
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the conversion of ethyl levulinate to γ-valerolactone over Ru/UiO-66 by introducing sulfonic groups into the framework†
Jie Yang *,
Wenjuan Huang,
Yongsheng Liu and
Tao Zhou
*,
Wenjuan Huang,
Yongsheng Liu and
Tao Zhou
School of Mathematics and Physics, Shanghai University of Electric Power, Shanghai, 200090, China. E-mail: yj_7667@aliyun.com
First published on 4th May 2018
Abstract
The conversion of ethyl levulinate (EL) to γ-valerolactone (GVL) is an important reaction in biomass conversion. This process undergoes two consecutive reactions: hydrogenation and transesterification of the intermediate compound, i.e. ethyl 4-hydroxypentanoate, which are catalyzed by metal nanoparticles and acid sites, respectively. In this study, we explored the catalytic activity of Ru supported on metal organic frameworks aiming to develop efficient metal–acid bifunctional catalysts for this green process. UiO-66 and its analogues with various substituted groups (–SO3H, –NH2 and –NO2) were employed in this study. The Ru particle size, oxidation state and reducibility were characterized by TEM, H2-TPR, and XPS. The results suggest that the introduction of functional groups reduces the hydrogenation activity of pristine Ru/UiO-66 to various extents. Catalyst modified with –SO3H group shows much higher acidic catalytic performance while showing hydrogenation activity towards C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, thus improving the overall transformation of EL to GVL due to the presence of strong Brønsted acid sites.
O bonds, thus improving the overall transformation of EL to GVL due to the presence of strong Brønsted acid sites.
1 Introduction
Global warming and depletion of fossil resources are two major issues concerning sustainable development.1 Biomass and its derivatives, as an abundant and inexpensive carbon source for the production of renewable bio-fuels and high value-added compounds, thus attract tremendous attention.2–5 γ-Valerolactone (GVL), as one of the versatile platform chemicals, can be applied as a fuel additive,6,7 a green solvent,8,9 and a precursor of value-added chemicals or liquid hydrocarbon fuels.8,10 GVL is usually produced by the hydrogenation and successive intramolecular dealcoholization of levulinic acid (LA)8,11 or its esters.12,13 Since LA is corrosive and needs formidable purification,14 employing esters of LA as reactants would be a promising green approach. Taking ethyl levulinate (EL) as an example, two steps are involved in the transformation from EL to GVL:8,15–17 hydrogenation of EL to ethyl 4-hydroxyvalerate (EHP) and transesterification of EHP to GVL. It is reported that metal catalysts, such as Pd,18,19 Pt,19 Ru,11,19,20 Au,21 Ni,22 Co,23 Mo,24 and Cu,25 are all active for hydrogenation of carbonyl groups when hydrogen is used. Among these metal catalysts, Ru has been reported to be the most active component.14 High catalytic activities of some Ru catalysts in the hydrogenation of LA into GVL have been reported by several research groups.26–29 For example, Ruppert et al.27 reported a LA conversion of 100% and a GVL selectivity of 100% in water at 70 °C, 5 MPa over 1 h when employing Ru nanoparticles supported on TiO2. Recently, Kuwahara et al.28 reported a GVL yield of 95.6% in aqueous medium at 70 °C, 0.5 MPa and 4 h by using Ru nanoparticles confined in Zr-containing spherical mesoporous silica. Galletti et al.30 investigated the catalytic activity of a ruthenium supported catalyst in combination with a heterogeneous acid co-catalyst in the process of hydrogenation of LA to GVL. The result reveals that the presence of the heterogeneous acid co-catalyst is favorable not only for the esterification step, but also for the activation of the carbonyl group, namely hydrogenation to the intermediate γ-hydroxyvaleric acid.Metal–organic frameworks (MOFs) as a new class of porous crystalline materials are composed by coordination between inorganic metal ions and organic linkers.30,31 Their tunable structures, physicochemical properties, and functionality32 make these materials attractive in the applications of catalysis,33–35 gas storage,36,37 separation,38,39 and sensing.40,41 Zirconium-based MOFs with higher stability in water and some organic solvents42 have attracted more attention in catalysis.43–46 Especially, Zr centers act as Lewis and/or Brønsted acid sites under certain reaction conditions, thus being widely applied in many acid-catalyzed reactions.46–51 UiO-66 (Zr6O4(OH)4(BDC)6 (BDC = 1,4-benezenedicarboxylate)), a perfect crystalline material consisting of Zr6O4(OH)4 octahedra featuring a 12-connected Zr6 cluster with BDC linkers,52 is an interesting Zr-based MOF due to its multiple acidic property. It is reported that missing linker defect sites always exist in UiO-66 crystals.53,54 Most likely, in aqueous phase, the defect sites bond with H2O and –OH groups to compensate the charge lost and there are at least three types of Brønsted protons in the UiO-66 framework present on the inorganic nodes: μ3-OH, –OH2 and –OH protons.55 Therefore, UiO-66 or organic-functionalized UiO-66 materials can serve as efficient supports with promising acidic catalytic function.
We herein introduced Ru nanoparticles into the frameworks of UiO-66 and its analogues modified with several commonly used functional groups including –NO2, –NH2, and –SO3H, aiming to reveal the effects of the substituted groups on the hydrogenation activity of Ru nanoparticles for the reaction of EL to EHP along with the acid-catalyzed conversion of EHP to GVL via transesterification. The results suggest that the Ru/UiO-66–SO3H catalyst shows the best performance in the overall reaction of converting EL to GVL via hydrogenation–transesterification pathways. These findings are helpful for developing efficient bifunctional catalysts for GVL production.
2 Experimental
2.1 Preparation of metal–organic frameworks
According to the literature,56,57 all of the MOFs were prepared under hydrothermal conditions. For instance, the synthesis of UiO-66–NH2 was performed by dissolving ZrCl4 (0.30 g) and 2-amino-1,4-benzenedicarboxylic acid (H2N–H2BDC) (0.31 g) in DMF (40 mL) at room temperature. The resulting mixture was placed in a Teflon-lined autoclave in a preheated oven at 120 °C for 36 hours. After the solution was cooled to room temperature in air, the resulting solid was filtered and repeatedly washed with absolute ethanol for 3 days while heated at 60 °C in an oil bath. The resulting powder was filtered and dried under vacuum at 50 °C.57 The functionalized UiO-66s were labelled UiO-66–X (X = –NO2, –NH2, –SO3H).2.2 Preparation of Ru/UiO-66–X
The supported Ru catalysts were prepared by a wet-impregnation method followed by deposition-reduction of Ru on UiO-66–X. Typically, 0.715 mL of RuCl3 solution (Ru: 21.60 mg mL−1) was impregnated on 0.5 g of UiO-66–X, the resulting sample being labelled RuCl3/UiO-66–X. After drying at 60 °C, RuCl3/UiO-66–X was immersed into 60 mL of methanol with stirring. The Ru precursor was reduced with NaBH4 methanol solution. The solid was filtered and dried at 50 °C under vacuum. The resulting products were denoted as Ru/UiO-66–X.2.3 Characterization
Powder X-ray diffraction (XRD) patterns were collected with a Rigaku Ultima IV X-ray diffractometer using Cu Kα radiation (λ = 1.5405 Å) operated at 35 kV and 25 mA. Scanning electron microscopy (SEM) was performed with a Hitachi S-4800 at 10 kV. Transmission electron microscopy (TEM) images were obtained with a FEI Tecnai G2 F30 microscope operated at 300 kV. Fourier transform infrared (IR) spectra were collected with a Nicolet Fourier transform infrared spectrometer (NEXUS 670). Liquid nitrogen adsorption was used to determine the BET surface areas and pore volumes. Prior to the adsorption measurements, the samples were degassed in situ under vacuum at 150 °C for 6 h. Inductively coupled plasma (ICP) analysis was used to quantify the Ru loading with a Thermo IRIS Intrepid II XSP atomic emission spectrometer. Samples were firstly dissolved in aqua regia and then diluted with deionized water. X-ray photoelectron spectroscopy (XPS) was conducted with an ESCALAB 250xi spectrometer, using a monochromated Al Kα X-ray source (hν = 1486.6 eV). High-resolution spectra were obtained using 40 eV pass energy and 0.1 eV step. Instrument base pressure was 8 × 10−10 Pa. All binding energies were referenced to the C 1s line at 284.6 eV. Due to the strong overlap between Ru 3d with C 1s, the Ru 3d5/2 peaks were analyzed. Before each measurement, the samples were pretreated by reducing in a hydrogen atmosphere at a reaction temperature (80 °C), stored in isopropanol and then transferred onto the sample holder in a glove box. To investigate the adsorption behavior of EL on supports, thermogravimetric analysis was performed with a NETZSCH STA449F3 TGA. Samples were heated from room temperature to 800 °C with a heating rate of 10 °C min−1 under an air flow. Temperature-programmed reduction (TPR) measurements were carried out using a TP-5080 (Tianjin Xianquan, China) equipped with a thermal conductivity detector. Prior to measurements, about 50 mg of sample was pretreated in a He stream, heated to 150 °C at a rate of 10 °C min−1, and held at this temperature for 1 h. After cooling down to room temperature, a gaseous mixture of 5% H2 in Ar was fed at a flow rate of about 30 mL min−1 and the sample was heated to 150 °C with a rate of 10 °C min−1.2.4 Catalytic activity measurements
The hydrogenation of EL in water was carried out in a Teflon-lined (120 mL) steel batch reactor. As-prepared catalysts were loaded into the reactor with 9.6 mL of deionized water and 340 μL of EL. After purging five times with H2, the reactor was pressurized with 0.5 MPa H2 and the rector was placed in an oil bath which was kept at 80 °C and the liquid was vigorously stirred to reduce the mass transfer effect. The transesterification of EHP in water was carried out in a glass pressure bottle. As-prepared catalysts were loaded into the bottle charged with 5 mL of deionized water and 200 μL of a mixture of EHP (84 mol%), GVL (5 mol%), and EL (11 mol%). The bottle was placed in an oil bath at 80 °C with stirring for 8 hours. All of the products were diluted with ethanol and analyzed with a Tianmei 7900 GC equipped with a DM-FFAP capillary column (30 m length, 0.25 μL film thickness and 0.25 mm internal diameter).3 Results and discussion
3.1 XRD, IR and BET characterization of UiO-66 and UiO-66–X
Fig. 1A displays the XRD patterns of the series of isostructural UiO-66. According to the XRD results, all materials show similar structure well matching the patterns in the literature.56,57 To confirm the successful incorporation of various substituents, i.e. –SO3H, –NH2 and –NO2, the IR spectra of UiO-66, UiO-66–NH2, UiO-66–SO3H and UiO-66–NO2 were compared as shown in Fig. 1B. In the case of UiO-66–SO3H, the bands at ca. 1080 and 1180 cm−1 are assigned to the stretching modes of –SO3H with double-bond character (OSO).44 The band at ca. 1025 cm−1 is due to the stretching vibration of the S–O single bond.44 The presence of –NH2 is confirmed by the presence of the bands at 3458 and 3353 cm−1 due to the N–H vibration,44 as well as the C–N stretching band at 1257 cm−1.57,58 The peak at 1540 cm−1, attributed to the asymmetric –NO2 stretching vibration, proves the existence of nitro groups in UiO-66–NO2.44 N2 physisorption was used to determine the BET surface areas (Fig. S1†) and the results are listed in Table 1. UiO-66 has a specific surface area (SSA) of 575 m2 g−1. The SSAs of UiO-66–NH2 and UiO-66–NO2 are 661 and 651 m2 g−1, respectively, which are ∼13% higher than that of UiO-66. In contrast, the SSA of UiO-66–SO3H (284 m2 g−1) is much lower than that of UiO-66, which is likely due to steric constraints and increased molecular weight resulting from the 2-substituted sulfonic group.56,57 A promising approach to achieve modulation of the physical properties of UiO-66–SO3H is to combine different ligands of the same denticity in different proportions, namely 1,4-benzenecarboxylic acid and 2-substituted 1,4-benzenecarboxylic acid.56 We cannot exactly determine the pore size distribution by N2 adsorption at this stage due to the microporous nature. The particle sizes of these materials range from 200 to 600 nm according to the SEM images (Fig. S2†).
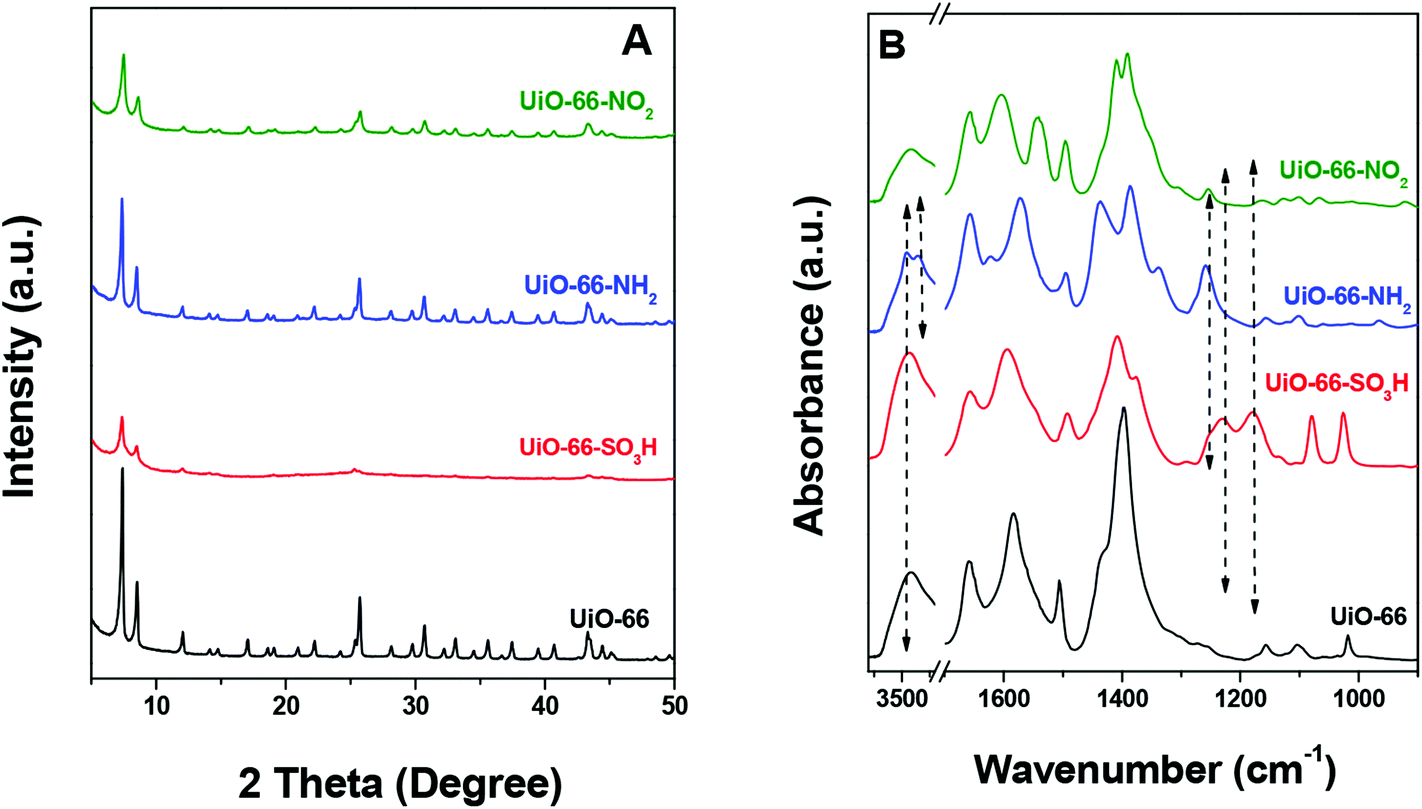 | ||
| Fig. 1 (A) XRD patterns of UiO-66 and UiO-66–X and (B) IR spectra of UiO-66 and UiO-66–X. | ||
3.2 Characterization of Ru/UiO-66 and Ru/UiO-66–X
Fig. 2 shows the XRD patterns of UiO-66 and UiO-66–X after loading with Ru nanoparticles. All Ru/UiO-66–X materials retained their crystalline structures, except for Ru/UiO-66–SO3H. The long-range order of Ru/UiO-66–SO3H collapsed after RuCl3 impregnation and reduction, indicating its unstable structure as reported in the literature.56 No diffraction peaks attributed to Ru nanoparticles were observed in the XRD patterns of Ru/UiO-66 and Ru/UiO-66–X, indicating the high dispersion of the Ru particles. N2 physisorption results of Ru/UiO-66 and Ru/UiO-66–X (Table 1 and Fig. S3†) reveal that the SSA of UiO-66 is retained after loading Ru nanoparticles. The SSA of Ru/UiO-66–NO2 is 489 m2 g−1, which is lower than that of the pristine material. However, after Ru loading, the SSAs of UiO-66–SO3H and UiO-66–NH2 dramatically decrease by 96% and 74% compared with the pristine materials, which could be explained by the compromised structures of UiO-66–SO3H and UiO-66–NH2 upon Ru loading.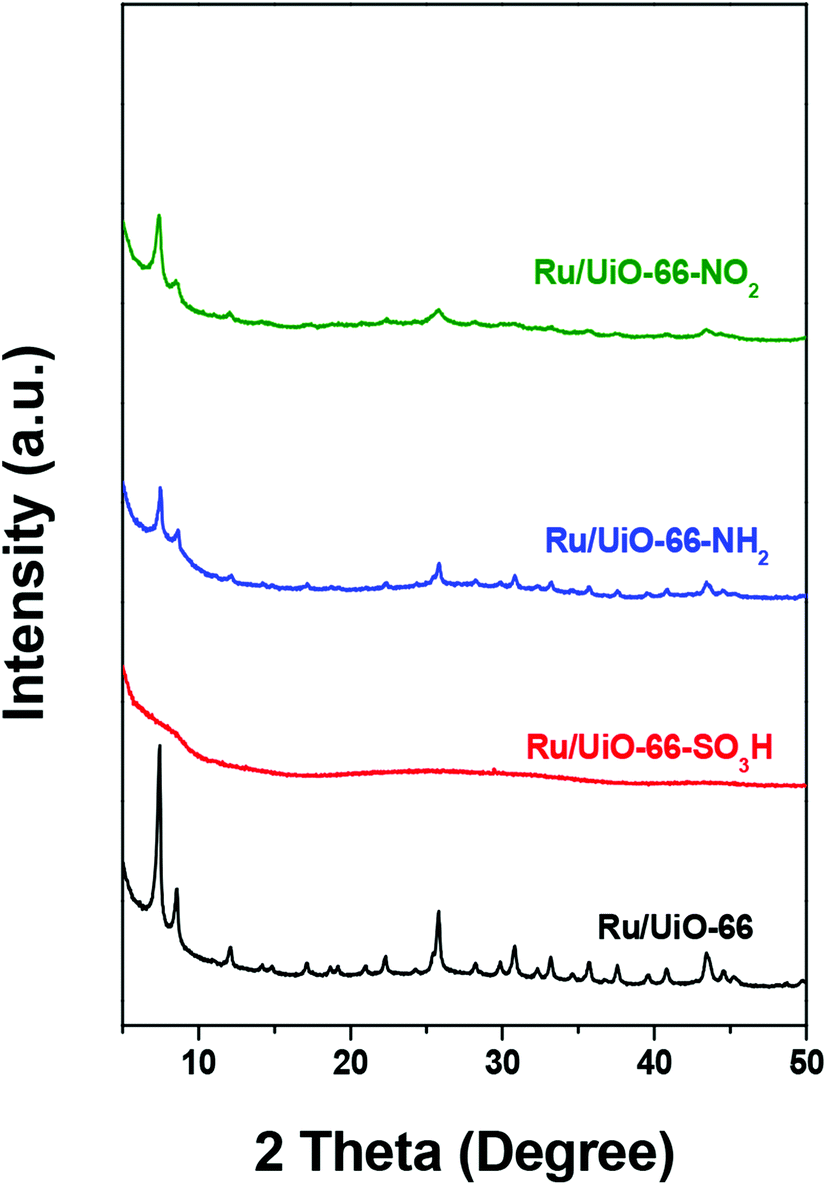 | ||
| Fig. 2 XRD patterns of Ru/UiO-66 and Ru/UiO-66–X. | ||
The Ru loadings were determined by ICP analysis and the results are listed in Table 1. The Ru loading is close to 2 wt% for the catalysts except for Ru/UiO-66–NH2. Ru loading of Ru/UiO-66–NH2 is about 4 wt% which is higher than that of the other Ru/UiO-66–X. This result suggests that the incorporation of Ru species is successful for Ru/UiO-66–NH2, which may be due to a strong coordination ability of –NH2 group with Ru species, leading to the high loading efficiency. The particle sizes of Ru for Ru/UiO-66–X were analyzed by high-resolution TEM (Fig. 3), the Ru particle sizes of all catalysts being in the range of 1–4 nm. It should be noted that the Ru particles of UiO-66–NH2 are smaller than those of the other catalysts.
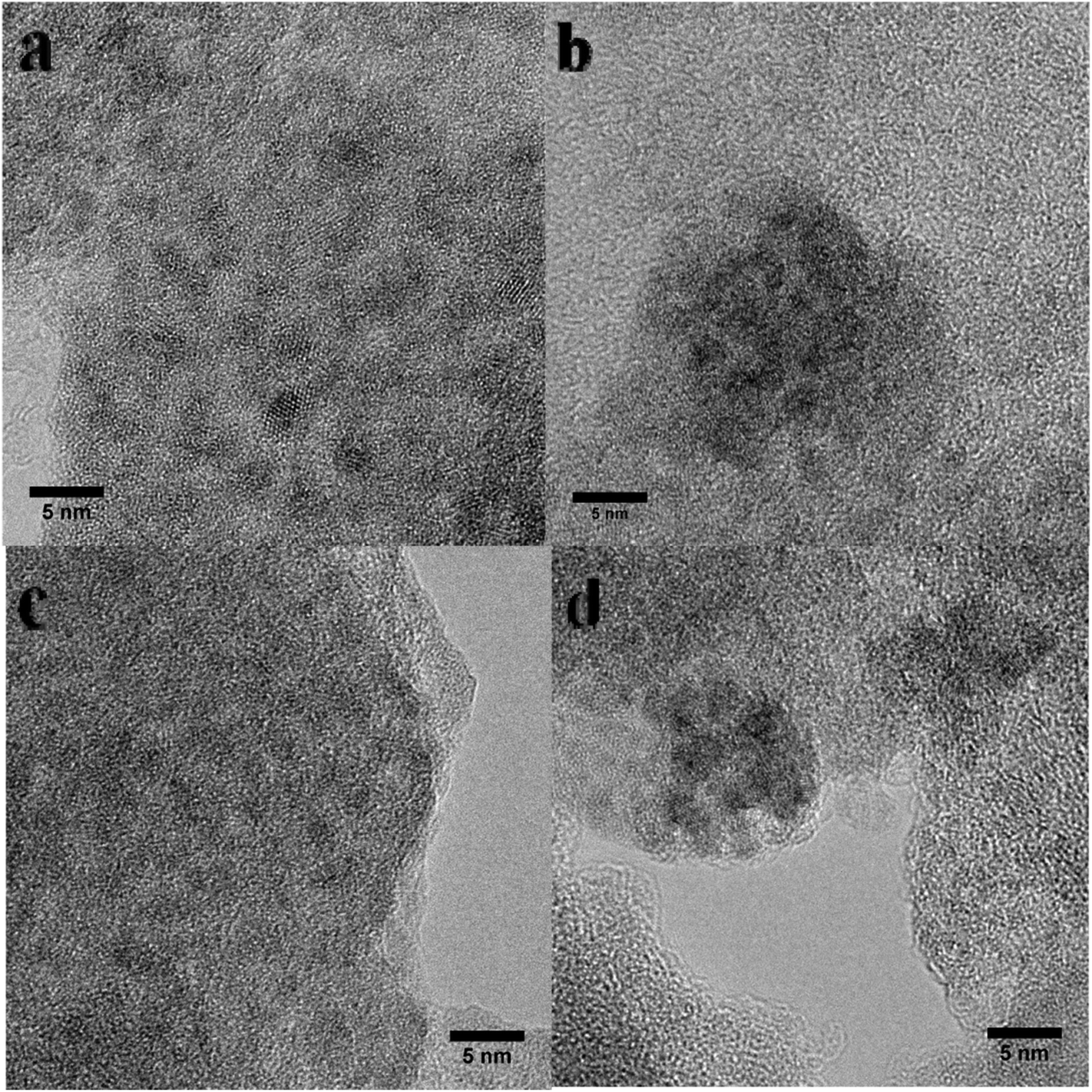 | ||
| Fig. 3 TEM images of (a) Ru/UiO-66, (b) Ru/UiO-66–SO3H, (c) Ru/UiO-66–NH2 and (d) Ru/UiO-66–NO2. | ||
The H2-TPR profiles of all catalysts are shown in Fig. 4. A significant H2 consumption peak ranging from 40 to 120 °C was observed for all catalysts, which is attributed to the reduction of oxidic Ru species below 150 °C.59 This result is consistent with the fact that Ru particles are prone to being oxidized when exposed to oxygen. We have roughly calculated the H/Ru ratio by normalizing on the amount of Ru loading. The H/Ru ratios of Ru/UiO-66, Ru/UiO-66–SO3H, Ru/UiO-66–NH2 and Ru/UiO-66–NO2 were found to be 0.89, 0.64, 0.16 and 0.47, respectively. One finding is that UiO-66–NH2 shows much lower H2 consumption compared with the other samples, which results from the stronger interaction between –NH2 and oxidic Ru species. In addition, the reduction of oxidic Ru takes place at 65 °C for Ru/UiO-66. The introduction of –SO3H group leads to a lower reduction temperature starting at 40 °C, while introducing –NO2 and –NH2 groups both leads to higher reduction temperature starting at about 80 °C. The result suggests that the reducibility of oxidic Ru species supported on UiO-66 is dependent on the organic groups introduced. The –SO3H functional group benefits the reduction of Ru species at lower temperatures, which plays a critical role in the hydrogenation process under mild conditions because metallic Ru species generally show better hydrogenation activity towards CO group than their oxidized counterparts.59
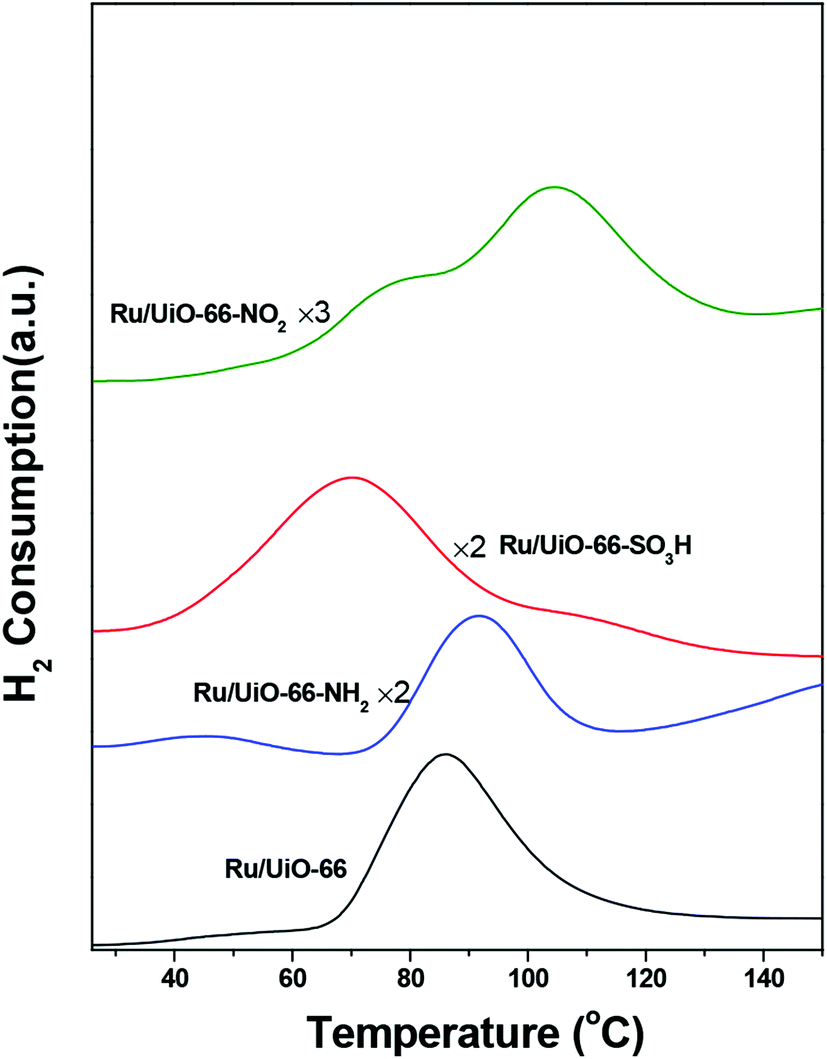 | ||
| Fig. 4 H2-TPR profiles of fresh Ru/UiO-66, Ru/UiO-66–NH2, Ru/UiO-66–SO3H and Ru/UiO-66–NO2. | ||
We also investigated the reduction degree of the supported Ru particles upon pretreatment under a H2 flow at 80 °C for 30 min by XPS. The fitted Ru 3d XPS spectra of the Ru/UiO-66 sample series and the corresponding results are shown in Fig. 5 and Table 2. Ru (3d) peaks are resolved as a doublet (3d5/2 and 3d3/2 at 279.90 and 284.07 eV, respectively) with an SOS of 4.17 eV.60 Because the 3d3/2 peak centered at ca. 284 eV is always overlapped by the C 1s peak, we herein focused on the Ru 3d5/2 peak which can be generally deconvoluted into three oxidation states: the feature at ca. 280 eV can be ascribed to metallic Ru, whereas the peaks at ca. 281 and 282 eV are ascribed to RuO2. In the cases of UiO-66 and UiO-66–SO3H, the peaks of Ru0 were clearly observed at about 280.2 eV upon flushing with H2 at 80 °C. It has been previously shown that the oxidized Ru particles on UiO-66 can be partially reduced by H2 at room temperature.59 The contents of metallic Ru on UiO-66 and UiO-66–SO3H were calculated to be ca. 71% and 60%, respectively. This result implies that both oxidic RuOx and metallic Ru are present in Ru/UiO-66 and UiO-66–SO3H at 80 °C in H2 atmosphere and the metallic Ru is present dominantly. For Ru/UiO-66–NH2, we only observed RuO2 from XPS, meaning that the Ruδ+ cations are strongly coordinated to –NH2 groups. Similar results have been previously reported by Wang et al. for a ruthenium-supported catalyst containing –NH2 groups.61 These results indicate that the functional groups on the ligands play a prominent role in the redox behavior of Ru species.
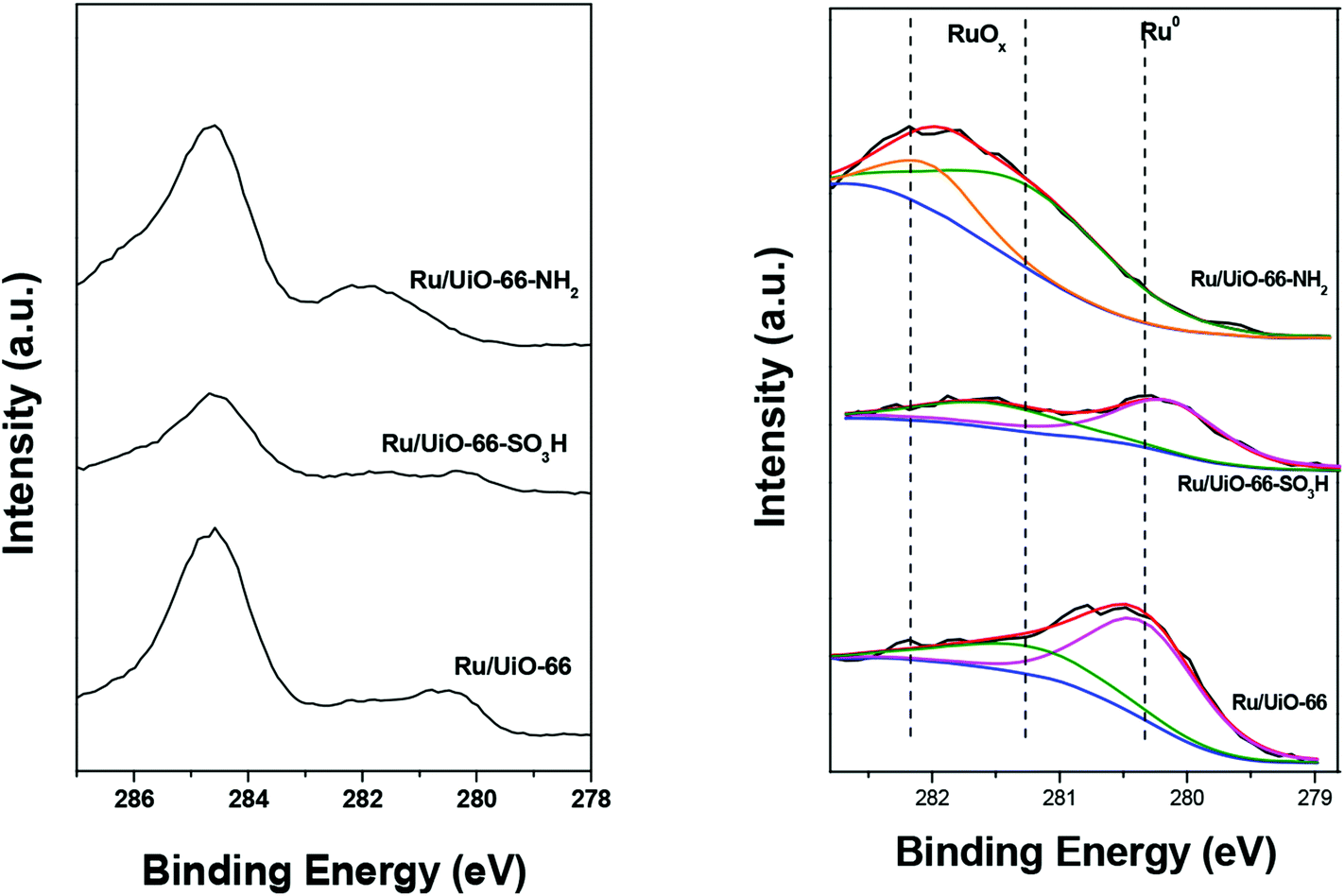 | ||
| Fig. 5 XPS spectra of Ru 3d C−1 1s region of Ru/UiO-66, Ru/UiO-66–NH2 and Ru/UiO-66–SO3H (left) and fitted XPS spectra of Ru 3d5/2 region of Ru/UiO-66, Ru/UiO-66–NH2 and Ru/UiO-66–SO3H (right). The dashed lines indicate the binding energies of the 3d5/2 peaks of reduced Ru (Ru0, magenta asymmetric curves) and oxidic Ru (green symmetric curves and orange symmetric curves) species. Red lines represent the overall fit of the data and blue lines represent background. | ||
| Sample | Ru0 3d5/2 BE (eV) | Ru3+ 3d5/2 BE (eV) | Ru4+ 3d5/2 BE (eV) | RD (3d5/2, %) | |
|---|---|---|---|---|---|
| Ru/UiO-66 | 280.3 | — | 281.2 | 71 | |
| Ru/UiO-66–SO3H | 280.1 | — | 281.5 | 60 | |
| Ru/UiO-66–NH2 | — | 282.0 | 281.2 | 0 | |
3.3 Catalytic activity measurements
The activities of Ru/UiO-66 and the three modified Ru/UiO-66 materials in the hydrogenation of EL were tested by using each catalyst of 100 mg in water at 80 °C for 1 h under 0.5 MPa of H2. The conversion of EL and selectivity of GVL and EHP after reaction for 1 h are listed in Table 3. It is clear that Ru/UiO-66 shows the highest hydrogenation activity with EL conversion of 100%. The conversions of EL on Ru/UiO-66–SO3H, Ru/UiO-66–NO2 and Ru/UiO-66–NH2 are 95, 82 and 37%, respectively. These results suggest that the introduction of functional groups reduces the hydrogenation activity of supported Ru nanoparticles to different extents. –SO3H groups do not reduce the hydrogenation activity markedly; whereas –NH2 groups may strongly coordinate with Ru surface sites thus hampering the activation of H2 and the adsorption of EL. The selectivity of GVL when using Ru/UiO-66, Ru/UiO-66–SO3H, Ru/UiO-66–NO2 and Ru/UiO-66–NH2 is 50, 61, 40 and 29%, respectively. UiO-66–SO3H containing strong Brønsted acidity shows the highest GVL selectivity. These results emphasize the importance of Brønsted acidity in transesterification reaction and prove the positive role of –SO3H in the transesterification of EHP. Prolonging the reaction period to 5 h leads to a full conversion of EL and 100% selectivity to GVL (Table S1†) on Ru/UiO-66 and Ru/UiO-66–SO3H. The above results reveal that introducing –SO3H groups does not reduce the activity of Ru/UiO-66 in the hydrogenation of EL, while enhancing the transesterification step, leading to higher activity in converting EL to GVL.| Catalyst | Catalyst loading (mg) | Conv. (%) | Sel. (%) | |
|---|---|---|---|---|
| GVL | EHP | |||
| a Reaction conditions: EL (0.34 mL), solvent (9.6 mL), 80 °C, 1 h, H2 (0.5 MPa). | ||||
| Ru/UiO-66 | 100 | 100 | 50 | 50 |
| 50 | 100 | 45 | 55 | |
| 25 | 98 | 39 | 61 | |
| Ru/UiO-66–SO3H | 100 | 95 | 61 | 39 |
| 50 | 72 | 52 | 48 | |
| 25 | 29 | 28 | 72 | |
| Ru/UiO-66–NH2 | 100 | 37 | 29 | 71 |
| Ru/UiO-66–NO2 | 100 | 82 | 40 | 60 |
| 50 | 27 | 53 | 47 | |
| 25 | 11 | 50 | 50 | |
To shed further light on the promotion effect of –SO3H groups on the overall reaction, the reactivities on Ru/UiO-66, Ru/UiO-66–NO2 and Ru/UiO-66–SO3H were studied in detail by employing varying amounts of the catalysts in water at 80 °C for 1 h under 0.5 MPa of H2. The results are listed in Table 3. 25 mg of the catalysts gave EL conversions of 98%, 11% and 29% for Ru/UiO-66, Ru/UiO-66–NO2 and Ru/UiO-66–SO3H, respectively. By increasing the amount of the catalysts to 50 mg, EL conversion on Ru/UiO-66 reaches 100% and EL conversions on Ru/UiO-66–SO3H and Ru/UiO-66–NO2 increase to 72% and 27% which are ∼24% and ∼67% lower than those obtained by employing 100 mg of the catalysts (95% for Ru/UiO-66–SO3H and 82% for Ru/UiO-66–NO2). The result indicates that the hydrogenation proceeds very fast in the presence of Ru/UiO-66 and the introduction of –NO2 group hampers the hydrogenation of EL, while –SO3H group plays a minor role in the hydrogenation of EL. With respect to transesterification of EHP, –SO3H and –NO2 groups play different roles in this step. The yields of GVL on Ru/UiO-66 for different dosages (25 mg, 50 mg and 100 mg) increase gradually from 39% to 45%, and to 50%, indicating a slow rate of transesterification of EHP. In the case of Ru/UiO-66–NO2, a slight increase was found in the yield of GVL on 50 mg of Ru/UiO-66–NO2 (53%) compared to the yield of GVL on 25 mg of Ru/UiO-66–NO2 (50%), but a decreased yield of GVL (40%) was obtained by further increasing the amount of Ru/UiO-66–NO2 to 100 mg, revealing that the introduction of –NO2 group not only impedes the hydrogenation of EL but also the transesterification of EHP. It is noted that the yield of GVL on Ru/UiO-66–SO3H increases significantly on increasing the dosage of Ru/UiO-66–SO3H. The yield of GVL on 25 mg of Ru/UiO-66–SO3H is 28% which is ∼28% lower than the yield found on 25 mg of UiO-66. When the amount of Ru/UiO-66–SO3H is increased to 50 mg, the yield of GVL increases to 52%, which is ∼16% higher than that on 50 mg of Ru/UiO-66 and ∼86% higher than that obtained on 25 mg of Ru/UiO-66–SO3H. Further employing 100 mg of Ru/UiO-66–SO3H, the yield of GVL is increased by ∼118% compared to the yield of GVL when using 25 mg of Ru/UiO-66–SO3H and is 22% higher than that obtained on 100 mg of Ru/UiO-66. The remarkable improvement indicates a faster transesterification of EHP and a positive role of –SO3H group in the transesterification of EHP catalyzed by Ru/UiO-66, which may be attributed to the Brønsted acidity of –SO3H.
4 Conclusions
In summary, the catalytic activity of Ru nanoparticles supported on Zr-UiO-66 and its analogues with various functional groups (–NO2, –SO3H and –NH2) for hydrogenation of EL was systematically studied. The introduction of functional groups –NH2 and –NO2 reduces the activity of UiO-66 in both the hydrogenation of EL and the transesterification of EHP. The poor activity of Ru/UiO-66–NH2 can be explained by the absence of metallic Ru particles, which is because of the strong coordination between Ru and –NH2 groups. The Lewis acidity of –NO2 results in the reduced activity of Ru/UiO-66 in the hydrogenation of EL. In contrast, –SO3H group plays a positive role in the transesterification of EHP, resulting in an increase of 22% in the yield of GVL, in spite of a slightly reduced activity (EL conversion: 100% for Ru/UiO-66 vs. 95% for Ru/UiO-66–SO3H). Reaction kinetics study further suggests that the introduction of –SO3H group is beneficial for the transesterification of EHP due to its Brønsted acidity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Science and Technology Commission of Shanghai Municipality (13ZR1417900), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, and the National Natural Science Foundation of China (grant no. 11674215 and 51402186).References
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- J. P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS PubMed.
- A. Villa, M. Schiavoni, C. E. Chan-Thaw, P. F. Fulvio, R. T. Mayes, S. Dai, K. L. More, G. M. Veith and L. Prati, ChemSusChem, 2015, 8, 2520–2528 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- C. E. Chan-Thaw, M. Marelli, R. Psaro, N. Ravasio and F. Zaccheria, RSC Adv., 2013, 3, 1302–1306 RSC.
- Z. Yan, L. Lin and S. Liu, Energy Fuels, 2009, 23, 3853–3858 CrossRef CAS.
- E. I. Gurbuz, D. M. Alonso, J. Q. Bond and J. A. Dumesic, ChemSusChem, 2011, 4, 357–361 CrossRef PubMed.
- A. H. Valekar, K.-H. Cho, S. K. Chitale, D.-Y. Hong, G.-Y. Cha, U. H. Lee, D. W. Hwang, C. Serre, J.-S. Chang and Y. K. Hwang, Green Chem., 2016, 18, 4542–4552 RSC.
- X. Tang, H. Chen, L. Hu, W. Hao, Y. Sun, X. Zeng, L. Lin and S. Liu, Appl. Catal., B, 2014, 147, 827–834 CrossRef CAS.
- K. Yan and A. Chen, Fuel, 2014, 115, 101–108 CrossRef CAS.
- A. S. Piskun, J. E. de Haan, E. Wilbers, H. H. van de Bovenkamp, Z. Tang and H. J. Heeres, ACS Sustainable Chem. Eng., 2016, 4, 2939–2950 CrossRef CAS.
- A. M. Hengne, N. S. Biradar and C. V. Rode, Catal. Lett., 2012, 142, 779–787 CrossRef CAS.
- F. Ye, D. Zhang, T. Xue, Y. Wang and Y. Guan, Green Chem., 2014, 16, 3951–3957 RSC.
- P. P. Upare, J.-M. Lee, D. W. Hwang, S. B. Halligudi, Y. K. Hwang and J.-S. Chang, J. Ind. Eng. Chem., 2011, 17, 287–292 CrossRef CAS.
- S. G. Wettstein, J. Q. Bond, D. M. Alonso, H. N. Pham, A. K. Datye and J. A. Dumesic, Appl. Catal., B, 2012, 117–118, 321–329 CrossRef CAS.
- M. L. Testa, L. Corbel-Demailly, V. La Parola, A. M. Venezia and C. Pinel, Catal. Today, 2015, 257, 291–296 CrossRef CAS.
- K.-I. Shimizu, S. Kanno and K. Kon, Green Chem., 2014, 16, 3899–3903 RSC.
- H. Zhou, J. Song, H. Fan, B. Zhang, Y. Yang, J. Hu, Q. Zhu and B. Han, Green Chem., 2014, 16, 3870–3875 RSC.
- D. Sun, A. Ohkubo, K. Asami, T. Katori, Y. Yamada and S. Sato, Mol. Catal., 2017, 437, 105–113 CrossRef CAS.
- E. F. Mai, M. A. Machado, T. E. Davies, J. A. Lopez-Sanchez and V. Teixeira da Silva, Green Chem., 2014, 16, 4092–4097 RSC.
- W. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. Bruijnincx and B. M. Weckhuysen, Nat. Commun., 2015, 6, 6540–6549 CrossRef PubMed.
- A. M. Ruppert, J. Grams, M. Jędrzejczyk, J. Matras-Michalska, N. Keller, K. Ostojska and P. Sautet, ChemSusChem, 2015, 8, 1538–1547 CrossRef CAS PubMed.
- Y. Kuwahara, Y. Magatani and H. Yamashita, Catal. Today, 2015, 258, 262–269 CrossRef CAS.
- Y. Wang, Z. Rong, Y. Wang, T. Wang, Q. Du, Y. Wang and J. Qu, ACS Sustainable Chem. Eng., 2016, 5, 1538–1548 CrossRef.
- A. M. R. Galletti, C. Antonetti, V. De Luise and M. Martinelli, Green Chem., 2012, 14, 688–694 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–794 CrossRef CAS PubMed.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- Y. Kuwahara, H. Kango and H. Yamashita, ACS Sustainable Chem. Eng., 2017, 5, 1141–1152 CrossRef CAS.
- A. Corma, H. García and F. X. Llabrési Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- J. B. DeCoste, M. H. Weston, P. E. Fuller, T. M. Tovar, G. W. Peterson, M. D. LeVan and O. K. Farha, Angew. Chem., Int. Ed., 2014, 53, 14092–14095 CrossRef CAS PubMed.
- M. Zhang, C. Chen, Q. Wang, W. Fu, K. Huang and W. J. Zhou, J. Mater. Chem. A, 2017, 5, 349–354 CAS.
- A. Chakraborty, A. Achari, M. Eswaramoorthy and T. K. Maji, Chem. Commun., 2016, 52, 11378–11381 RSC.
- J. Fu, S. Das, G. Xing, T. Ben, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2016, 138, 7673–7680 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- J. J. Gassensmith, J. Y. Kim, J. M. Holcroft, O. K. Farha, J. F. Stoddart, J. T. Hupp and N. C. Jeong, J. Am. Chem. Soc., 2014, 136, 8277–8282 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- Y. R. Lee, Y. M. Chung and W. S. Ahn, RSC Adv., 2014, 4, 23064–23067 RSC.
- R. Ghorbani-Vaghei, D. A. Davood Azarifar, S. Daliran and A. R. Oveisi, RSC Adv., 2016, 6, 29182–29189 RSC.
- U. S. F. Arrozi, H. W. Wijaya, A. Patah and Y. Permana, Appl. Catal., A, 2015, 506, 77–84 CrossRef CAS.
- J. F. Blandez, A. Santiago-Portillo, S. Navalón, M. Giménez-Marqués, M. Álvaro, P. Horcajada and H. García, J. Mol. Catal. A: Chem., 2016, 425, 332–339 CrossRef CAS.
- Y. Guo, Y. Li, J. Chen and L. Chen, Catal. Lett., 2016, 146, 2041–2052 CrossRef CAS.
- Z. Zhang, J. Chen, Z. Bao, G. Chang, H. Xing and Q. Ren, RSC Adv., 2015, 5, 79355–79360 RSC.
- T. L. Doan, T. Q. Dao, H. N. Tran, P. H. Tran and T. N. Le, Dalton Trans., 2016, 45, 7875–7880 RSC.
- N. E. Thornburg, Y. Liu, P. Li, J. T. Hupp, O. K. Farha and J. M. Notestein, Catal. Sci. Technol., 2016, 6, 6480–6484 CAS.
- B. Van de Voorde, I. Stassen, B. Bueken, F. Vermoortele, D. De Vos, R. Ameloot, J. –C. Tan and T. D. Bennett, J. Mater. Chem. A, 2015, 3, 1737–1742 CAS.
- M. J. Katz, Z. J. Brown, Y. J. Colon, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS PubMed.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 CAS.
- M. Lin Foo, S. Horike, T. Fukushima, Y. Hijikata, Y. Kubota, M. Takata and S. Kitagawa, Dalton Trans., 2012, 41, 13791–13794 RSC.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- J. Hou, Y. Luan, J. Tang, A. M. Wensley, M. Yang and Y. Lu, J. Mol. Catal. A: Chem., 2015, 407, 53–59 CrossRef CAS.
- Q. Yuan, D. Zhang, L. V. Haandel, F. Ye, T. Xue, E. J. M. Hensen and Y. Guan, J. Mol. Catal. A: Chem., 2015, 406, 58–64 CrossRef CAS.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- S. Wang, Z. Zhang, B. Liu and J. Li, Ind. Eng. Chem. Res., 2014, 53, 5820–5827 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01314d |
| This journal is © The Royal Society of Chemistry 2018 |