DOI:
10.1039/C8RA01288A
(Paper)
RSC Adv., 2018,
8, 12752-12759
Synthesis and self-assembly of a novel amphiphilic diblock copolymer consisting of isotactic polystyrene and 1,4-trans-polybutadiene-graft-poly(ethylene oxide)
Received
9th February 2018
, Accepted 29th March 2018
First published on 3rd April 2018
Abstract
Herein, a novel amphiphilic diblock copolymer consisting of isotactic polystyrene (iPS) and 1,4-trans-polybutadiene-graft-poly(ethylene oxide) (1,4-trans-PBD-g-PEO), iPS-b-(1,4-trans-PBD-g-PEO), was synthesized by the combination of living coordination copolymerization and graft copolymerization. iPS-b-1,4-trans-PBD was firstly synthesized via sequential monomer addition in the presence of 1,4-dithiabutandiyl-2,2′-bis(6-cumenyl-4-methylphenoxy) titanium dichloride (complex 1) activated by triisobutyl aluminum modified methylaluminoxane (MMAO). Moreover, hydroboration of double bonds in the 1,4-trans-PBD blocks were performed with 9-borabicyclo[3.3.1]nonane (9-BBN) and subsequent oxidation by NaOH/H2O2 to form hydroxyls. Consequently, PEO was grafted into the hydroxylated 1,4-trans-PBD block in terms of ring-opening polymerization of ethylene oxide with potassium/naphthalide as initiatior. We also described solvent-evaporation-induced self-assembly of iPS-b-1,4-trans-PBD in n-dodecane and iPS-b-1,4-trans-PBD-g-PEO in aqueous solution, which were selective solvent for 1,4-trans-PBD and for 1,4-trans-PBD-g-PEO blocks, respectively. In these cases, tetrahydrofuran (THF) was used as good and volatile solvent. These resultant iPS-containing diblock copolymers could self-assemble into spherical nano-micelles with an iPS core as amorphous agglomeration or a very low degree of crystallinity resulting from slow crystallization rate and nanoconfinement. In addition, after isothermal crystallization of iPS in the micellar cores self-assembled in n-dodecane at 120 °C for 3 hours, the micellar morphology changed from sphere-like to platelet-like. It was believed that isothermal crystallization of iPS induced the deformation of the micelles.
Introduction
Isotactic polystyrene (iPS) is an important stereoregular polymer with a good application foreground because of its unique physical and chemical properties such as high melting temperature, high heat deflection temperature and good corrosion resistance. iPS was first synthesized by the Ziegler-Natta catalyst in the 1950s with coordination polymerization. However, the living stereospecific living polymerization of styrene and 1,3-butadiene were achieved in 2010 by titanium complexes containing a tetradentate [OSSO]-type bis(phenolato) ligand ([OSSO]-Ti) and the polymerisates were respectively iPS and 1,4-trans-polybutadiene (1,4-trans-PBD) with narrow molecular weight distribution. Furthermore, the diblock copolymer consisting of iPS and 1,4-trans-PBD was synthesized by this [OSSO]-Ti complex via sequential monomer addition,1 which makes it possible for the research into the self-assembly of iPS-core diblock copolymers in selective solvent.2
The self-assembly of crystalline-coil diblock copolymers has drawn general attention at recent years because of their remarkable advantages.3–10 The insoluble crystalline block undergoes microphase separation and aggregation to form the core of micelle-like objects, surrounded by a solvent-swollen corona that provides colloidal stability. Crystalline-coil diblock copolymers can enrich the nano structures of the micelles as some complex architectures may be obtained by controlling the crystallization pathway.11–13 There are several strategies for the crystallization-driven self-assembly of the crystalline-coil block copolymers, that are, thermally controlled crystallization,14 morphological transitions,15 hierarchical assembly,16 and “living” crystallization.17 In most cases, the crystalline-coil diblock copolymers contain a crystalline core-forming block such as polyethylene (PE),18–20 isotactic (iPP) or syndiotactic polypropylene (sPP),21,22 poly(ferrocenyldimethylsilane) (PFDMS),23–25 polyethylene oxide (PEO),26–28 poly(L-lactide) (PLLA) or poly(D-lactide) (PDLA),29,30 poly(ε-caprolactone) (PCL),31–35 polythiophene (PTh),36,37 polyacrylonitrile (PAN).38,39 iPS was considered as a semi-crystalline polymer characterized by slow crystallization rates and low crystallinity.40–42 Therefore, the solution crystallization and self-assembly of iPS-core block copolymers in selective solvent have caused great interest.43
Recently, we have reported the self-assembly of iPS-b-PEO in N,N-dimethylformamide (DMF) with iPS as core.2 To the best of our knowledge, there is no example for the investigation of the self-assembly of iPS-b-1,4-trans-PBD in the nonpolar solvent such as n-heptane or n-dodecane.
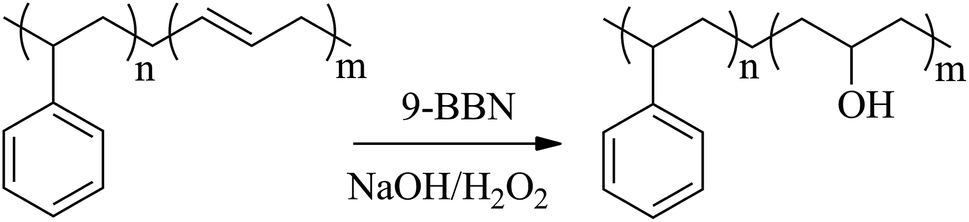
Note that there are a lot of double bonds in the PBD block of the PBD-containing diblock copolymers, which is easy to be hydroxylated. After hydroxylation, the hydroxyl connected to the backbone can initiate the ring-opening polymerization of ethylene oxide using the initiator of naphthalene/potassium to form new diblock copolymers. Undoubtedly, the graft of poly(ethylene oxide) (PEO) can change the polarity of the PBD block and makes it possible to self-assemble in the polar solvent such as water. In this article, the catalytic system [OSSO]-Ti/isobutyl modified methylaluminoxane (MMAO) was used to synthesize the diblock copolymers containing isotactic polystyrene segment and trans-1,4-polybutadiene segment. Furthermore, the iPS-b-1,4-trans-PBD-g-PEO was synthesized and characterized. The self-assembly behaviors of iPS-b-1,4-trans-PBD in n-dodecane and iPS-b-1,4-trans-PBD-g-PEO in aqueous solution were explored.
Experimental
Materials
All reagents were purchased from Aladdin and J&K Scientific. Styrene was dried with calcium hydride and distilled on a vacuum line. 1,3-butadiene was dried with calcium hydride at −40 °C and distilled at room temperature to form a toluene solution. Tetrahydrofuran (THF) was dried by distilling from potassium with benzophenone as an indicator. Toluene was dried by distilling from sodium with benzophenone as an indicator. 9-Borabicyclo[3.3.1]nonane (9-BBN) (Aladdin, 0.5 M in THF) was used as received. NaOH and H2O2 (30 wt%) were used as received. Naphthalene/potassium was prepared using dry THF, naphthalene (Aladdin, ≥ 99%), and excess potassium metal. Ethylene oxide (EO) was distilled and collected on the vacuum line and kept as a liquid at −34 °C. 1.9 M solution of triisobutyl aluminum modified methylalumoxane (MMAO) in heptane was used as received.
Synthesis of isotactic polystyrene-block-1,4-trans-polybutadiene (iPS-b-1,4-trans-PBD). The iPS-b-1,4-trans-PBD was synthesized as literature.1 A typical example was as follows. A 100 mL flask equipped with a magnetic bar was charged with 6.3 mL of a 1.9 M solution of MMAO in n-heptane, 8.7 mL toluene, and 5.0 mL of a solution of 10 μmol [OSSO]-Ti complex in toluene. The mixture was stirred for 5 min at room temperature. The polymerization was started by injecting 0.7 mL styrene. After 2 h, 4.6 mL of a 4.3 M solution of 13-butadiene in toluene was then rapidly added to this flask. The copolymerization continuously proceeded for an additional 2 h. The reaction mixture was terminated by introducing 2 mL ethanol containing the antioxidant. The polymer was precipitated in 200 mL ethanol acidified with 20 mL concentrated hydrochloric acid, recovered by filtration, washed with an excess of ethanol, and dried to constant weight under vacuum at room temperature.
Preparation of isotactic polystyrene-block-hydroxylated 1,4-trans-polybutadiene. The hydroboration of iPS-b-1,4-trans-PBD was performed in a Schlenk flask equipped with a condenser. iPS-b-1,4-trans-PBD was dissolved in dry THF and then 9-BBN was added dropwise at room temperature. After hydroboration at 75 °C for 48 h, the reaction solution was cooled to 0 °C. The required amount of 6 M NaOH (aq.) and then 30 wt% H2O2 (aq.) was added dropwise. After oxidation at 50 °C for 24 h, the iPS-b-hydroxylated 1,4-trans-PBD was precipitated into water and filtered. The resultant polymer was washed with methanol for several times to remove traces of boric acid.
Preparation of isotactic polystyrene-block-(1,4-trans-polybutadiene-graft-poly(ethylene oxide)) (iPS-b-(1,4-trans-PBD-g-PEO)). iPS-b-(1,4-trans-PBD-g-PEO) was prepared by ring-opening graft copolymerization of ethylene oxide on hydroxylated 1,4-trans-PBD block initiated by naphthalene/potassium. The iPS-b-1,4-trans-PBD and dry THF was placed in a Schlenk flask at room temperature. Graft copolymerization of ethylene oxide was initiated by a dark green THF solution of naphthalene/potassium at – 40 °C for 5 min. Heating temperature to 25 °C, the reaction was carried out for more than 24 h until the solution turned to light yellow. The copolymerization solution was quenched with concentrated hydrochloric acid acidic methanol, and then the polymer was precipitated in diethyl ether. After isolation by filtration, the polymer was purified by removing poly(ethylene oxide) homopolymer through dialyzed against deionized water. The insoluble fraction of the iPS-b-(1,4-trans-PBD-g-PEO) was dried under vacuum.
The self-assembly of iPS-b-1,4-trans-PBD in n-dodecane. 1,4-trans-PBD can rapidly dissolve in n-dodecane after slightly heated, while iPS can not dissolve in n-dodecane after heated to 150 °C, which demonstrate that n-dodecane is a selective solvent for iPS-b-1,4-trans-PBD. Moreover, tetrahydrofuran (THF) is a good solvent for both iPS and 1,4-trans-PBD. The solvent-induced self-assembly of iPS-b-1,4-trans-PBD can be generally proceeding as follows. Taking the preparation of self-assembled solution of 0.5 mg mL−1 for example, 5.0 mg of iPS-b-1,4-trans-PBD was dissolved in about 2 mL THF and the solution was added to 10 mL n-dodecane dropwise to obtain a light blue solution indicating the self-assembly of iPS-b-1,4-trans-PBD in n-dodecane. Then the THF was completely removed by vacuum evaporation for several hours proved by 1H NMR.
The self-assembly of iPS-b-(1,4-trans-PBD-g-PEO) in aqueous solution. Water is a suitable selective solvent for the self-assembly of iPS-b-(PBD-g-PEO). The solvent-induced self-assembly of iPS-b-(PBD-g-PEO) is generally proceeding as follows. 5.0 mg of iPS-b-(1,4-trans-PBD-g-PEO) was dissolved in about 2 mL of THF and the solution was added to 10 mL of deionized water to obtain a light blue solution, implying the self-assembly of iPS-b-(1,4-trans-PBD-g-PEO) in aqueous solution. Then the THF was completely removed by dialyzed against deionized water for a week proved by 1H NMR.
Measurements
1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded in CDCl3 at 25 °C on a Varian Unity Inova 400 spectrometer. Molecular weight and molecular weight distribution (Mw/Mn) were determined by gel permeation chromatography (GPC) against narrow molecular weight distribution polystyrene standards on a Waters 2414 refractive index detector at 40 °C with THF as the eluent.
The morphology of the micelles was observed by scanning electron microscope (SEM) (Hitachi S-4800) with an accelerating voltage of 10.0 kV and transmission electron microscope (TEM) (JEM 1400Plus) with an accelerating voltage of 120 kV. For the SEM observation, a drop from the previously prepared micellar solution was deposited onto a mica plate. The mica plate was dried at room temperature for 12 hours before examination by SEM. For the TEM observation, a drop from the previously prepared micellar solution was deposited onto a carbon-coated copper grid.
Dynamic light scattering (DLS) measurements were conducted on a Brookhaven BI-200SM apparatus with a BI-9000AT digital correlator and a He–Ne laser at 632 nm. Data were analyzed by the CONTIN algorithm, while the average hydrodynamic diameter (Dh) and size polydispersity of the particles were obtained by a cumulant analysis of the experimental correlation function.
DSC experiments were carried out on a TA Instruments (New Castle, DE, USA) Q10 calorimeter. Calibration was performed with indium and tin, and all tests were run employing ultrapure nitrogen as the purge gas. DSC heating and cooling scans were performed at 10 °C min−1 over the temperature range 0 °C to 250 °C for standard measurements.
Results and discussion
Synthesis and characterization of iPS-Containing diblock copolymers
In recent years, [OSSO]-based titanium complexes have attracted much attention because of the isospecific polymerization of styrene and α-olefins.1,44,45 In 2003, Okuda et al.44 first reported that [OSSO]-Ti complexes could promote polymerization of styrene upon activation with methylaluminoxane (MAO) to give iPS.46–48 Subsequently, the living stereospecific polymerization of styrene and 1,3-butadiene catalyzed by 1,4-dithiabutandiyl-2,2′-bis(6-cumenyl-4-methylphenoxy) titanium dichloride (complex 1) activated with MAO was reported by Capacchione in 2010.1 In this case, we synthesized the diblock copolymers of iPS-b-1,4-trans-PBD with complex 1 and MMAO used as cocatalyst via sequential monomer addition as shown in Scheme 1 and Table 1.
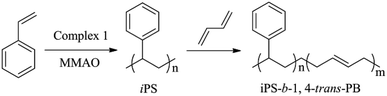 |
| | Scheme 1 The synthesis of iPS-b-14-trans-PBD by complex 1/MMAO via sequential monomer addition. | |
Table 1 Block copolymerization of styrene with 1,3-butadiene catalyzed by complex 1/MMAO via sequential monomer additiona
| Entry |
St/BD in feed (mmol mmol−1) |
CPBDb (%) |
Ctransc (%) |
Mnd × 10−4 |
Mw/Mnd |
TmPS (°C)e |
| Polymerization conditions: Ti = 1.0 μmol; [Al]/[Ti] = 1200; T = 25 °C; Vtot = 20 mL (styrene + toluene); polymerization time, 2 h (styrene) + 2 h (butadiene). PBD content in copolymers, determined from 1H NMR spectra. trans-1,4-regularities in PBD block, determined from 1H NMR spectra. Determined by GPC with THF as the eluent. Melting temperature relative to the isotactic polystyrene block in the resulting iPS-b-1,4-trans-PBD, determined by 10 °C min−1 heating rate DSC scans. |
| 1 |
0/8.0 |
100 |
95 |
4.6 |
1.30 |
— |
| 2 |
1.3/8.0 |
78.7 |
96 |
7.0 |
1.36 |
222 |
| 3 |
2.0/8.0 |
71.4 |
96 |
7.9 |
1.39 |
220 |
| 4 |
3.4/8.0 |
57.1 |
96 |
10.0 |
1.30 |
222 |
| 5 |
8.0/18.7 |
54.1 |
95 |
15.8 |
1.27 |
220 |
| 6 |
8.0/8.0 |
39.1 |
95 |
14.6 |
1.20 |
220 |
| 7 |
8.0/3.4 |
28.8 |
97 |
13.1 |
1.25 |
221 |
| 8 |
8.0/0 |
0 |
— |
11.3 |
1.20 |
221 |
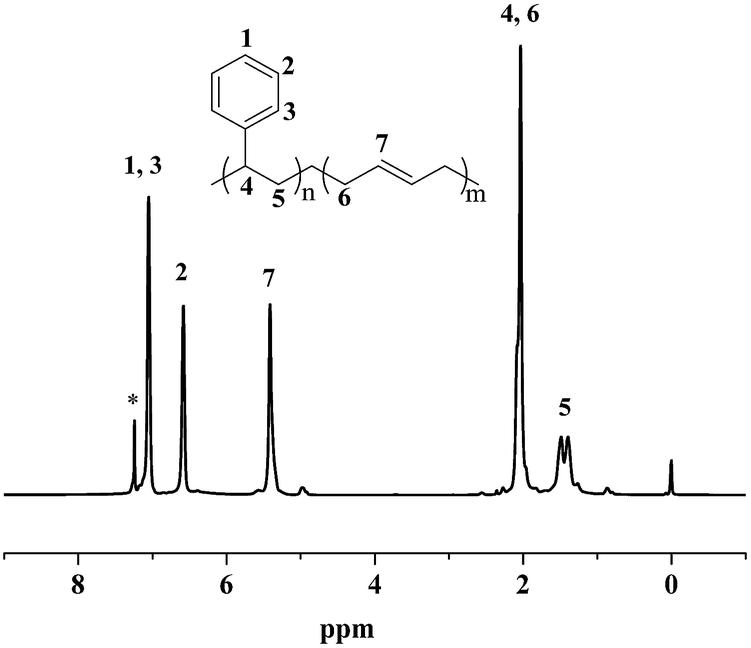
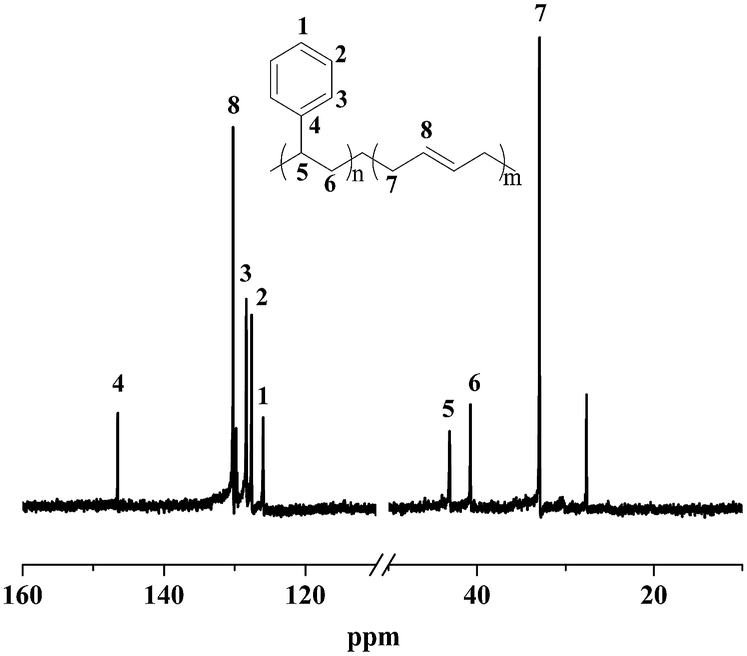
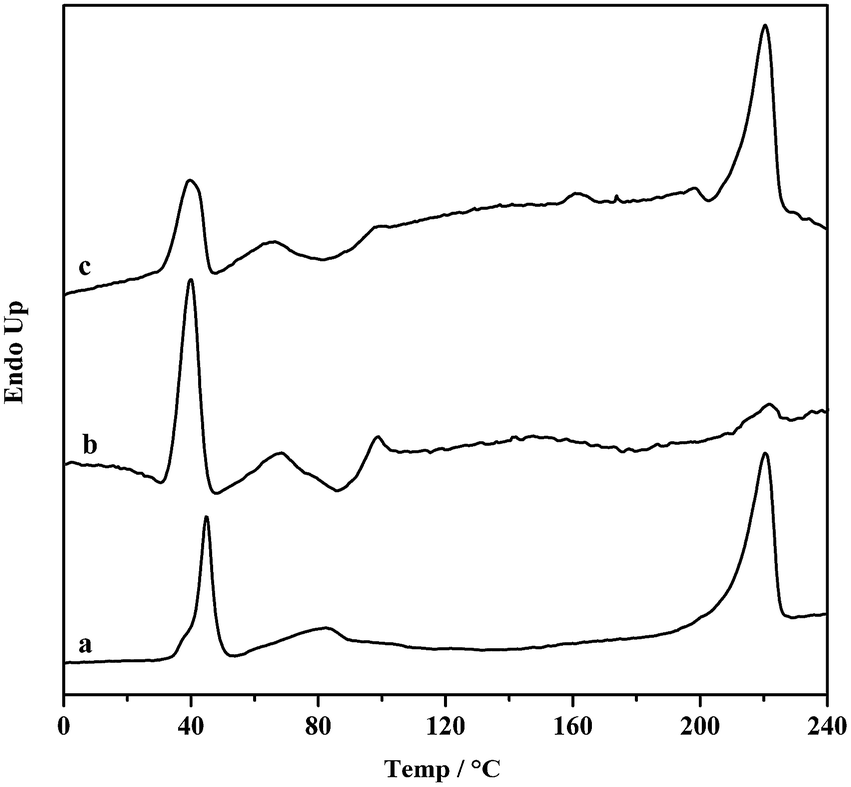
Moreover, the resultant diblock copolymers were characterized by GPC, NMR and DSC. The 1H NMR and 13C NMR (Fig. 1 and 2) shows that the PS block is iPS (mmm ≥ 95%) and the PBD block is 1,4-trans-PBD (1,4-trans addition ≥ 95%). In addition, the relative length of the iPS block and the 1,4-trans-PBD block could be calculated from the 1H NMR. To study the thermal properties, one of the prepared diblock copolymers of iPS-b-1,4-trans-PBD with no other heat treatment or mechanical treatment was used for DSC scan at a heating rate of 10 °C min−1 (the first heating curve for solution crystallization, as shown in Fig. 3a), which exhibits that the melting temperature (Tm) of iPS block (Mn = 9.2 × 104) is 220 °C and 1,4-trans-PBD block (Mn = 2.9 × 104) is 45 °C/82 °C, indicating these two blocks were crystalline. After cooling at a rate of 10 °C min−1, this copolymer was heating again at a rate of 10 °C min−1 (the second heating curve, as shown in Fig. 3b). In this case, the area of crystal-melting peak of iPS at 220 °C was much smaller than that in the first heating curve, suggesting the very slow crystallization rate of iPS. To confirm this, the third heating curve at a rate of 10 °C min−1 as shown in Fig. 3c was displayed after the isothermal crystallization of this sample at 120 °C for 3 hours. As might be expected, the area of crystal-melting peak of iPS at 220 °C greatly increased, which is even bigger than that in the first heating curve.
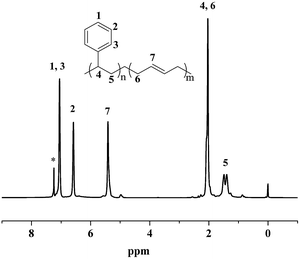 |
| | Fig. 1 1H NMR spectrum of iPS-b-1,4-trans-PBD (Entry 6 in Table 1) synthesized by complex 1/MMAO via sequential monomer addition. | |
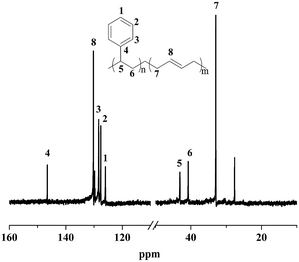 |
| | Fig. 2 13C NMR spectrum of iPS-b-1,4-trans-PBD (Entry 6 in Table 1) synthesized by complex 1/MMAO via sequential monomer addition. | |
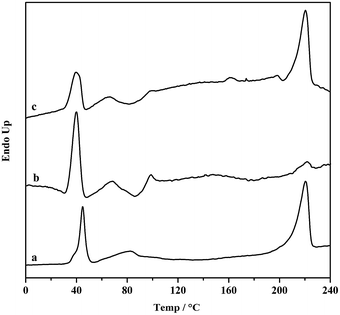 |
| | Fig. 3 DSC scans of iPS-b-1,4-trans-PBD with Mn = 1.21 × 105 at 10 °C min−1 heating rate, (a) the first heating curve of the newly prepared sample; (b) the second heating curve after cooling at a rate of 10 °C min−1; (c) the third heating curve after isothermal crystallization at 120 °C for 3 h. | |
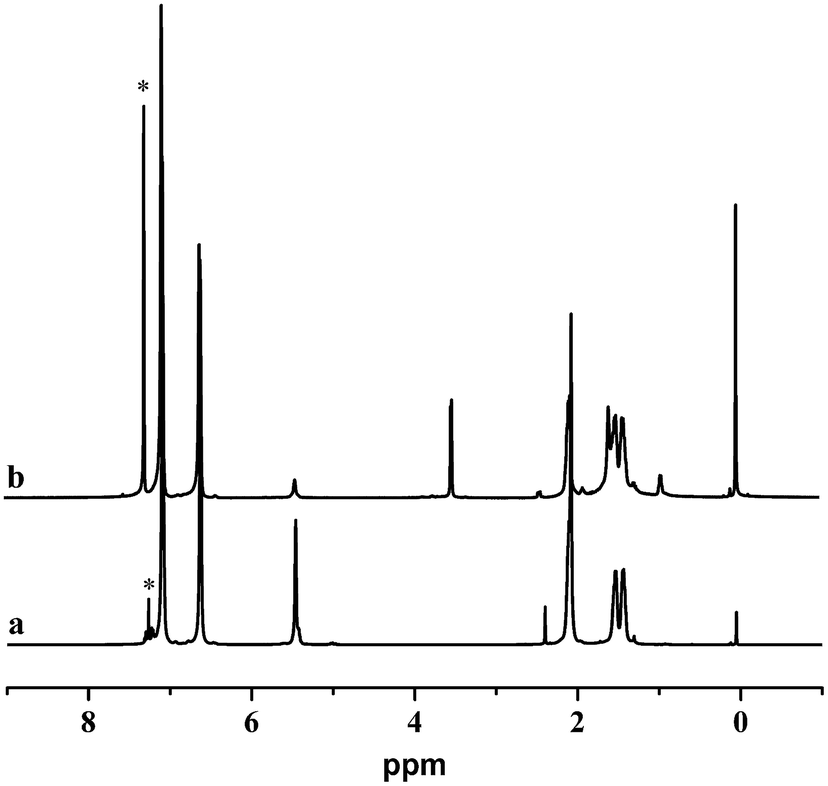
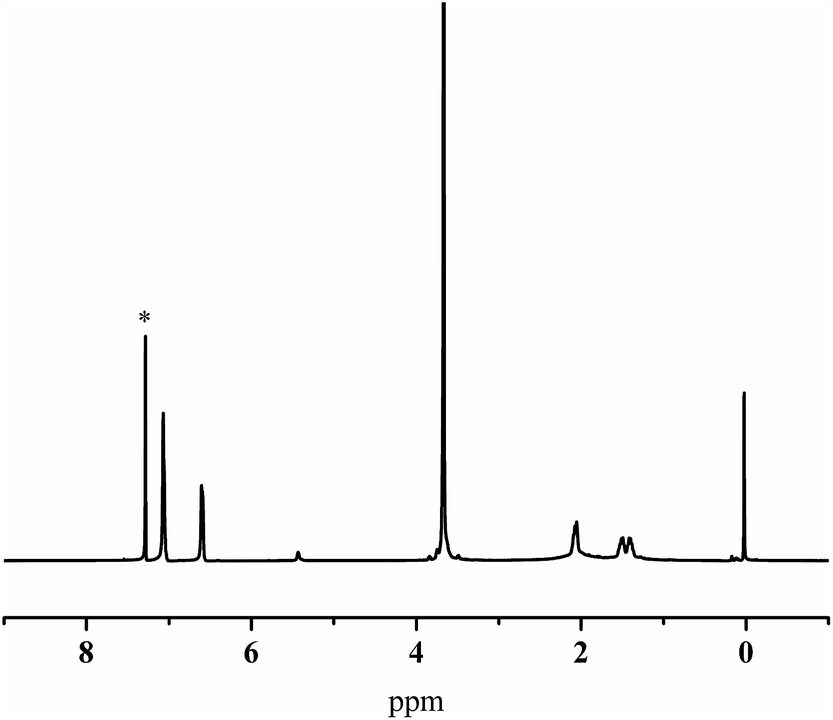
The hydroxylation of the 1,4-trans-PBD block was carried out as procedures reported.49,50 Note that the hydroboration of the 1,4-microstructure of polybutadiene was more difficult than the 1,2-microsture.49 Moreover, as reported by Ramakrishnan,50 83% 1,4-cis-polybutadiene synthesized by the ring-opening metathesis polymerization of 1,5-cyclooctadiene can be easily hydroborated at around 60 °C. However, in this case, more than 95% 1,4-trans-PBD block in the copolymer can handly be hydroborated on the same conditions. Therefore, the temperature of hydroboration was raised to 75 °C and the 1,4-trans-PBD block was almost completely hydroxylated. As shown in Fig. 4, the 1,4-trans-PBD block was almost completely hydroxylated. Compared with the 1H NMR spectrum of the 1,4-trans-PBD block before hydroxylation, the corresponding peak at 5.44 ppm assigned to the chemical shift of unsaturated bond in 1,4-trans-PBD block almost disappears and the characteristic peak at 3.52 ppm of the methine proton of CHOH appears after hydroxylation.
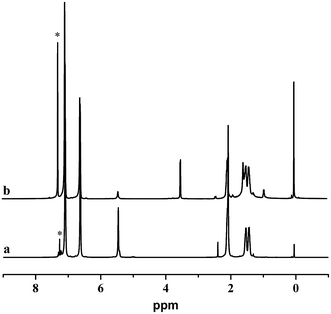 |
| | Fig. 4 1H NMR spectra of iPS-b-1,4-trans-PBD (Entry 6 in Table 1) before (a) and after (b) hydroxylation. | |
The graft of PEO into 1,4-trans-PBD block was performed using naphthalene/potassium as initiator via anion ring-opening polymerization of ethylene oxide (as shown in Scheme 2). To remove as completely as possible the PEO homopolymer, the graft copolymerization products were dispersed in water by stirring and dialyzed against deionized water with a dialysis bag molecular weight cut off of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for a week. The GPC results indicated that the PEO homopolymer was almostly removed by dialysis against deionized water. Fig. 5 shows a representative 1H NMR spectrum of iPS-b-(1,4-trans-PBD-g-PEO) prepared from Entry 6 in Table 1. The high and shark peak at 3.67 ppm is the characteristic chemical shift of PEO and the characteristic peak of the methine proton of CHOH at 3.52 ppm almost disappears. These results demonstrated that the iPS-b-(1,4-trans-PBD-g-PEO) was successfully prepared.
000 for a week. The GPC results indicated that the PEO homopolymer was almostly removed by dialysis against deionized water. Fig. 5 shows a representative 1H NMR spectrum of iPS-b-(1,4-trans-PBD-g-PEO) prepared from Entry 6 in Table 1. The high and shark peak at 3.67 ppm is the characteristic chemical shift of PEO and the characteristic peak of the methine proton of CHOH at 3.52 ppm almost disappears. These results demonstrated that the iPS-b-(1,4-trans-PBD-g-PEO) was successfully prepared.
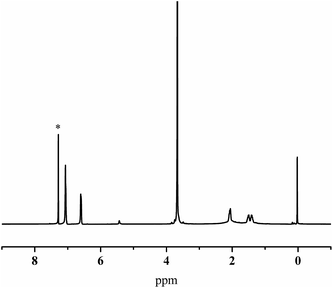 |
| | Fig. 5 1H NMR spectrum of iPS-b-(1,4-trans-PBD-g-PEO) prepared from Entry 6 in Table 1 by graft copolymerization. | |
 |
| | Scheme 2 The hydroxylation of iPS-b-1,4-trans-PBD synthesized by complex 1/MMAO via sequential monomer addition. | |
The self-assembly of iPS-b-1,4-trans-PBD in n-dodecane
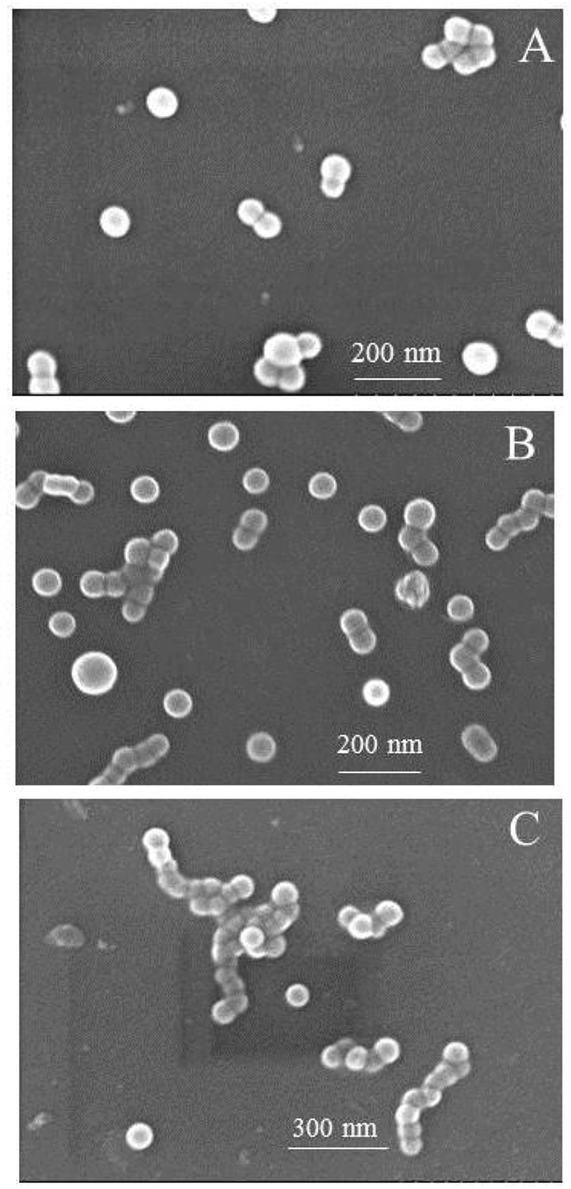
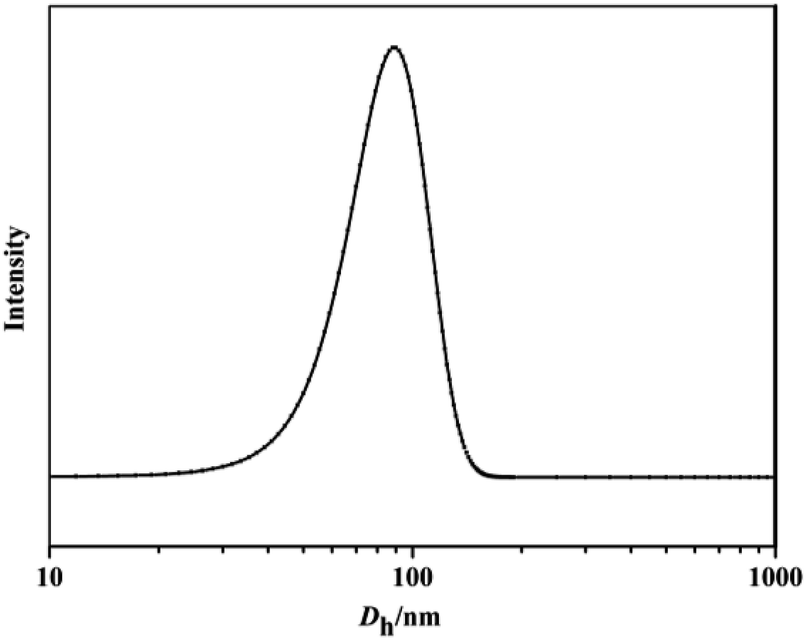
The self-assembly of iPS-b-1,4-trans-PBD was performed in n-dodecane, a selective solvent for 1,4-trans-PBD. A solution of the iPS-b-1,4-trans-PBD in THF was added dropwise to n-dodecane to form a light blue solution, indicating the self-assembly process of the iPS-b-1,4-trans-PBD (Entry 6 in Table 1). Then the THF was completely removed by vacuum evaporation. DLS measurement displays that the average hydrodynamic diameter (Dh) of the self-assembled micelles of iPS-b-1,4-trans-PBD in n-dodecane were around 80 nm as shown in Fig. 6, which is much larger than that of the individual chain in THF (∼35 nm), confirming the self-assembling of iPS-b-1,4-trans-PBD in n-dodecane. Fig. 7 shows the SEM micrographs of the self-assembled micelles of iPS-b-1,4-trans-PBD in n-dodecane after prepared for 0 day, 7 days and 14 days, respectively. Spherical micelles with a diameter of about 60 nm were observed when the micelles were newly prepared. After prepared for two weeks, the micelles aggregated but keep their morphology to be spherical and identical diameter of about 60 nm, which suggests that the iPS core did not crystallize at room temperature as aging. To explain these outcomes, we suppose that the core-forming iPS block could not crystallize or have a very low degree of crystallinity in nanoconfined space of micelles when the micelles were newly formed by removing the THF. After aged for a period at room temperature, the core-forming iPS block still could not crystallize as a result of the room temperature much lower than the glass transition temperature. The TEM images of self-assembled micelles of iPS-b-1,4-trans-PBD (as shown in Fig. 8) in n-dodecane also suggested the formation of spherical micelles.
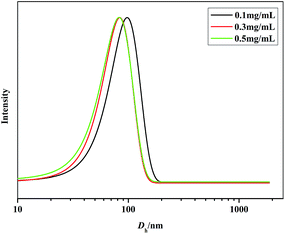 |
| | Fig. 6 Dh distributions of the self-assembled micelles of iPS-b-1,4-trans-PBD (Entry 6 in Table 1) in n-dodecane (0.1 mg mL−1, 0.5 mg mL−1, 1.0 mg mL−1, respectively). | |
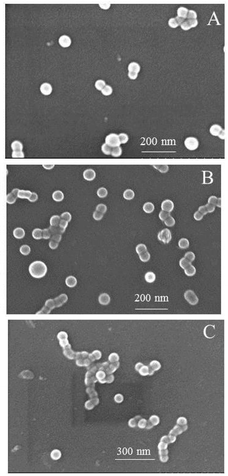 |
| | Fig. 7 SEM microscopy of the self-assembled micelles of iPS-b-1,4-trans-PBD (Entry 6 in Table 1) in n-dodecane after prepared for, respectively, (A). 0 day, (B). 7 day, (C). 14 day. | |
 |
| | Fig. 8 TEM images of the self-assembled micelles of iPS-b-(1,4-trans-PBD) prepared from Entry 6 in Table 1 in n-dodecane. | |
To confirm our speculation, the prepared micellar solution was heated to 120 °C for 3 h to promote the crystallization of the core-forming iPS block. Fig. 9 shows the SEM microscopy of the self-assembled micelles of iPS-b-1,4-trans-PBD (Entry 6 in Table 1) before and after isothermal crystallization. The micellar morphology changed from sphere-like to platelet-like and the diameter changed larger after isothermal crystallization. The TEM images of the self-assembled micelles before and after isothermal crystallization are exhibited in Fig. 10, which also suggests that the micellar morphology changed from sphere-like to bigger platelet-like induced by the crystallization of the iPS core.
 |
| | Fig. 9 SEM microscopy of the self-assembled micelles of iPS-b-1,4-trans-PBD before (A) and after (B) isothermal crystallization at 150 °C for 3 hours. | |
 |
| | Fig. 10 TEM images of the self-assembled micelles of iPS-b-1,4-trans-PBD (Entry 6 in Table 1) before (left) and after (right) isothermal crystallization at 120 °C for 3 hours. | |
The self-assembly of iPS-b-(1,4-trans-PBD-g-PEO) in aqueous solution
The self-assembly of iPS-b-(1,4-trans-PBD-g-PEO) was performed in aqueous solution, which is a selective solvent for PEO. A solution of the iPS-b-(1,4-trans-PBD-g-PEO) in THF was added dropwise to deionized water to form a light blue solution, indicating the self-assembly process of the iPS-b-(1,4-trans-PBD-g-PEO). After the THF was completely removed by dialysis against deionized water, the solution was detected by DLS. As exhibited in Fig. 11, the average hydrodynamic diameter (Dh) of the representative self-assembled micelles of iPS-b-(1,4-trans-PBD-g-PEO) prepared from Entry 6 in Table 1 were around 90 nm with narrow distribution, confirming the self-assembling of iPS-b-(1,4-trans-PBD-g-PEO) in aqueous solution. In addition, the self-assembled micellar solution was so stable that no obvious precipitation could be observed after prepared for a month. Fig. 12 shows the SEM microscopy of the self-assembled micelles of iPS-b-(1,4-trans-PBD-g-PEO) in aqueous solution after prepared for, 0 day, 15 day and 30 day.
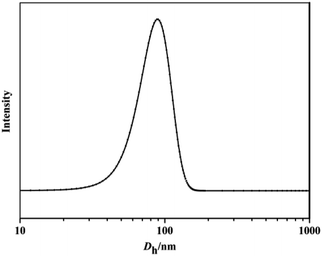 |
| | Fig. 11 Dh distributions of the self-assembled micelles of iPS-b-(1,4-trans-PBD-g-PEO) prepared from Entry 6 in Table 1 in aqueous solution. | |
 |
| | Fig. 12 SEM microscopy of the self-assembled micelles of iPS-b-(1,4-trans-PBD-g-PEO) prepared from Entry 6 in Table 1 in aqueous solution after prepared for, respectively, (A). 0 day, (B). 15 day, (C). 30 day. | |
Spherical micelles with a diameter of around 70 nm were observed and keep unchanged as time went on. These results were consistent with the DLS detect and its self-assembly behavior was similar to that of iPS-b-1,4-trans-PBD in n-dodecane, suggesting the formation of an amorphous iPS core or with low degree of crystallinity in the micelles as a result of slow crystallization rate of iPS and confined crystallization in nano-space. Fig. 13 displays the TEM images of self-assembled micelles of iPS-b-(1,4-trans-PBD-g-PEO) in aqueous solution. The uniform round dark domains in the images confirm that spherical micelles but not vesicles were formed and the iPS core was amorphous or low crystalline.
 |
| | Fig. 13 TEM image of the self-assembled micelles of iPS-b-(1,4-trans-PBD-g-PEO) prepared from Entry 6 in Table 1 in aqueous solution. | |
Conclusions
iPS-containing diblock copolymers including iPS-b-1,4-trans-PBD and a novel amphiphilic diblock copolymer of iPS-b-(1,4-trans-PBD-g-PEO) with controlled molecular weight, block ratio and narrow molecular weight distribution were synthesized in terms of living coordination polymerization and graft copolymerization. Moreover, this work also demonstrates the morphology and morphology transition of self-assembled micelles of iPS-b-1,4-trans-PBD in n-dodecane and iPS-b-(1,4-trans-PBD-g-PEO) in aqueous solution. These iPS-containing diblock copolymers can all self-assemble into spherical micelles in n-dodecane and in aqueous solution, respectively, implying that the core-forming iPS block could not crystallize or have a very low degree of crystallinity in nanoconfined space of micelles. It is interesting that heating the micellar solution of iPS-b-1,4-trans-PBD in n-dodecane to 120 °C and keeping the constant temperature for 3 hours, the micellar morphology changed from sphere-like to platelet-like as a result of the isothermal crystallization of the core-forming iPS block. We suggest that isothermal crystallization would induce the deformation of the micelles.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21174167, 51573212) and the NSF of Guangdong Province (S2013030013474, 2014A030313178).
Notes and references
- A. Proto, A. Avagliano, D. Saviello, R. Ricciardi and C. Capacchione, Macromolecules, 2010, 43, 5919 CrossRef CAS.
- Z. Y. Li, R. Liu, B. Y. Mai, F. Feng, Q. Wu, G. D. Liang, H. Y. Gao and F. M. Zhu, Polym. Chem., 2013, 4, 954 RSC.
- N. Hadjichristidis, H. Iatou and M. Pitsikalis, Adv. Polym. Sci., 2005, 189, 1 CrossRef CAS.
- W. Zheng, N. Yan, Y. Zhu, W. Zhao, C. Zhang, H. Zhang, C. Bai, Y. Hu and X. Zhang, Polym. Chem., 2015, 6, 6088 RSC.
- N. Yan, Y. Zhang, Y. He, Y. Zhu and W. Jiang, Macromolecules, 2017, 50, 6771 CrossRef CAS.
- Y. Han, H. Yu, H. Du and W. Jiang, J. Am. Chem. Soc., 2010, 132, 1144 CrossRef CAS PubMed.
- N. Yan, H. Liu, Y. Zhu, W. Jiang and Z. Dong, Macromolecules, 2015, 48, 5980 CrossRef CAS.
- Y. Zhang, Y. He, N. Yan, Y. Zhu and Y. Hu, J. Phys. Chem. B, 2017, 121, 8417 CrossRef CAS PubMed.
- M. Wu, Y. Zhu and W. Jiang, ACS Macro Lett., 2016, 5, 1212 CrossRef CAS.
- Y. He, Y. Zhang, N. Yan, Y. Zhu, W. Jiang and D. Shi, Nanoscale, 2017, 9, 15056 RSC.
- W. N. He and J. T. Xu, Prog. Polym. Sci., 2012, 37, 1350 CrossRef CAS.
- J. Schmelz, F. H. Schacher and H. Schmalz, Soft Matter, 2013, 9, 2101 RSC.
- J. J. Crassous, P. Schurtenberger, M. Ballauff and A. M. Mihut, Polymer, 2015, 62, A1 CrossRef CAS.
- A. M. Mihut, J. J. Crassous, H. Schmalz, M. Drechsler and M. Ballauff, Soft Matter, 2012, 8, 3163 RSC.
- J. Raez, J. P. Tomba, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2003, 125, 9546 CrossRef CAS PubMed.
- J. Raez, I. Manners and M. A. Winnik, Langmuir, 2002, 18, 7229 CrossRef CAS.
- S. F. M. Yusoff, J. B. Gilroy, G. Cambridge, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 11220 CrossRef CAS PubMed.
- E. K. Lin and A. P. Gast, Macromolecules, 1996, 29, 4432 CrossRef CAS.
- H. Schmalz, J. Schmelz, M. Drechsler, J. Yuan, A. Walther, K. Schweimer and A. M. Mihut, Macromolecules, 2008, 41, 3235 CrossRef CAS.
- T. Li, W. J. Wang, R. Liu, W. H. Liang, G. F. Zhao, Z. Y. Li, Q. Wu and F. M. Zhu, Macromolecules, 2009, 42, 3804 CrossRef CAS.
- R. Liu, Z. Y. Li, B. Y. Mai, Q. Wu, G. D. Liang, H. Y. Gao and F. M. Zhu, J. Polym. Res., 2013, 20, 64 CrossRef.
- A. Radulescu, R. T. Mathers, G. W. Coates, D. Richter and L. J. Fetters, Macromolecules, 2004, 7, 6962 CrossRef.
- J. Massey, K. N. Power, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 1998, 20, 9533 CrossRef.
- G. Guerin, J. Raez, I. Manners and M. A. Winnik, Macromolecules, 2005, 38, 7819 CrossRef CAS.
- X. S. Wang, K. Liu, A. C. Arsenault, D. A. Rider, G. A. Ozin, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2007, 129, 5630 CrossRef CAS PubMed.
- J. T. Xu, J. P. A. Fairclough, S. M. Mai and A. J. Ryan, J. Mater. Chem., 2003, 13, 2740 RSC.
- J. T. Xu, G. D. Liang, S. M. Mai and A. J. Ryan, Chem. J. Chin. Univ. Chin., 2004, 25, 1978 CAS.
- A. M. Mihut, M. Drechsler, M. Moller and M. Ballauff, Macromol. Rapid Commun., 2010, 31, 449 CrossRef CAS PubMed.
- D. Portinha, F. Boue, L. Bouteiller, G. Carrot, C. Chassenieux, S. Pensec and G. Reiter, Macromolecules, 2007, 40, 4037 CrossRef CAS.
- J. Fu, B. Luan, X. Yu, Y. Cong, J. Li, C. Y. Pan, Y. C. Han, Y. M. Yang and B. Y. Li, Macromolecules, 2004, 37, 976 CrossRef CAS.
- K. Rajagopal, A. Mahmud, D. A. Christian, J. D. Pajerowski, A. E. X. Brown, S. M. Loverde and D. E. Discher, Macromolecules, 2010, 43, 9736 CrossRef CAS PubMed.
- Z. X. Du, J. T. Xu and Z. Q. Fan, Macromolecules, 2007, 40, 7633 CrossRef CAS.
- Z. X. Du, J. T. Xu and Z. Q. Fan, Macromol. Rapid Commun., 2008, 29, 467 CrossRef CAS.
- W. N. He, J. T. Xu, B. Y. Du, Z. Q. Fan and X. S. Wang, Macromol. Chem. Phys., 2010, 211, 1909 CrossRef CAS.
- S. C. Chan, S. W. Kuo, C. H. Lu, H. F. Lee and F. C. Chang, Polymer, 2007, 48, 5059 CrossRef CAS.
- Z. C. Li, R. J. Ono, Z. Q. Wu and C. W. Bielawski, Chem. Commun., 2011, 47, 197 RSC.
- A. C. Kamps, M. Fryd and S. J. Park, ACS Nano, 2012, 6, 2844 CrossRef CAS PubMed.
- M. Lazzari, D. Scalarone, C. E. Hoppe, C. Vazquez-Vazquez and M. A. Lopez-Quintela, Chem. Mater., 2007, 19, 5818 CrossRef CAS.
- M. Lazzari, D. Scalarone, C. Vazquez-Vazquez and M. A. Lopez-Quintela, Macromol. Rapid Commun., 2008, 29, 352 CrossRef CAS.
- G. Natta, P. Corradini and I. W. Bassi, Nuovo Cimento, 1960, 15, 68 CrossRef CAS.
- P. J. Lemstra and G. Challa, J. Polym. Sci., Part B: Polym. Lett., 1975, 13, 1809 CAS.
- P. J. Lemstra, J. Postma and G. Challa, Polymer, 1974, 15, 757 CrossRef CAS.
- G. Xue, Y. Wang, X. Gu and Y. Lu, Macromolecules, 1994, 27, 4016 CrossRef CAS.
- C. Capacchione, A. Proto, H. Ebeling, R. Mulhaupt, K. Moller, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2003, 125, 4964 CrossRef CAS PubMed.
- K. Beckerle, R. Manivannan, T. P. Spaniol and J. Okuda, Organometallics, 2006, 25, 3019 CrossRef CAS.
- C. Capacchione, R. Manivannan, M. Barone, K. Beckerle, R. Centore, L. Oliva, A. Proto, A. Tuzi, T. P. Spaniol and J. Okuda, Organometallics, 2005, 24, 2971 CrossRef CAS.
- A. Rodrigues, E. Kirillov, T. Roisnel, A. Razavi, B. Vuillemin and J. F. Carpentier, Angew. Chem., Int. Ed., 2007, 46, 7240 CrossRef CAS PubMed.
- T. Kawasaki, C. Hohberger, Y. Araki, K. Hatase, K. Beckerle, J. Okuda and K. Soai, Chem. Commun., 2009, 5621 RSC.
- T. C. Chung, M. Raate, M. Berluche and D. M. Schulz, Macromolecules, 1988, 21, 1903 CrossRef CAS.
- S. Ramakrishnan, Macromolecules, 1991, 24, 3753 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab and
FangMing Zhu
ab and
FangMing Zhu