.
- Q. Sun, Z. F. Dai, X. L. Liu, N. Sheng, F. Deng, X. J. Meng and F. S. Xiao, J. Am. Chem. Soc., 2015, 137, 5204–5209 CrossRef CAS PubMed.
- M. Kumar, R. V. Chaudhari, B. Subramaniam and T. A. Jackson, Organometallics, 2014, 33, 4183–4191 CrossRef CAS.
- W. R. Jackson, P. Parlmutter and G. H. Suh, Chem. Commun., 1987, 10, 724–725 RSC.
- E. R. Nelsen, A. C. Brezny and C. R. Landis, J. Am. Chem. Soc., 2015, 137, 14208–14219 CrossRef CAS PubMed.
- C. Z. Li, E. Widjaja and M. Garland, Organometallics, 2004, 23, 4131–4138 CrossRef CAS.
- K. Tomishige, I. Furikado, T. Yamagishi, S. Ito and K. Kunimori, Catal. Lett., 2005, 103, 15–21 CrossRef CAS.
- Y. Ma, J. Fu and Z. Gao, Catalysts, 2017, 7, 103 CrossRef.
- C. Z. Li, F. Gao, S. Y. Cheng, M. Tjahjono, M. V. Meurs, B. Y. Tay, C. Jacob, L. F. Guo and M. Garland, Organometallics, 2011, 30, 4292–4296 CrossRef CAS.
- J. Li, Y. Chen, Y. J. Jiang, B. L. Zhu, S. M. Zhang and W. P. Huang, J. Mol. Catal., 2016, 30, 505–514 CAS.
- Y. Suzuki and S. Yoshikawa, J. Mater. Res., 2004, 19, 982–985 CrossRef CAS.
- X. J. Hu, Y. K. Shi, Y. J. Zhang, B. L. Zhu, S. M. Zhang and W. P. Huang, Catal. Commun., 2015, 59, 45–49 CrossRef CAS.
- K. Vinokurov, Y. Bekenstein, V. Gutkin, I. Popov, O. Millobc and U. Banin, CrystEngComm, 2014, 16, 9506–9512 RSC.
- X. Y. Zhao, J. Hrbek and J. A. Rodriguez, Surf. Sci., 2005, 575, 115–124 CrossRef CAS.
- J. J. Song, B. L. Zhu, W. L. Zhao, X. J. Hu, Y. K. Shi and W. P. Huang, J. Nanopart. Res., 2013, 15, 1494 CrossRef.
- G. A. Flores-Escamilla and J. C. Fierro-Gonzalez, Catal. Sci. Technol., 2015, 5, 843–850 CAS.
- S. Kraft, K. Ryan and R. B. Kargbo, J. Am. Chem. Soc., 2017, 139, 11630–11641 CrossRef CAS PubMed.
- J. P. Dunne, D. Blazina, S. Aiken, H. A. Carteret, S. B. Duckett, J. A. Jones, R. Poli and A. C. Whitwood, Dalton Trans., 2004, 21, 3616–3628 RSC.
- C. Kubis, I. Profir, I. Fleischer, W. Baumann, D. Selent, C. Fischer, A. Spannenberg, R. Ludwig, D. Hess, R. Franke and A. Börner, Chem.–Eur. J., 2016, 22, 2746–2757 CrossRef CAS PubMed.
- B. D. Rowsell, R. McDonald and M. Cowie, Organometallics, 2004, 23, 3873–3883 CrossRef CAS.
- C. P. Casey, ChemCatChem, 2010, 2, 1209–1211 CrossRef.
- K. Takahashi, M. Yamashita, T. Ichihara, K. Nakano and K. Nozaki, Angew. Chem., 2010, 122, 4590–4592 CrossRef.
- K. Takahashi, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 18746–18757 CrossRef CAS PubMed.
- Y. Yuki, K. Takahashi, Y. Tanaka and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 17393–17400 CrossRef CAS PubMed.
- A. A. Dabbawala, R. V. Jasra and H. C. Bajaj, Catal. Commun., 2010, 11, 616–619 CrossRef CAS.
- A. A. Dabbawala, H. C. Bajaj, G. V. S. Rao and S. H. R. Abdi, Appl. Catal., A, 2012, 419–420, 185–193 CrossRef CAS.
- E. R. Nelsen, A. C. Brezny and C. R. Landis, J. Am. Chem. Soc., 2015, 137, 14208–14219 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01399c |
| This journal is © The Royal Society of Chemistry 2018 |