
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancements in six-membered cyclic carbonate (1,3-dioxan-2-one) synthesis utilizing carbon dioxide as a C1 source
Akram Hosseinian
a,
Sepideh Farshbaf
b,
Robab Mohammadi
*c,
Aazam Monfared
c and
Esmail Vessally
 c
c
aDepartment of Engineering Science, College of Engineering, University of Tehran, P. O. Box 11365-4563, Tehran, Iran
bDepartment of Chemistry, Center for Photochemical Sciences, Bowling Green State University, Bowling Green, Ohio 43403, USA
cDepartment of Chemistry, Payame Noor University, P. O. Box 19395-3697, Tehran, Iran. E-mail: mohammadi_rb@yahoo.com
First published on 16th May 2018
Abstract
This review article surveys literature methods for the synthesis of six-membered cyclic carbonates using various substrates in the presence of CO2 with special emphasis on the mechanistic aspects of the reactions. We have classified these reactions based on the type of starting material.
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from University of Tehran, Iran, and her M.S. degree in inorganic chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed her PhD degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now she is working at University of Tehran as Associate Professor. Her research interests include inorganic and organic synthesis, new methodologies in nano material synthesis. |
 Sepideh Farshbaf | Sepideh Farshbaf was born in Sari, Iran and graduated from Kharazmi University, Tehran, with a bachelor degree in applied chemistry. In 2009, she obtained her M.Sc. degree in organic chemistry from K. N. Toosi University of Technology, Tehran, Iran, where she carried out research with Prof. Movassagh. Sepideh has been teaching various courses as a university lecturer at Payame Noor University, Karaj, Iran (2011–2015). In 2015, she has started her PhD program in the center for photochemical sciences at Bowling Green State University, Ohio, USA, under the supervision of Prof. Anzenbacher. Her field of study is organic synthesis and supramolecular materials for molecular sensing. |
 Robab Mohammadi | Robab Mohammadi was born in Tabriz, Iran, in 1979. She received her B.S. degree in Pure Chemistry from University of Tabriz, Iran, and her M.S. degree in applied chemistry from Islamic Azad University, Tabriz Branch, Tabriz, Iran, in 2008 under the supervision of Dr L. Edjlali. She received her PhD degree in 2012 under the supervision of Prof. M. Rabani and Prof. B. Massoumi from Islamic Azad University, Tehran Shomal Branch, Tehran, Iran. Now she is working at Payame Noor University of Tabriz as Associate Professor. Her research interests include nano materials synthesis and new methodologies in material synthesis. |
 Aazam Monfared | Aazam Monfared was born in Tehran, Iran, in 1965. She received her B.S. degree in Pure Chemistry from University of Shahid Beheshti, Tehran, Iran, and her M.S. degree in Organic chemistry from Shahid Beheshti University, Tehran, Iran, in 1991 under the supervision of Prof. A. Rustaiyan. She received her PhD degree in 1999 under the supervision of Prof. A. Rustaiyan in Shahid Beheshti University, Tehran, Iran. Now, she is working at Payame Noor University of Tehran as Associate Professor. Her research interests include organic synthesis, phytochemistry, drug synthesis, nano chemistry, methodologies and theoretical chemistry. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as full Professor of Organic Chemistry. His research interests include Theoretical Organic Chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
Needless to say that the global warming trend and climate change are caused by the emission of greenhouse gases into the Earth's atmosphere. Interestingly, ∼72% of the total emitted greenhouse gases is carbon dioxide (CO2) and every year, human activity releases ∼38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 million tons of this gas into the atmosphere.1 Growing environmental concerns in recent years for global warming, climate change, and ocean acidification have motivated research activities toward developing efficient and green strategies to capture, store, and utilize COa.1,2 On the other hand, from the viewpoint of chemistry, CO2 is considered as an abundant, inexpensive, nontoxic, nonflammable, and renewable carbon feedstock for producing value-added organic compounds (e.g. alcohols, carboxylic acids, esters, aldehydes, amides, urethanes, ureas, quinazoline-2,4(1H,3H)-diones, and carbonates).3–13 However, most reactions involving CO2 require the use of stoichiometric amounts of catalysts and/or drastic reaction conditions.14,15 Therefore, development of more-efficient and improved synthetic protocols for chemical transformation of CO2 is a very attractive topic in modern organic synthesis.
000 million tons of this gas into the atmosphere.1 Growing environmental concerns in recent years for global warming, climate change, and ocean acidification have motivated research activities toward developing efficient and green strategies to capture, store, and utilize COa.1,2 On the other hand, from the viewpoint of chemistry, CO2 is considered as an abundant, inexpensive, nontoxic, nonflammable, and renewable carbon feedstock for producing value-added organic compounds (e.g. alcohols, carboxylic acids, esters, aldehydes, amides, urethanes, ureas, quinazoline-2,4(1H,3H)-diones, and carbonates).3–13 However, most reactions involving CO2 require the use of stoichiometric amounts of catalysts and/or drastic reaction conditions.14,15 Therefore, development of more-efficient and improved synthetic protocols for chemical transformation of CO2 is a very attractive topic in modern organic synthesis.
1,3-Dioxan-2-one is a cyclic carbonate ester having the chemical formula C4H6O3. This potential monomer continues to play an overwhelming role in the synthesis of aliphatic polycarbonates and polyurethanes, which are of interest for biomedical applications because of their low toxicity and biodegradability.16–19 The most common preparation of 1,3-dioxan-2-one (six-membered cyclic carbonate) monomers involves the reaction of 1,3-diols with phosgene derivatives20,21 or carbon monoxide.22 However, both phosgene and CO are very toxic, which extremely limits their employment in academic and industrial research. Over the past few years, CO2 has emerged as an alternative to phosgene and CO in carbonate synthesis.23,24 Along this line, various synthetic organic chemists have put forward considerable efforts in the synthesis of five- and six-membered cyclic carbonates using various substrates in the presence of CO2. Due to the inherent thermodynamic instability of six-membered cyclic carbonates (Scheme 1), synthesis of these compounds utilizing CO2 as a C1 source is much less studied than the corresponding five-membered cycles.
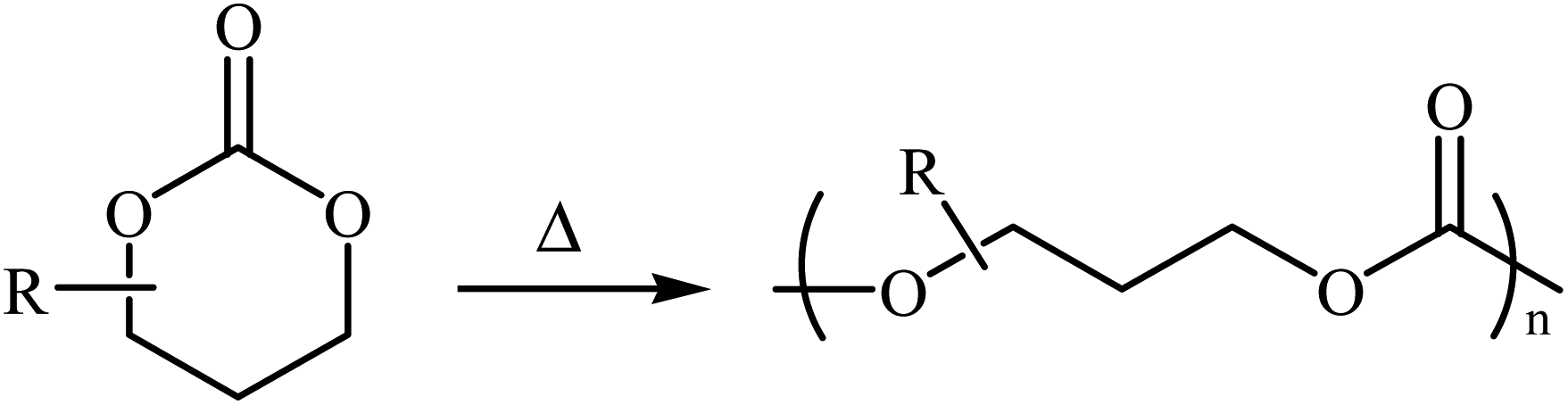 | ||
| Scheme 1 The ring-opening polymerization of 1,3-dioxan-2-ones is thermodynamically favored at all temperatures.25 | ||
As a part of our continual review papers on the chemical conversion of CO2 into value-added organic compound,15,26 herein we will highlight the most important advances on the synthesis of six-membered cyclic carbonates using various substrates in the presence of CO2 (Fig. 1), by hoping that it will stimulate researchers to develop more-efficient and improved methods for the synthesis of these extremely important potential monomers with sustainable and green chemistry perspectives. It should be mentioned that special emphasis is laid on mechanistic aspects of the reactions.
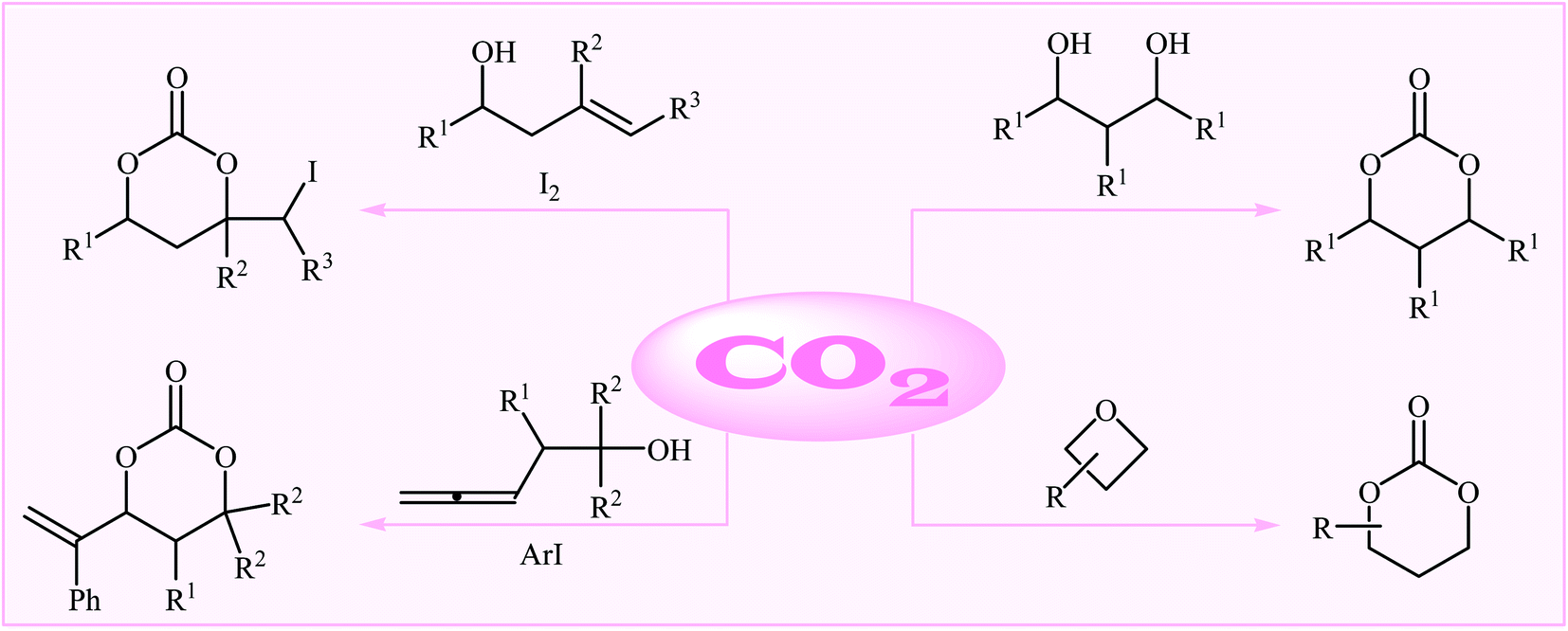 | ||
| Fig. 1 Synthesis of six-membered cyclic carbonates using various substrates in the presence of CO2. | ||
2. From 1,3-diols
Chemical fixation of carbon dioxide to 1,2-diols is a well-known synthetic route to five-membered cyclic carbonates and has been the subject of numerous research studies and reviews.27 In contrary, direct six-membered cyclic carbonates synthesis from carbon dioxide and 1,3-diols has been seldom addressed in the literature. One of the earliest general methods for six-membered cyclic carbonates synthesis through carboxylative cyclization of 1,3-diols with CO2 was published in 2014, by Honda et al.28 They showed that treatment of diols 1 with CeO2 as a catalyst and 2-cyanopyridine as a dehydrating agent under the CO2 atmosphere (5 MPa) furnished corresponding six-membered cyclic carbonates 2 in good to excellent yields (Scheme 2). It is noted that the presence of 2-cyanopyridine was crucial to the success of this reaction. In the absence of 2-cyanopyridine very low yield of desired product was observed, which is due to the equilibrium limitation. The mechanistic pathway by the authors for this cyclization starts with the formation of cerium alkoxide species A from the adsorption of one OH group of diol 1 to Lewis acid sites of CeO2, followed by insertion of CO2 into the Ce–O bond in intermediate A to give alkyl carbonate species B. Next, intramolecular nucleophile attack of the other OH group to the carbonyl carbon in intermediate B leads to the cyclic carbonate 2 and one molecule of water with concomitant regeneration of CeO2. Finally, hydration of 2-cyanopyridine with the produced water affords corresponding amide (Scheme 3). | ||
| Scheme 2 CeO2-catalyzed synthesis of six-membered cyclic carbonates 2 from 1,3-diols 1 and CO2 developed by Honda et al.28 | ||
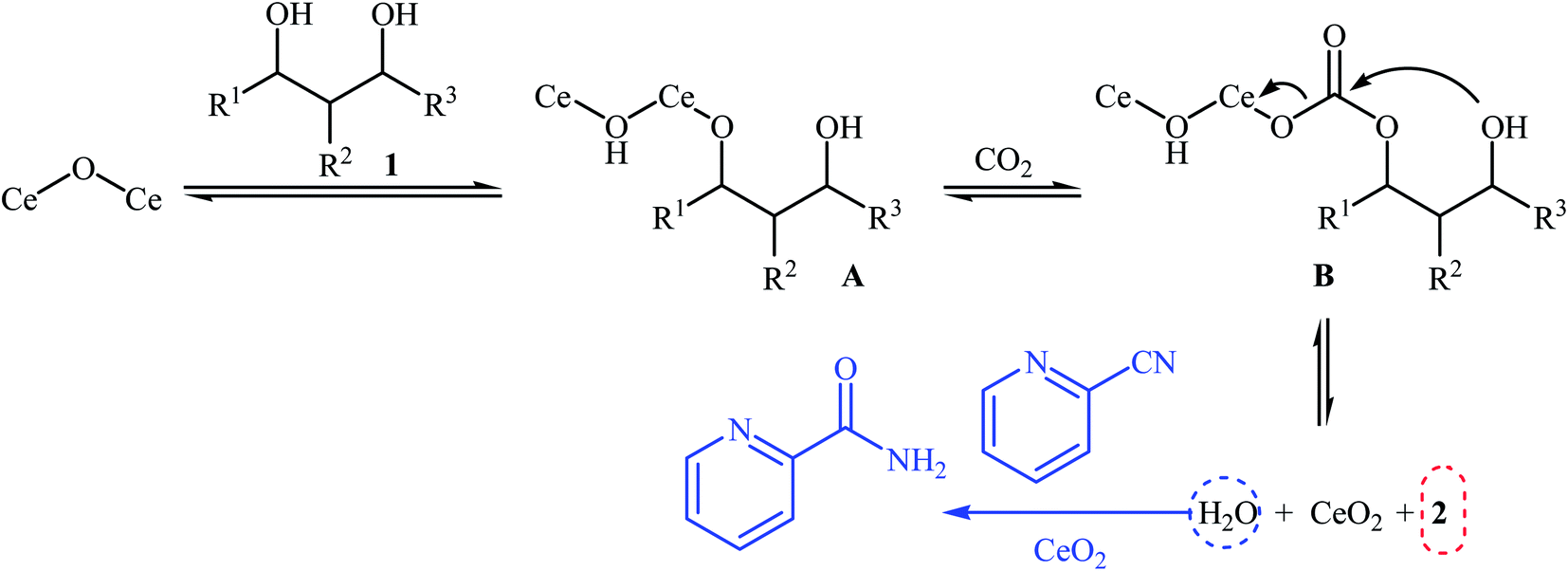 | ||
| Scheme 3 Mechanism that accounts for the formation of carbonates 2. | ||
Subsequently, with the objective of designing a milder procedure to six-membered cyclic carbonates via the carboxylative cyclization of 1,3-diols with CO2, Gregory, Ulmann and Buchard were able to demonstrate that a series of substituted carbonates 4 could be obtained in satisfactory yields from the reaction of corresponding diols 3 with CO2 under very mild conditions (room temperature and atmospheric pressure) employing 1,8-diazabicyclo-[5.4.0]-undec-7-ene (DBU) as a promotor and CHCl3 as the solvent (Scheme 4). Important functional groups such as ester and ether functionalities that would allow further elaboration of the products were well tolerated under this reaction conditions. The authors investigated the detailed reaction mechanism with density functional theory (DFT) calculations. As shown in Fig. 2 this reaction proceeds through an addition/elimination pathway.29
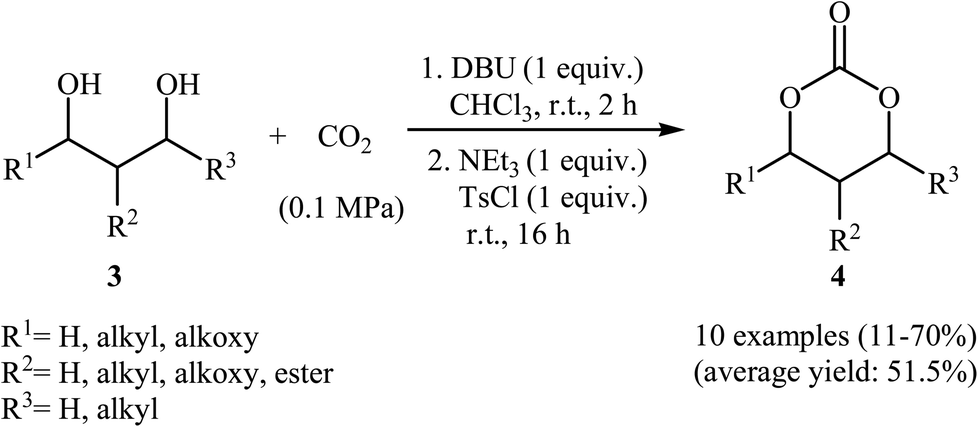 | ||
| Scheme 4 Buchard's synthesis of six-membered cyclic carbonates 4. | ||
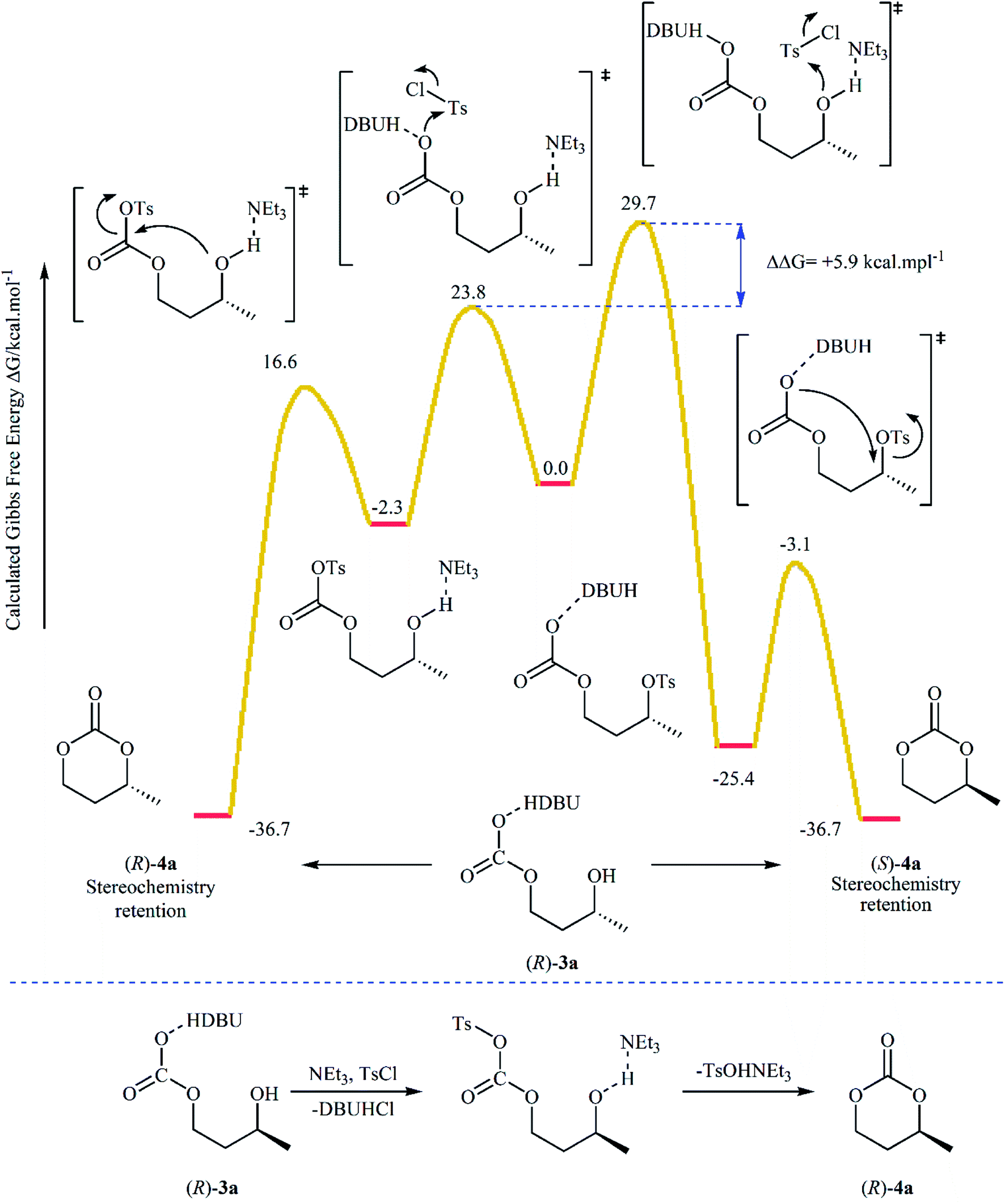 | ||
| Fig. 2 DFT computed pathways and mechanism for the cyclization of 3a to 4a. | ||
Very recently, the same research team prepared a novel D-mannose-based cyclic carbonate 6 from natural sugar D-mannose 5 and CO2 using their standard reaction conditions and successfully applied this monomer in the synthesis of polycarbonates 7 via a controlled organocatalytic ring-opening polymerization (Scheme 5).30,31
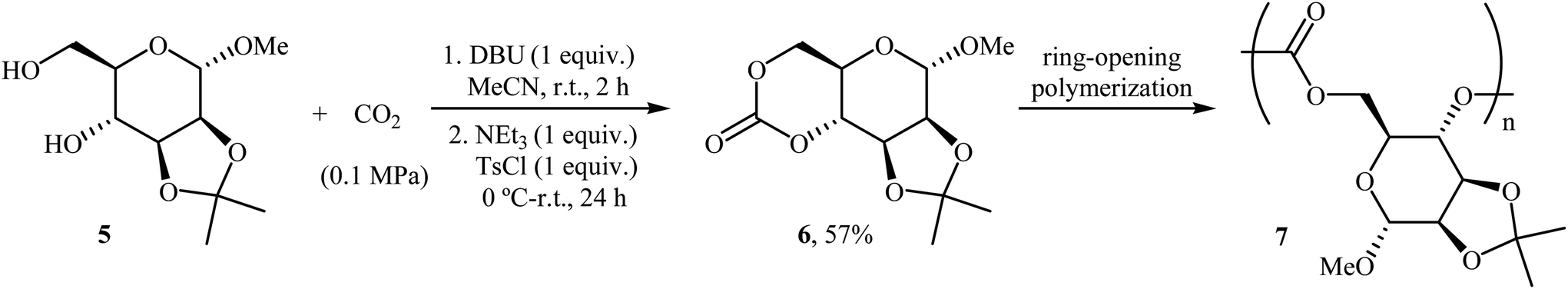 | ||
| Scheme 5 Synthesis and polymerization of a D-mannose-based cyclic carbonate 6. | ||
In 2016, Bobbink and co-workers reported an innovative example of six-membered cyclic carbonate 9 preparation through a carbene-catalyzed fixation of CO2 onto corresponding 1,3-diol 8. The reaction was performed at the atmospheric pressure of CO2 in the presence of over stoichiometric amounts of nBuBr and Cs2CO3 to produce expected carbonate 2 in yield of 51% (Scheme 6). It is noted that both the base and alkyl halide are essential in the reaction, no carbonate was formed in their absence. Based on labeling studies and other experiments the mechanism shown in Scheme 7 was proposed for this reaction.32
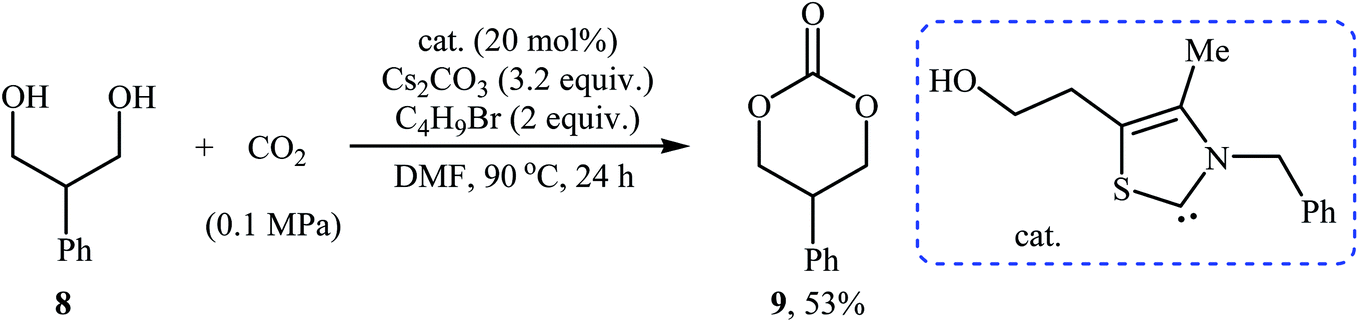 | ||
| Scheme 6 Synthesis of six-membered cyclic carbonate 2 through carbene-catalyzed fixation of CO2 onto 1,3-diols 8. | ||
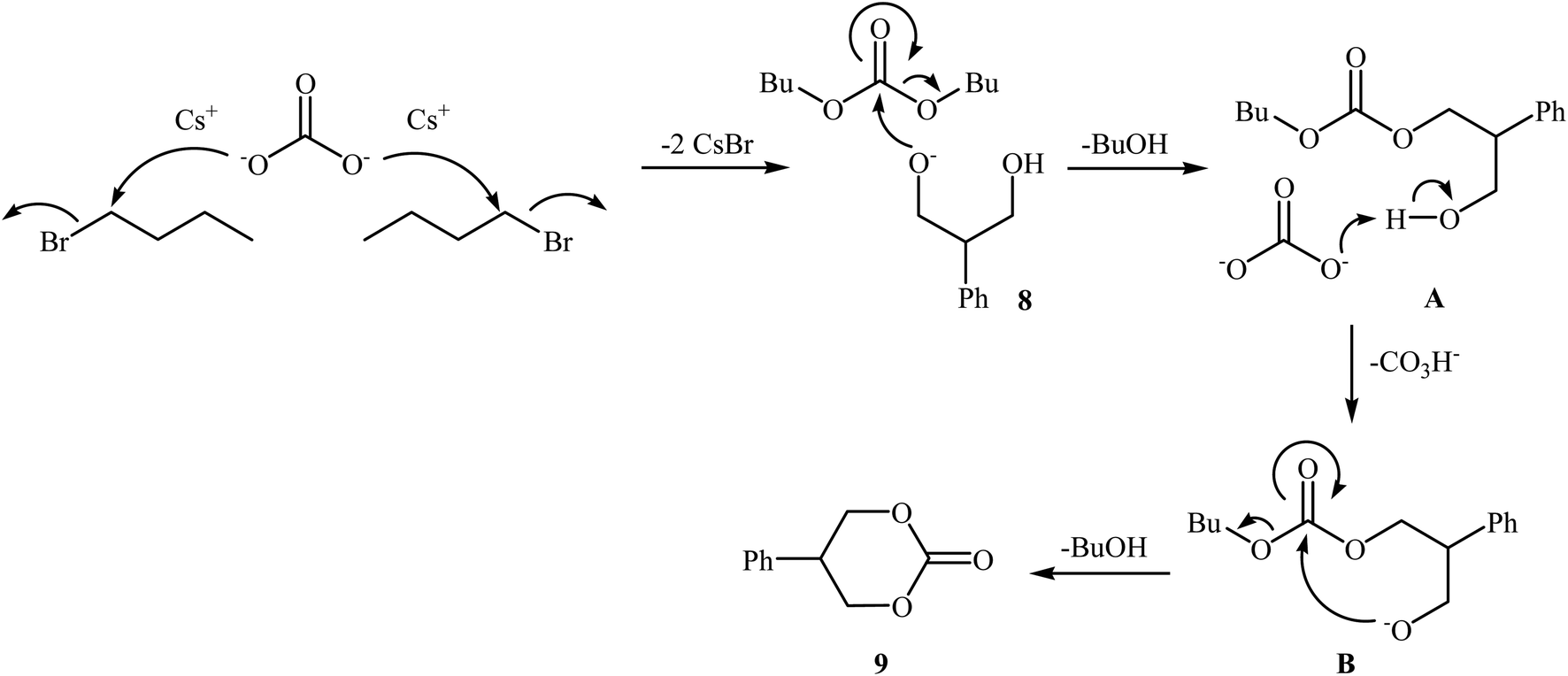 | ||
| Scheme 7 Mechanistic proposal for the reaction in Scheme 6. | ||
3. From homoallylic alcohols
In 1981, in a beautiful approach, Cardillo and co-workers disclosed a one-pot, three-component reaction between homoallylic alcohols 10, carbon dioxide, and molecular iodine for the regio- and stereo-selective synthesis of six-membered cyclic carbonates 11 using nBuLi as a base in dry THF (Scheme 8). The authors suggested that the formation of 11 occurs via addition of CO2 to the alkoxide group in intermediate A, followed by a formal electrophilic iodocyclization sequence. They also successfully extended this chemistry to synthesis of five-membered cyclic carbonates via the reaction of corresponding allylic alcohols with CO2 and I2 under standard conditions.33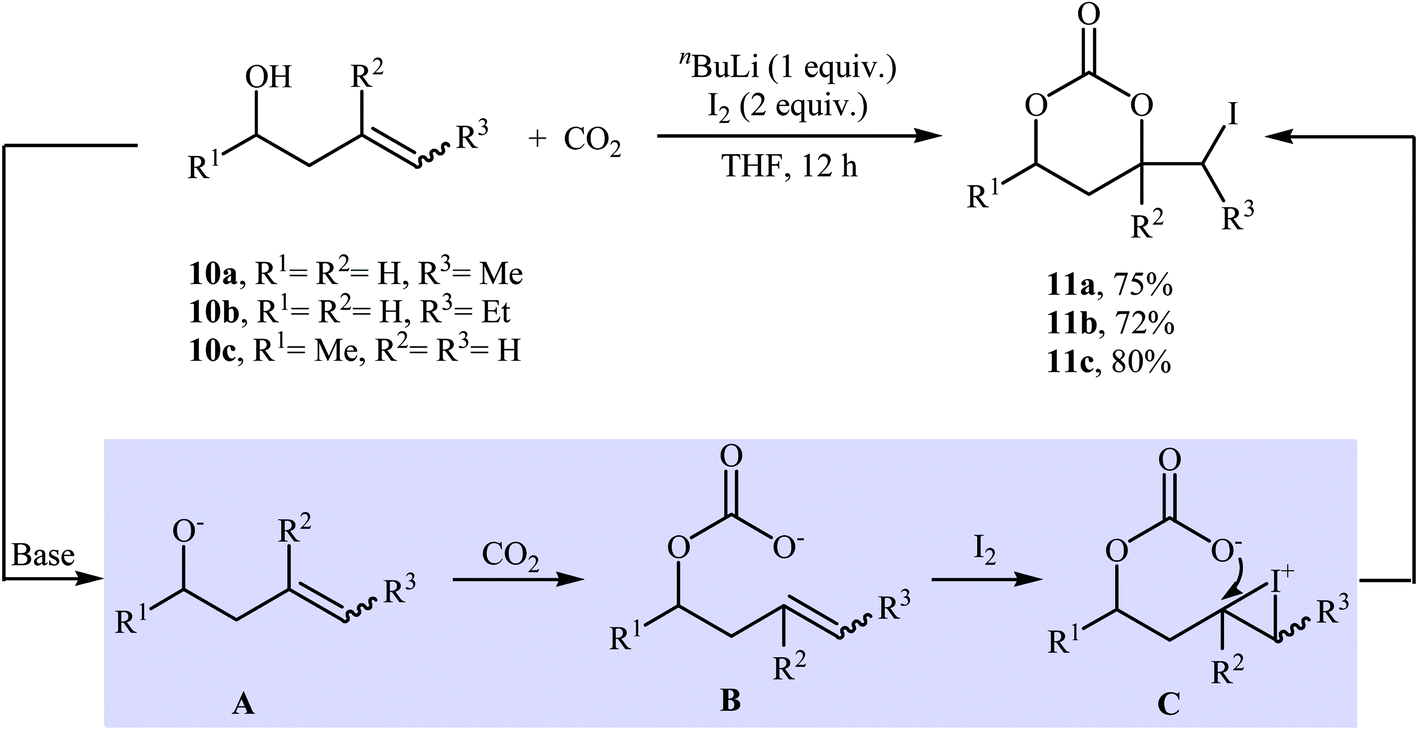 | ||
| Scheme 8 Base-mediated three-component reactions of homoallylic alcohols 10, CO2, and I2 developed by Cardillo. | ||
Three decades later, an interesting study about metal-free and base-free atmospheric CO2 fixation by homoallylic alcohols 12 using tert-butyl hypoiodite (tBuOI) as a reactive iodonium source, was published by Minakata, Sasaki, and Ide. The reactions proceeded cleanly at −20 °C in THF (or MeCN) to afford the corresponding iodinated six-membered cyclic carbonates 13 in high yields (Scheme 9a). According to the author proposed mechanism, the key step of the reaction involve generation of an O-iodinated intermediate A by iodination of the in situ generated homoallyl carbonic acid with tBuOI. They also found that butynyl alcohol 14 was converted to the corresponding six-membered cyclic carbonate 15, via treatment with tBuOI under the CO2 atmosphere at −20 °C (Scheme 9b). To the best of our knowledge, this is the only example of synthesis of cyclic carbonates from butynyl alcohols reported so far.34
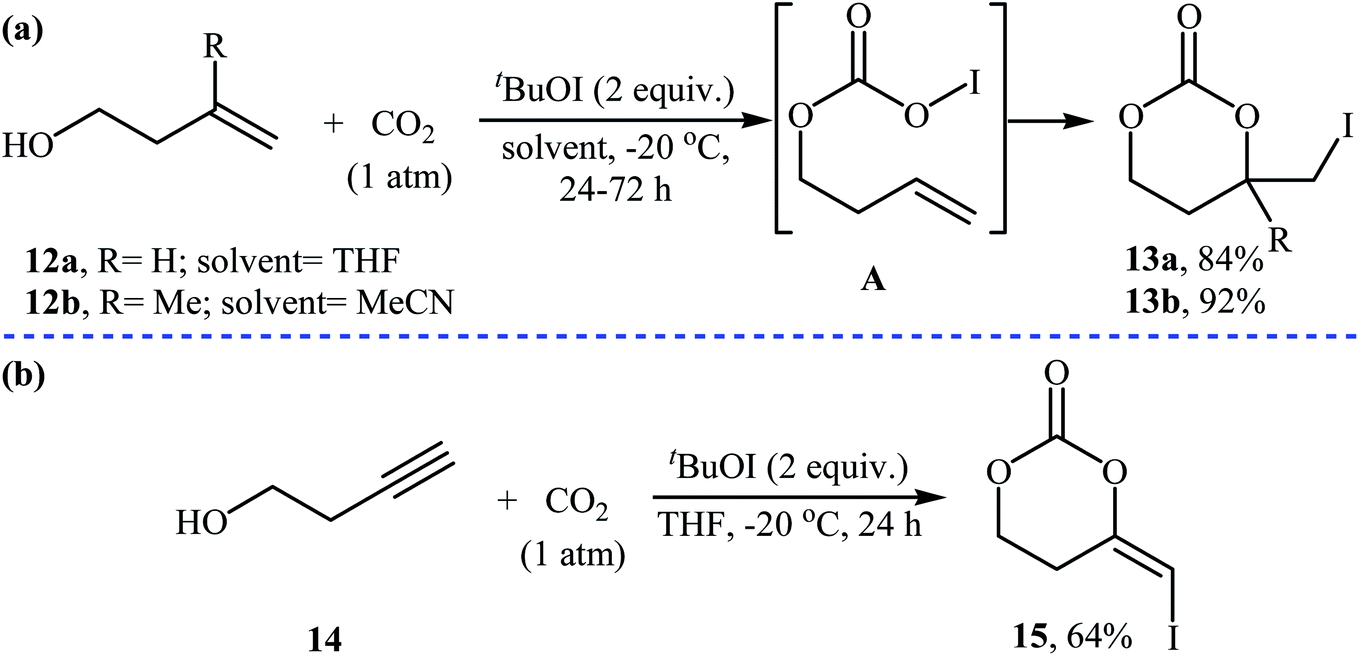 | ||
| Scheme 9 (a) Atmospheric CO2 fixation with homoallylic alcohols 12 and tBuOI reported by Minakata; (b) CO2 fixation with butynyl alcohol 14 and tBuOI. | ||
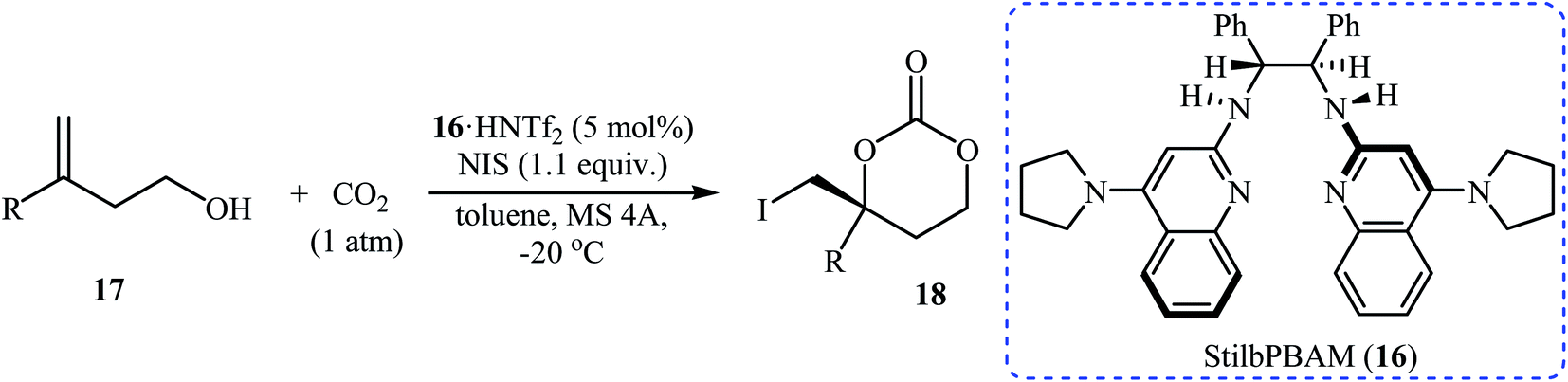
Inspired by these works, Johnston and co-workers developed a beautiful one-pot procedure for the enantioselective synthesis of six-membered cyclic carbonates via a dual Brønsted acid/base organocatalyzed three component reaction between homoallylic alcohols, carbon dioxide, and N-iodosuccinimide. Under the optimized conditions [StilbPBAM 16 (5 mol%), HNTf2 (5 mol%), NIS (1.1 equiv.) toluene, −20 °C, 0.1 MPa] various homoallylic alcohols 17 react to give moderate to excellent yields of the corresponding carbonates 18 in high enantioselectivity (Table 1). The reaction showed good functional group diversity, including fluoro, chloro, bromo, and alkoxy functionalities that would allow further elaboration of the products to structurally diverse carbonates. However, substitution near the alkene moiety was not tolerated and no desired product was obtained in these cases. The authors investigated the synthetic utility of prepared carbonates (Scheme 10a) and found that hydrolysis of 18a with a basic resin in methanol provided the corresponding epoxide 19 in a yield of 90%. Full reduction employing a stronger reducing agent (LiAlH4) gave tertiary alcohol 20 in a yield of 71% and reduction by stannane provided carbonate 21 in 86% yield. They also showed that carboxylation/alkene functionalization reaction of 2-cyclohexenylethanol 22 under the standard reaction conditions provided a useful route to enantio-rich spirocyclic carbonate 23 (Scheme 10b).35
|
|||||
|---|---|---|---|---|---|
| Entry | R | Product | Time (h) | ee (%) | Yield (%) |
| a It was noted that purified 18e was prone to decomposition.b 10 mol% catalyst loading. | |||||
| 1 | Ph | 18a | 48 | 91 | 95 |
| 2 | 4-Me–C6H4 | 18b | 48 | 91 | 96 |
| 3 | 3-Me–C6H4 | 18c | 48 | 93 | 96 |
| 4 | 2-Naphthyl | 18d | 48 | 90 | 88 |
| 5 | 4-OMe–C6H4 | 18e | 48 | 80 | 26a |
| 6 | 3-OMe–C6H4 | 18f | 48 | 90 | 97 |
| 7 | 4-Br–C6H4 | 18g | 48 | 90 | 65 |
| 8b | 3-Cl–C6H4 | 18h | 120 | 87 | 44 |
| 9b | 3-F–C6H4 | 18i | 96 | 89 | 40 |
| 10 | 4-F–C6H4 | 18j | 48 | 90 | 54 |
| 11 | 4-(Me3C)–C6H4 | 18k | 48 | 91 | 99 |
| 12 | PhCH2CH2 | 18l | 72 | 67 | 71 |
| 13 | Me | 18m | 48 | 68 | 72 |
| 14b | Cy | 18n | 48 | 74 | 76 |
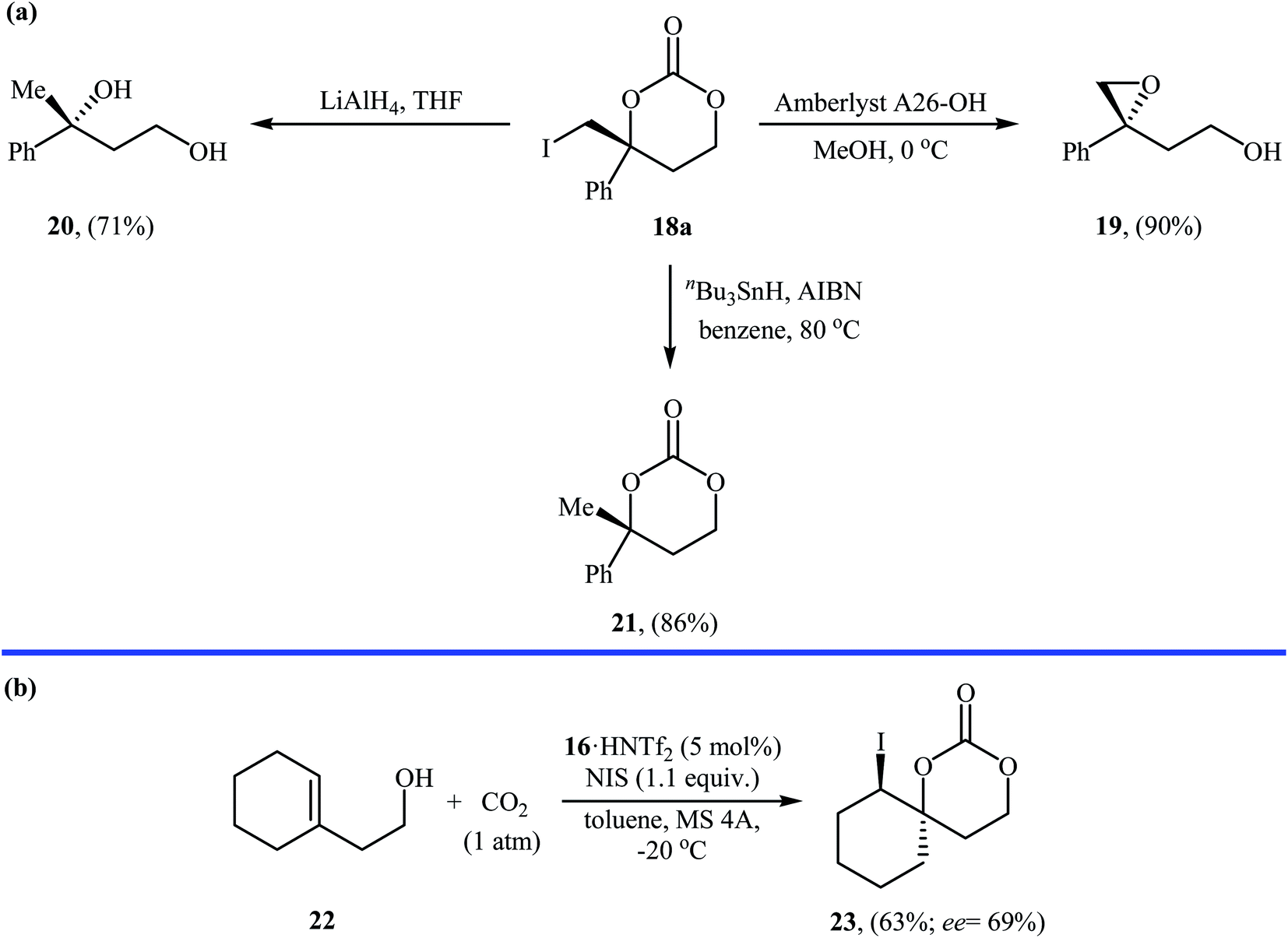 | ||
| Scheme 10 (a) Conversions of carbonate 18a to various value-added organic compounds; (b) Vara's synthesis of spirocyclic carbonate 23. | ||
4. From oxetanes
The synthesis of five-membered cyclic carbonates through the ring-expansion of oxiranes with CO2 is a well-established reaction,36 whereas the synthesis of six-membered cyclic carbonates from oxetanes and CO2 is considerably more challenging,37 and few efficient catalysts are available. In 1985, Baba, Kashiwagi and Matsuda reported the first example of such a reactions. They showed that unsubstituted oxetane 24 underwent ring-expansion with CO2 (4.9 MPa) in the presence of 2 mol% of Ph4SbI as a catalyst under solvent-free conditions and gave corresponding carbonate 25 in a yield of 96% (Table 2, entry 1). The authors found that the yield in this system was strongly dependent on the temperature. The best results were obtained at 100 °C. It is interesting to note that neither Ph3SbI2 nor Ph4SbBr could promote the reaction under the similar conditions.39 Two years later, the same authors improved the efficiency of this cycloaddition in the term of yield by performing the reaction in the presence of a combination of Bu3SnI and HMPA as a catalytic system and obtained desired carbonate in a quantitative yield (Table 2, entry 2).39 Later, the group of Darensbourg showed that heating of the same starting materials at 60 °C in the presence of VO(acac)2/n-Bu4NBr system in toluene gave expected carbonate 25 in a yield of 95% (Table 2, entry 3).40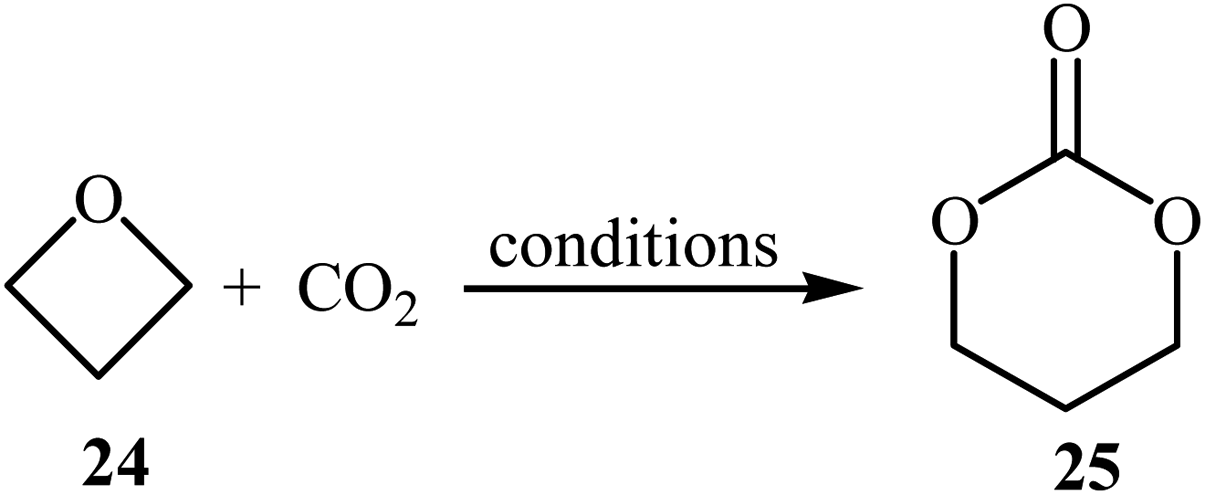
In 1989, the group of Baba applied their methodology to the synthesis of substituted six-membered cyclic carbonates. Thus, a series of mono- and di-substituted carbonates 27 were synthesized via the Ph4SbI-catalyzed reaction of corresponding oxetanes 26 and CO2 under solvent-free conditions (Scheme 11). The results demonstrated that mono-substituted oxetanes gave much higher yields than di-substituted oxetanes under this reaction conditions. The reaction is noteworthy in that both alkyl and aryl substituted oxetanes are well tolerated.41
 | ||
| Scheme 11 Ph4SbI-catalyzed coupling of oxetanes 26 and CO2. | ||
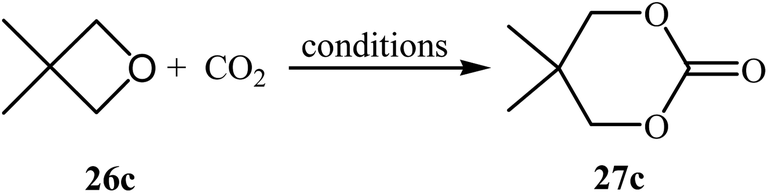
Following this work, several catalytic systems have been developed over the last several years towards the improvement of the yields of di-substituted six-membered cyclic carbonates under this synthetic protocol. In 2012, the group of Kleij reported the preparation of 5,5-dimethyl-1,3-dioxan-2-one 27c via ring-expansion of 3,3-dimethyloxetane 26c with CO2 using only 0.5 mol% of [Fe(TPhOA)]2 as a catalyst and 5 mol% of Bu4NI as a co-catalyst in methyl ethyl ketone at 85 °C. The reaction was carried out under 0.2 MPa pressure of CO2 to afford within 66 h the corresponding carbonate in 28% yield (Table 3, entry 1).42 In their subsequent study, they reinvestigated the same reaction by replacing [Fe(TPhOA)]2 with an Al triphenolate catalyst. However, the process resulted in a reduced yield (Table 3, entry 2).43 In 2015, Buckley, Patel, and Wijayantha were able to improve this reaction with a considerable increase in yield by performing the process under electrosynthesis conditions using an electrochemical cell containing a copper cathode and a magnesium anode and 1.0 equiv. of Bu4NI as a supporting electrolyte in MeCN (Table 3, entry 3).44 High yield and mild reaction condition (50 °C, 0.1 MPa) were the advantages mentioned for this catalyst-free synthetic approach. The author proposed mechanism for this reaction is shown in Scheme 12.
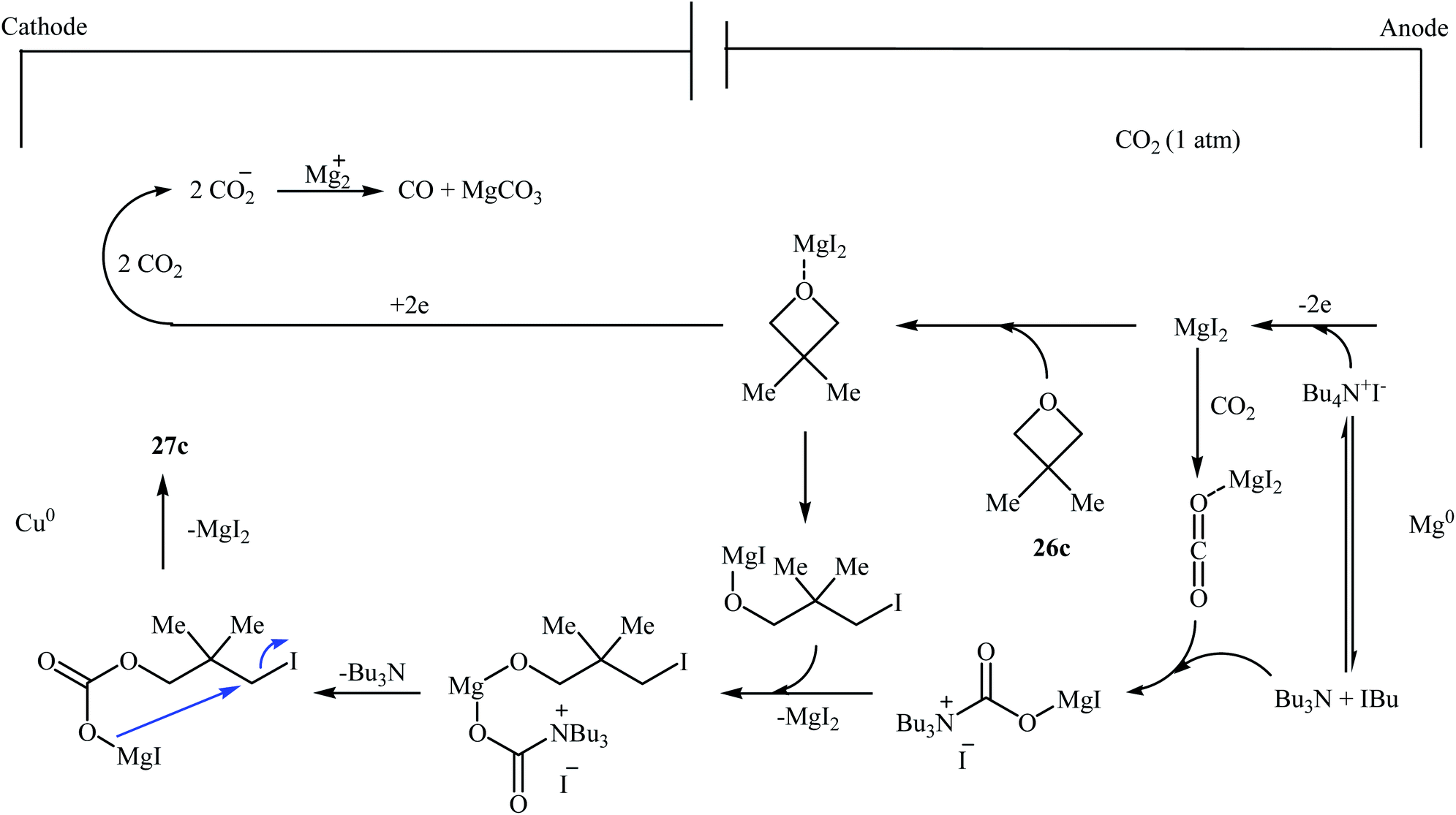 | ||
| Scheme 12 Proposed mechanism for the formation of 27c under electrosynthesis conditions. | ||
Recently, Kleij's group presented one of the most striking examples of the synthesis of substituted six-membered cyclic carbonates 29 via an Al(III)-catalyzed coupling of corresponding oxetanes 28 with carbon dioxide (Scheme 13). Under the optimized conditions (AlIII-aminotriphenolate complex 30, tetrabutylammonium bromide (TBAB), methyl ethyl ketone, 1–4 MPa, 75 °C) various 2-, 3-, and 3,3-disubstituted oxetanes react to give moderate to almost quantitative yields of the corresponding carbonates. The reaction tolerated a variety of sensitive functional groups, including bromo, hydroxy, alkoxy, and ester functionalities that would allow further elaboration of the products. Interestingly, when similar reaction conditions were applied to the oxetane precursors having pendent hydroxyl or amino substituents 31, the formation of five-membered heterocycles 32 were obtained in moderate to high yields, without any of expected six-membered cyclic carbonates (Scheme 14a). According to the author proposed mechanism this reaction proceeds via the initial formation of carbonic or carbamic acid derivative A from the starting oxetane 31 and CO2. Next, coordination of the catalyst to the intermediate A, leading to the formation of complex B, which then undergoes an intramolecular SN2 reaction to produce intermediate C. Finally, reductive elimination of aluminum affords the observed carbonates/carbamates 32 (Scheme 14b).45
 | ||
| Scheme 13 Al(III)-catalyzed coupling of oxetanes 28 with CO2 reported by Kleij. | ||
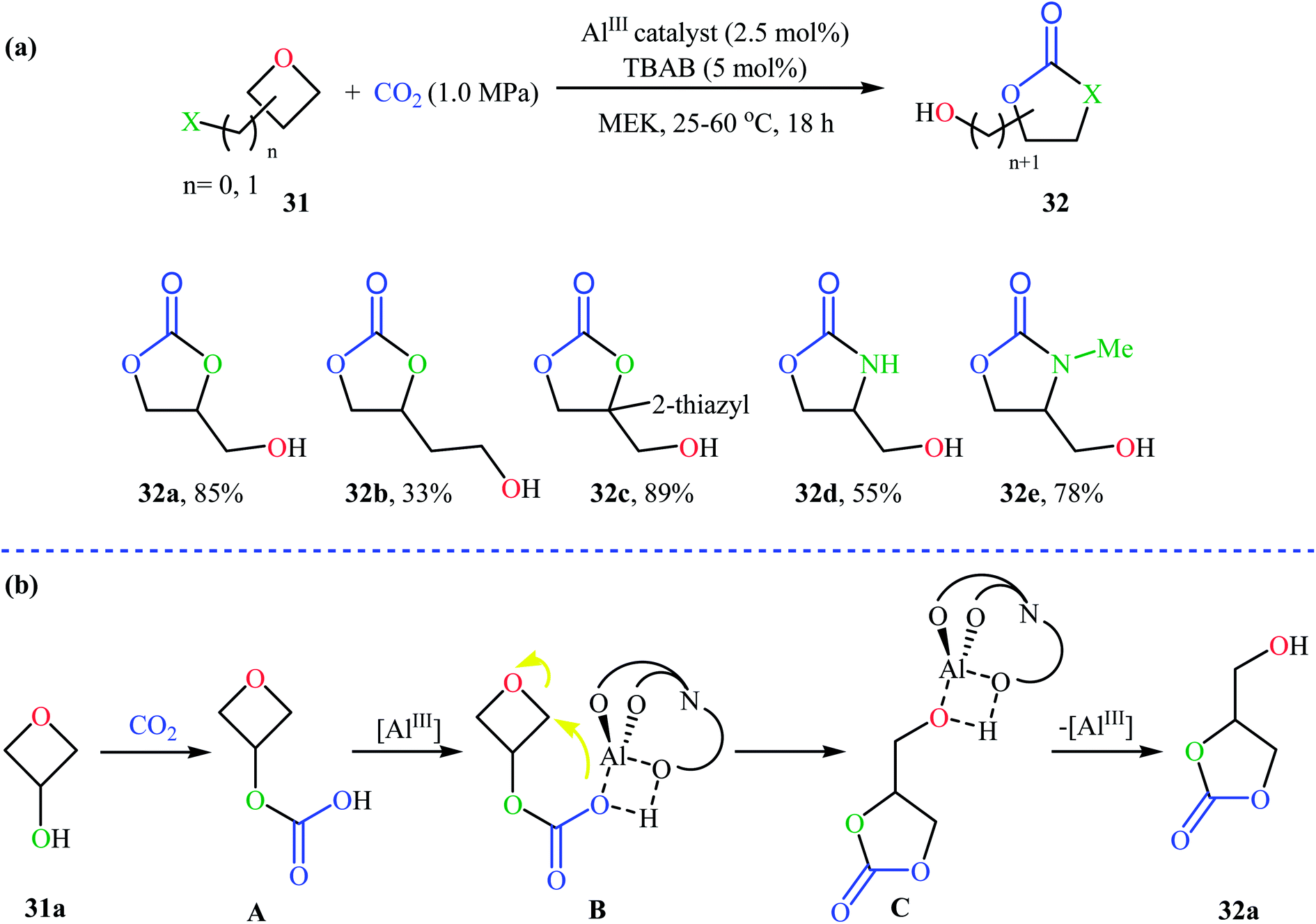 | ||
| Scheme 14 (a) Synthesis of five-membered heterocycles 32 through Al(III)-catalyzed coupling of amino- and hydroxy-substituted oxetanes 31 with CO2; (b) mechanism proposed to explain the synthesis of 32. | ||
Very recently, the same research team extended this methodology to the high yielding synthesis of highly functionalized acyclic carbamates 35 via a one-pot three-component coupling between oxetanes 33, amines 34, and CO2 under their optimized conditions (Scheme 15). According to mechanistic studies, it proceeds through the formation of a six-membered cyclic carbonate from the starting oxetanes and CO2, followed by an in situ aminolysis of these carbonates. It should be mentioned that the authors successfully showed the application of this methodology for the high yielding synthesis of carisoprodol, a muscle relaxer drug, and the mono-carbamate of Felbatol, an anticonvulsant drug.46
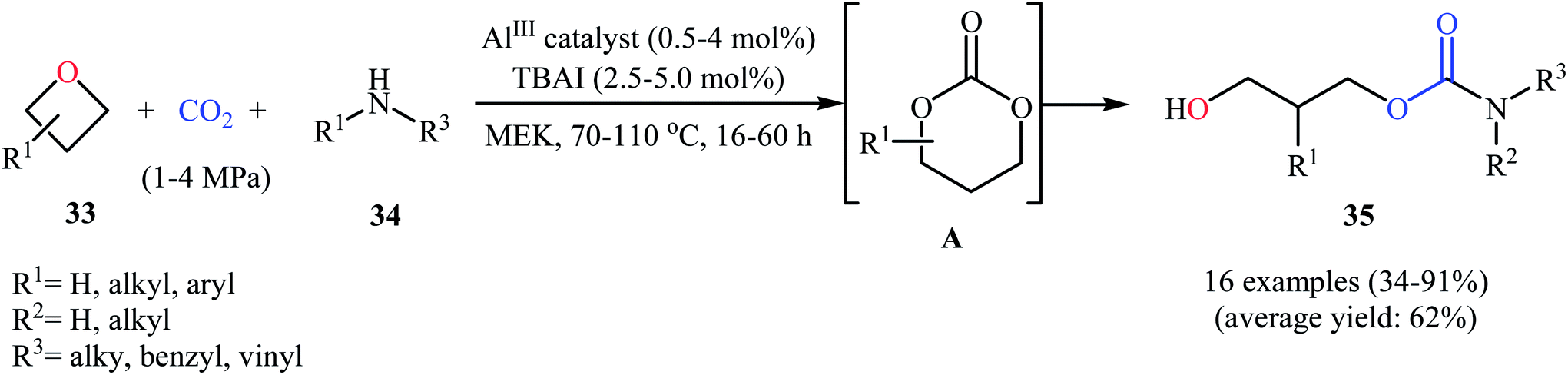 | ||
| Scheme 15 Al(III)-catalyzed synthesis of acyclic carbamates 35 from the reaction between oxetanes 33, amines 34, and CO2. | ||
5. Miscellanies reactions
In 1999, Uemura and co-workers developed an efficient palladium catalyzed three-component reaction of 3,4-alkadienols 36, phenyl iodide, and CO2, which allowed for the synthesis of highly substituted six-membered cyclic carbonates 37 in moderate yields (Scheme 16a). The authors also successfully applied this methodology in the synthesis of a variety of five-membered cyclic carbonates via the coupling of 3,4-alkadienols with aryl/vinyl halides and CO2. According to the author proposed mechanism, this reaction starts with the formation of phenylpalladium-(II) species A via oxidative addition of phenyl iodide to palladium(0), and then coordination of A to the allenic bond of alcohol 36 to give π-allylic palladium species B, which undergoes an intramolecular carbopalladation to produce intermediate C. Next, reaction of CO2 with alkoxide group of C forms carbonate species D. Finally, an intramolecular cyclization of D, followed by a reductive elimination affords the observed products 37 (Scheme 16b).47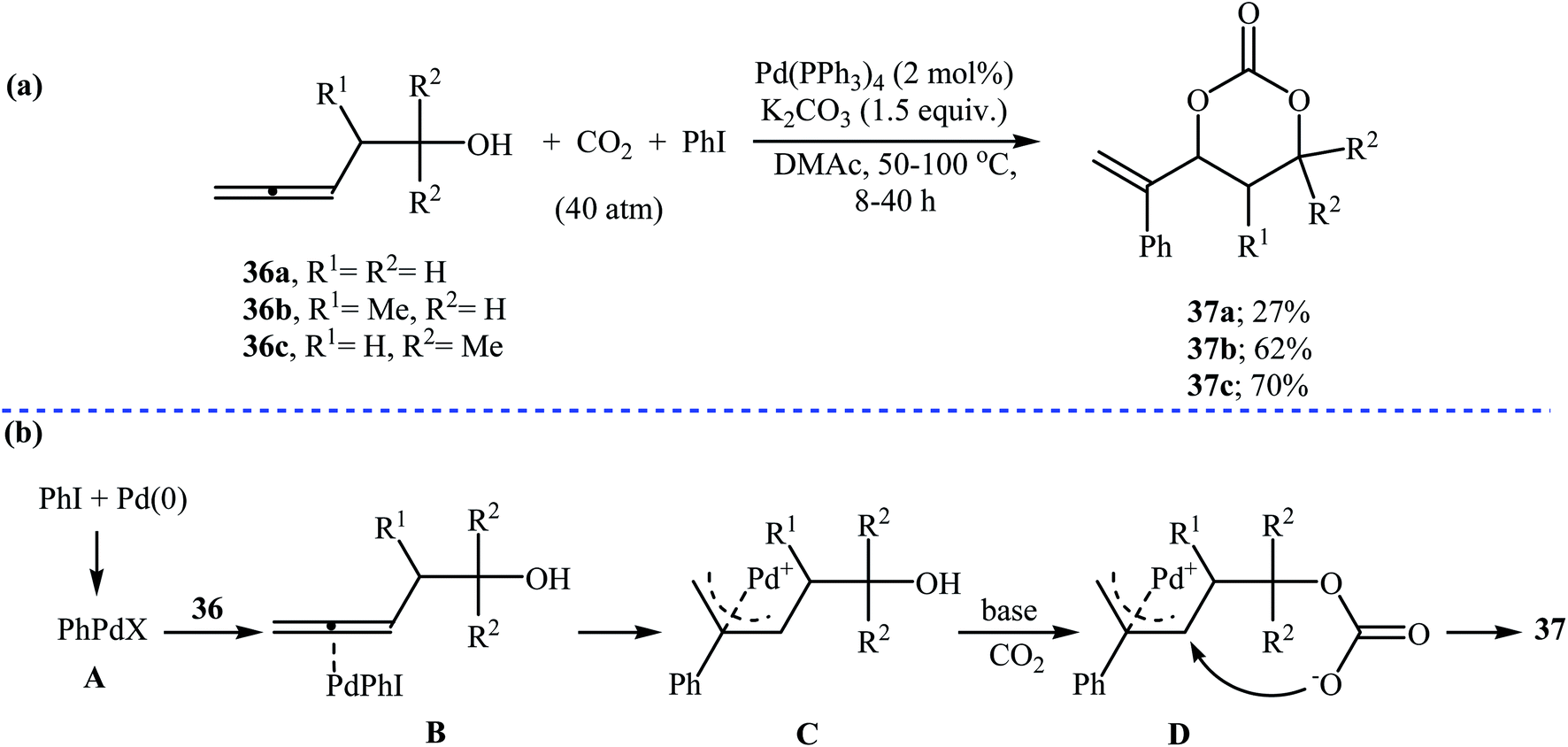 | ||
| Scheme 16 (a) synthesis of highly substituted six-membered cyclic carbonates 37 via a Pd-catalyzed three-component reaction of 3,4-alkadienols 36, phenyl iodide, and CO2; (b) mechanism that accounts for the formation of 37. | ||
Recently, the group of Zhang reported an efficient protocol for the synthesis of six-membered cyclic carbonates 39 from the reaction between commercially available 3-(pseudo)halo-1-propanols 38 and carbon dioxide through a Cs2CO3-catalyzed intermolecular ring closing strategy using mild reaction conditions. The corresponding cyclic carbonates were obtained in good to high yields (Scheme 17). Noteworthy, other bases such as K2CO3 and tBuOK were also found to promote the reaction; however, in lower yields. Under optimized conditions, the reaction also tolerated 2-(pseudo)halo-1-ethanols and gave corresponding five-membered cyclic carbonates in good to high yields, but the extension of the reaction to the synthesis of seven- and eight-membered rings was failed.48
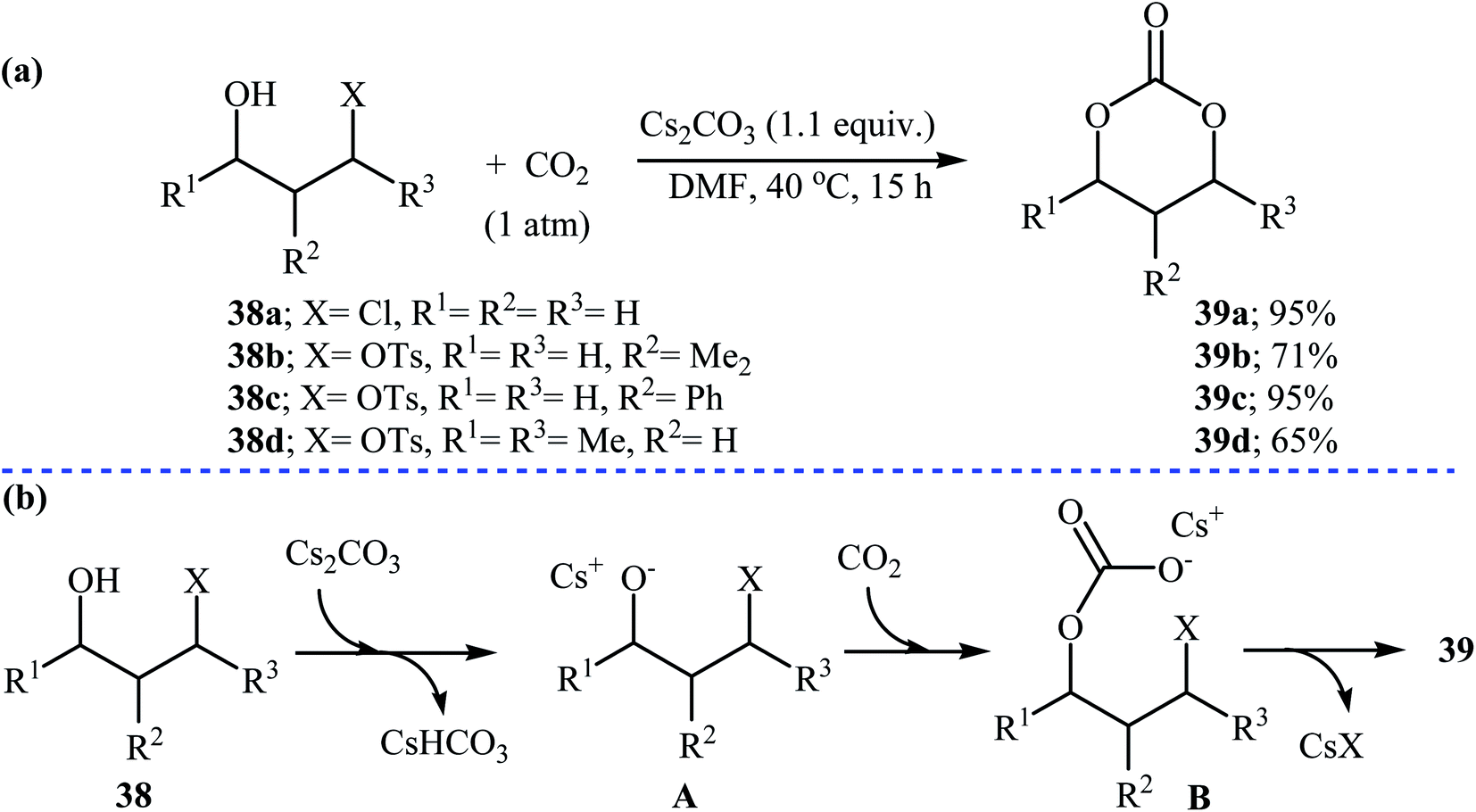 | ||
| Scheme 17 (a) Synthesis of six-membered cyclic carbonates 39 from 3-(pseudo)halo-1-propanols 38 and CO2; (b) plausible mechanism for the formation of 39. | ||
6. Conclusion
Carbon dioxide is a promising and sustainable carbon feedstock for producing value-added organic compounds. New catalytic protocols for efficient incorporation of CO2 into acyclic and five-membered cyclic carbonates are continuously being reported. However, little progress has been made in the synthesis of six-membered cyclic carbonates in the presence of CO2. As illustrated, synthesis of titled compounds using various substrates in the presence of CO2 have gained a lot of interest in recent years as useful alternative procedures. Interestingly, some of the conversion of CO2 covered in this mini-review could be easily achieved under ambient conditions. This results clearly show the potential application of these reactions in industry. Despite all these successes, the number of reported examples in this interesting field is limited. There is still further need to study the scope and limitations of this approach for the preparation of various six-membered cyclic carbonates.Conflicts of interest
There are no conflicts to declare.References
- A. Samanta, A. Zhao, G. K. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2011, 51, 1438–1463 CrossRef.
- A. J. Hunt, E. H. Sin, R. Marriott and J. H. Clark, ChemSusChem, 2010, 3, 306–322 CrossRef PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef PubMed.
- Z.-Z. Yang, L.-N. He, J. Gao, A.-H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 Search PubMed.
- S. Pulla, C. M. Felton, P. Ramidi, Y. Gartia, N. Ali, U. B. Nasini and A. Ghosh, J. CO2 Util., 2013, 2, 49–57 CrossRef.
- B. Yu and L. N. He, ChemSusChem, 2015, 108, 52–62 CrossRef PubMed.
- J. Rintjema and A. W. Kleij, Synthesis, 2016, 48, 3863–3878 CrossRef.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- J. E. Gomez and A. W. Kleij, Curr. Opin. Green Sustain. Chem., 2017, 3, 55–60 CrossRef.
- G. Yuan, C. Qi, W. Wu and H. Jiang, Curr. Opin. Green Sustain. Chem., 2017, 3, 22–27 CrossRef.
- L. Zhang and Z. Hou, Curr. Opin. Green Sustain. Chem., 2017, 3, 17–21 CrossRef.
- X.-D. Lang, X. He, Z.-M. Li and L.-N. He, Curr. Opin. Green Sustain. Chem., 2017, 7, 31–38 CrossRef.
- X.-F. Wu and F. Zheng, Top. Curr. Chem., 2017, 375, 4 CrossRef PubMed.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- E. Vessally, M. Babazadeh, A. Hosseinian, S. Arshadi and L. Edjlali, J. CO2 Util., 2017, 21, 491–502 CrossRef.
- G. Rokicki, T. Kowalczyk and M. Glinski, Polym. J., 2000, 32, 381–390 CrossRef.
- F. He, Y.-P. Wang, G. Liu, H.-L. Jia, J. Feng and R.-X. Zhuo, Polymer, 2008, 49, 1185–1190 CrossRef.
- J. Feng, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2012, 37, 211–236 CrossRef.
- R. H. Lambeth, S. M. Mathew, M. H. Baranoski, K. J. Housman, B. Tran and J. M. Oyler, J. Appl. Polym. Sci., 2017, 134, 44941–44947 CrossRef.
- (a) S.-H. Pyo, P. Persson, S. Lundmark and R. Hatti-Kaul, Green Chem., 2011, 13, 976–982 RSCPlease note that the industrial preparation of dialkyl carbonates typically requires toxic reagents such as phosgene and/ or harsh reaction conditions, for example see: (b) N. Keller, G. Rebmann and V. Keller, Catalysts, J. Mol. Catal. A: Chem., 2010, 317, 1–18 CrossRef.
- M. Tryznowski, Z. Żołek-Tryznowska, A. Świderska and P. Parzuchowski, Green Chem., 2016, 18, 802–807 RSC.
- B. Gabriele, R. Mancuso, G. Salerno, G. Ruffolo, M. Costa and A. Dibenedetto, Tetrahedron Lett., 2009, 50, 7330–7332 CrossRef.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- C. Martin, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef.
- (a) S. Arshadi, E. Vessally, A. Hosseinian, S. Soleimani-amiri and L. Edjlali, J. CO2 Util., 2017, 21, 108–118 CrossRef; (b) S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 20–129 Search PubMed; (c) E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2017, 21, 342–352 CrossRef; (d) E. Vessally, K. Didehban, M. Babazadeh, A. Hosseinian and L. Edjlali, J. CO2 Util., 2017, 21, 480–490 CrossRef; (e) E. Vessally, A. Hosseinian, M. Babazadeh, R. Hosseinzadeh-Khanmiri and L. Edjlali, Curr. Org. Chem., 2018, 22, 315–322 CrossRef; (f) K. Didehban, E. Vessally, M. Salary, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 23, 42–50 CrossRef; (g) S. Farshbaf, L. Zare Fekri, M. Nikpassand, R. Mohammadi and E. Vessally, J. CO2 Util., 2018, 25, 194–204 CrossRef; (h) E. Vessally, R. Mohammadi, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 24, 361–368 CrossRef.
- N. Kindermann, T. Jose and A. W. Kleij, Top. Curr. Chem., 2017, 375, 1–28 CrossRef PubMed.
- M. Honda, M. Tamura, K. Nakao, K. Suzuki, Y. Nakagawa and K. Tomishige, ACS Catal., 2014, 4, 1893–1896 CrossRef.
- G. L. Gregory, M. Ulmann and A. Buchard, RSC Adv., 2015, 5, 39404–39408 RSC.
- G. L. Gregory, L. M. Jenisch, B. Charles, G. Kociok-Kohn and A. Buchard, Macromolecules, 2016, 49, 7165–7169 CrossRef.
- G. L. Gregory, G. Kociok-Kohn and A. Buchard, Polym. Chem., 2017, 8, 2093–2104 RSC.
- F. D. Bobbink, W. Gruszka, M. Hulla, S. Das and P. J. Dyson, Chem. Commun., 2016, 52, 10787–10790 RSC.
- G. Cardillo, M. Orena, G. Porzi and S. Sandri, Chem. Commun., 1981, 465–466 RSC.
- S. Minakata, I. Sasaki and T. Ide, Angew. Chem., Int. Ed., 2010, 122, 1331–1333 CrossRef.
- B. A. Vara, T. J. Struble, W. Wang, M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2015, 137, 7302–7305 CrossRef PubMed.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef.
- (a) D. J. Darensbourg and A. I. Moncada, Macromolecules, 2010, 43, 5996–6003 CrossRef; (b) D. J. Darensbourg and A. I. Moncada, Macromolecules, 2009, 42, 4063–4070 CrossRef; (c) M. Alves, B. Grignard, A. Boyaval, R. Mereau, J. De Winter, P. Gerbaux, C. Detrembleur, T. Tassaing and C. Jerome, ChemSusChem, 2017, 10, 1128–1138 CrossRef PubMed.
- A. Baba, H. Kashiwagi and H. Matsuda, Tetrahedron Lett., 1985, 26, 1323–1324 CrossRef.
- A. Baba, H. Kashiwagi and H. Matsuda, Organometallics, 1987, 6, 137–140 CrossRef.
- D. J. Darensbourg, A. Horn Jr and A. I. Moncada, Green Chem., 2010, 12, 1376–1379 RSC.
- M. Fujiwara, A. Baba and H. Matsuda, J. Heterocycl. Chem., 1989, 26, 1659–1663 CrossRef.
- C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469–476 CrossRef.
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adan, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef PubMed.
- B. R. Buckley, A. P. Patel and K. Wijayantha, Eur. J. Org. Chem., 2015, 474–478 CrossRef PubMed.
- J. Rintjema, W. Guo, E. Martin, E. C. Escudero-Adan and A. W. Kleij, Chem. - Eur. J., 2015, 21, 10754–10762 CrossRef PubMed.
- W. Guo, V. Laserna, J. Rintjema and A. W. Kleij, Adv. Synth. Catal., 2016, 358, 1602–1607 CrossRef.
- K. Uemura, D. Shiraishi, M. Noziri and Y. Inoue, Bull. Chem. Soc. Jpn., 1999, 72, 1063–1069 CrossRef.
- M. R. Reithofer, Y. N. Sum and Y. Zhang, Green Chem., 2013, 15, 2086–2090 RSC.
| This journal is © The Royal Society of Chemistry 2018 |